Pd-Catalyzed Mizoroki-Heck Reactions Using Fluorine-Containing Agents as the Cross-Coupling Partners



Abstract
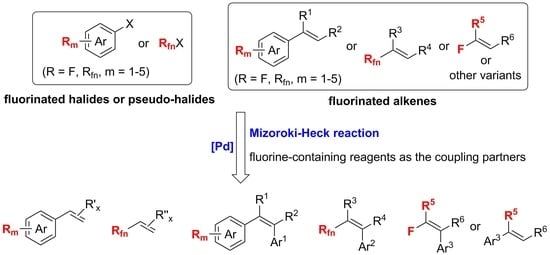
:
{kind=link}
{kind=link}
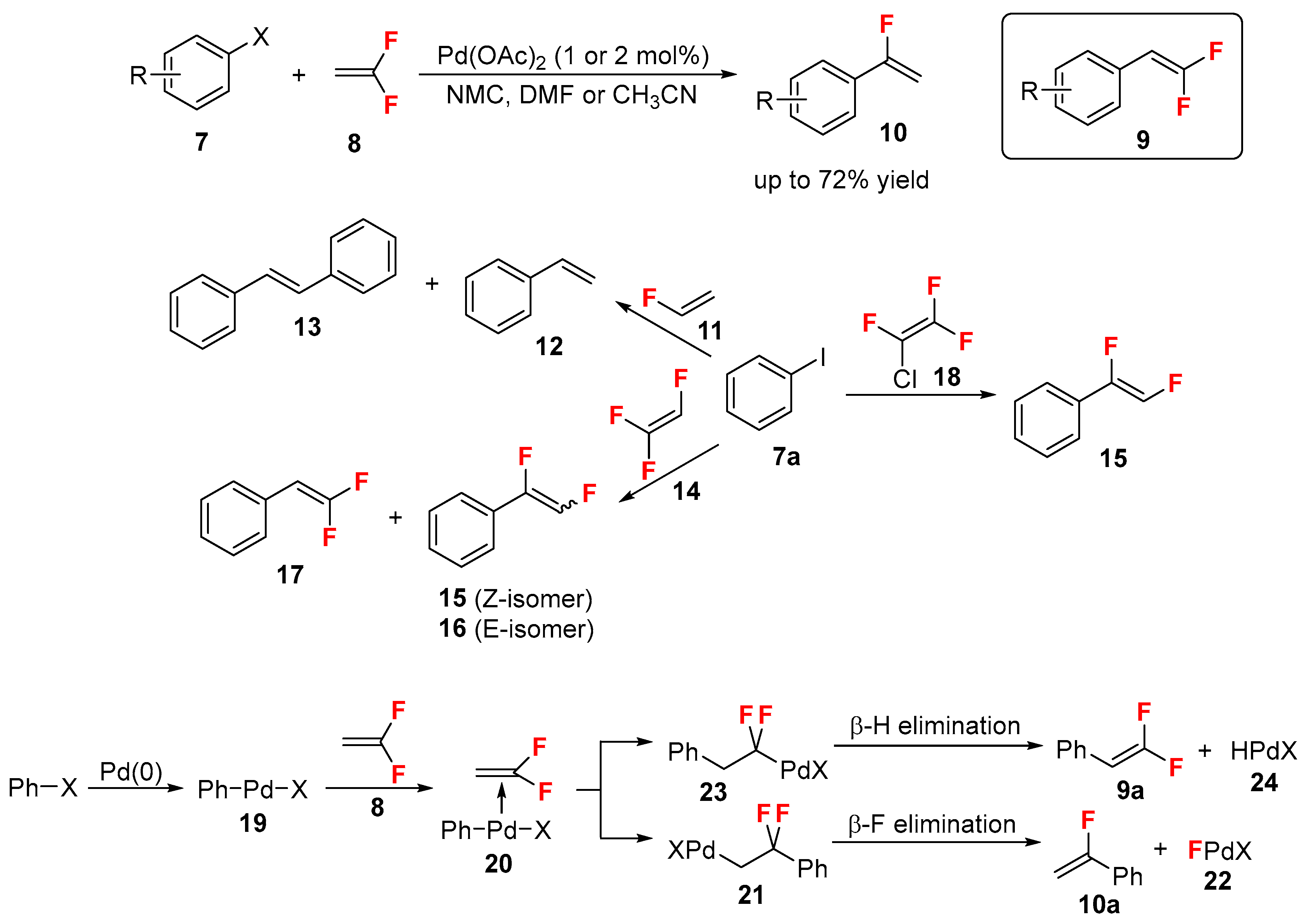{kind=link}
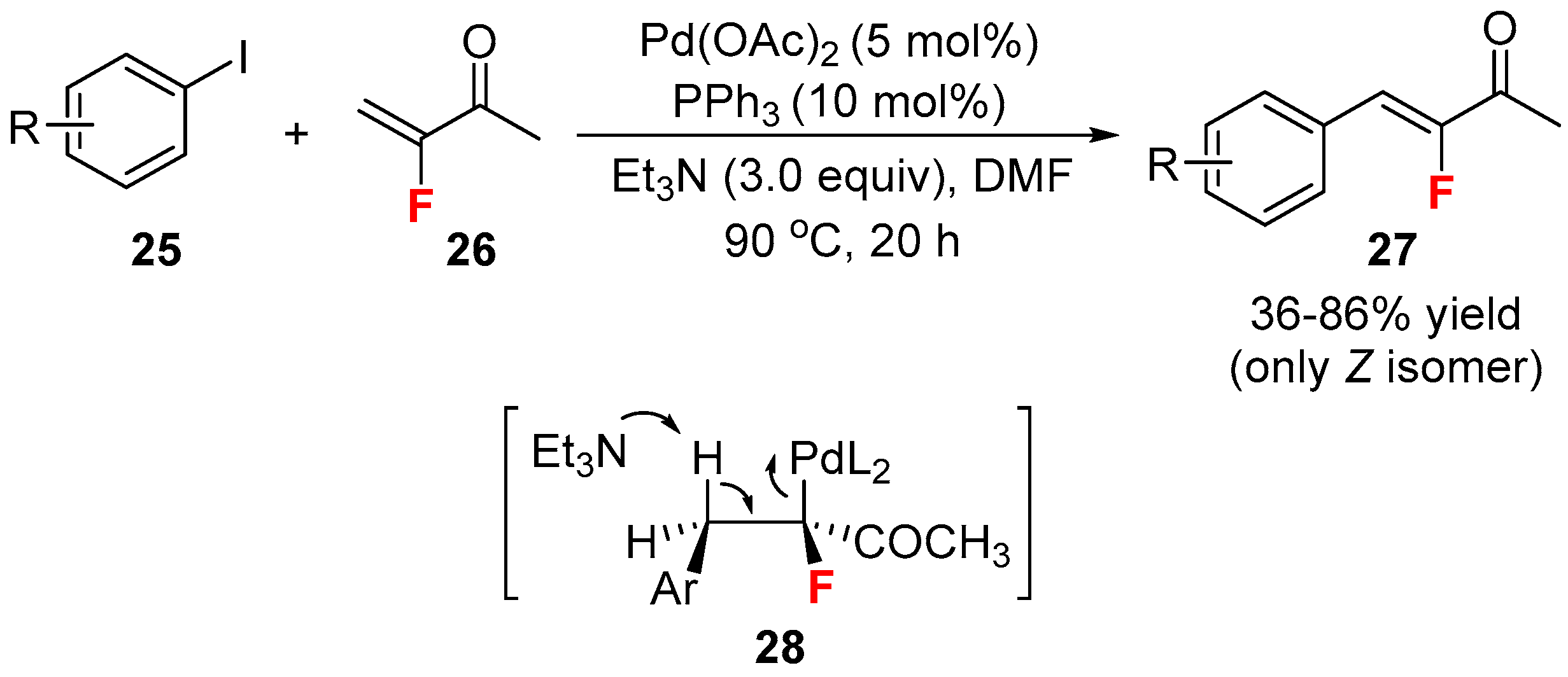{kind=link}
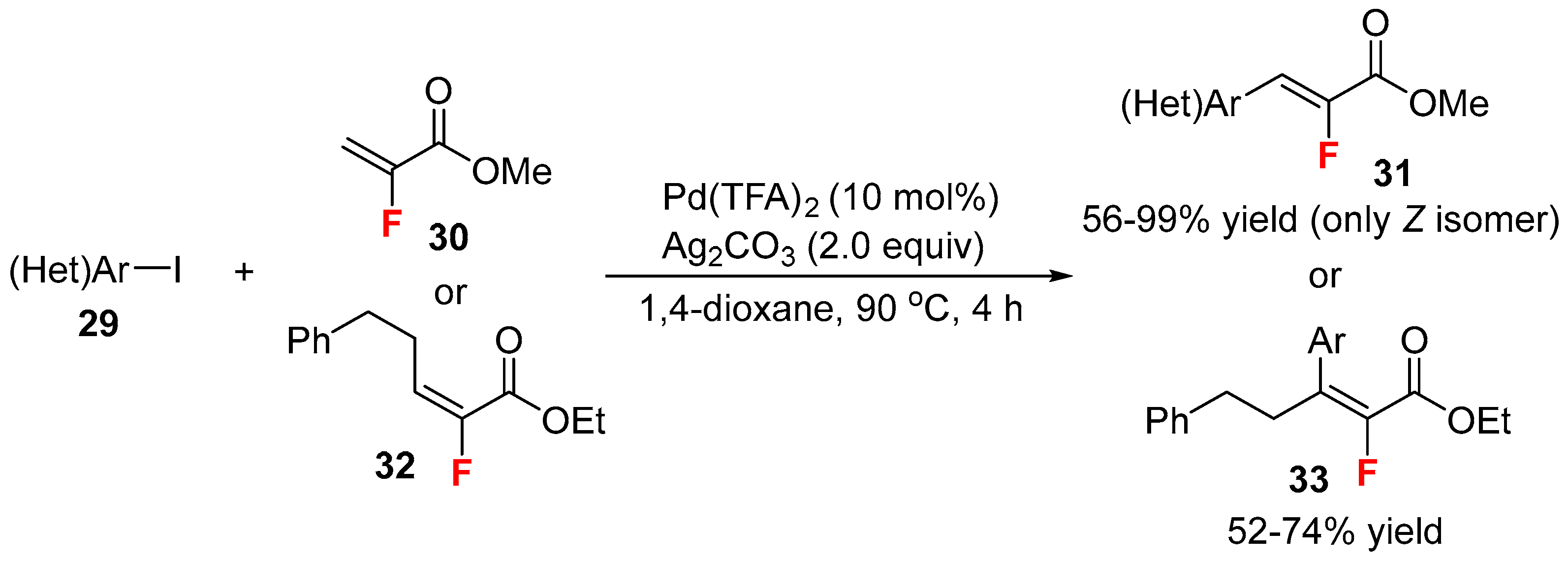{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
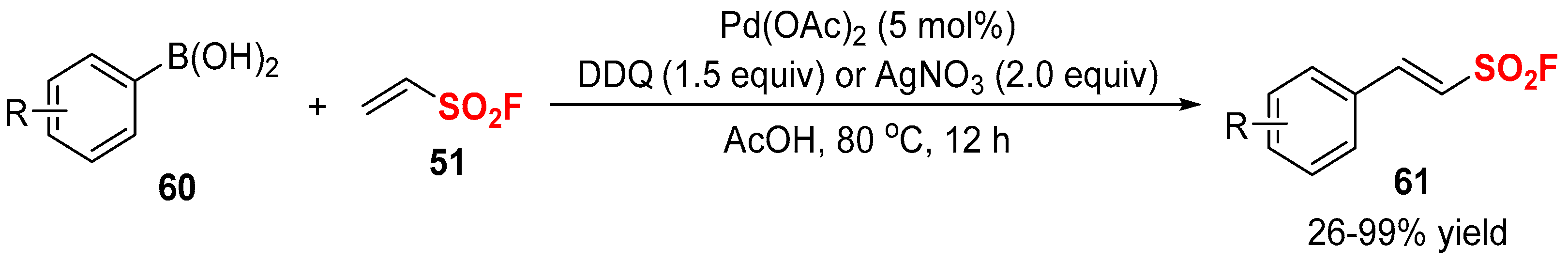{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
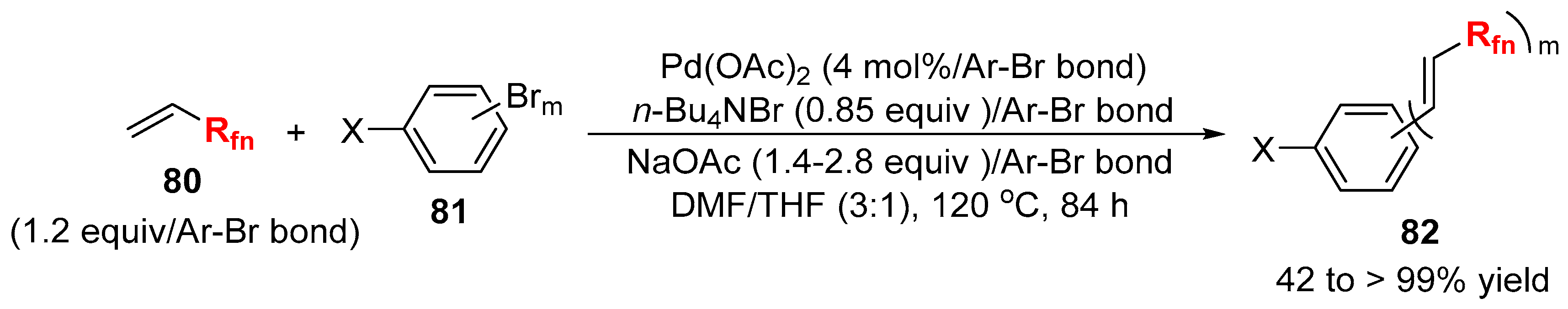{kind=link}
{kind=link}
{kind=link}
{kind=link}
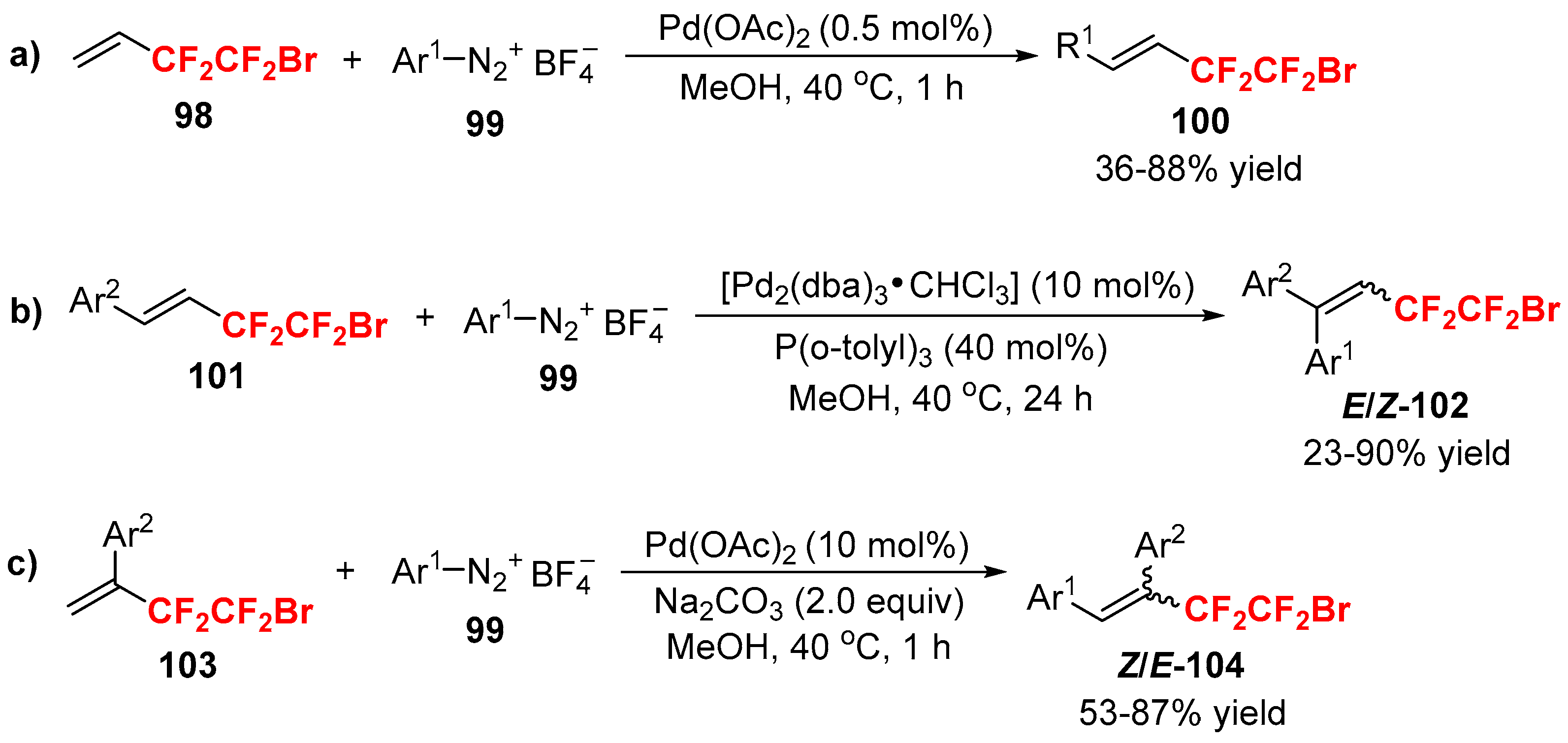{kind=link}
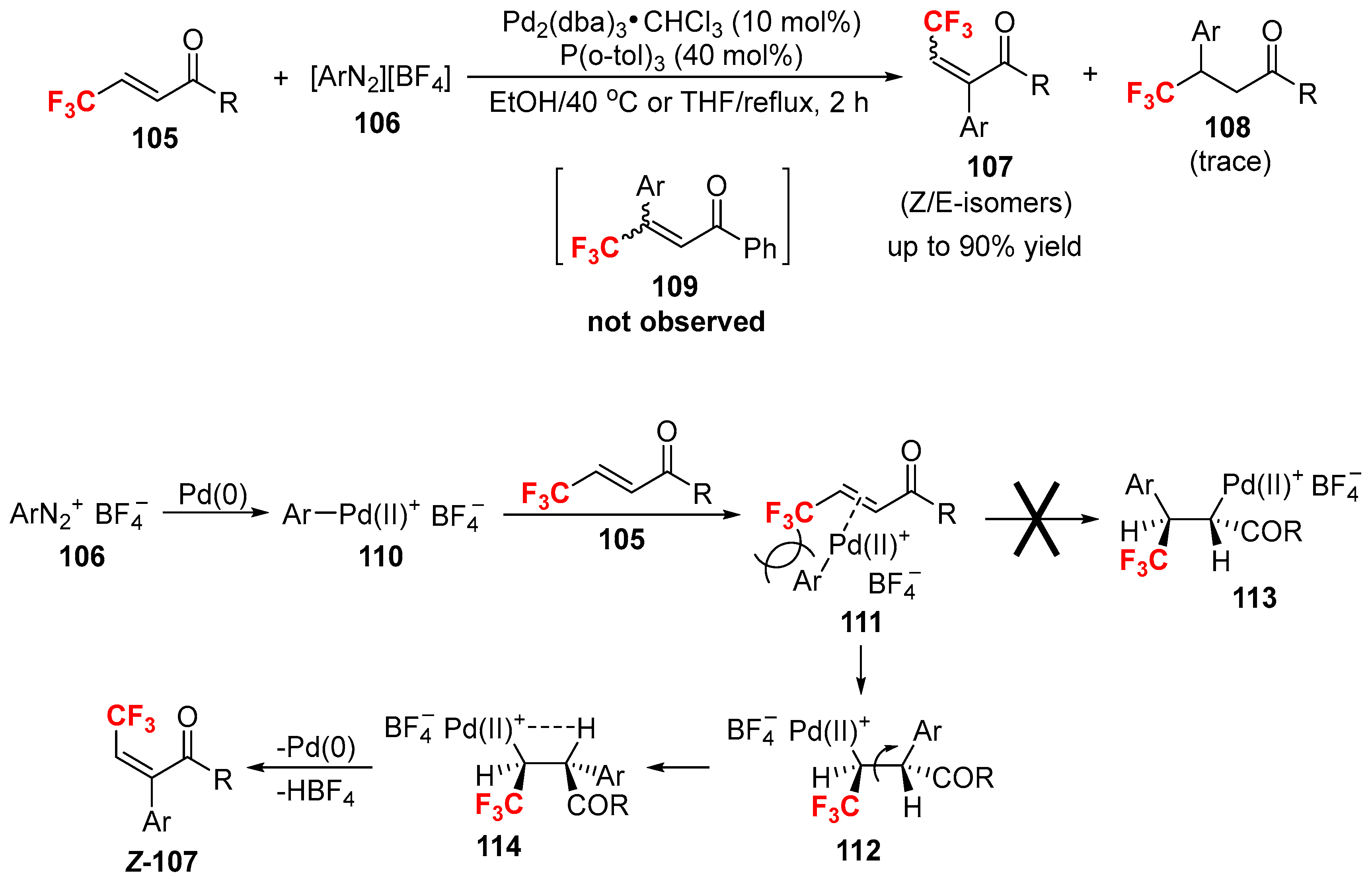{kind=link}
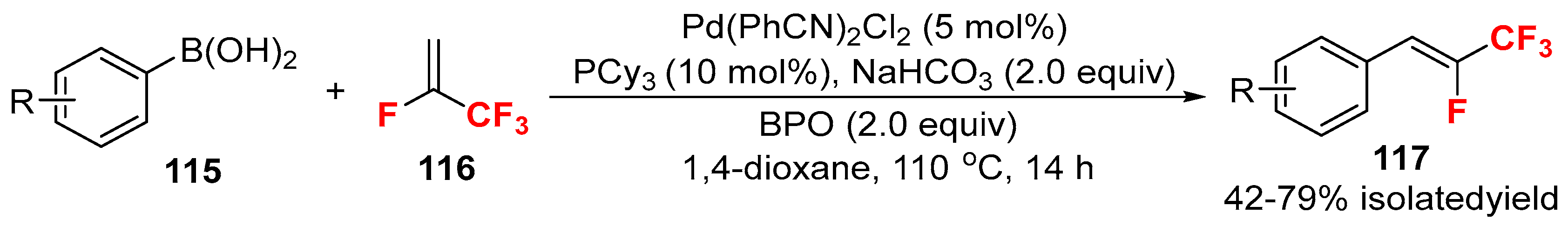{kind=link}
{kind=link}
{kind=link}
{kind=link}
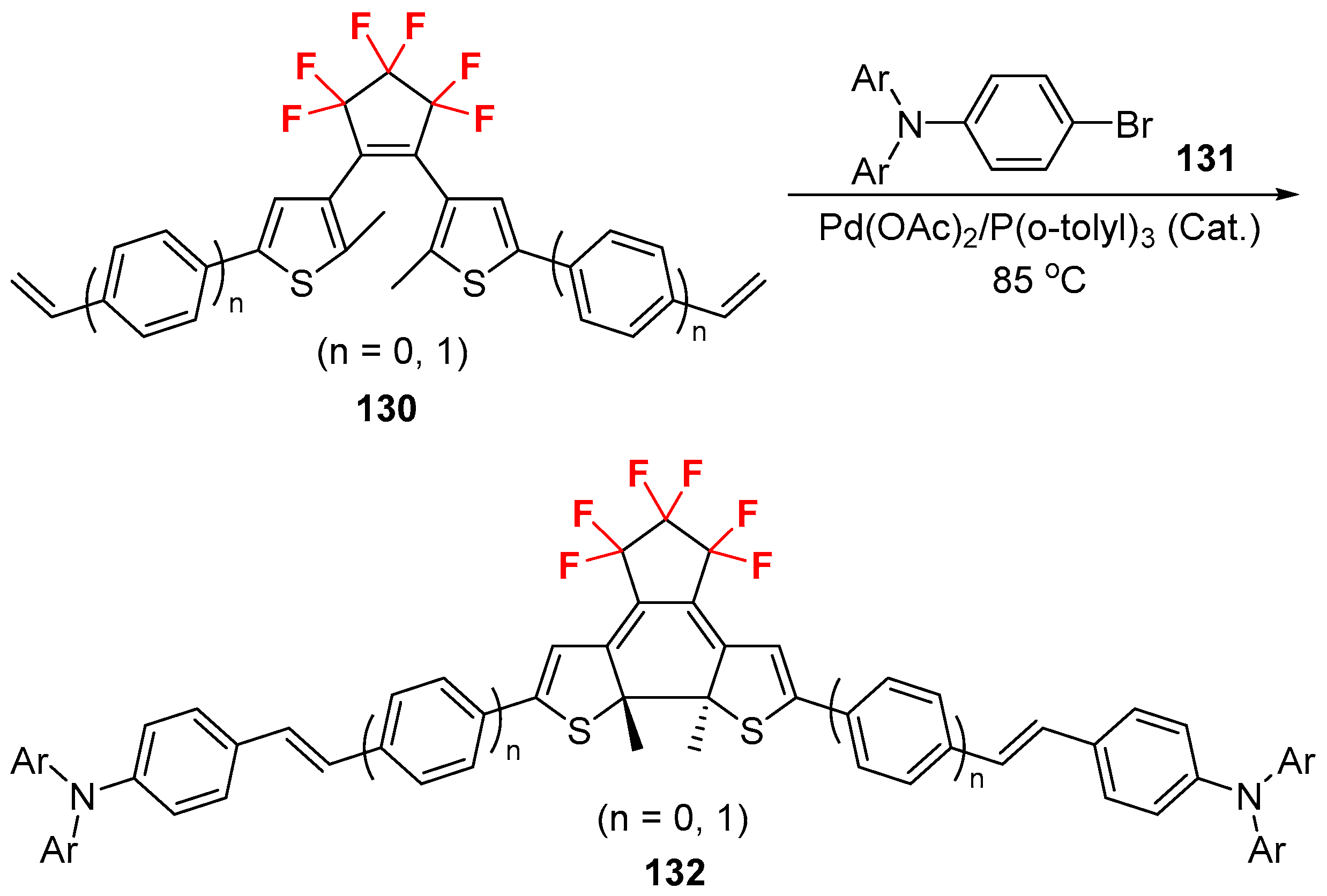{kind=link}
{kind=link}
{kind=link}
{kind=link}
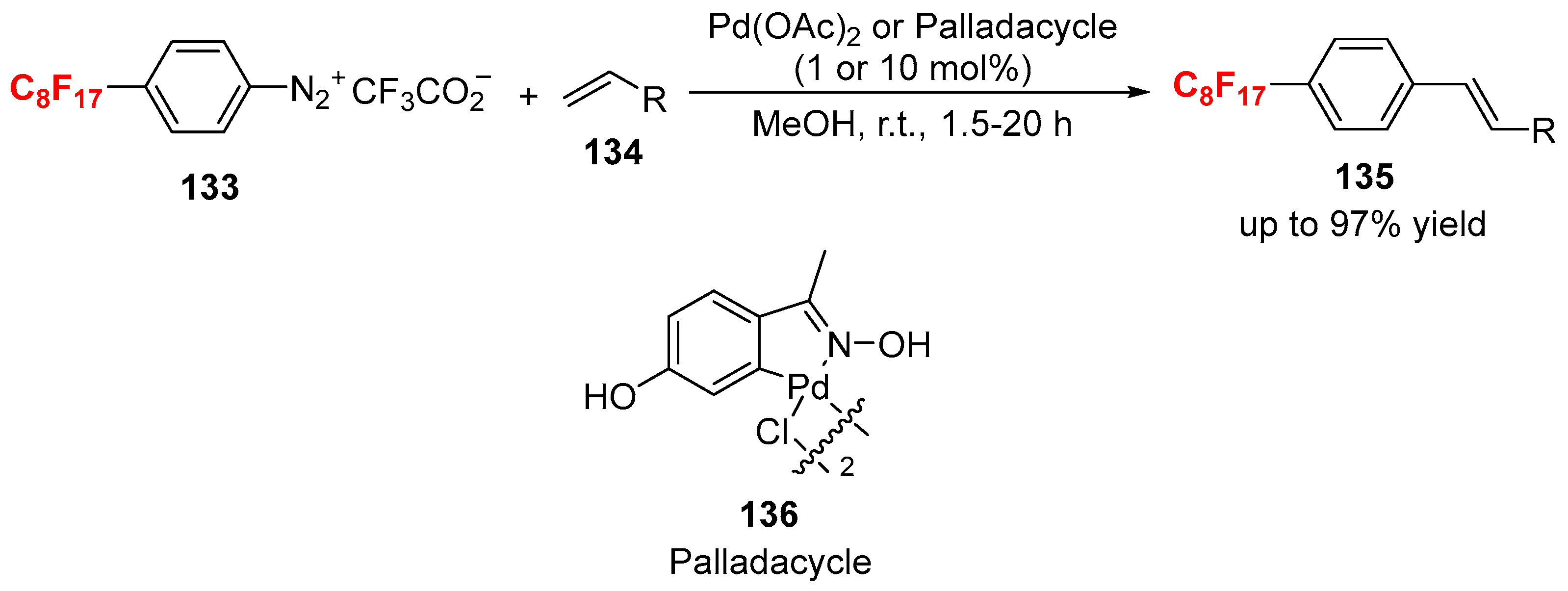
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
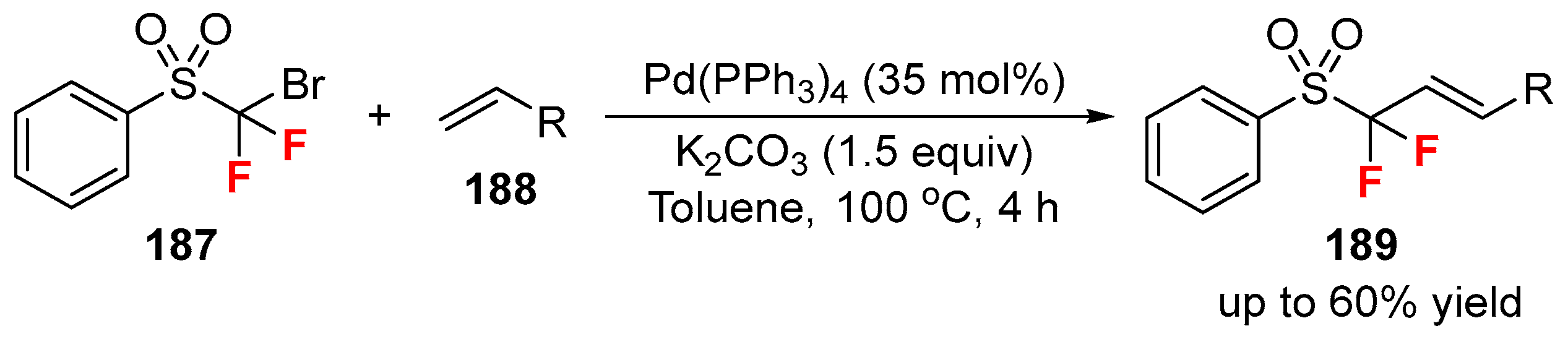{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
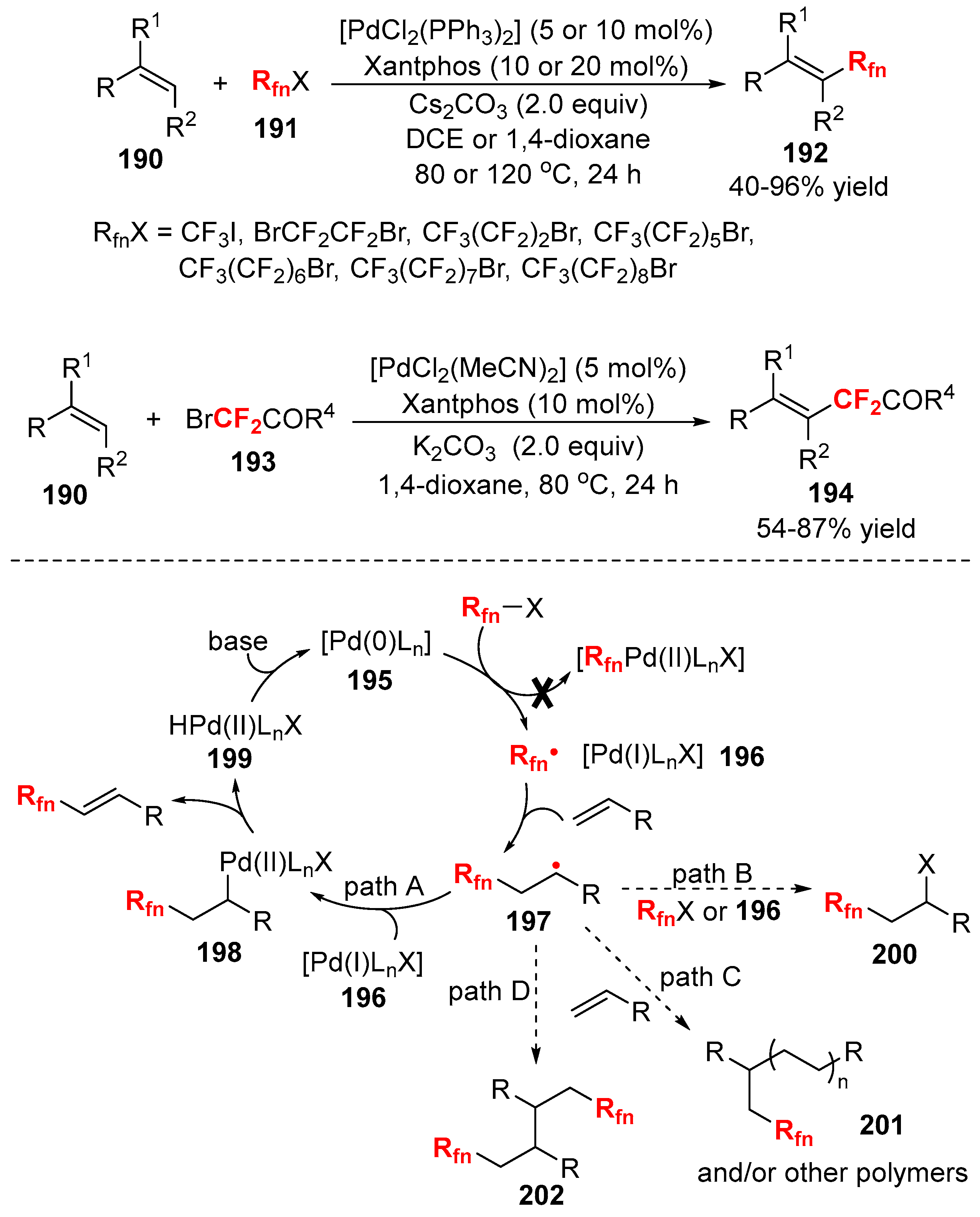
{kind=link}
{kind=link}
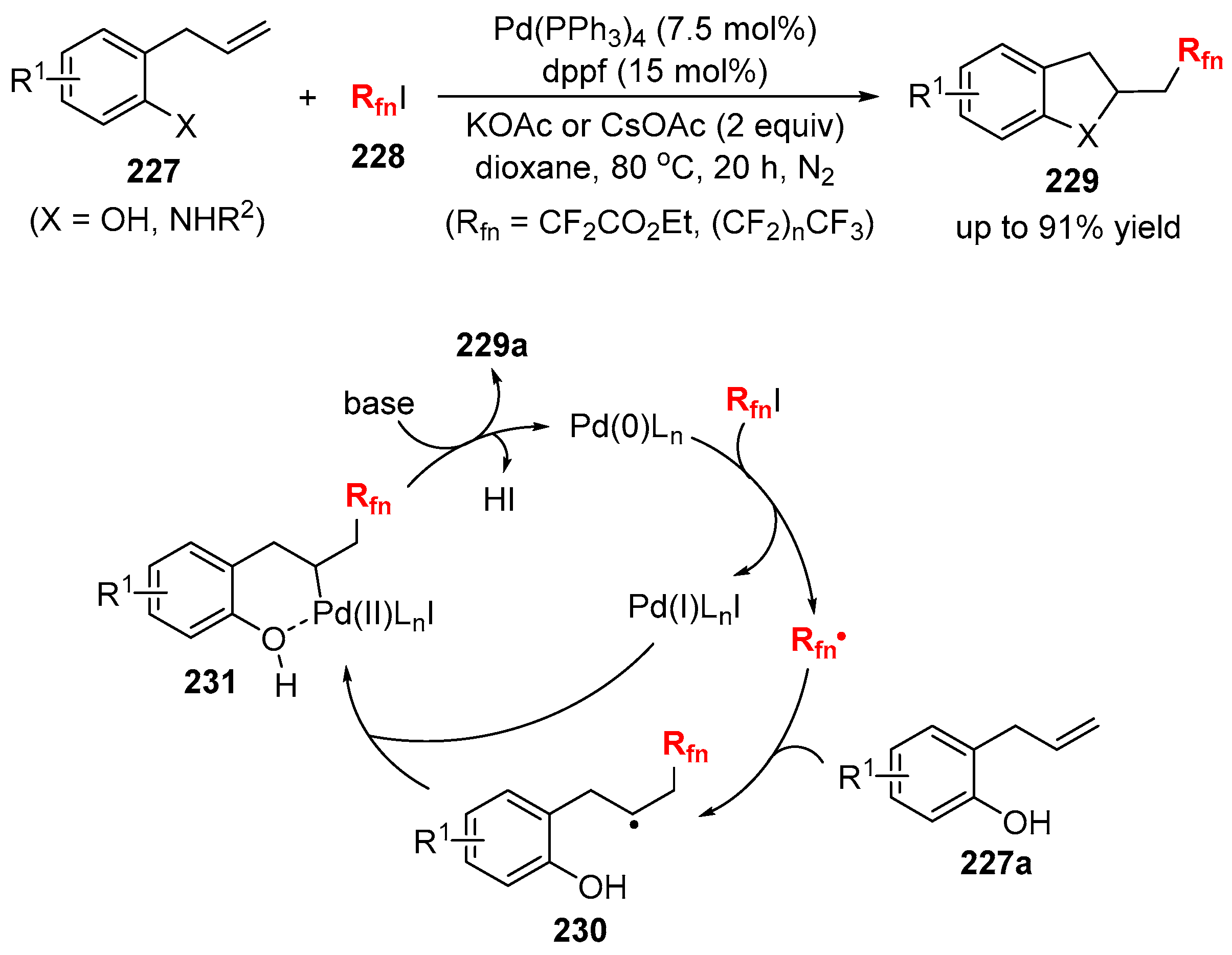
{kind=link}
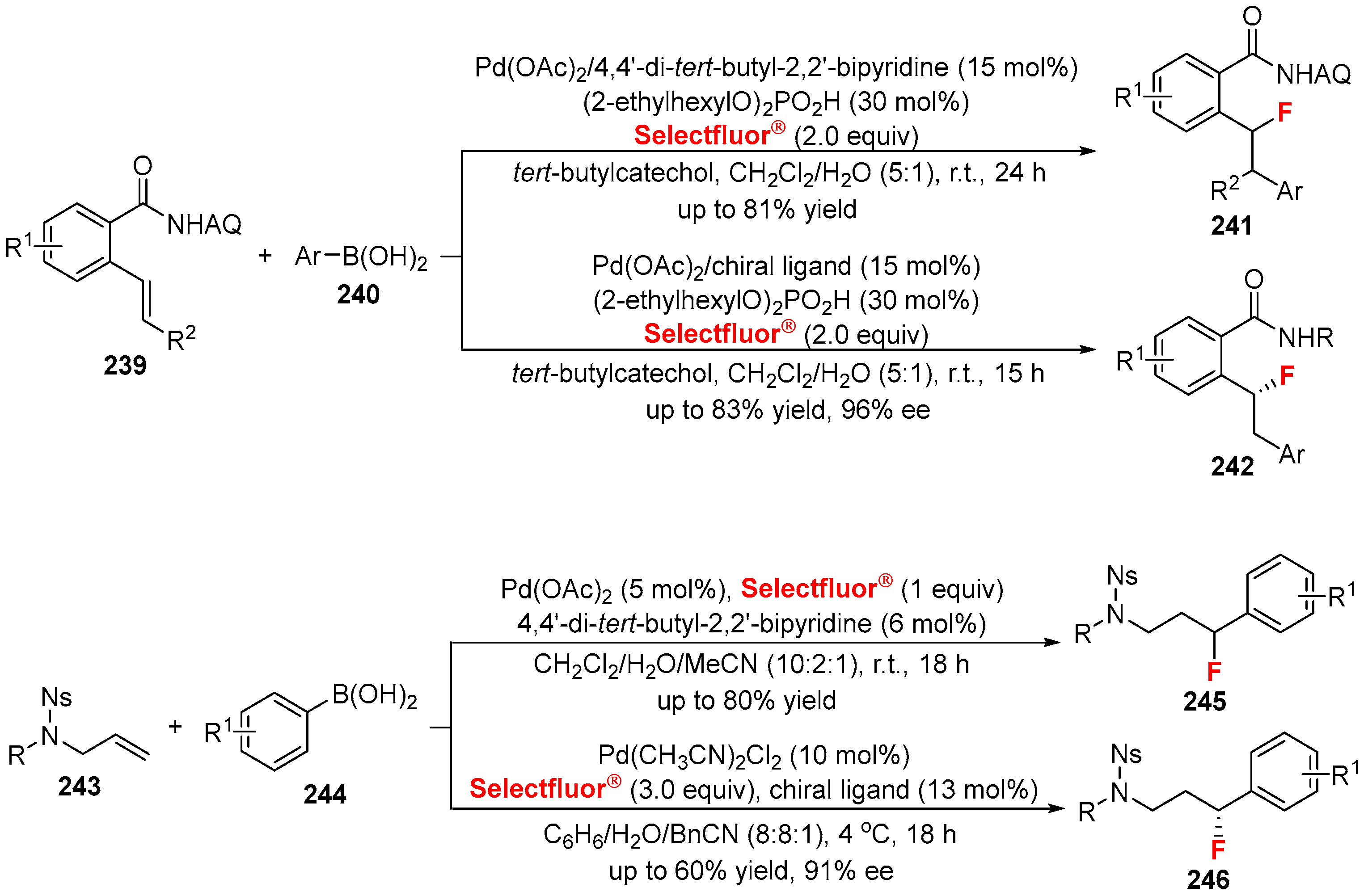
{kind=link}
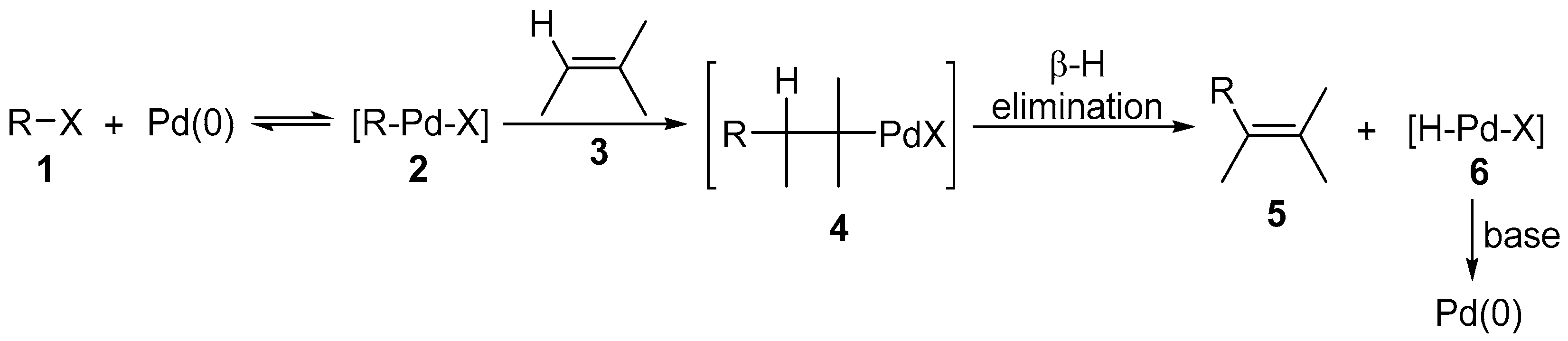
1. Introduction
2. Mizoroki-Heck Reactions of Aryl Halides or Pseudo-Halides with Fluorine-Containing Alkenes
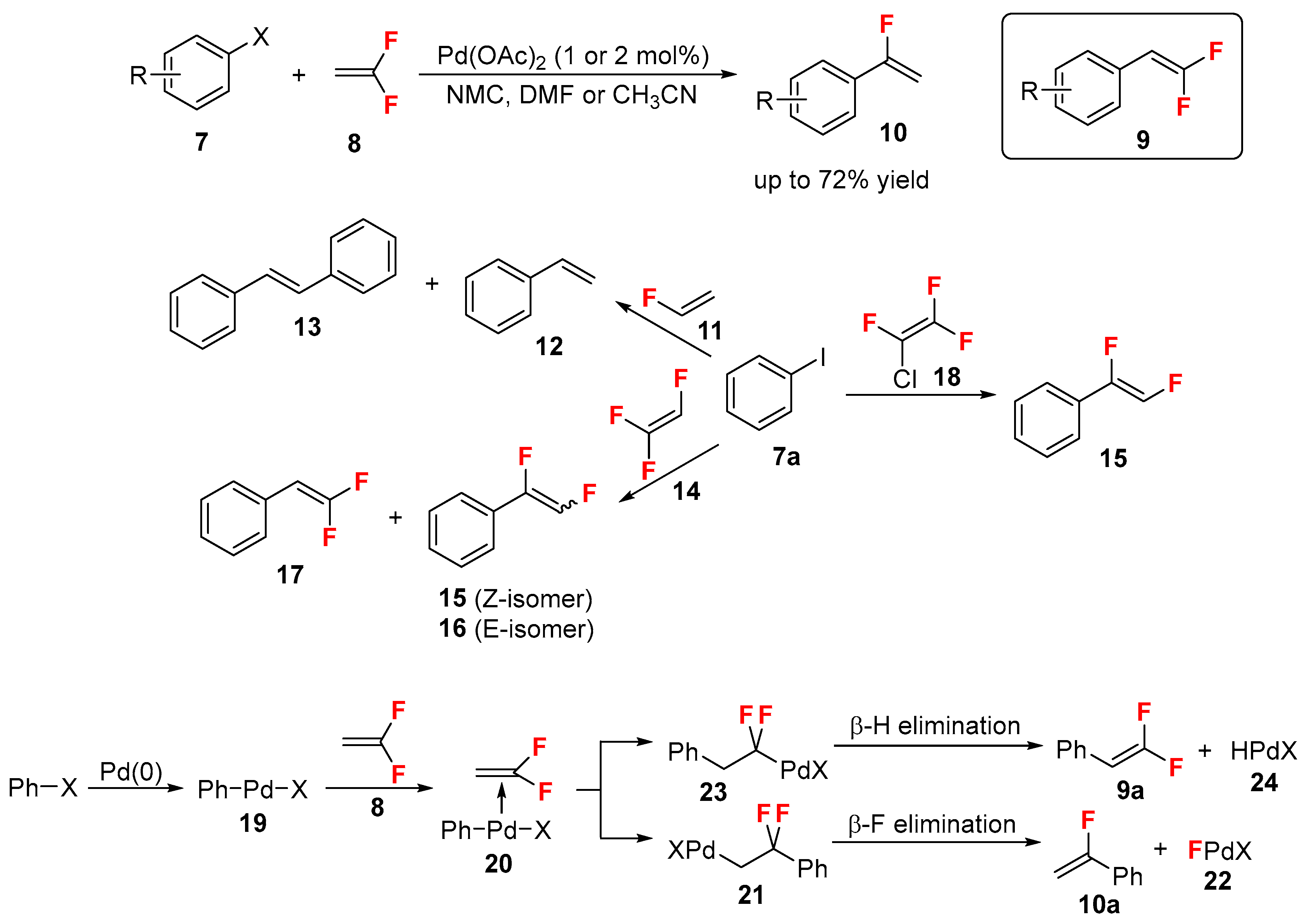
2.1. Fluoroalkenes as Cross-Coupling Participants
2.2. Fluorine-Containing Vinyl Sulfur Compounds as the Cross-Coupling Participants
2.3. Fluoroalkylated Alkenes as the Cross-Coupling Partners
2.4. Other Fluorine-Containing Alkenes as the Cross-Coupling Reagents
3. Mizoroki-Heck Reactions of Fluorinated Halides or Pseudo-Halides with Alkenes
3.1. Fluorinated Aryl Halides or Pseudo-Halides as the Coupling Agents
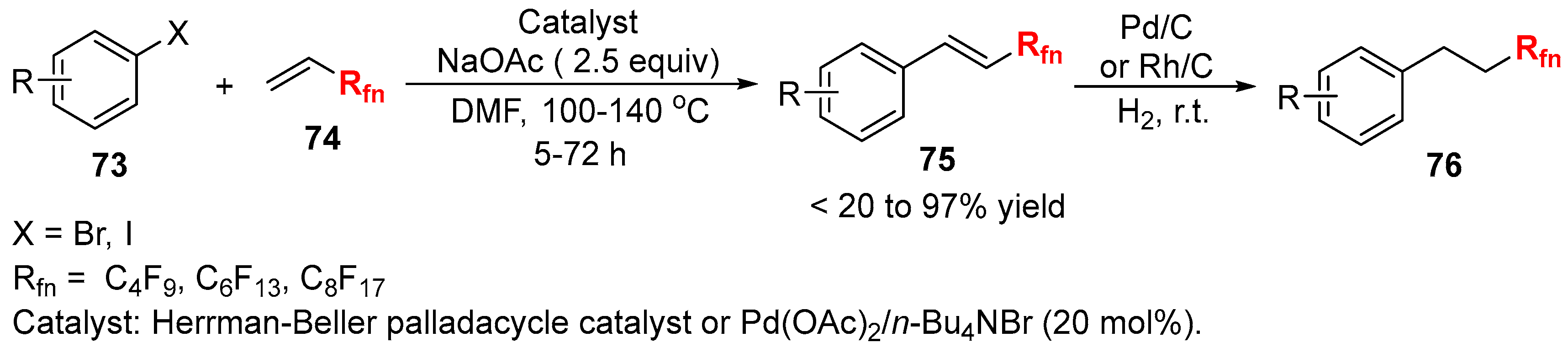
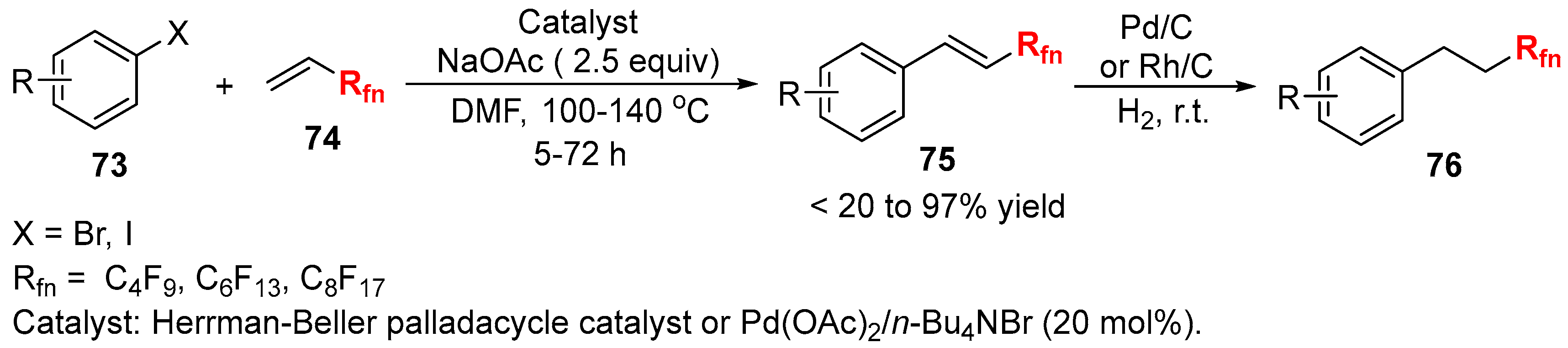
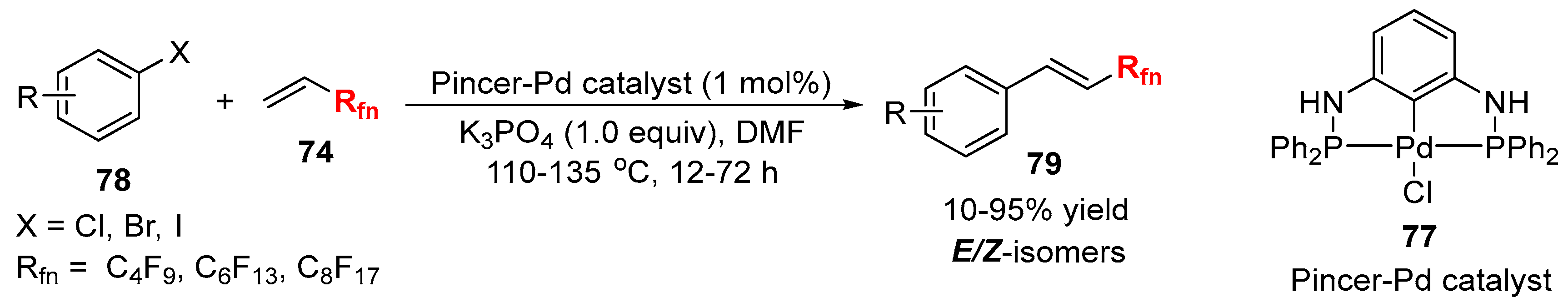
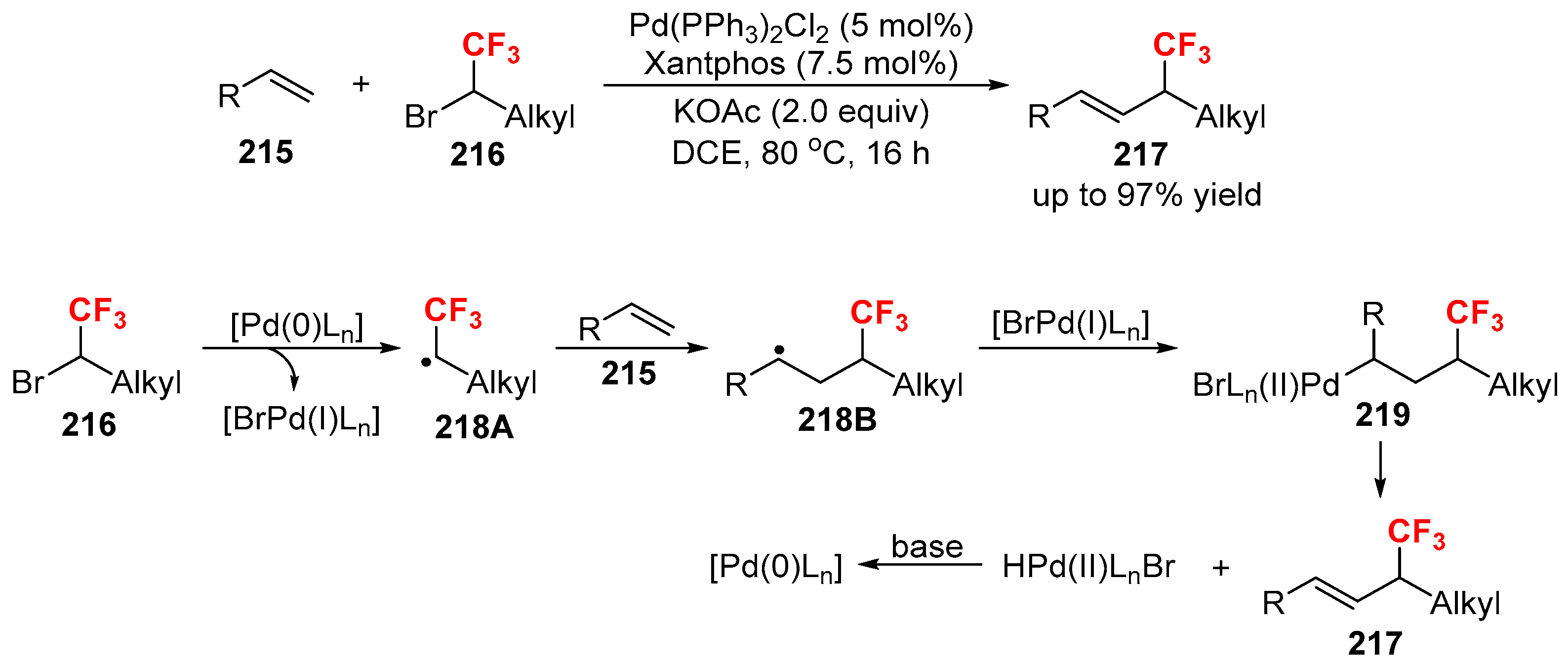
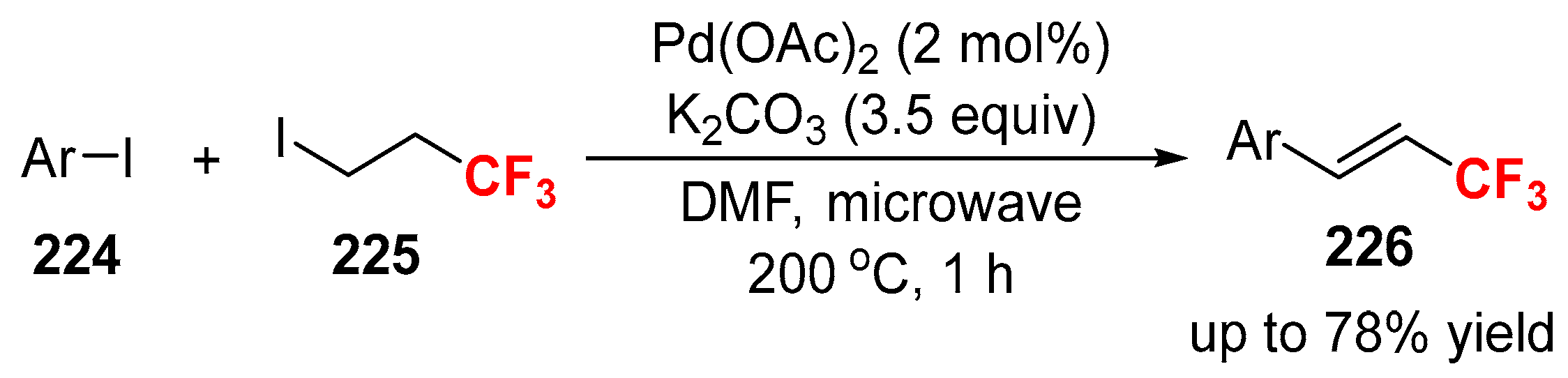
3.2. RCF2X and RfnX as the Cross-Coupling Reagents
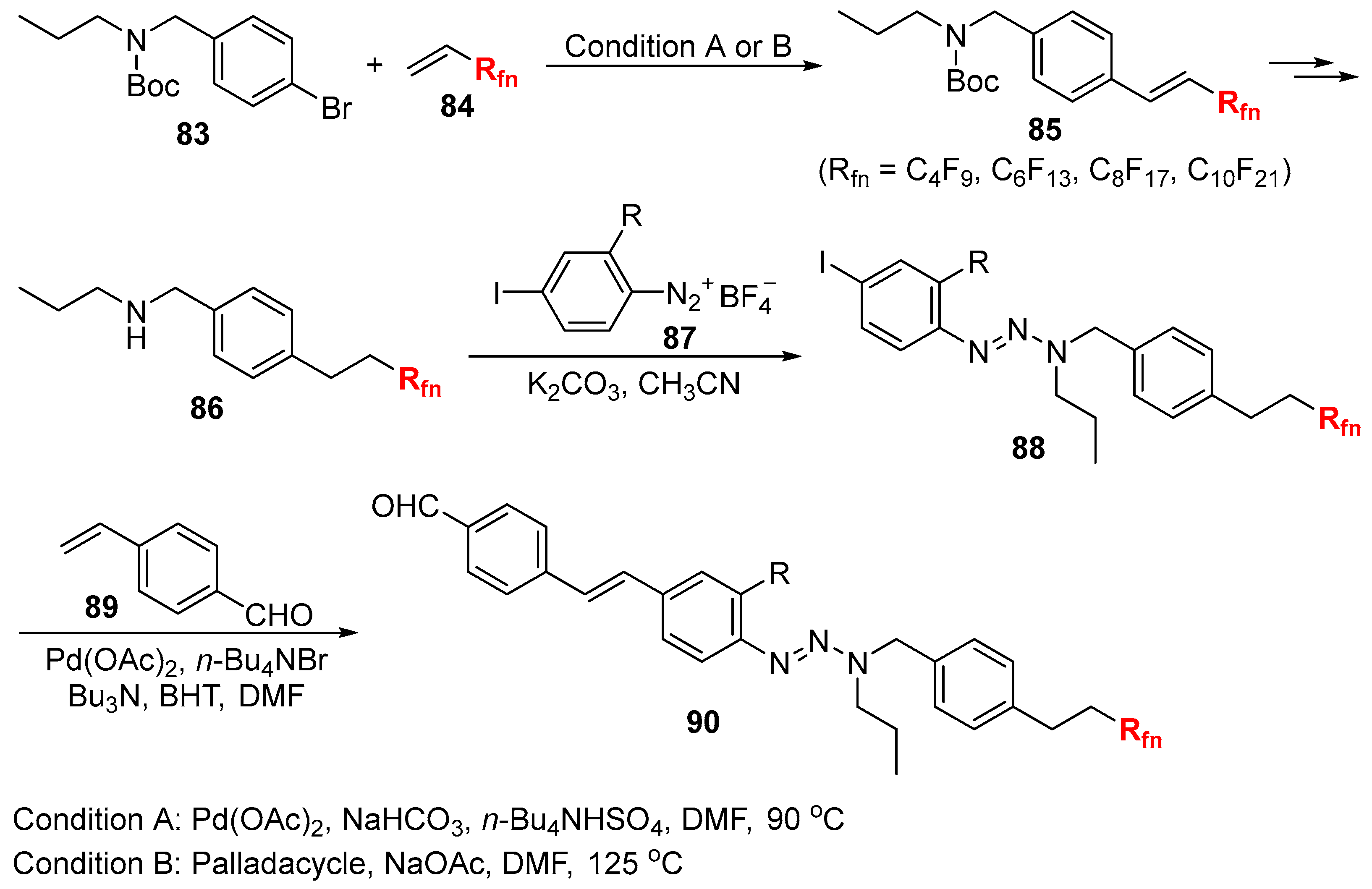
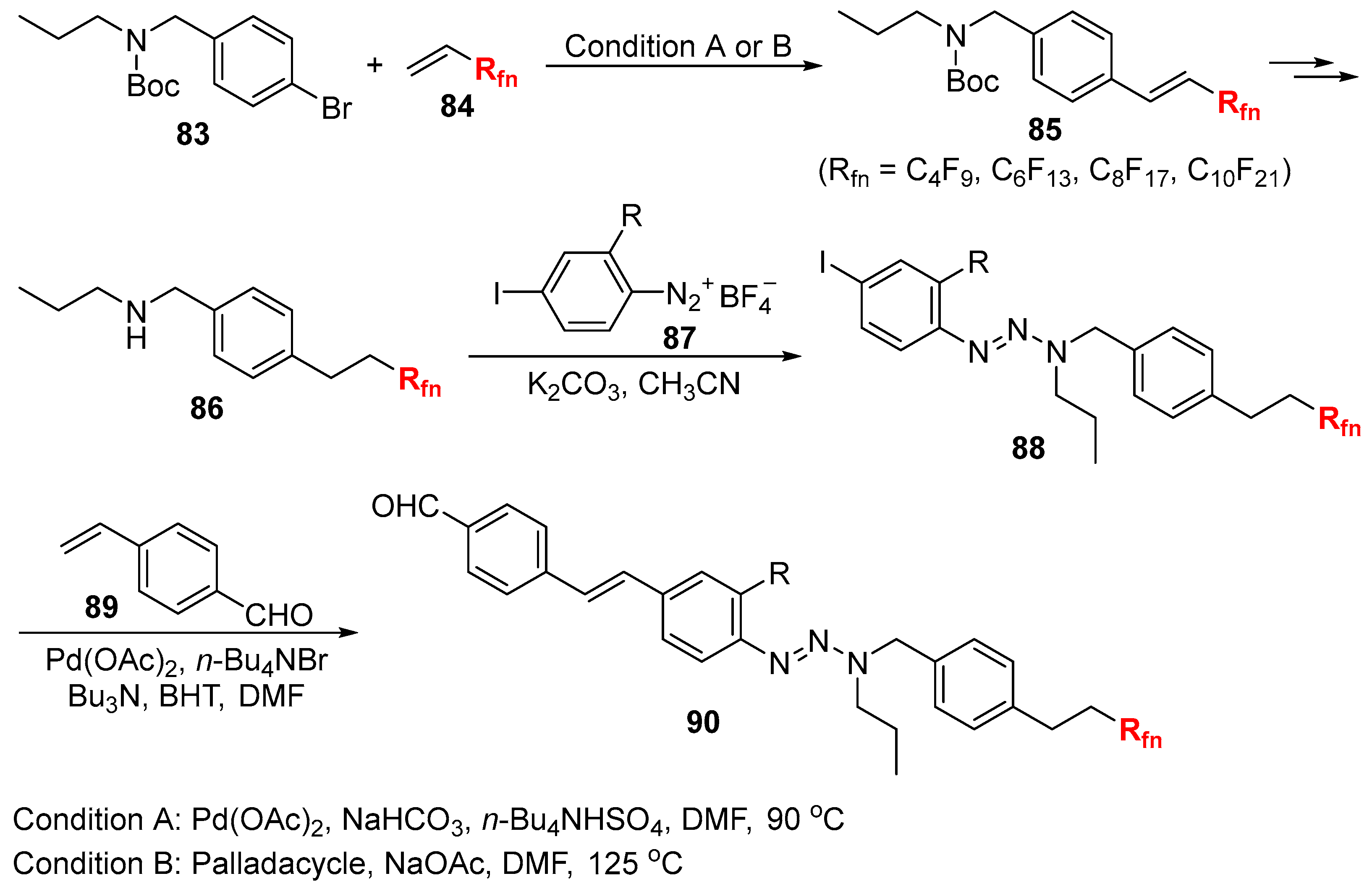
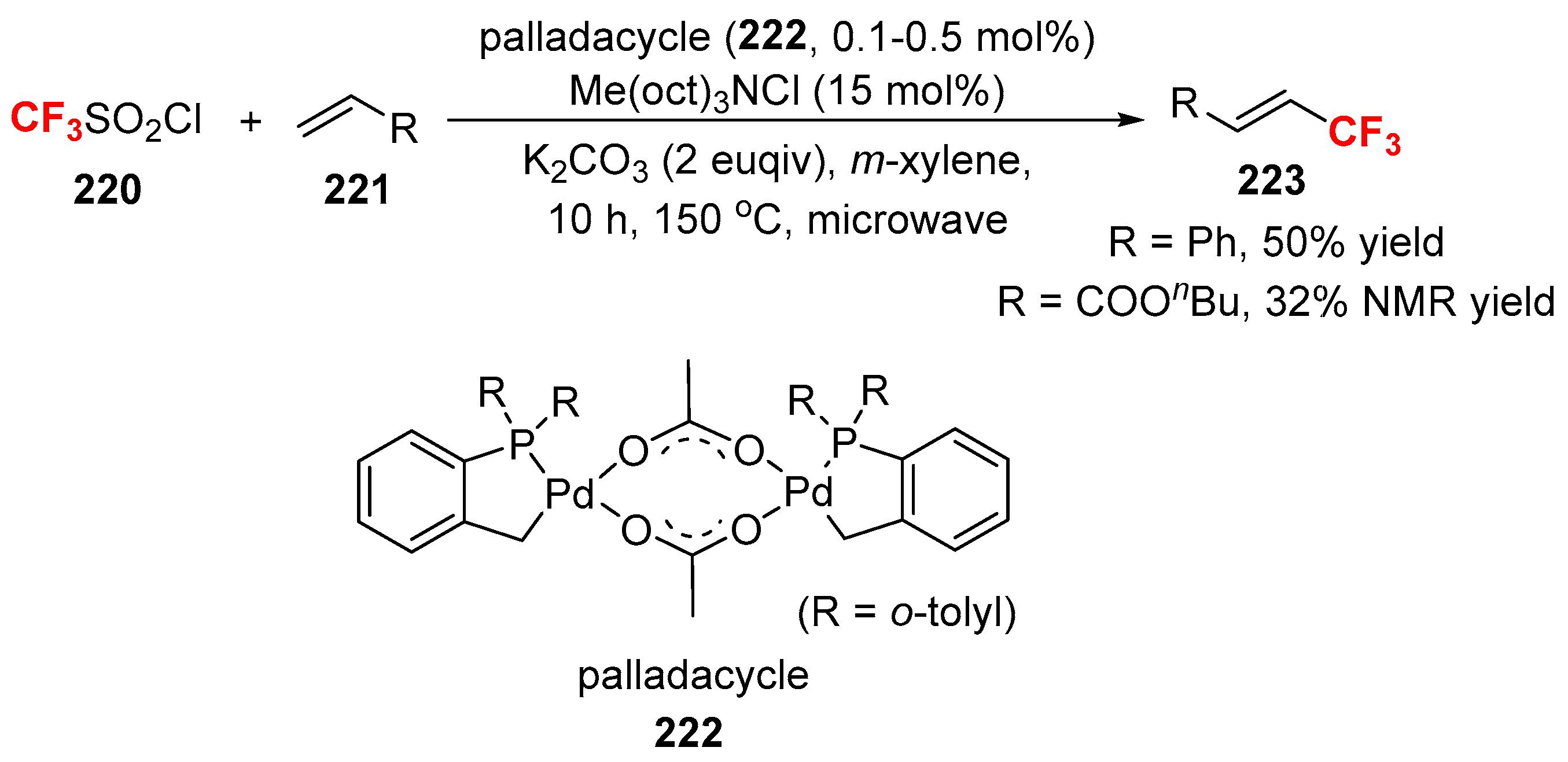
3.3. Fluoroalkylation Reagents as the Precursors of the Cross-Coupling Participants
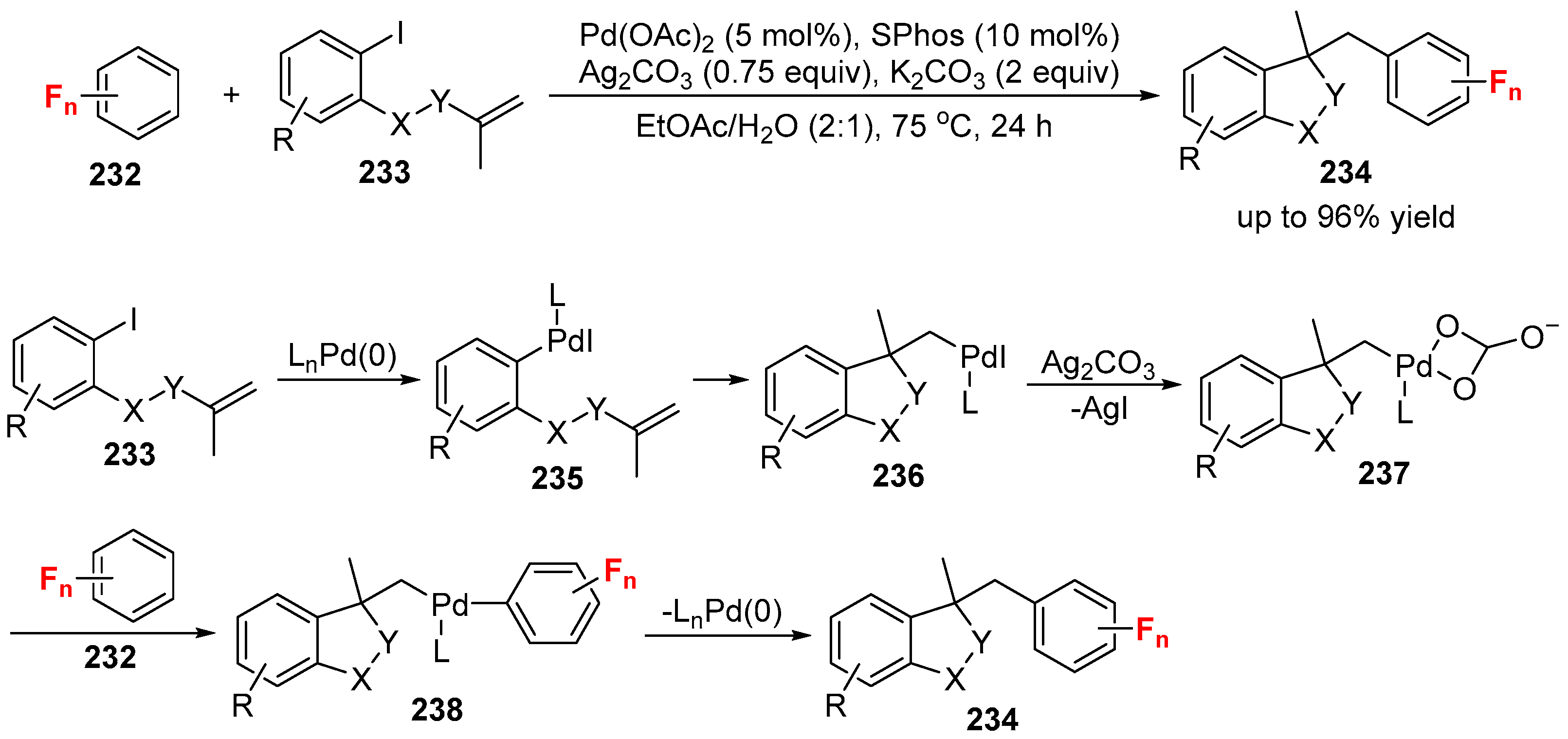
4. Variants of the Mizoroki-Heck Reactions with the Fluorine-Containing Reagents
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Beletskaya, I.P.; Cheprakov, A.V. The Heck reaction as a sharpening stone of palladium catalysis. Chem. Rev. 2000, 100, 3009–3066. [Google Scholar] [CrossRef] [PubMed]
- Whitcombe, N.J.; Hii, K.K.; Gibson, S.E. Advances in the Heck chemistry of aryl bromides and chlorides. Tetrahedron 2001, 57, 7449–7476. [Google Scholar] [CrossRef]
- Farina, V. High-turnover palladium catalysts in cross-coupling and Heck chemistry: A critical overview. Adv. Synth. Catal. 2004, 346, 1553–1582. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Bulger, P.G.; Sarlah, D. Palladium-catalyzed cross-coupling reactions in total synthesis. Angew. Chem. Int. Ed. 2005, 44, 4442–4489. [Google Scholar] [CrossRef] [PubMed]
- Phan, N.T.S.; Van Der Sluys, M.; Jones, C.W. On the nature of the active species in palladium catalyzed Mizoroki-Heck and Suzuki-Miyaura couplings-homogeneous or heterogeneous catalysis, a critical review. Adv. Synth. Catal. 2006, 348, 609–679. [Google Scholar] [CrossRef]
- Li, H.; Johansson, S.; Carin, C.C.; Colacot, T.J. Development of preformed Pd Catalysts for cross-coupling reactions, beyond the 2010 Nobel Prize. ACS Catal. 2012, 2, 1147–1164. [Google Scholar] [CrossRef]
- Heck, R.F. Acylation, methylation, and carboxyalkylation of olefins by group VIII metal derivatives. J. Am. Chem. Soc. 1968, 90, 5518–5526. [Google Scholar] [CrossRef]
- Mizoroki, T.; Mori, K.; Ozaki, A. Arylation of olefin with aryl iodide catalyzed by palladium. Bull. Chem. Soc. Jpn. 1971, 44, 581. [Google Scholar] [CrossRef]
- Heck, R.F.; Nolley, J.P. Palladium-catalyzed vinylic hydrogen substitution reactions with aryl, benzyl, and styryl halides. J. Org. Chem. 1972, 37, 2320–2322. [Google Scholar] [CrossRef]
- Cacchi, S.; Morera, E.; Ortar, G. Palladium-catalysed vinylation of enol triflates. Tetrahedron Lett. 1984, 25, 2271–2274. [Google Scholar] [CrossRef]
- Nilsson, P. Reaction with nonaromatic alkenyl halides or alkenyl sulfonates. Sci. Synth. 2013, 3, 285–302. [Google Scholar]
- Weimar, M.; Fuchter, M.J. Reaction with nonaromatic halides, sulfonates, or related compounds. Sci. Synth. 2013, 3, 137–168. [Google Scholar]
- Miura, M.; Hashimoto, H.; Itoh, K.; Nomura, M. Palladium-catalyzed desulfonylative coupling of arylsulfonyl chlorides with acrylate esters under solid-liquid phase transfer conditions. Tetrahedron Lett. 1989, 30, 975–976. [Google Scholar] [CrossRef]
- Dubbaka, S.R.; Vogel, P. Organosulfur compounds: Electrophilic reagents in transition-metal-catalyzed carbon-carbon bond-forming reactions. Angew. Chem. Int. Ed. 2005, 44, 7674–7684. [Google Scholar] [CrossRef] [PubMed]
- Dubbaka, S.R.; Zhao, D.; Fei, Z.; Vollaa, C.M.R.; Dyson, P.J.; Vogel, P. Palladium-catalyzed desulfitative Mizoroki-Heck coupling reactions of sulfonyl chlorides with olefins in a nitrile-functionalized ionic liquid. Synlett 2006, 3155–3157. [Google Scholar] [CrossRef]
- Blaser, H.-U.; Spencer, A. The palladium-catalysed arylation of activated alkenes with aroyl chlorides. J. Organomet. Chem. 1982, 233, 267–274. [Google Scholar] [CrossRef]
- Liu, C.; Meng, G.; Szostak, M. N-Acylsaccharins as amide-based arylating reagents via chemoselective N–C cleavage: Pd-catalyzed decarbonylative Heck reaction. J. Org. Chem. 2016, 81, 12023–12030. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.F.; Kurokhtina, A.A.; Larina, E.V.; Yarosh, E.V.; Lagoda, N.A. Direct kinetic evidence for the active anionic palladium(0) and palladium(II) intermediates in the ligand-free Heck reaction with aromatic carboxylic anhydrides. Organometallics 2017, 36, 3382–3386. [Google Scholar] [CrossRef]
- Masllorens, J.; Moreno-Manas, M.; Pla-Quintana, A.; Roglans, A. First Heck reaction with arenediazonium cations with recovery of Pd-triolefinic macrocyclic catalyst. Org. Lett. 2003, 5, 1559–1561. [Google Scholar] [CrossRef] [PubMed]
- Felpin, F.-X.; Nassar-Hardy, L.; Le Callonnec, F.; Fouquet, E. Recent advances in the Heck-Matsuda reaction in heterocyclic chemistry. Tetrahedron 2011, 67, 2815–2831. [Google Scholar] [CrossRef]
- Oger, N.; d’Halluin, M.; Le Grognec, E.; Felpin, F.-X. Using aryl diazonium salts in palladium-catalyzed reactions under safer conditions. Org. Process Res. Dev. 2014, 18, 1786–1801. [Google Scholar] [CrossRef]
- Moriarty, R.M.; Epa, W.R.; Awasthi, A.K. Palladium-catalyzed coupling of alkenyl iodonium salts with olefins: A mild and stereoselective Heck-type reaction using hypervalent iodine. J. Am. Chem. Soc. 1991, 113, 6315–6317. [Google Scholar] [CrossRef]
- Szabó, K.J. Mechanism of the oxidative addition of hypervalent iodonium salts to palladium(II) pincer-complexes. J. Mol. Catal. A Chem. 2010, 324, 56–63. [Google Scholar] [CrossRef]
- Hwang, L.K.; Na, Y.; Lee, J.; Do, Y.; Chang, S. Tetraarylphosphonium halides as arylating reagents in Pd-catalyzed Heck and cross-coupling reactions. Angew. Chem. Int. Ed. 2005, 44, 6166–6169. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.-Y.; Hu, Y.-T.; Teng, H.-B.; Zhang, C.-P. Application of arylsulfonium salts as arylation reagents. Tetrahedron Lett. 2018, 59, 299–309. [Google Scholar] [CrossRef]
- Tasker, S.Z.; Gutierrez, A.C.; Jamison, T.F. Nickel-catalyzed Mizoroki-Heck reaction of aryl sulfonates and chlorides with electronically unbiased terminal olefins: High selectivity for branched products. Angew. Chem. Int. Ed. 2014, 53, 1858–1861. [Google Scholar] [CrossRef] [PubMed]
- Maity, S.; Dolui, P.; Kancherla, R.; Maiti, D. Introducing unactivated acyclic internal aliphatic olefins into a cobalt catalyzed allylic selective dehydrogenative Heck reaction. Chem. Sci. 2017, 8, 5181–5185. [Google Scholar] [CrossRef] [PubMed]
- Phipps, R.J.; McMurray, L.; Ritter, S.; Duong, H.A.; Gaunt, M.J. Copper-catalyzed alkene arylation with diaryliodonium salts. J. Am. Chem. Soc. 2012, 134, 10773–10776. [Google Scholar] [CrossRef] [PubMed]
- Dughera, S.; Barbero, M. Gold catalyzed Heck-coupling of arenediazonium o-Benzenedisulfonimides. Org. Biomol. Chem. 2018, 16, 295–301. [Google Scholar]
- Zhu, K.; Dunne, J.; Shaver, M.P.; Thomas, S.P. Iron-catalyzed Heck-type alkenylation of functionalized alkyl bromides. ACS Catal. 2017, 7, 2353–2356. [Google Scholar] [CrossRef]
- Wang, S.-S.; Yang, G.-Y. Recent developments in low-cost TM-catalyzed Heck-type reactions (TM = transition metal, Ni, Co, Cu, Fe). Catal. Sci. Technol. 2016, 6, 2862–2876. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Cheprakov, A.V. Modern Heck reactions. RSC Catal. Ser. 2015, 21, 355–478. [Google Scholar]
- Hajipour, A.R.; Rezaei, F.; Khorsandi, Z. Pd/Cu-free Heck and Sonogashira cross-coupling reaction by Co nanoparticles immobilized on magnetic chitosan as reusable catalyst. Green Chem. 2017, 19, 1353–1361. [Google Scholar] [CrossRef]
- Kurandina, D.; Parasram, M.; Gevorgyan, V. Visible light-induced room-temperature Heck reaction of functionalized alkyl halides with vinyl arenes/heteroarenes. Angew. Chem. Int. Ed. 2017, 56, 14212–14216. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.-Z.; Shang, R.; Cheng, W.-M.; Fu, Y. Irradiation-induced Heck reaction of unactivated alkyl halides at room temperature. J. Am. Chem. Soc. 2017, 139, 18307–18312. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D. Understanding organofluorine chemistry. An introduction to the C–F bond. Chem. Soc. Rev. 2008, 39, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Eisenstein, O.; Milani, J.; Perutz, R.N. Selectivity of C–H activation and competition between C–H and C–F bond activation at fluorocarbons. Chem. Rev. 2017, 117, 8710–8753. [Google Scholar] [CrossRef] [PubMed]
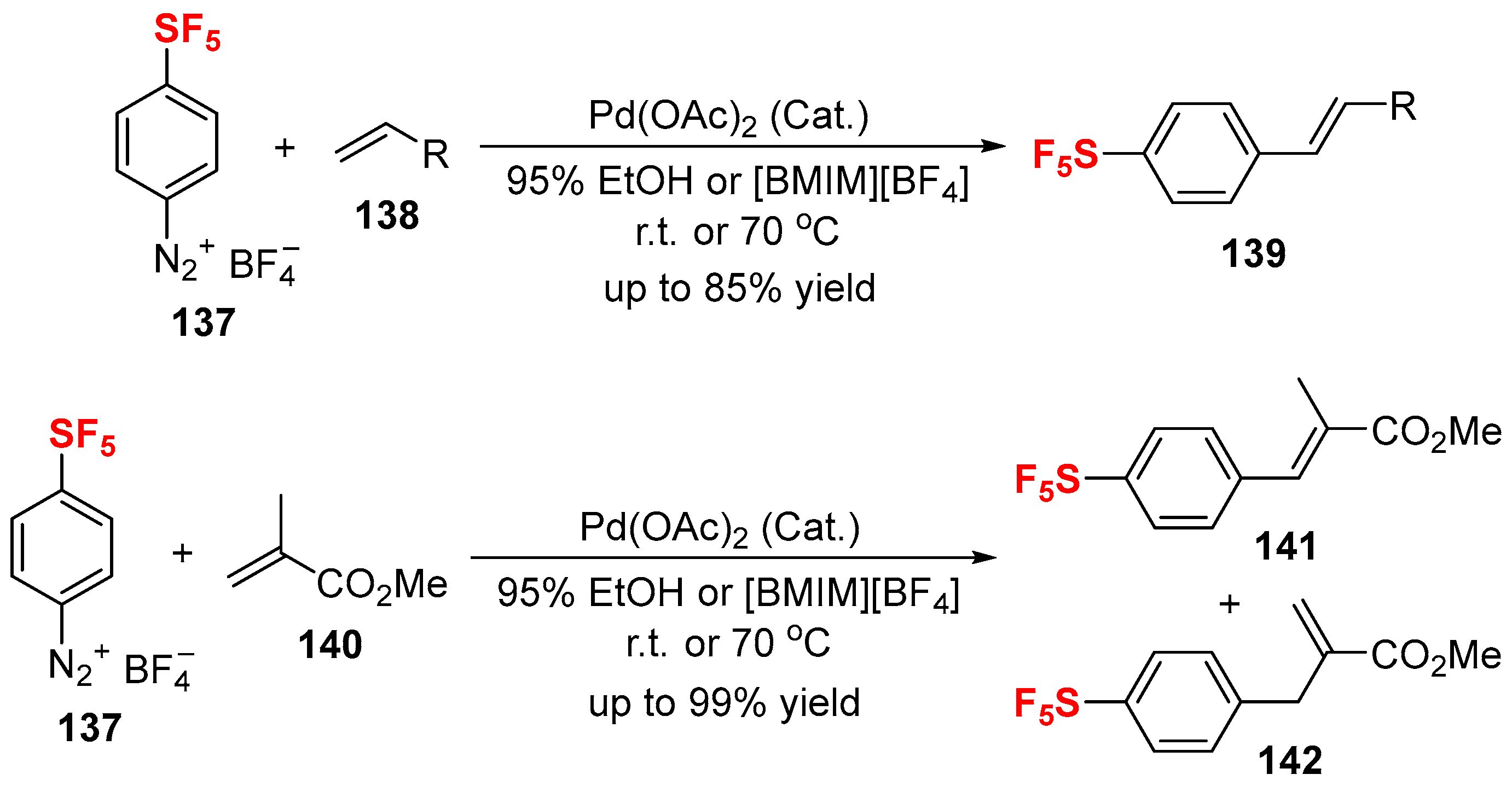
- Sowaileh, M.F.; Hazlitt, R.A.; Colby, D.A. Application of the pentafluorosulfanyl group as a bioisosteric replacement. ChemMedChem 2017, 12, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Hiyama, T.; Kanie, K.; Kusumoto, T.; Morizawa, Y.; Shimizu, M. Organofluorine Compounds: Chemistry and Applications; Springer: Berlin, Germany, 2000. [Google Scholar]
- Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceñ, J.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next generation of fluorine-containing pharmaceuticals, compounds currently in phase II–III clinical trials of major pharmaceutical companies: New structural trends and therapeutic areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.A.; Völler, J.-S.; Budisa, N.; Koksch, B. Deciphering the fluorine code-the many hats fluorine wears in a protein environment. Acc. Chem. Res. 2017, 50, 2093–2103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Kelly, M.A.; Bauer, N.; You, W. The curious case of fluorination of conjugated polymers for solar cells. Acc. Chem. Res. 2017, 50, 2401–2409. [Google Scholar] [CrossRef] [PubMed]
- Kusoglu, A.; Weber, A.Z. New insights into perfluorinated sulfonic-acid ionomers. Chem. Rev. 2017, 117, 987–1104. [Google Scholar] [CrossRef] [PubMed]
- Amii, H.; Uneyama, K. C–F Bond activation in organic synthesis. Chem. Rev. 2009, 109, 2119–2183. [Google Scholar] [CrossRef] [PubMed]
- Yerien, D.E.; Bonesi, S.; Postigo, A. Fluorination methods in drug discovery. Org. Biomol. Chem. 2016, 14, 8398–8427. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Dong, T.; Revankar, H.M.; Zhang, C.-P. Recent progress on fluorination in aqueous media. Green Chem. 2017, 19, 3951–3992. [Google Scholar] [CrossRef]
- Preshlock, S.; Tredwell, M.; Gouverneur, V. 18F-Labeling of arenes and heteroarenes for applications in positron emission tomography. Chem. Rev. 2016, 116, 719–766. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, B.; Walkowiak-Kulikowsk, J.; Koroniak, H. On the halofluorination reactions of olefins as selective, and an efficient methodology for the introduction of fluorine into organic molecules. J. Fluor. Chem. 2017, 203, 47–61. [Google Scholar] [CrossRef]
- Song, X.; Xu, C.; Wang, M. Transformations based on ring-opening of gem-difluorocyclopropanes. Tetrahedron Lett. 2017, 58, 1806–1816. [Google Scholar] [CrossRef]
- Zhang, C.-P.; Chen, Q.-Y.; Guo, Y.; Xiao, J.-C.; Gu, Y.-C. Progress in fluoroalkylation of organic compounds via sulfinatodehalogenation initiation system. Chem. Soc. Rev. 2012, 41, 4536–4559. [Google Scholar] [CrossRef] [PubMed]
- Heitz, W.; Knebelkamp, A. Synthesis of fluorostyrenes via palladium-catalyzed reactions of aromatic halides with fluoroolefins. Makromol. Rapid Commun. 1991, 12, 69–75. [Google Scholar] [CrossRef]
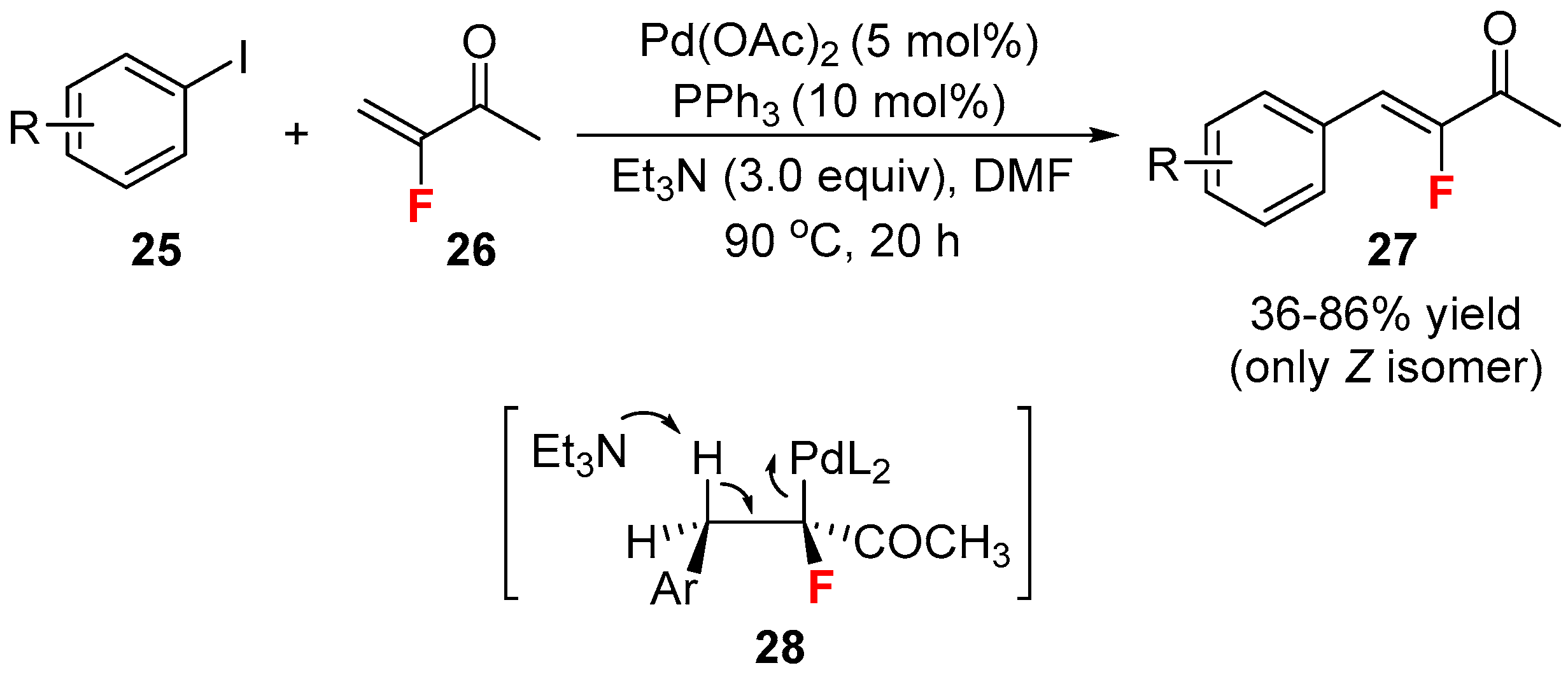
- Patrick, T.B.; Agboka, T.Y.; Gorrell, K. Heck reaction with 3-fluoro-3-buten-2-one. J. Fluor. Chem. 2008, 129, 983–985. [Google Scholar] [CrossRef]
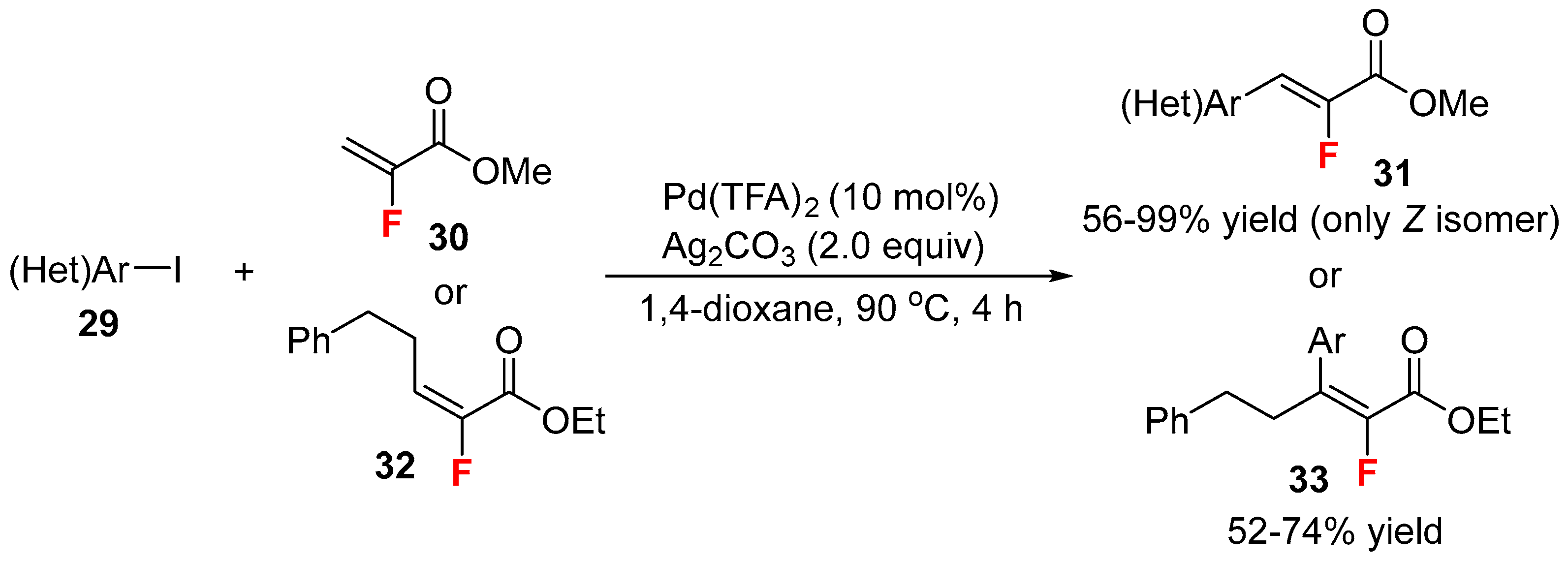
- Rousee, K.; Bouillon, J.P.; Couve-Bonnaire, S.; Pannecoucke, X. Stereospecific synthesis of tri- and tetrasubstituted α-fluoroacrylates by Mizoroki-Heck reaction. Org. Lett. 2016, 18, 540–543. [Google Scholar] [CrossRef] [PubMed]
- Hirotaki, K.; Hanamoto, T. Mizoroki-Heck reaction of (1-fluorovinyl)methyldiphenylsilane with aryl iodides. J. Org. Chem. 2011, 76, 8564–8568. [Google Scholar] [CrossRef] [PubMed]
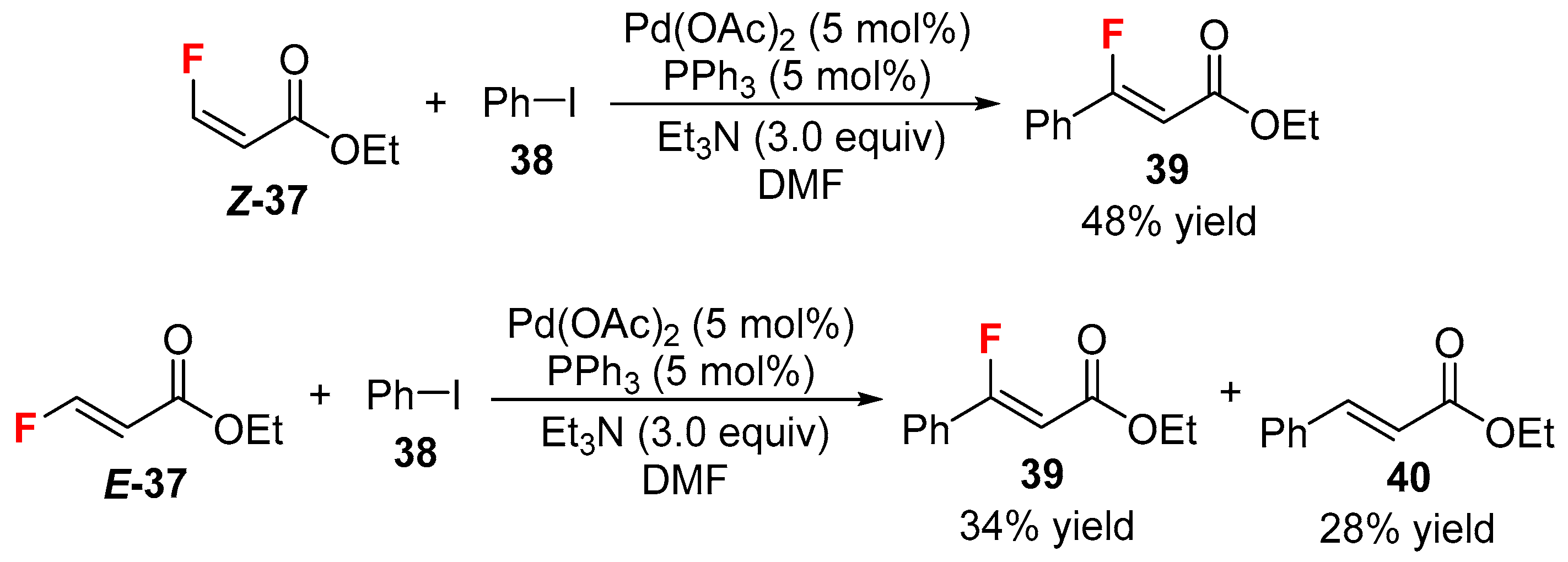
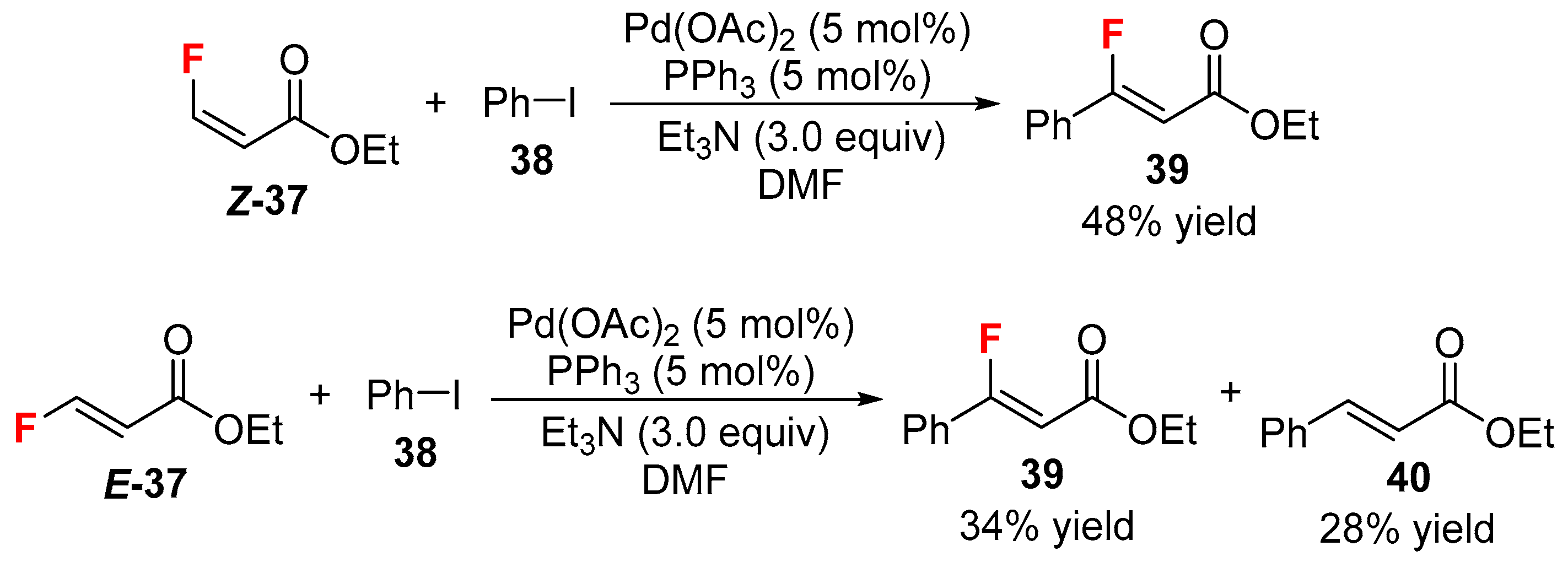
- Patrick, T.B.; Blay, A.A. Heck reaction with ethyl (E)- and (Z)-3-fluoropropenoate. J. Fluor. Chem. 2016, 189, 68–69. [Google Scholar] [CrossRef]
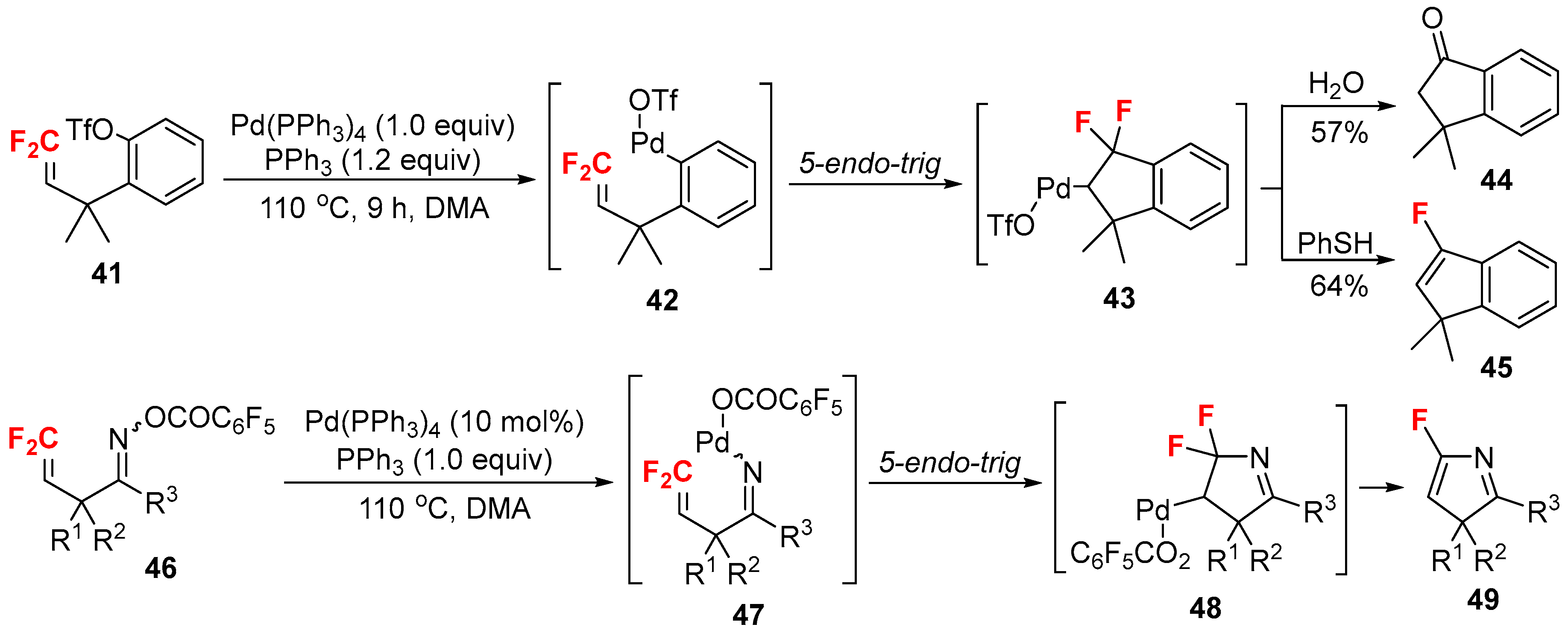
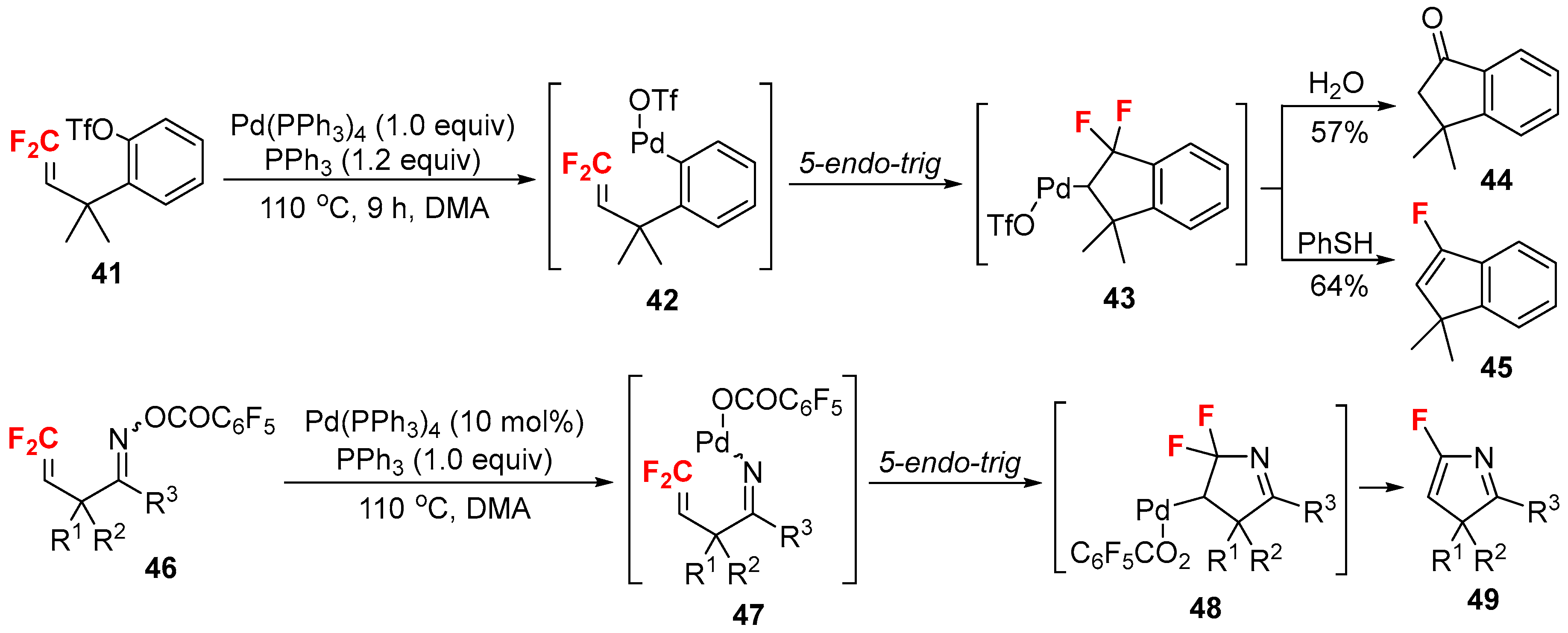
- Ichikawa, J.; Sakoda, K.; Mihara, J.; Ito, N. Heck-type 5-endo-trig cyclizations promoted by vinylic fluorines: Ring-fluorinated indene and 3H-pyrrole syntheses from 1,1-difluoro-1-alkenes. J. Fluor. Chem. 2006, 127, 489–504. [Google Scholar] [CrossRef]
- Sakoda, K.; Mihara, J.; Ichikawa, J. Heck-type 5-endo-trig cyclization promoted by vinylic fluorines: Synthesis of 5-fluoro-3H-pyrroles. Chem. Commun. 2005, 0, 4684–4686. [Google Scholar] [CrossRef] [PubMed]
- Krutak, J.J.; Burpitt, R.D.; Moore, W.H.; Hyatt, J.A. Chemistry of ethenesulfonyl fluoride. Fluorosulfonylethylation of organic compounds. J. Org. Chem. 1979, 44, 3847–3858. [Google Scholar] [CrossRef]
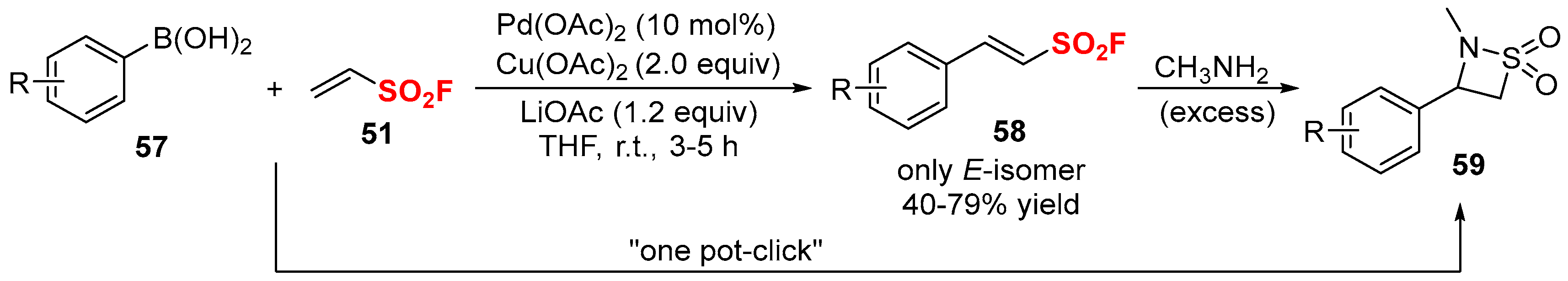
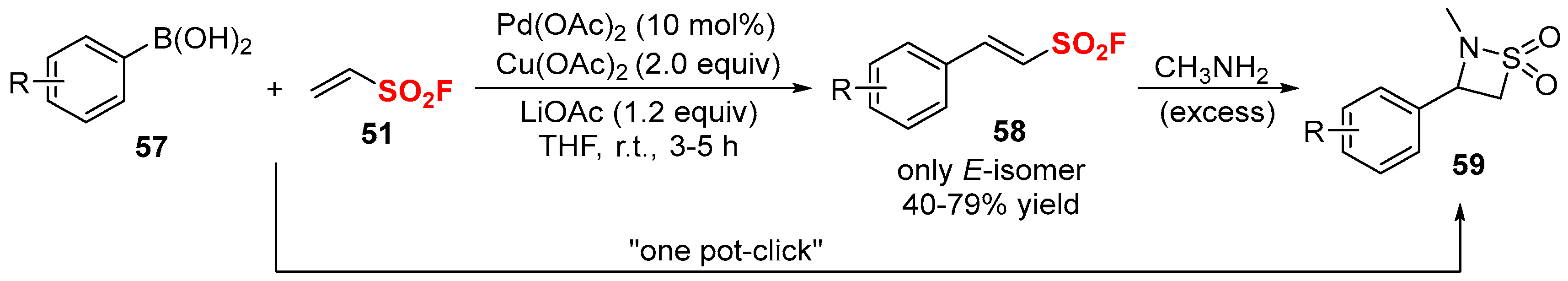
- Qin, H.-L.; Zheng, Q.; Bare, G.A.L.; Wu, P.; Sharpless, K.B. A Heck–Matsuda process for the synthesis of β-arylethenesulfonyl fluorides: Selectively addressable bis-electrophiles for sufex click chemistry. Angew. Chem. Int. Ed. 2016, 55, 14155–14158. [Google Scholar] [CrossRef] [PubMed]
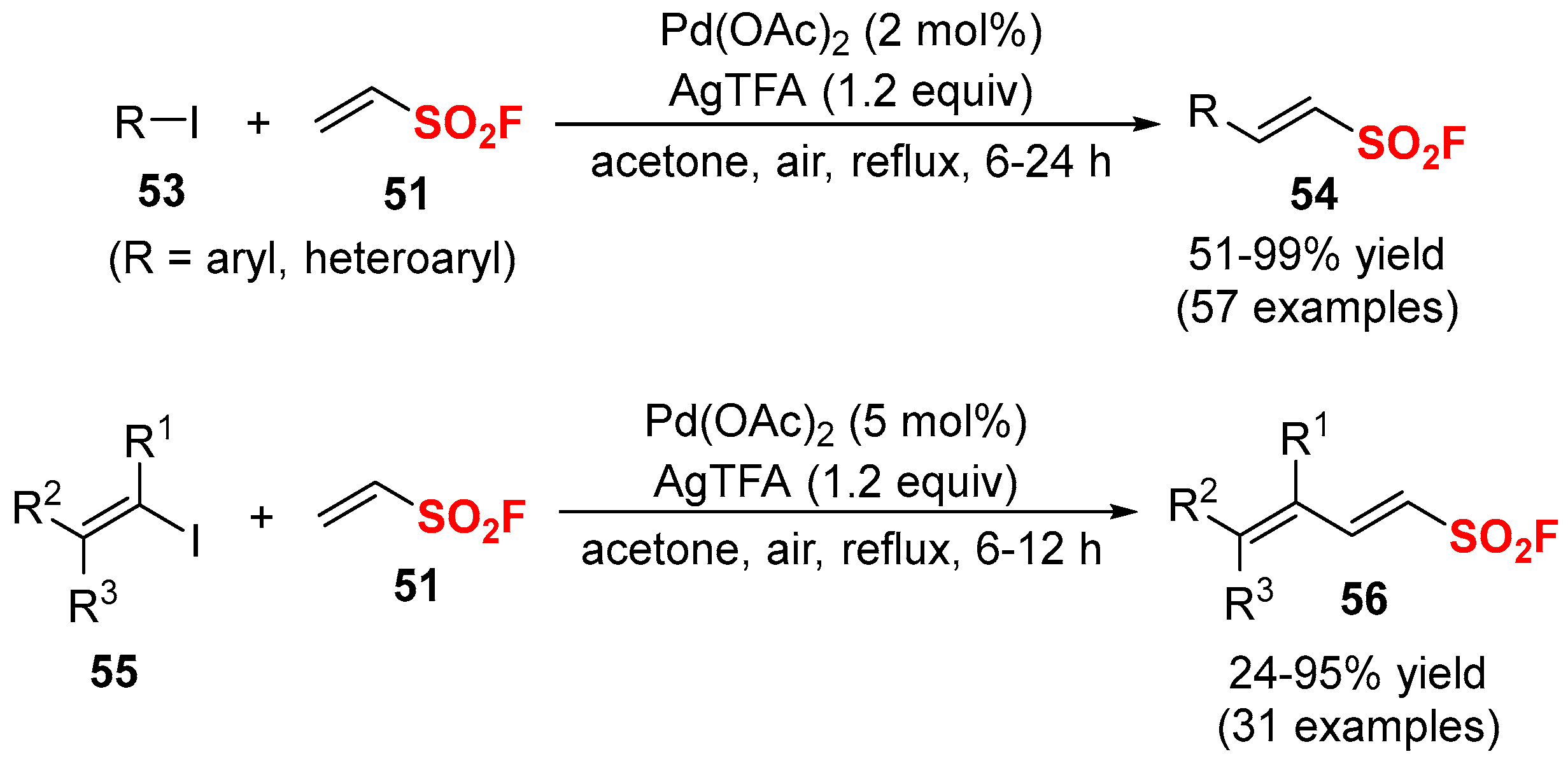
- Zha, G.F.; Zheng, Q.; Leng, J.; Wu, P.; Qin, H.L.; Sharpless, K.B. Palladium-catalyzed fluorosulfonylvinylation of organic iodides. Angew. Chem. Int. Ed. 2017, 56, 4849–4852. [Google Scholar] [CrossRef] [PubMed]
- Karimi, B.; Behzadnia, H.; Elhamifar, D.; Akhavan, P.F.; Esfahani, F.K.; Zamani, A. Transition-metal-catalyzed oxidative Heck reactions. Synthesis 2010, 2010, 1399–1427. [Google Scholar] [CrossRef]
- Lee, A.L. Enantioselective oxidative boron Heck reactions. Org. Biomol. Chem. 2016, 14, 5357–5366. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Gandeepan, P.; Li, J.; Ackermann, L. Recent advances in positional-selective alkenylations: Removable guidance for twofold C–H activation. Org. Chem. Front. 2017, 4, 1435–1467. [Google Scholar] [CrossRef]
- Chinthakindi, P.K.; Govender, K.B.; Kumar, A.S.; Kruger, H.G.; Govender, T.; Naicker, T.; Arvidsson, P.I. A synthesis of “dual warhead” β-aryl ethenesulfonyl fluorides and one-pot reaction to beta-sultams. Org. Lett. 2017, 19, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Zha, G.-F.; Bare, G.A.L.; Leng, J.; Shang, Z.-P.; Luo, Z.; Qin, H.-L. Gram-scale synthesis of β-(hetero)arylethenesulfonyl fluorides via a Pd(OAc)2 catalyzed oxidative Heck process with DDQ or AgNO3 as an oxidant. Adv. Synth. Catal. 2017, 359, 3237–3242. [Google Scholar] [CrossRef]
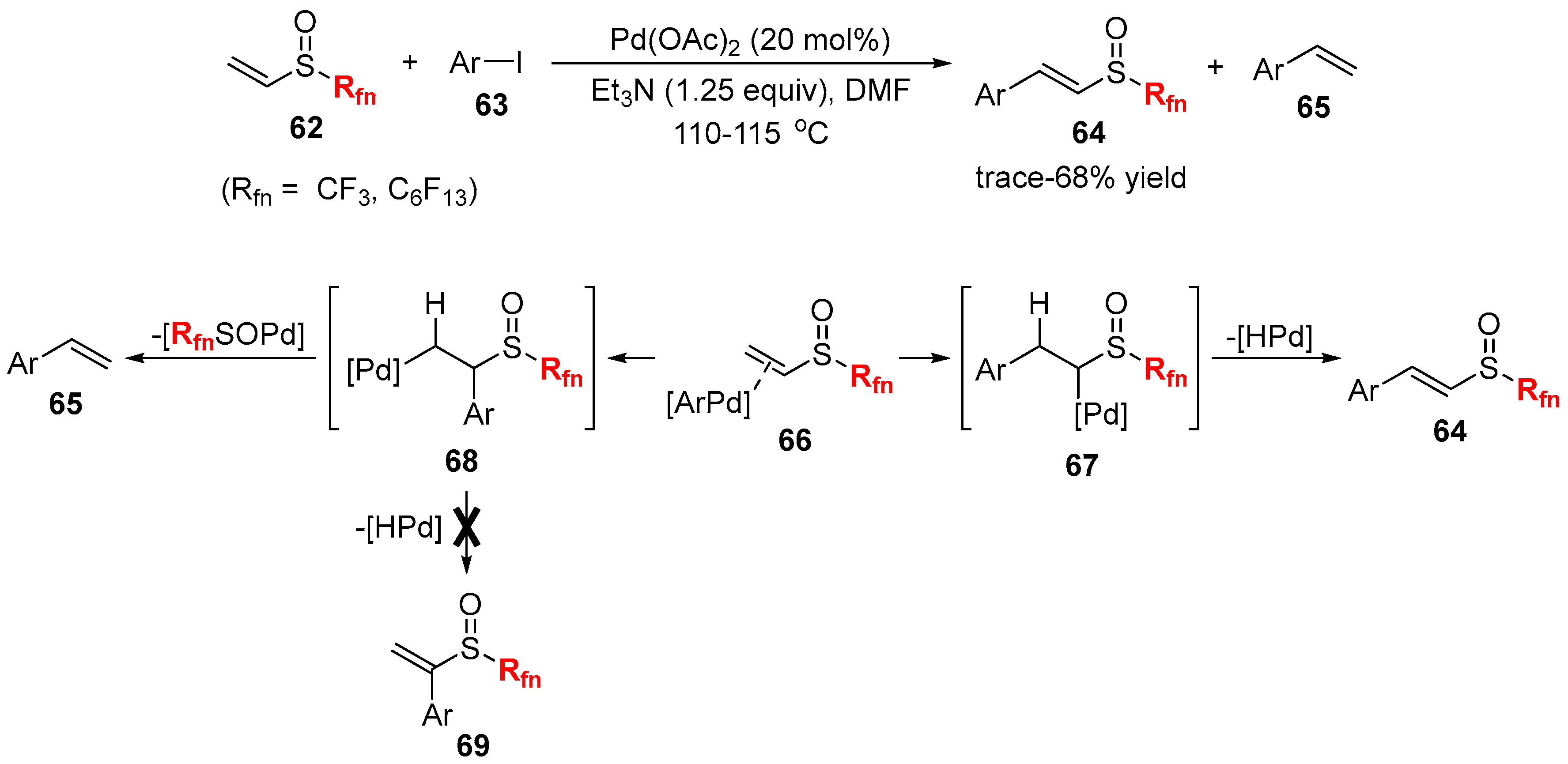
- Sokolenko, L.V.; Yagupolskii, Y.L.; Vlasenko, Y.G.; Babichenko, L.N.; Lipetskij, V.O.; Anselmi, E.; Magnier, E. Arylation of perfluoroalkyl vinyl sulfoxides via the Heck reaction. Tetrahedron Lett. 2015, 56, 1259–1262. [Google Scholar] [CrossRef]
- Fuchikami, T.; Yatabe, M.; Ojima, I. A facile synthesis of trans-β-trifluoromethylstyrene, trans-2,3,4,5,6-pentafluorostilbene and their derivatives. Synthesis 1981, 1981, 365–366. [Google Scholar] [CrossRef]
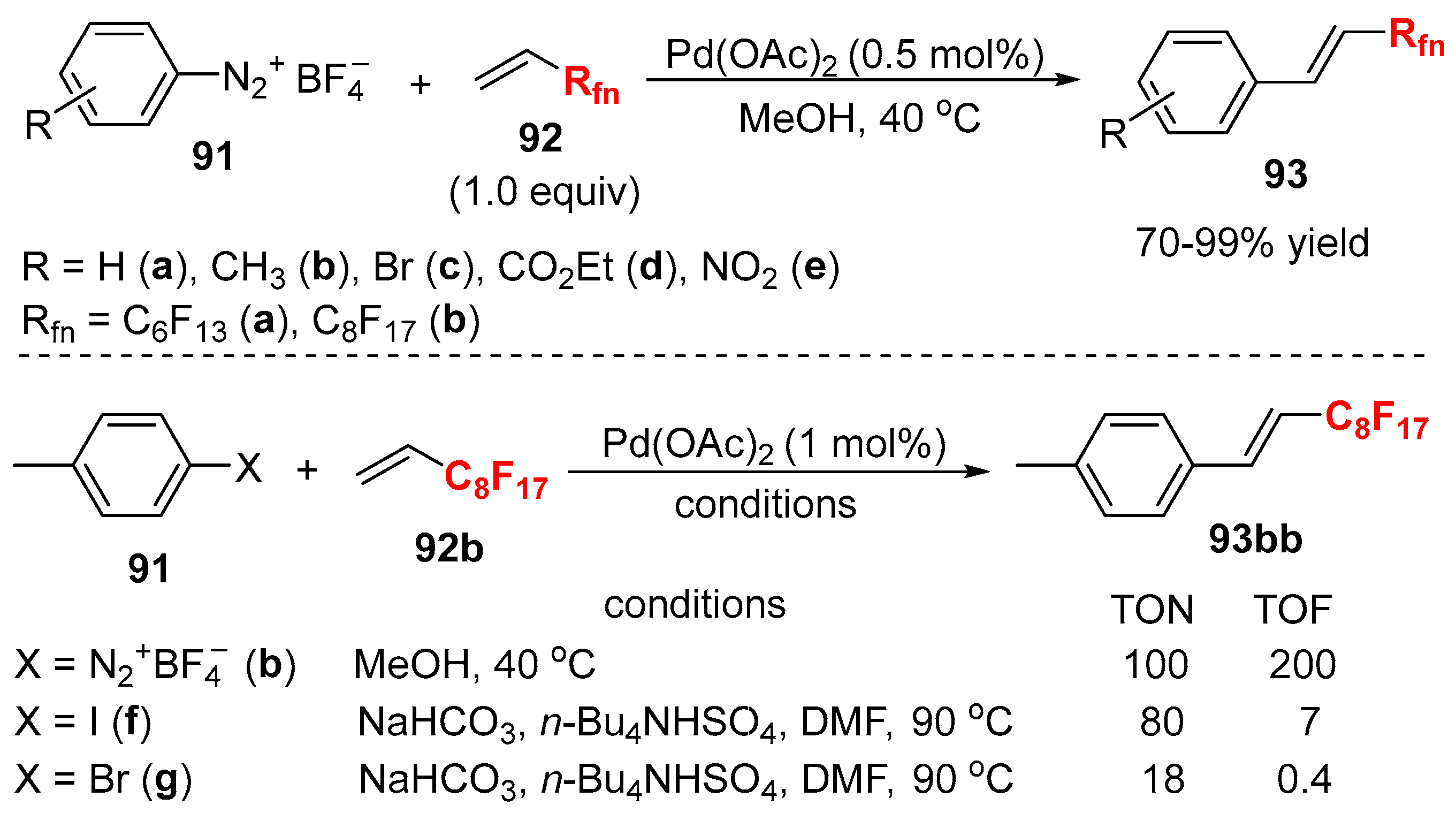
- Chen, W.; Xu, L.; Xiao, J. A general method to fluorous ponytail-substituted aromatics. Tetrahedron Lett. 2001, 42, 4275–4278. [Google Scholar] [CrossRef]
- Birdsall, D.J.; Hope, E.G.; Stuart, A.M.; Chen, W.; Hu, Y.; Xiao, J. Synthesis of fluoroalkyl-derivatised BINAP ligands. Tetrahedron Lett. 2001, 42, 8551–8553. [Google Scholar] [CrossRef]
- Feng, J.; Cai, C. An efficient synthesis of perfluoroalkenylated aryl compounds via pincer-Pd catalyzed Heck couplings. J. Fluor. Chem. 2013, 146, 6–10. [Google Scholar] [CrossRef]
- Su, H.L.; Balogh, J.; Al-Hashimi, M.; Seapy, D.G.; Bazzi, H.S.; Gladysz, J.A. Convenient protocols for Mizoroki-Heck reactions of aromatic bromides and polybromides with fluorous alkenes of the formula H2C=CH(CF2)n−1CF3 (n = 8, 10). Org. Biomol. Chem. 2016, 14, 10058–10069. [Google Scholar] [CrossRef] [PubMed]
- Jian, H.; Tour, J.M. Preparative fluorous mixture synthesis of diazonium-functionalized oligo(phenylenevinylene)s. J. Org. Chem. 2005, 70, 3396–3424. [Google Scholar] [CrossRef] [PubMed]
- Darses, S.; Pucheault, M.; Genêt, J.P. Efficient access to perfluoroalkylated aryl compounds by Heck reaction. Eur. J. Org. Chem. 2001, 2001, 1121–1128. [Google Scholar] [CrossRef]
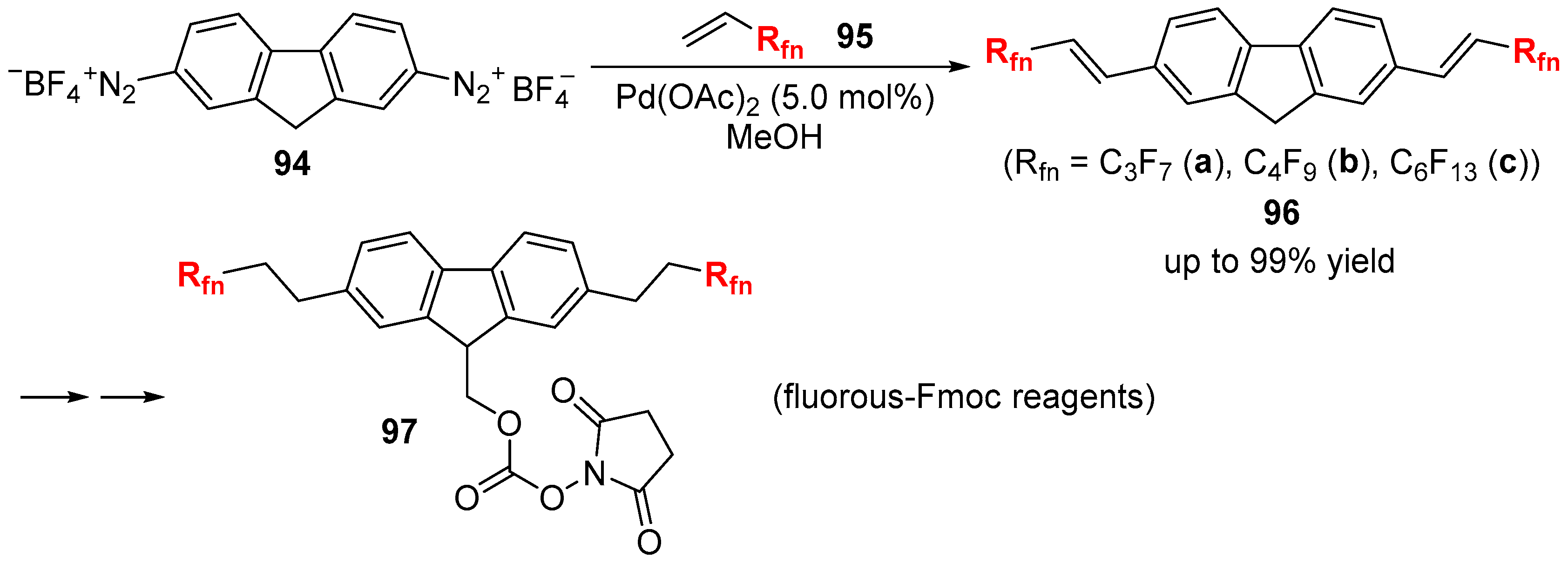
- Sugiyama, Y.; Endo, N.; Ishihara, K.; Kobayashi, Y.; Hamamoto, H.; Shioiri, T.; Matsugi, M. Development of efficient processes for multi-gram scale and divergent preparation of fluorous-Fmoc reagents. Tetrahedron 2015, 71, 4958–4966. [Google Scholar] [CrossRef]
- Sakaguchi, Y.; Yamada, S.; Konno, T.; Agou, T.; Kubota, T. Stereochemically defined various multisubstituted alkenes bearing a tetrafluoroethylene (-CF2CF2-) fragment. J. Org. Chem. 2017, 82, 1618–1631. [Google Scholar] [CrossRef] [PubMed]
- Konno, T.; Yamada, S.; Tani, A.; Miyabe, T.; Ishihara, T. A novel synthesis of trifluoromethylated multi-substituted alkenes via regio- and stereoselective heck reaction of (E)-4,4,4-trifluoro-1-phenyl-2-buten-1-one. Synlett 2006, 2006, 3025–3028. [Google Scholar] [CrossRef]
- Konno, T.; Yamada, S.; Tani, A.; Nishida, M.; Miyabe, T.; Ishihara, T. Unexpected high regiocontrol in Heck reaction of fluorine-containing electron-deficient olefins—Highly regio- and stereoselective synthesis of β-fluoroalkyl-α-aryl-α,β-unsaturated ketones. J. Fluor. Chem. 2009, 130, 913–921. [Google Scholar] [CrossRef]
- Li, Y.; Tu, D.-H.; Gu, Y.-J.; Wang, B.; Wang, Y.-Y.; Liu, Z.-T.; Liu, Z.-W.; Lu, J. Oxidative Heck reaction of fluorinated olefins with arylboronic acids by palladium catalysis. Eur. J. Org. Chem. 2015, 2015, 4340–4343. [Google Scholar] [CrossRef]
- Ichikawa, J.; Nadano, R.; Ito, N. 5-Endo Heck-type cyclization of 2-(trifluoromethyl)allyl ketone oximes: Synthesis of 4-difluoromethylene-substituted 1-pyrrolines. Chem. Commun. 2006, 4425–4427. [Google Scholar] [CrossRef] [PubMed]
- Renak, M.L.; Bartholomew, G.P.; Wang, S.; Ricatto, P.J.; Lachicotte, R.J.; Bazan, G.C. Fluorinated distyrylbenzene chromophores: Effect of fluorine-regiochemistry on molecular properties and solid-state organization. J. Am. Chem. Soc. 1999, 121, 7787–7799. [Google Scholar] [CrossRef]
- Bernier, D.; Brückner, R. Novel synthesis of naturally occurring pulvinones: A Heck coupling, transesterification, and dieckmann condensation strategy. Synthesis 2007, 2249–2272. [Google Scholar] [CrossRef]
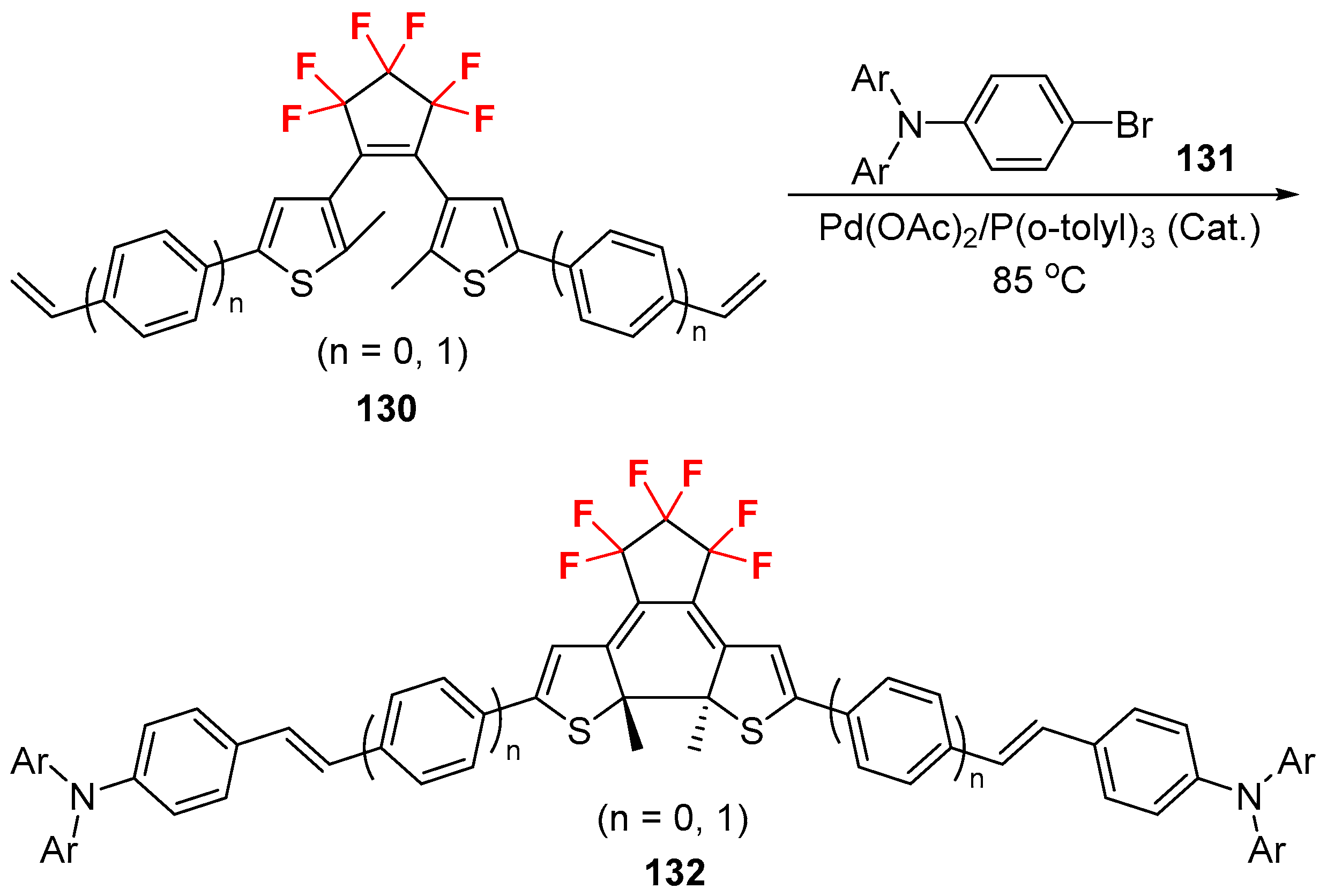
- Fan, C.; Yang, P.; Wang, X.; Liu, G.; Jiang, X.; Chen, H.; Tao, X.; Wang, M.; Jiang, M. Synthesis and organic photovoltaic (OPV) properties of triphenylamine derivatives based on a hexafluorocyclopentene “core”. Sol. Energy Mater. Sol. Cells 2011, 95, 992–1000. [Google Scholar] [CrossRef]
- Salabert, J.; Sebastián, R.M.; Vallribera, A.; Roglans, A.; Nájera, C. Fluorous aryl compounds by Matsuda-Heck reaction. Tetrahedron 2011, 67, 8659–8664. [Google Scholar] [CrossRef]
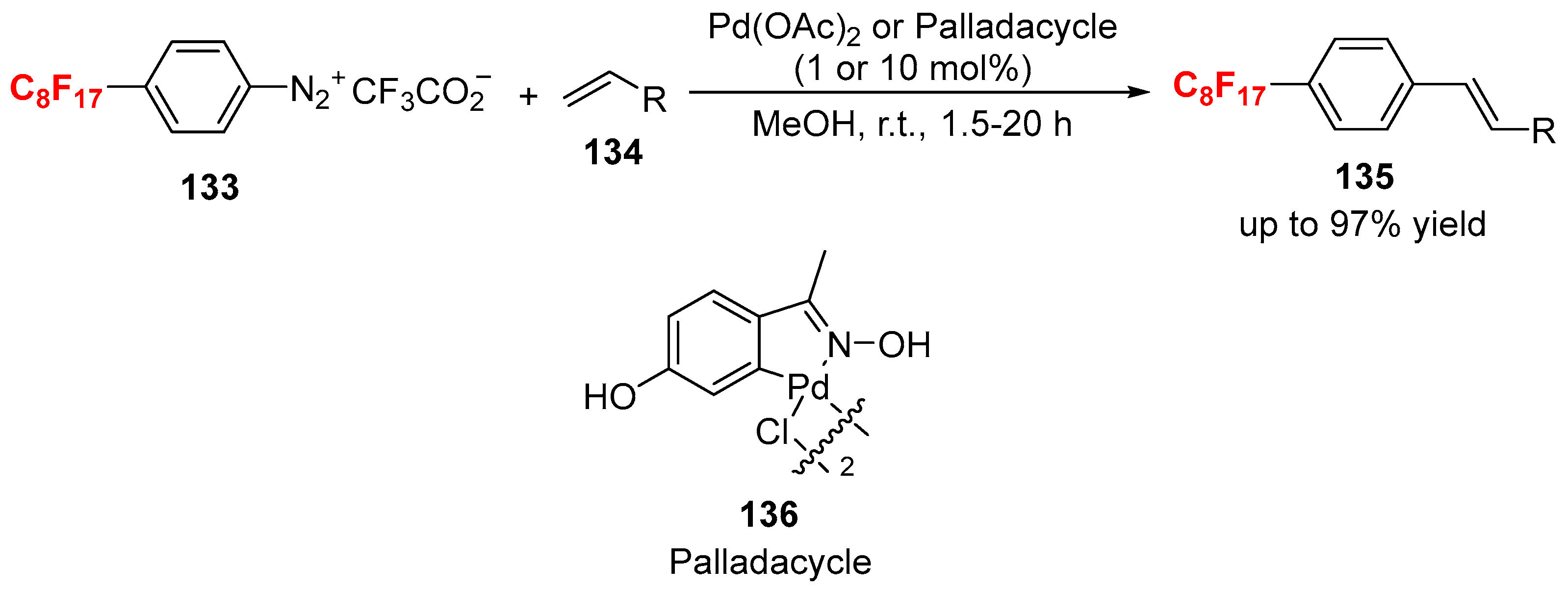
- Okazaki, T.; Laali, K.K.; Bunge, S.D.; Adas, S.K. 4-(Pentafluorosulfanyl)benzenediazonium tetrafluoroborate: A versatile launch pad for the synthesis of aromatic SF5 compounds via cross coupling, azo coupling, homocoupling, dediazoniation, and click chemistry. Eur. J. Org. Chem. 2014, 1630–1644. [Google Scholar] [CrossRef]
- Albéniz, A.C.; Espinet, P.; Martínruiz, B.; Milstein, D. Catalytic system for Heck reactions involving insertion into Pd-(perfluoro-organyl) bonds. J. Am. Chem. Soc. 2001, 123, 11504–11505. [Google Scholar] [CrossRef] [PubMed]
- Albeniz, A.C.; Espinet, P.; Martin-Ruiz, B.; Milstein, D. Catalytic system for the Heck reaction of fluorinated haloaryls. Organometallics 2005, 24, 3679–3684. [Google Scholar] [CrossRef]
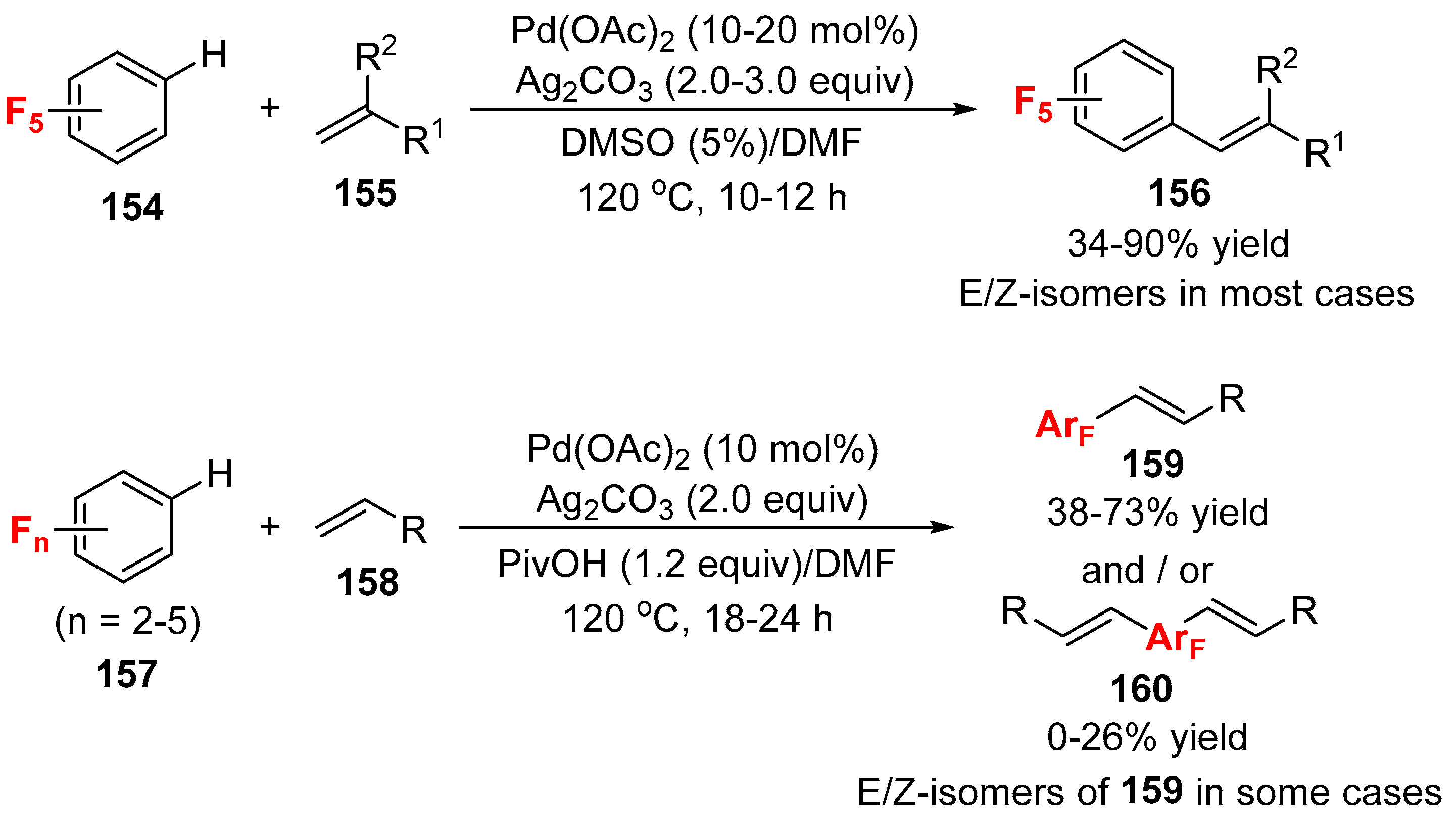
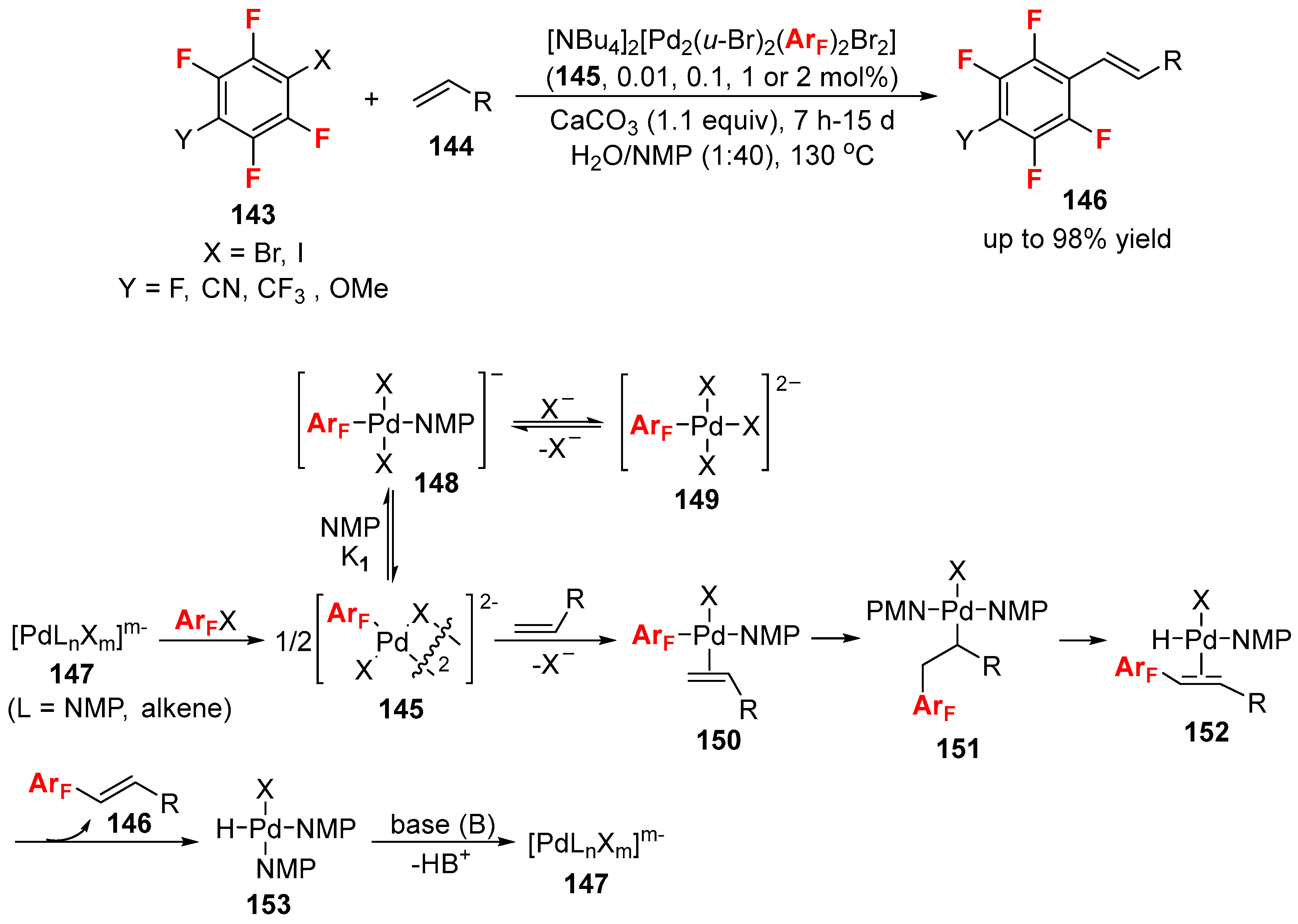
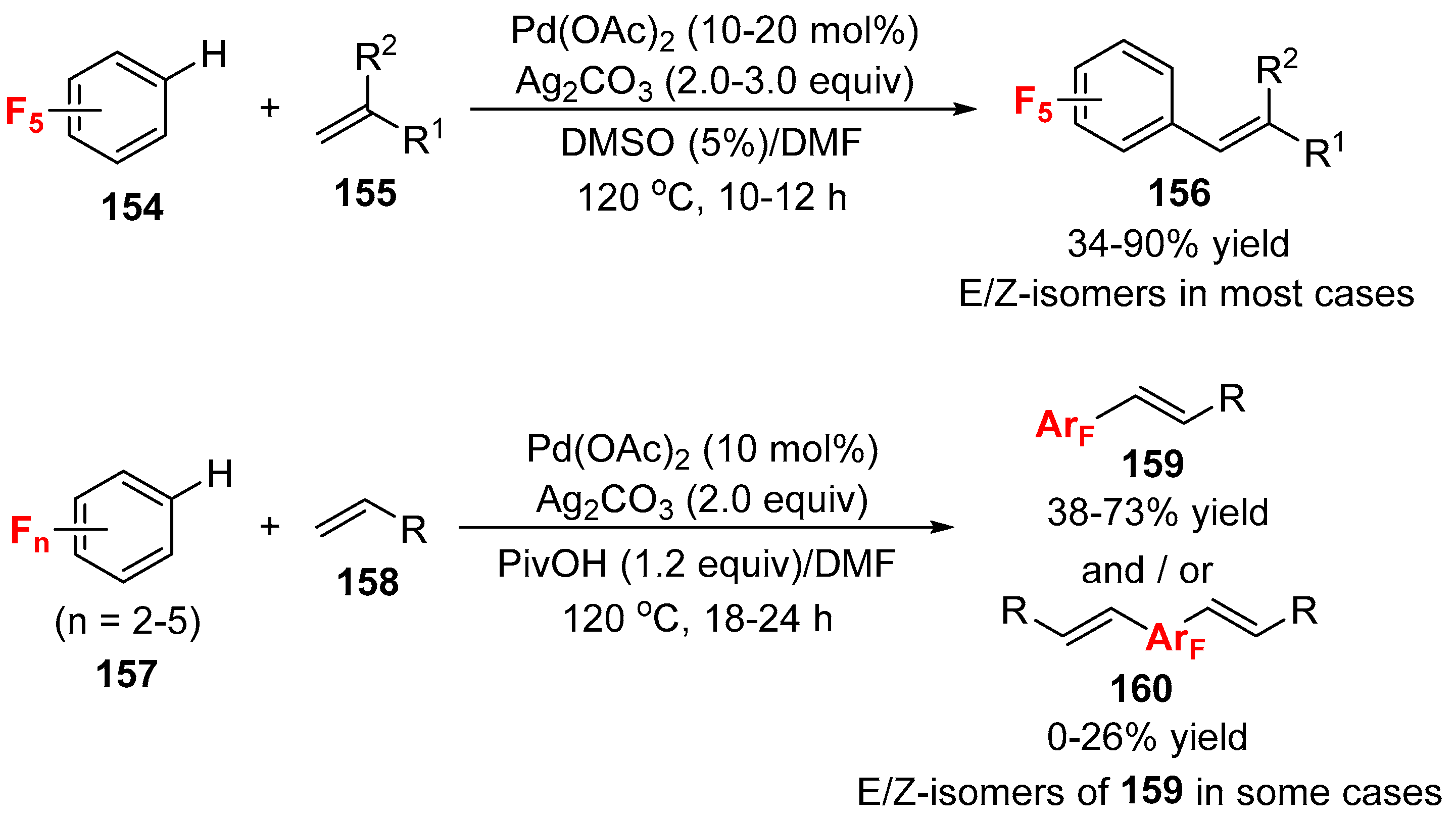
- Zhang, X.; Fan, S.; He, C.Y.; Wan, X.; Min, Q.Q.; Yang, J.; Jiang, Z.X. Pd(OAc)2 catalyzed olefination of highly electron-deficient perfluoroarenes. J. Am. Chem. Soc. 2010, 132, 4506–4507. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Z.; He, C.Y.; Huang, Y.; Zhang, X. Thioether-promoted direct olefination of polyfluoroarenes catalyzed by palladium. Org. Lett. 2013, 15, 5266–5269. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.-L.; Zhang, B.; He, C.-Y.; Zhang, X. Direct olefination of fluorinated benzothiadiazoles: A new entry to optoelectronic materials. Chem. Eur. J. 2014, 20, 4532–4536. [Google Scholar] [CrossRef] [PubMed]
- He, C.-Y.; Qing, F.-L.; Zhang, X. Pd-catalyzed aerobic direct olefination of polyfluoroarenes. Tetrahedron Lett. 2014, 55, 2962–2964. [Google Scholar] [CrossRef]
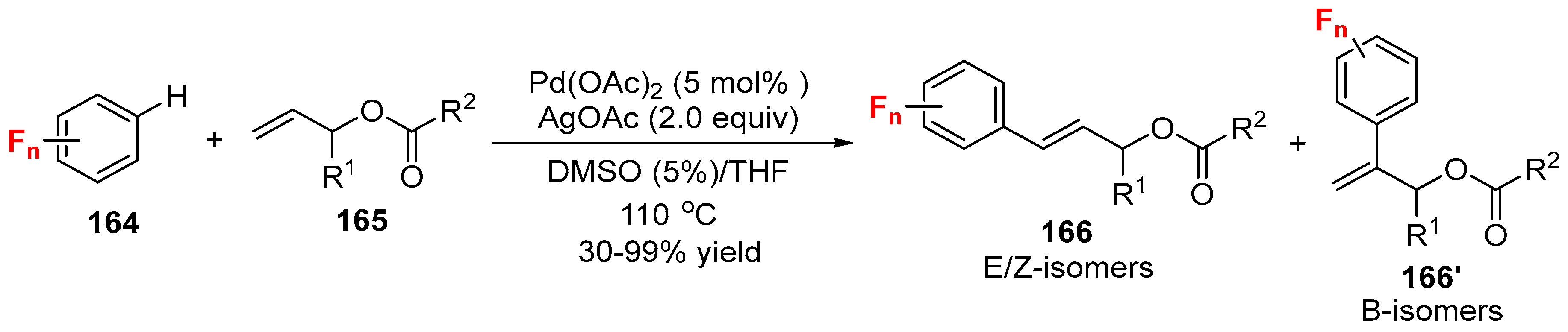
- Li, Z.; Zhang, Y.; Liu, Z.-Q. Pd-catalyzed olefination of perfluoroarenes with allyl esters. Org. Lett. 2012, 14, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-M.; Song, H.-X.; Wang, X.-Y.; Liu, N.; Qin, H.-L.; Zhang, C.-P. Palladium-catalyzed Mizoroki-Heck-type reactions of [Ph2SRfn][OTf] with alkenes at room temperature. Chem. Commun. 2016, 52, 11893–11896. [Google Scholar] [CrossRef] [PubMed]
- Dolci, L.; Dolle, F.; Valette, H.; Vaufrey, F.; Fuseau, C.; Bottlaender, M.; Crouzel, C. Synthesis of a fluorine-18 labeled derivative of epibatidine for in vivo nicotinic acetylcholine receptor PET imaging. Bioorg. Med. Chem. 1999, 7, 467–479. [Google Scholar] [CrossRef]
- Quandt, G.; Höfner, G.; Wanner, K.T. Synthesis and evaluation of N-substituted nipecotic acid derivatives with an unsymmetrical bis-aromatic residue attached to a vinyl ether spacer as potential GABA uptake inhibitors. Bioorg. Med. Chem. 2013, 21, 3363–3378. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Ahmed, S.; McLaughlin, L.W. Syntheses of pyridine C-nucleosides as analogues of the natural nucleosides dC and dU. J. Org. Chem. 2006, 71, 2922–2925. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-D.; Chow, T.J. Fluorine substituent effect on organic dyes for sensitized solar cells. J. Photochem. Photobiol. A Chem. 2012, 230, 47–54. [Google Scholar] [CrossRef]
- Belfield, K.D.; Najjar, O.; Sriram, S.R. Synthesis of polyurethanes and polyimides for photorefractive applications. Polymer 2000, 41, 5011–5020. [Google Scholar] [CrossRef]
- Belfield, K.D.; Chinna, C.; Najjar, O.; Sriram, S.; Schafer, K.J. Methodology for the synthesis of new multifunctional polymers for photorefractive applications. ACS Symp. Ser. 1999, 726, 250–263. [Google Scholar]
- Surapanich, N.; Kuhakarn, C.; Pohmakotr, M.; Reutrakul, V. Palladium-mediated Heck-type reactions of [(bromodifluoromethyl)sulfonyl]benzene: Synthesis of α-alkenyl- and α-heteroaryl-substituted α,α-difluoromethyl phenyl sulfones. Eur. J. Org. Chem. 2012, 2012, 5943–5952. [Google Scholar] [CrossRef]
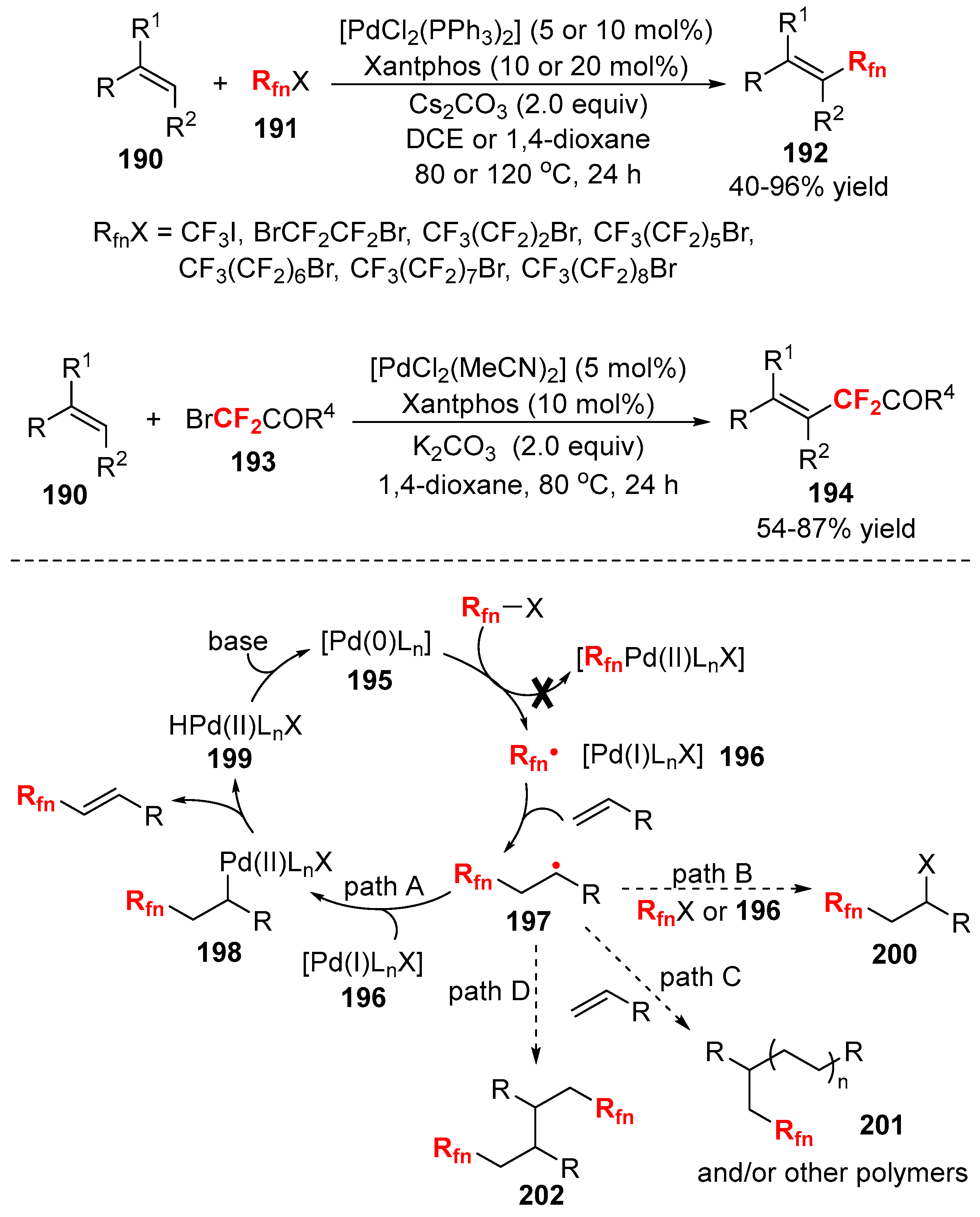
- Feng, Z.; Min, Q.Q.; Zhao, H.Y.; Gu, J.W.; Zhang, X. A general synthesis of fluoroalkylated alkenes by palladium-catalyzed Heck-type reaction of fluoroalkyl bromides. Angew. Chem. Int. Ed. 2015, 54, 1270–1274. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.-Y.; Yang, Z.-Y.; Zhao, C.-X.; Qiu, Z.-M. Studies on fluoroalkylation and fluoroalkoxylation. Part 28. Palladium(0)-induced addition of fluoroalkyl iodides to alkenes: An electron transfer process. J. Chem. Soc. Perkin Trans. 1 1988, 0, 563–567. [Google Scholar] [CrossRef]
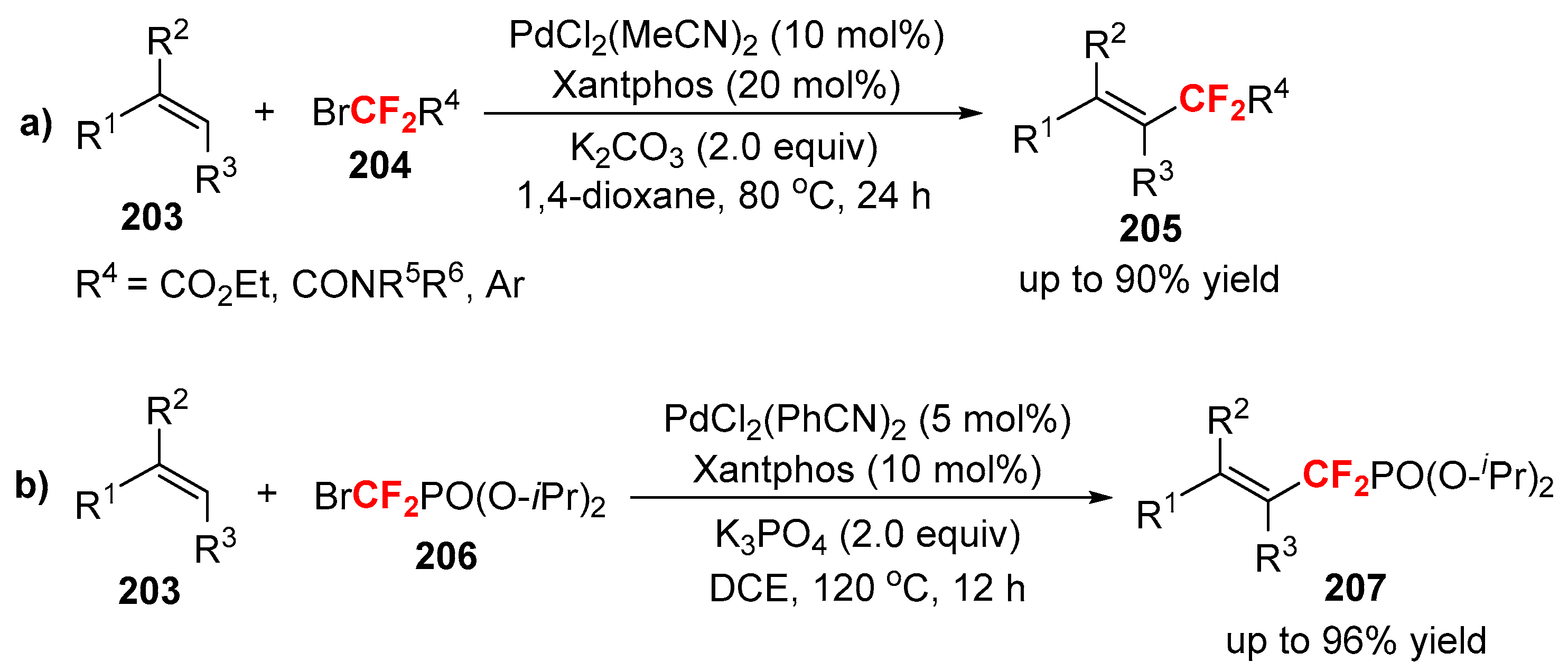
- Zhang, X.; Zhang, F.; Min, Q.-Q. Palladium-catalyzed Heck-type difluoroalkylation of alkenes with functionalized difluoromethyl bromides. Synthesis 2015, 47, 2912–2923. [Google Scholar] [CrossRef]
- Feng, Z.; Xiao, Y.-L.; Zhang, X. Palladium-catalyzed phosphonyldifluoromethylation of alkenes with bromodifluoromethylphosphonate. Org. Chem. Front. 2016, 3, 466–469. [Google Scholar] [CrossRef]
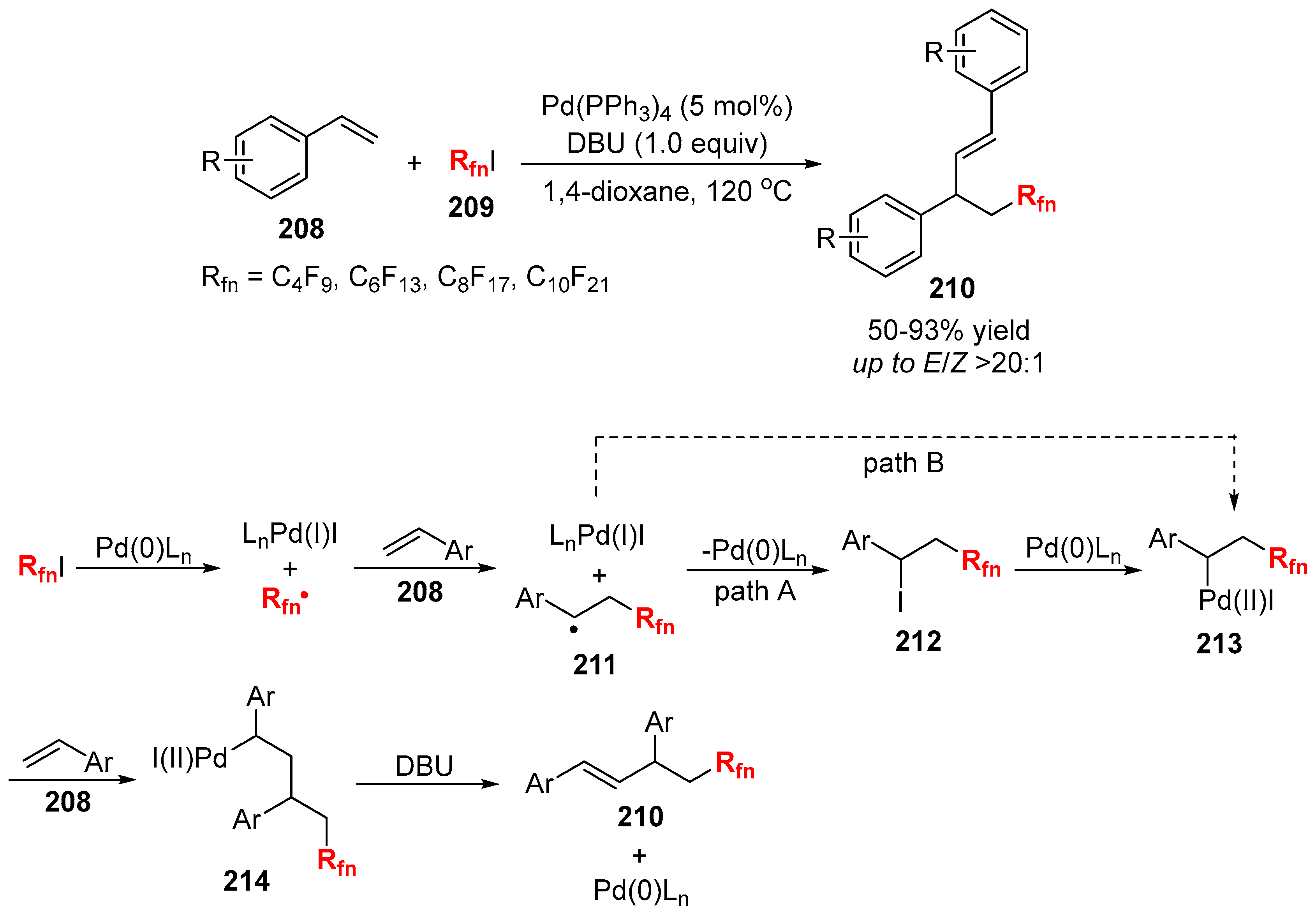
- Ai, H.-J.; Cai, C.-X.; Qi, X.; Peng, J.-B.; Zheng, F.; Wu, X.-F. Palladium-catalyzed Heck reaction of in-situ generated benzylic iodides and styrenes. Tetrahedron Lett. 2017, 58, 3846–3850. [Google Scholar] [CrossRef]
- Fan, T.; Meng, W.-D.; Zhang, X. Palladium-catalyzed Heck-type reaction of secondary trifluoromethylated alkyl bromides. Beilstein J. Org. Chem. 2017, 13, 2610–2616. [Google Scholar] [CrossRef] [PubMed]
- Dubbaka, S.R.; Vogel, P. Palladium-catalyzed desulfitative mizoroki-heck couplings of sulfonyl chlorides with mono- and disubstituted olefins: Rhodium-catalyzed desulfitative Heck-type reactions under phosphine- and base-free conditions. Chem. Eur. J. 2005, 11, 2633–2641. [Google Scholar] [CrossRef] [PubMed]
- Prakash, G.K.S.; Krishnan, H.S.; Jog, P.V.; Iyer, A.P.; Olah, G.A. A domino approach of Heck coupling for the synthesis of β-trifluoromethylstyrenes. Org. Lett. 2012, 14, 1146–1149. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Fan, L.; Guo, W.; Zhang, Z.; Li, J.; Zhu, C.; Ren, Y.; Wu, W.; Jiang, H. Palladium-catalyzed fluoroalkylative cyclization of olefins. Org. Lett. 2017, 19, 1008–1011. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-X.; Chen, W.-L.; Shen, Y.; Chen, S.; Xu, P.-F.; Liang, Y.-M. Palladium-catalyzed domino Heck/intermolecular C–H bond functionalization: Efficient synthesis of alkylated polyfluoroarene derivatives. Org. Lett. 2016, 18, 1784–1787. [Google Scholar] [CrossRef] [PubMed]
- Talbot, E.P.A.; Fernandes, T.d.A.; McKenna, J.M.; Toste, F.D. Asymmetric palladium-catalyzed directed intermolecular fluoroarylation of styrenes. J. Am. Chem. Soc. 2014, 136, 4101–4104. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Yang, Z.; Thornbury, R.T.; Toste, F.D. Palladium-catalyzed enantioselective 1,1-fluoroarylation of aminoalkenes. J. Am. Chem. Soc. 2015, 137, 12207–12210. [Google Scholar] [CrossRef] [PubMed]
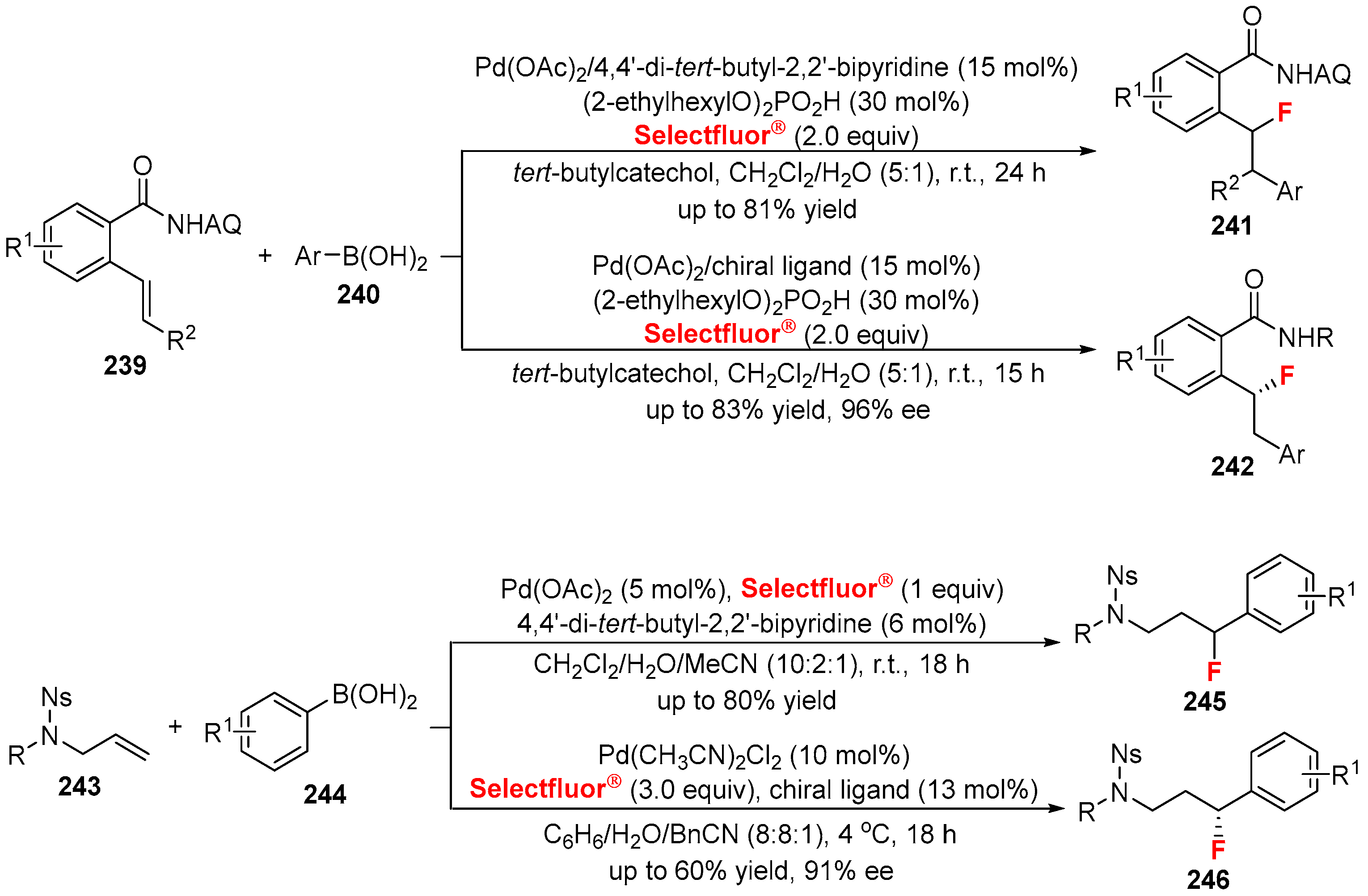
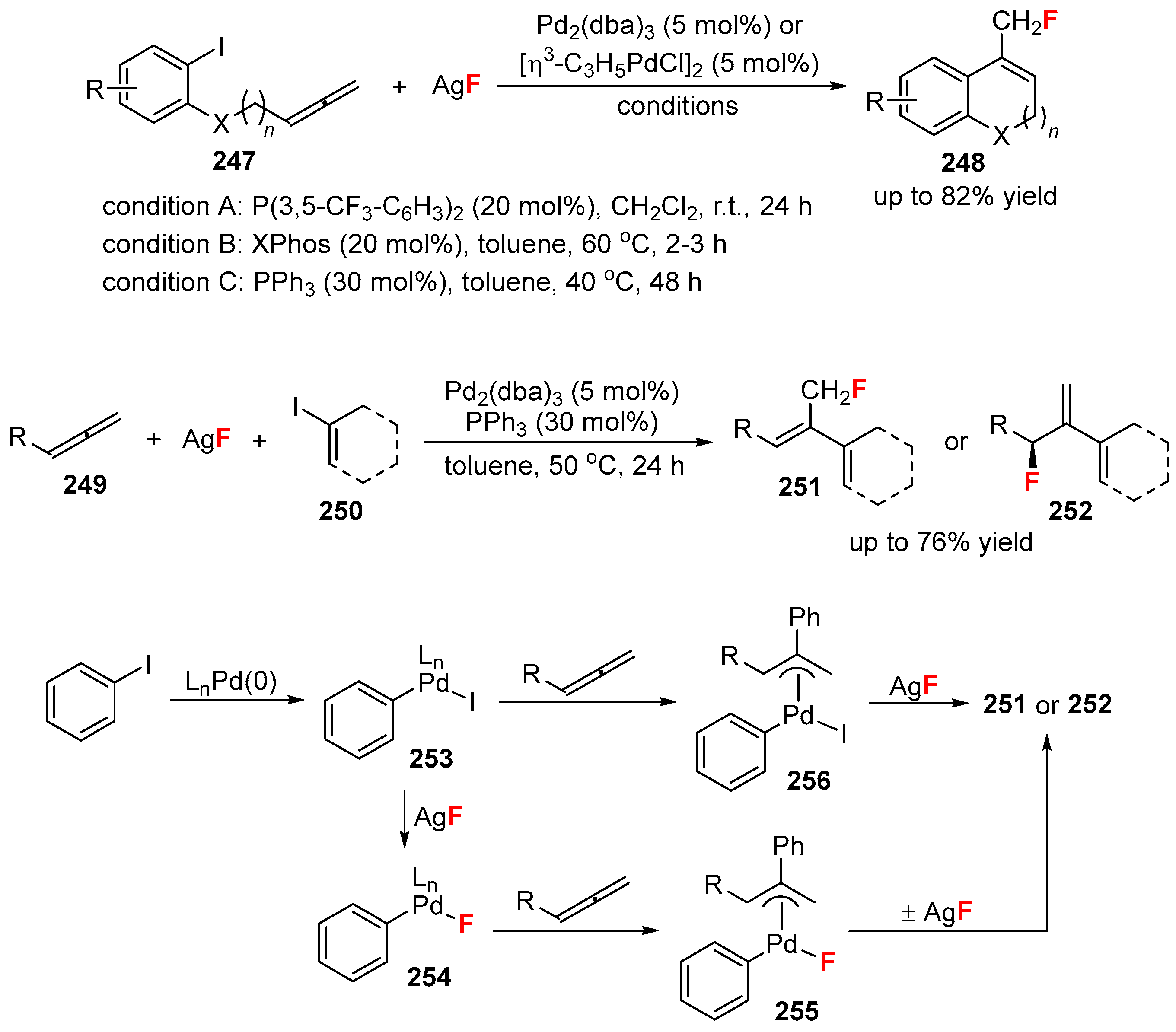
- Braun, M.-G.; Katcher, M.H.; Doyle, A.G. Carbofluorination via a palladium-catalyzed cascade reaction. Chem. Sci. 2013, 4, 1216–1220. [Google Scholar] [CrossRef]
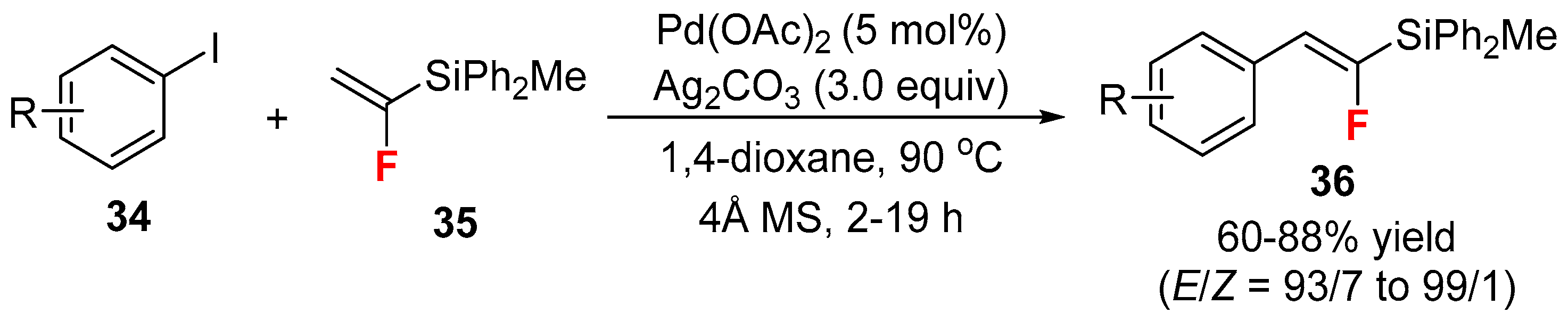
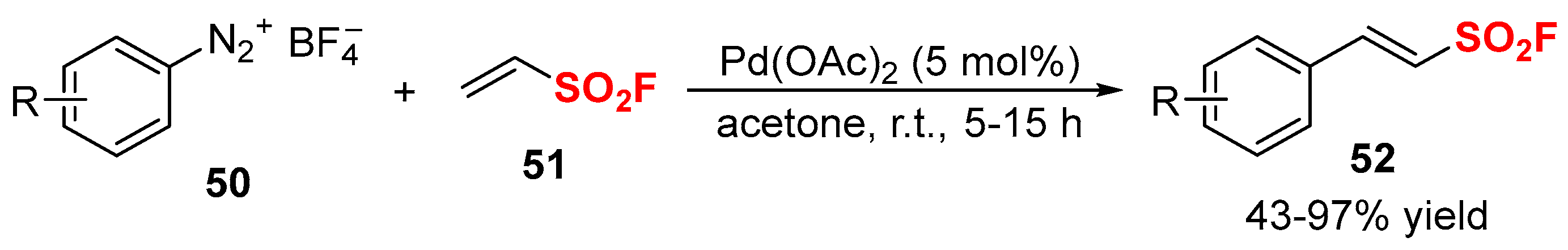

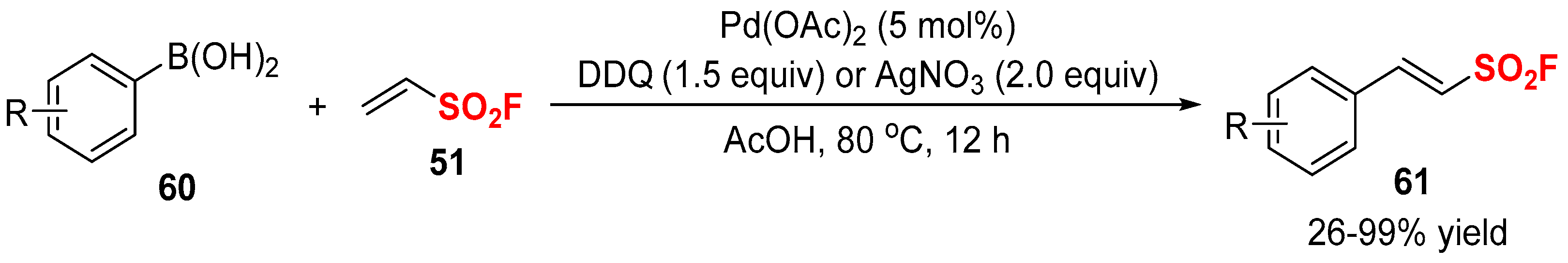
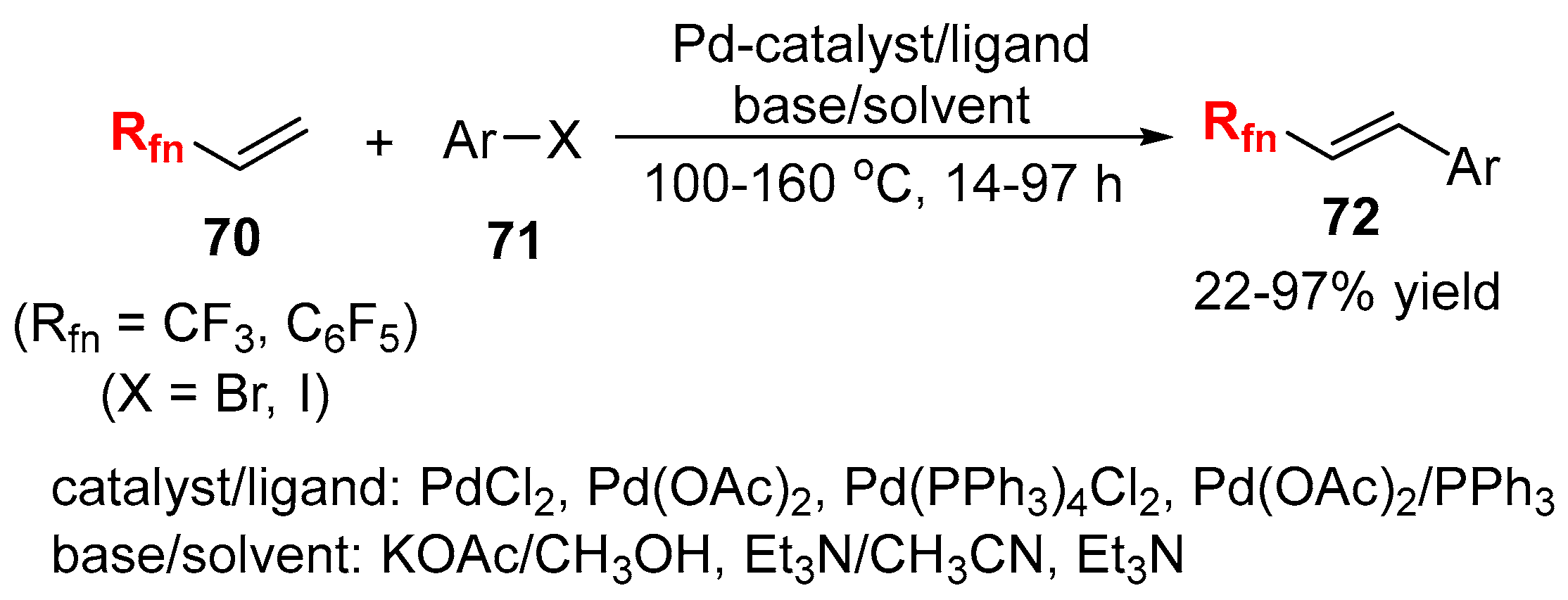
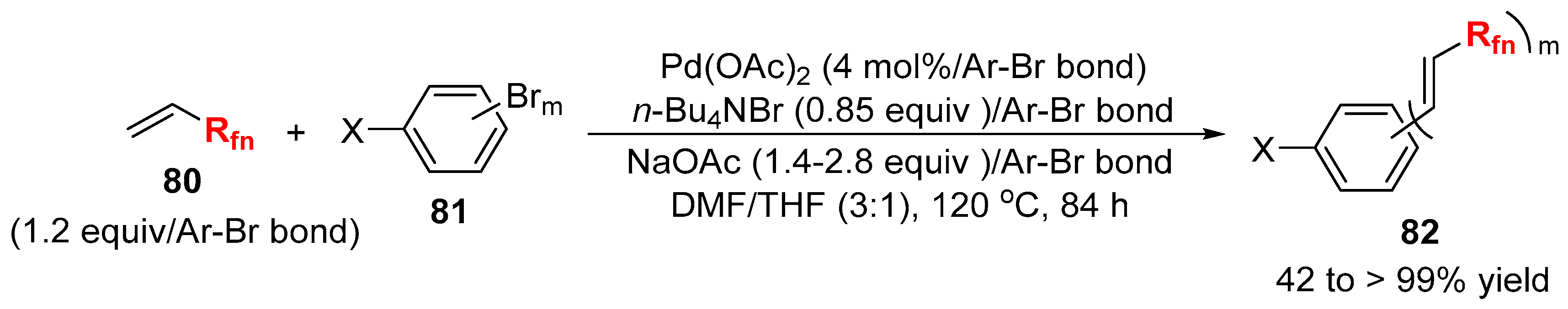
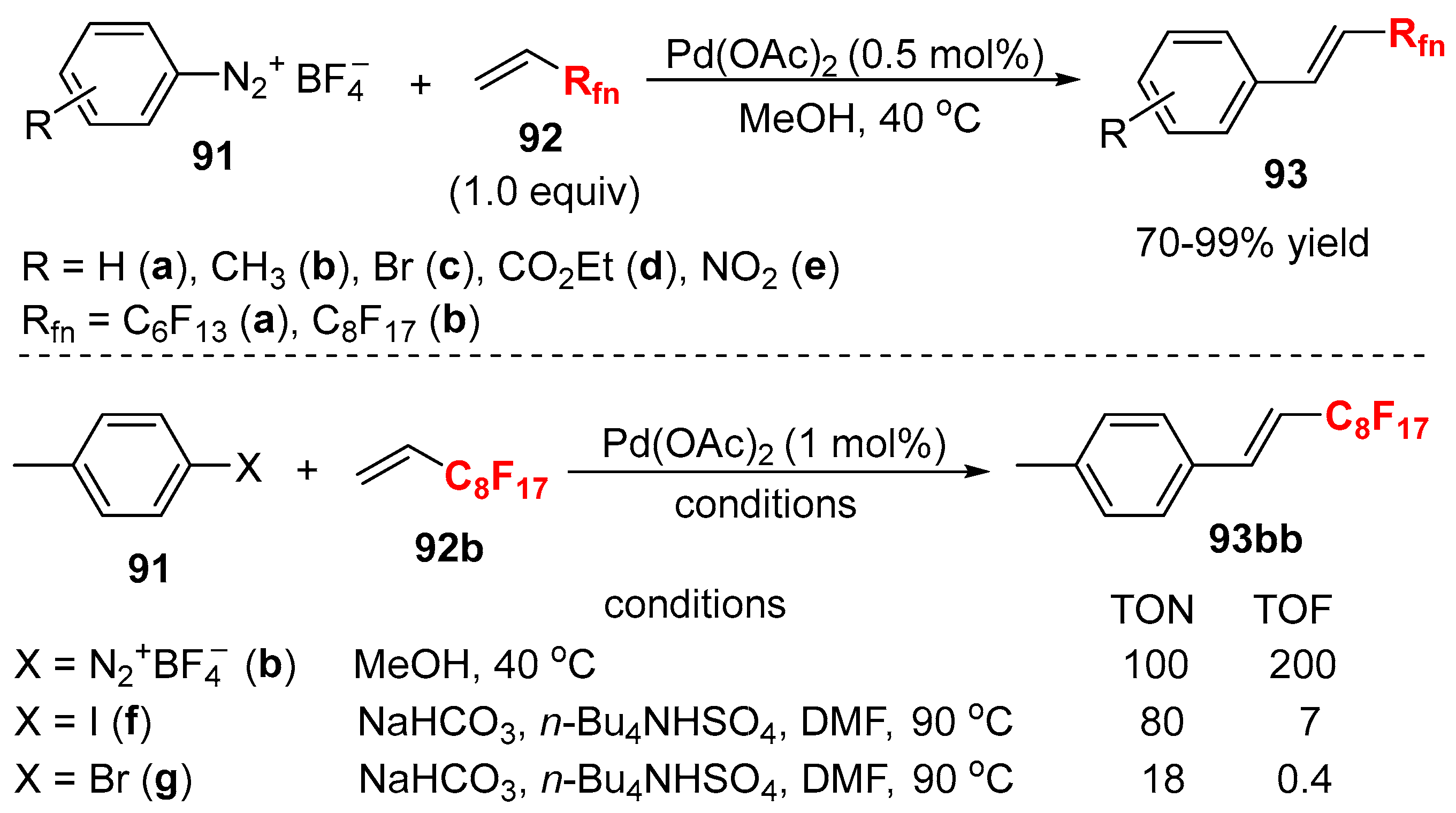
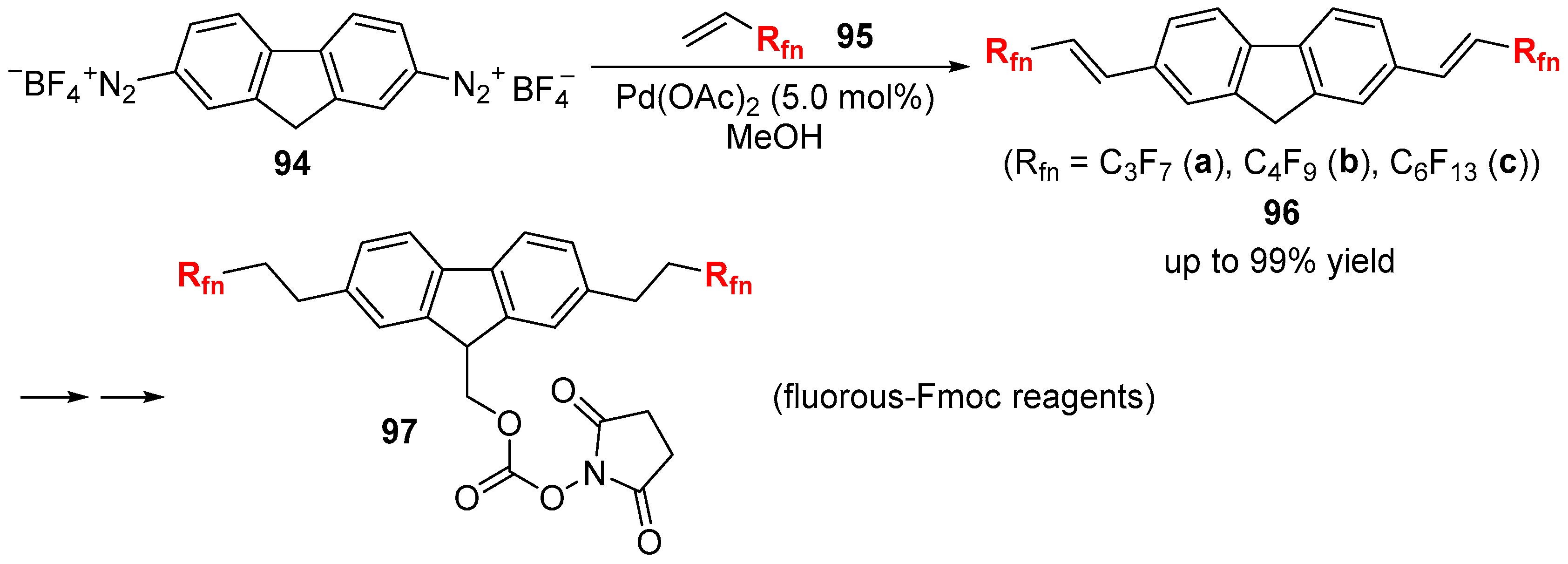
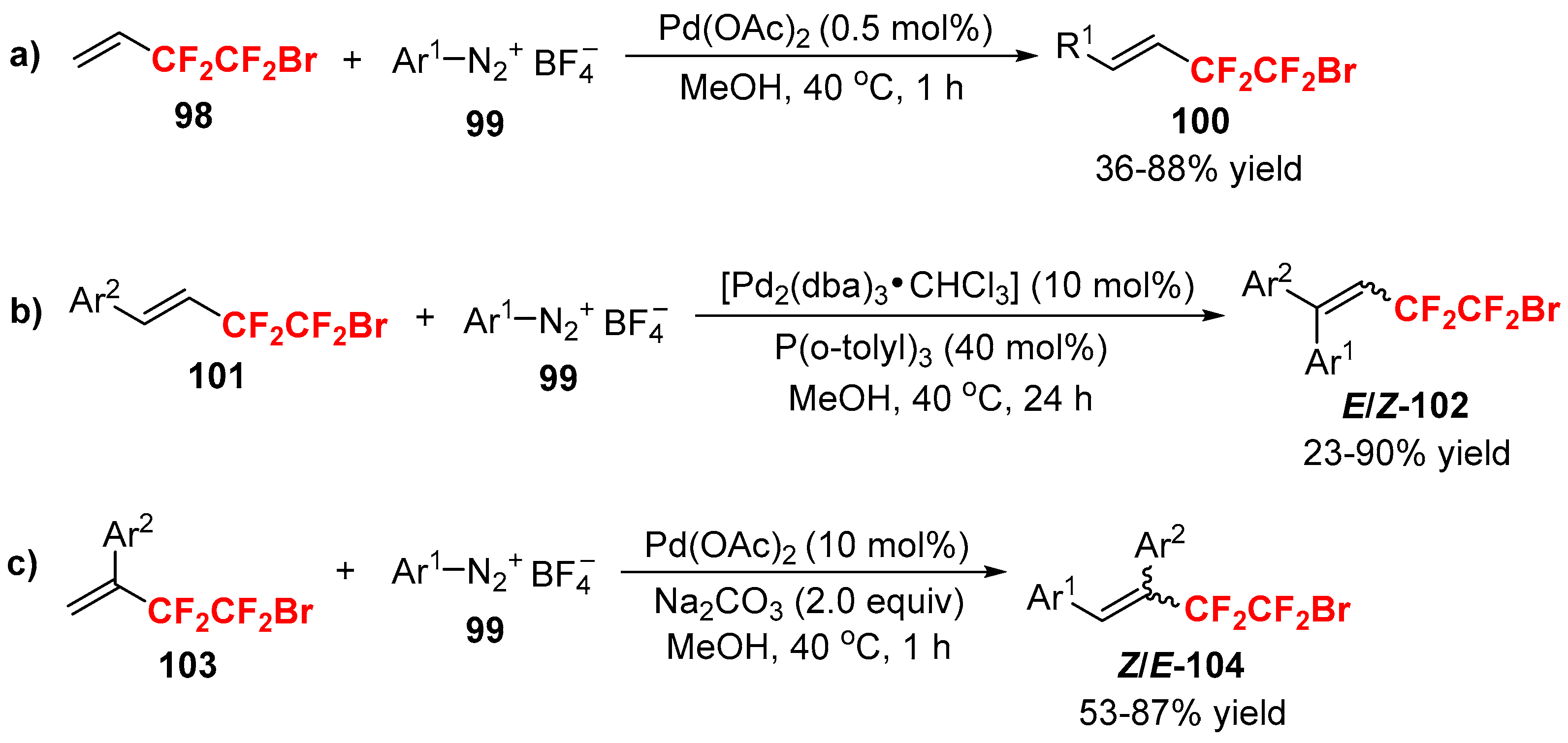
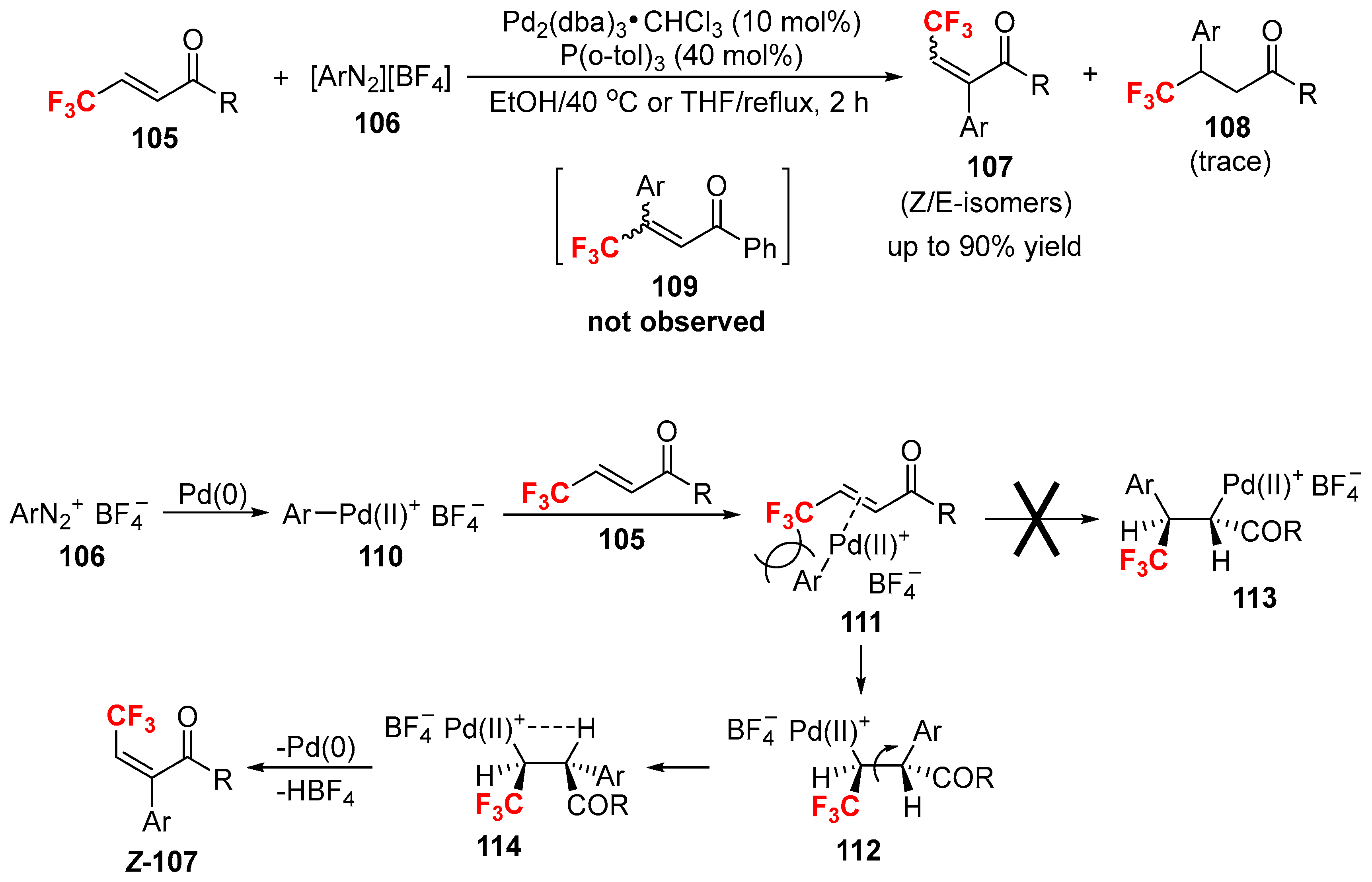
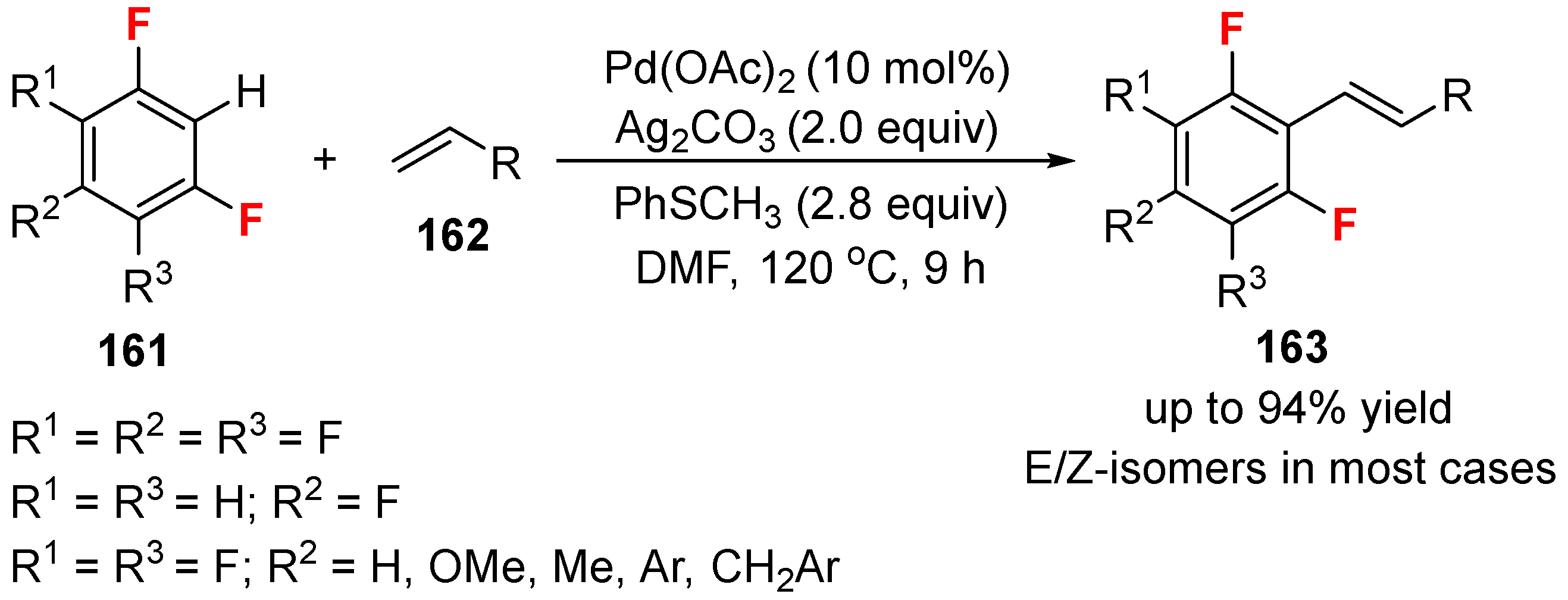
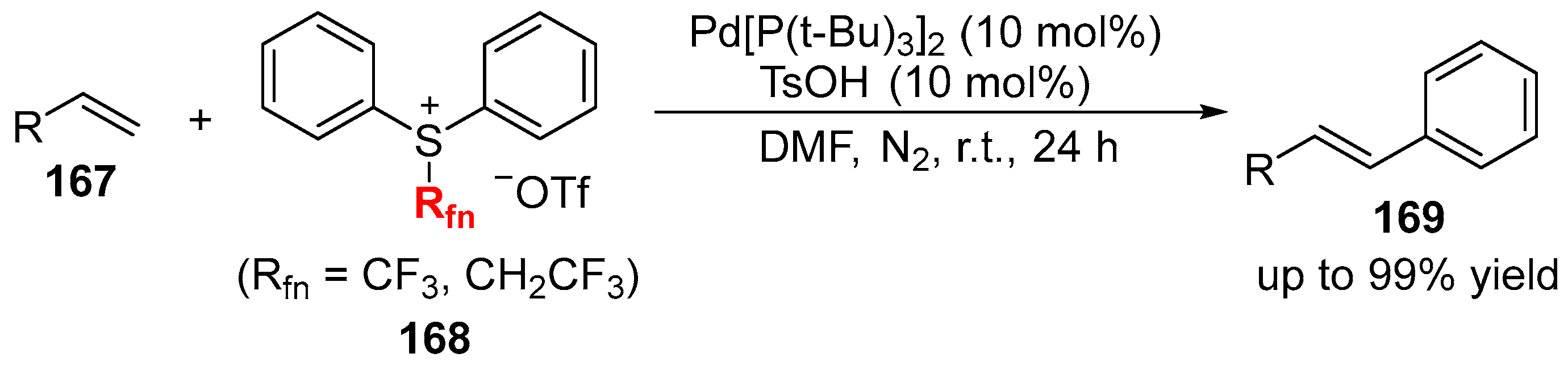

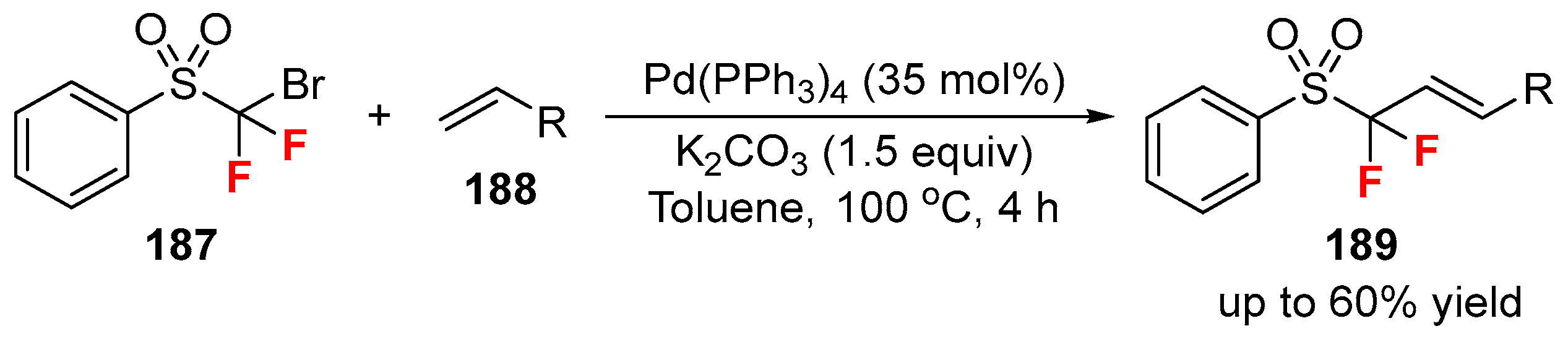

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Zhao, H.-W.; He, J.; Zhang, C.-P. Pd-Catalyzed Mizoroki-Heck Reactions Using Fluorine-Containing Agents as the Cross-Coupling Partners. Catalysts 2018, 8, 23. https://doi.org/10.3390/catal8010023
Yang J, Zhao H-W, He J, Zhang C-P. Pd-Catalyzed Mizoroki-Heck Reactions Using Fluorine-Containing Agents as the Cross-Coupling Partners. Catalysts. 2018; 8(1):23. https://doi.org/10.3390/catal8010023
Chicago/Turabian StyleYang, Jing, Hua-Wen Zhao, Jian He, and Cheng-Pan Zhang. 2018. "Pd-Catalyzed Mizoroki-Heck Reactions Using Fluorine-Containing Agents as the Cross-Coupling Partners" Catalysts 8, no. 1: 23. https://doi.org/10.3390/catal8010023
APA StyleYang, J., Zhao, H.-W., He, J., & Zhang, C.-P. (2018). Pd-Catalyzed Mizoroki-Heck Reactions Using Fluorine-Containing Agents as the Cross-Coupling Partners. Catalysts, 8(1), 23. https://doi.org/10.3390/catal8010023