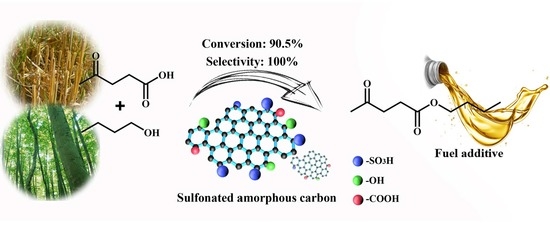
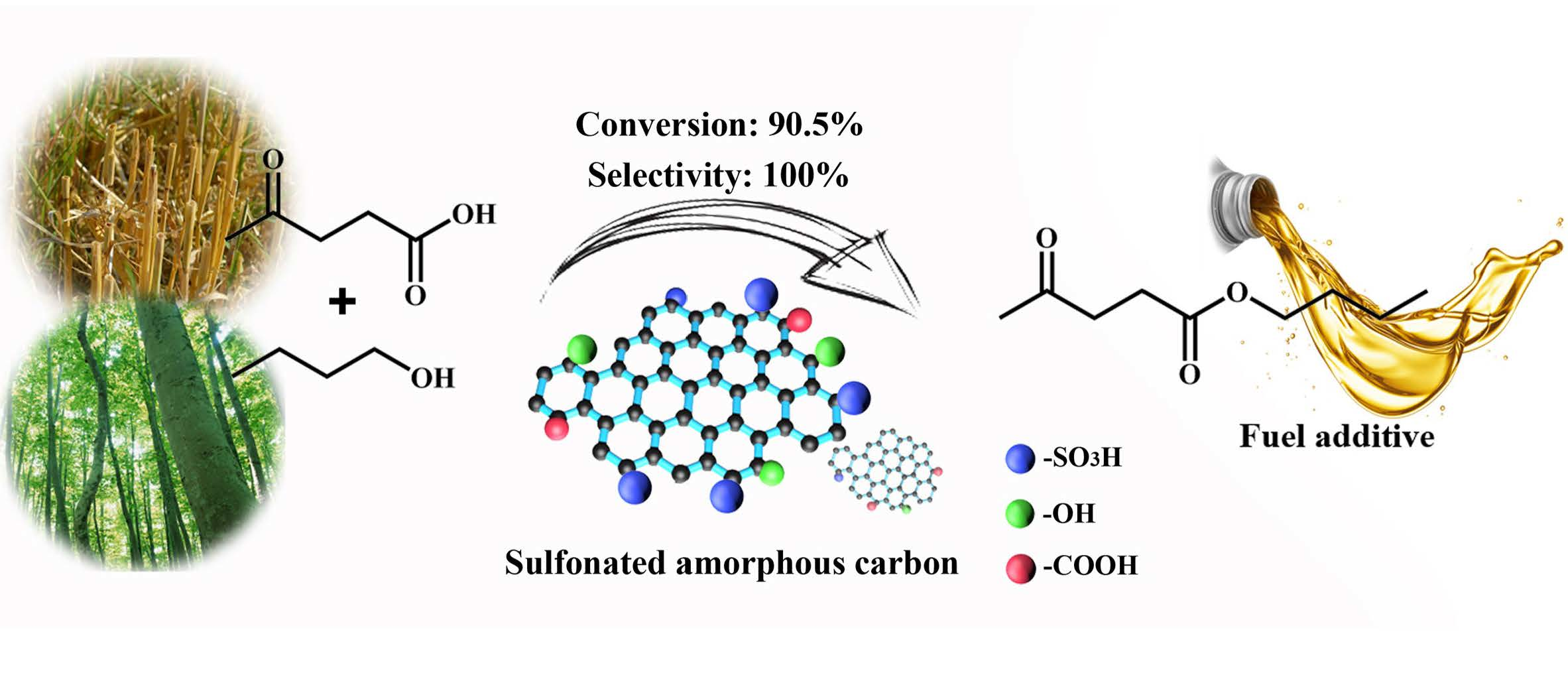
Efficient Production of N-Butyl Levulinate Fuel Additive from Levulinic Acid Using Amorphous Carbon Enriched with Oxygenated Groups

Abstract
:
1. Introduction
2. Results and Discussion
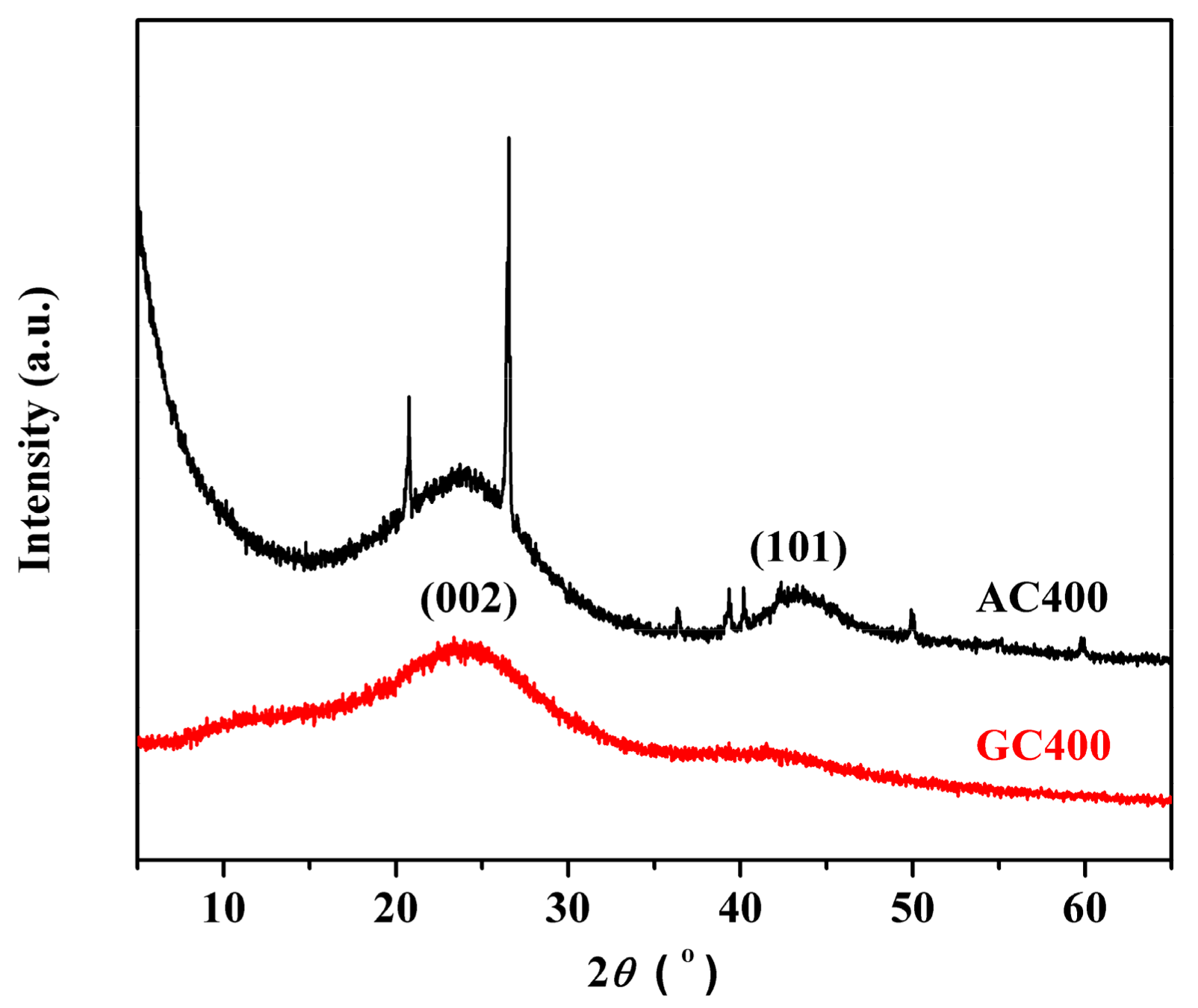
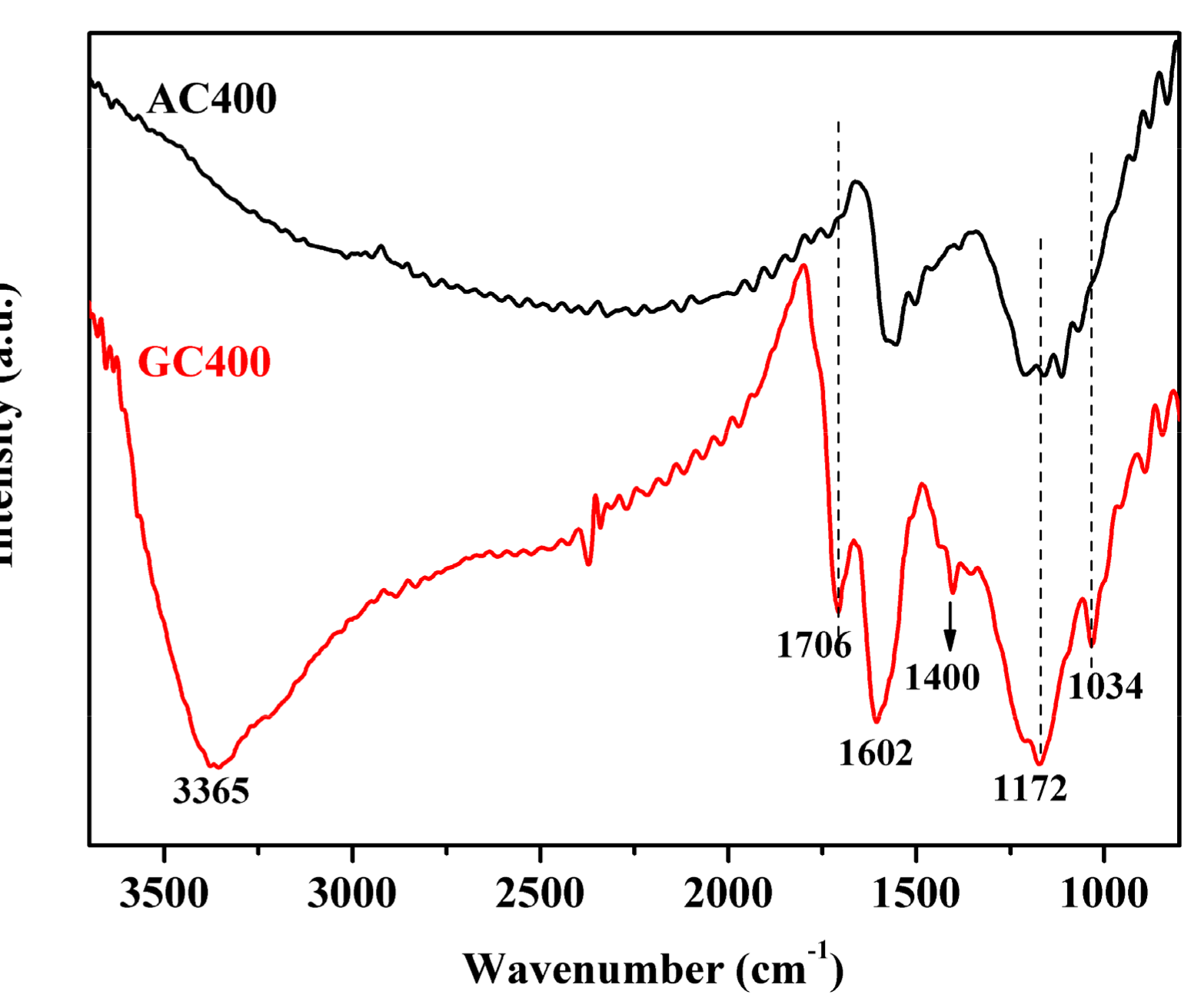
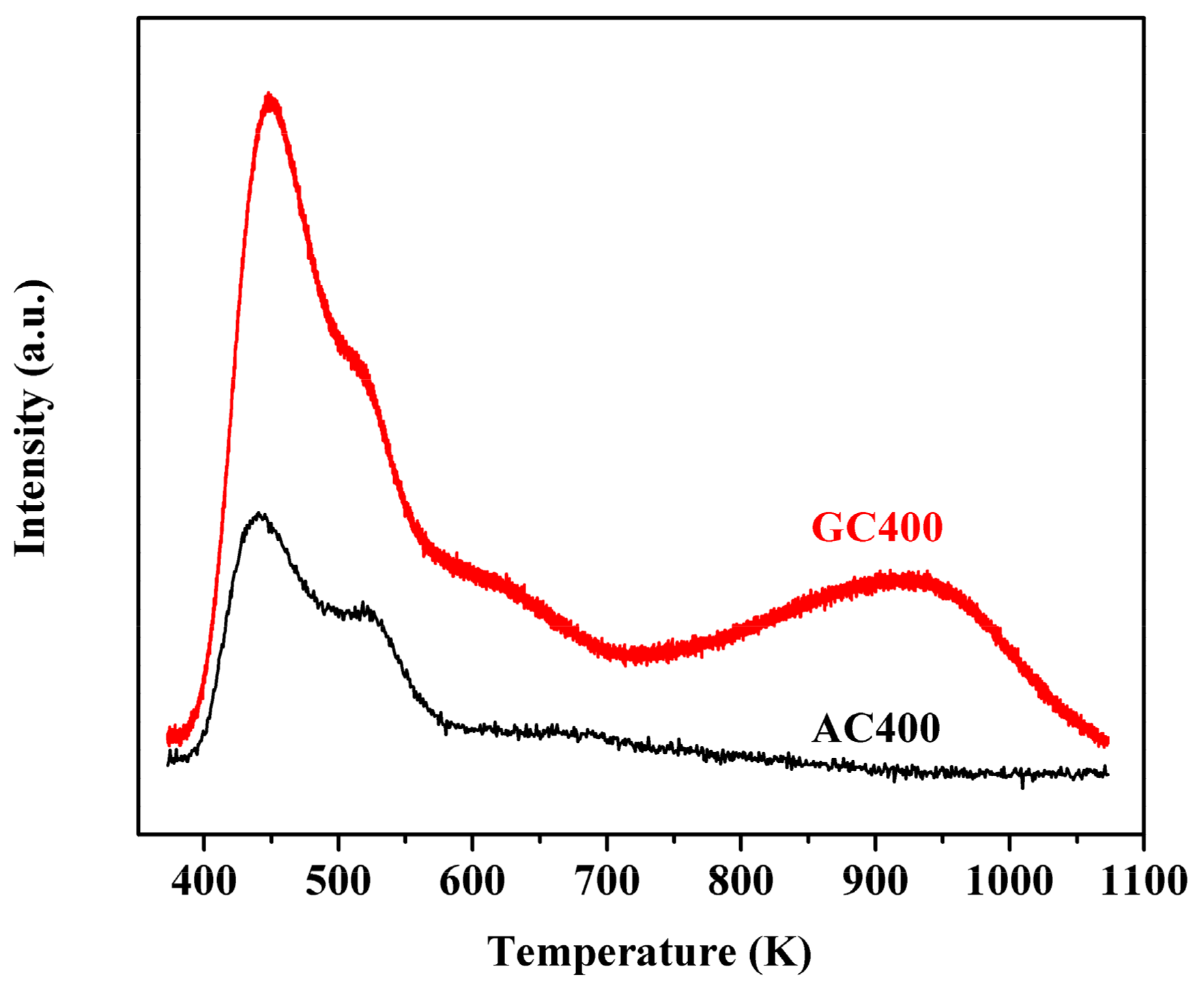
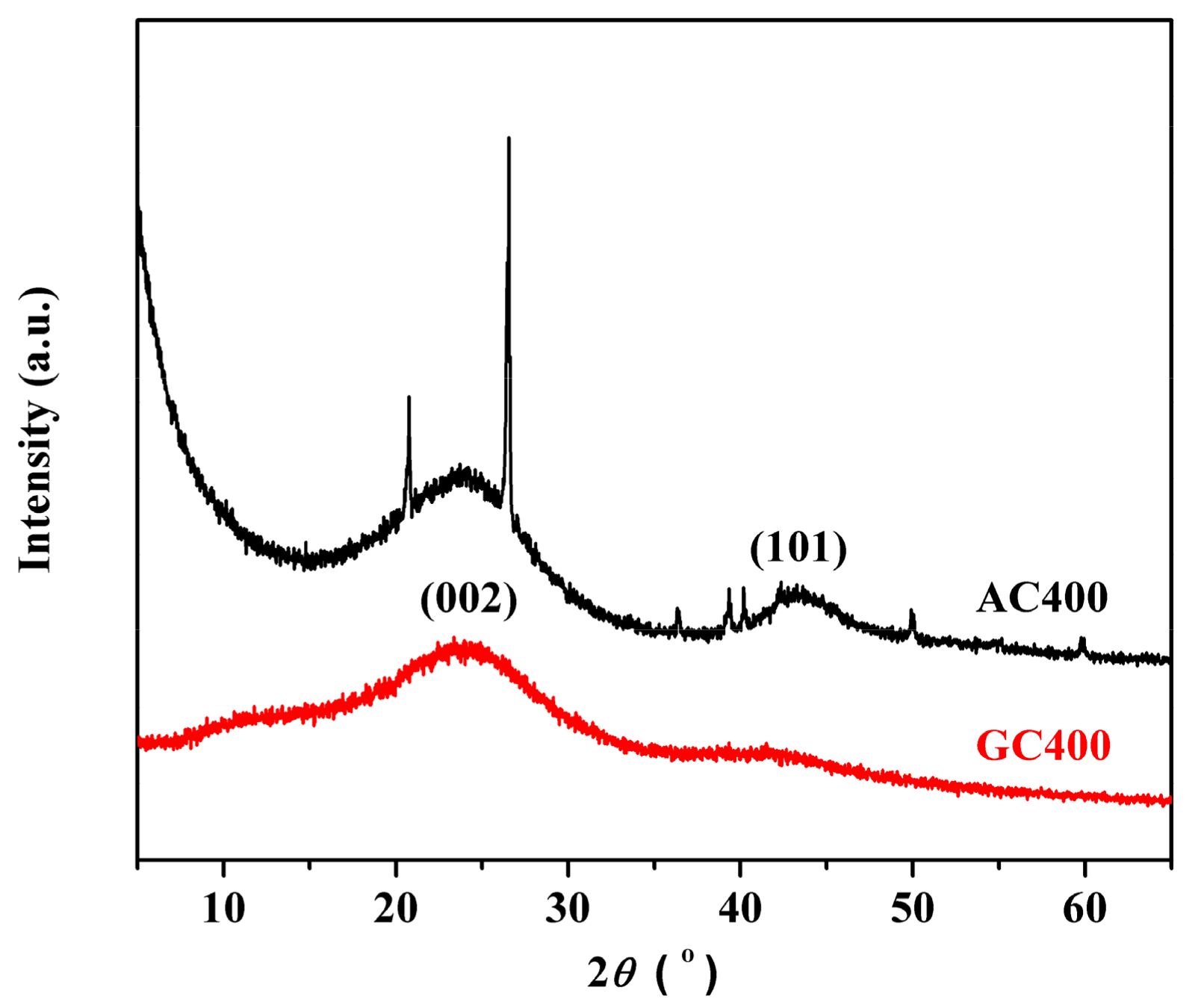
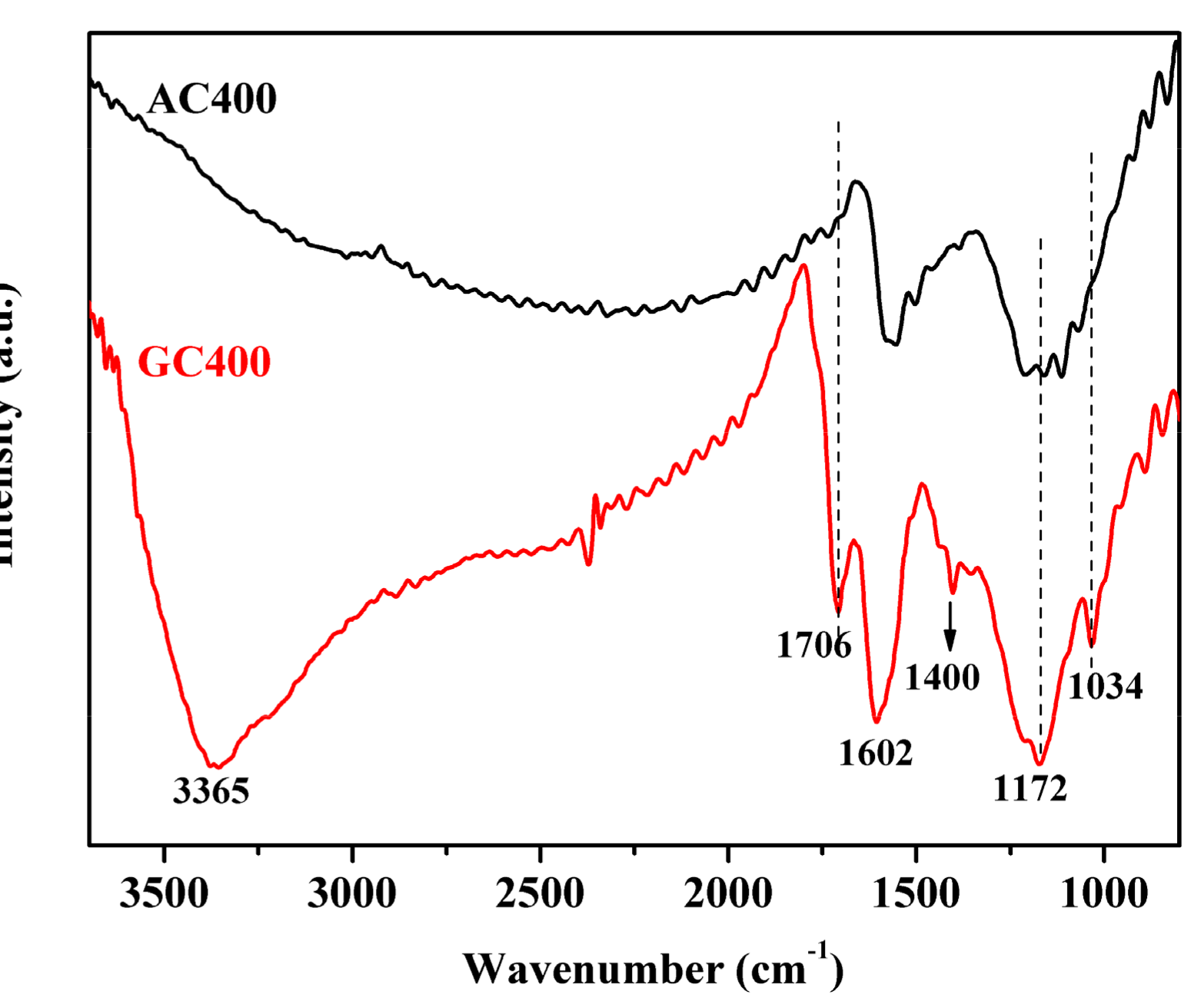
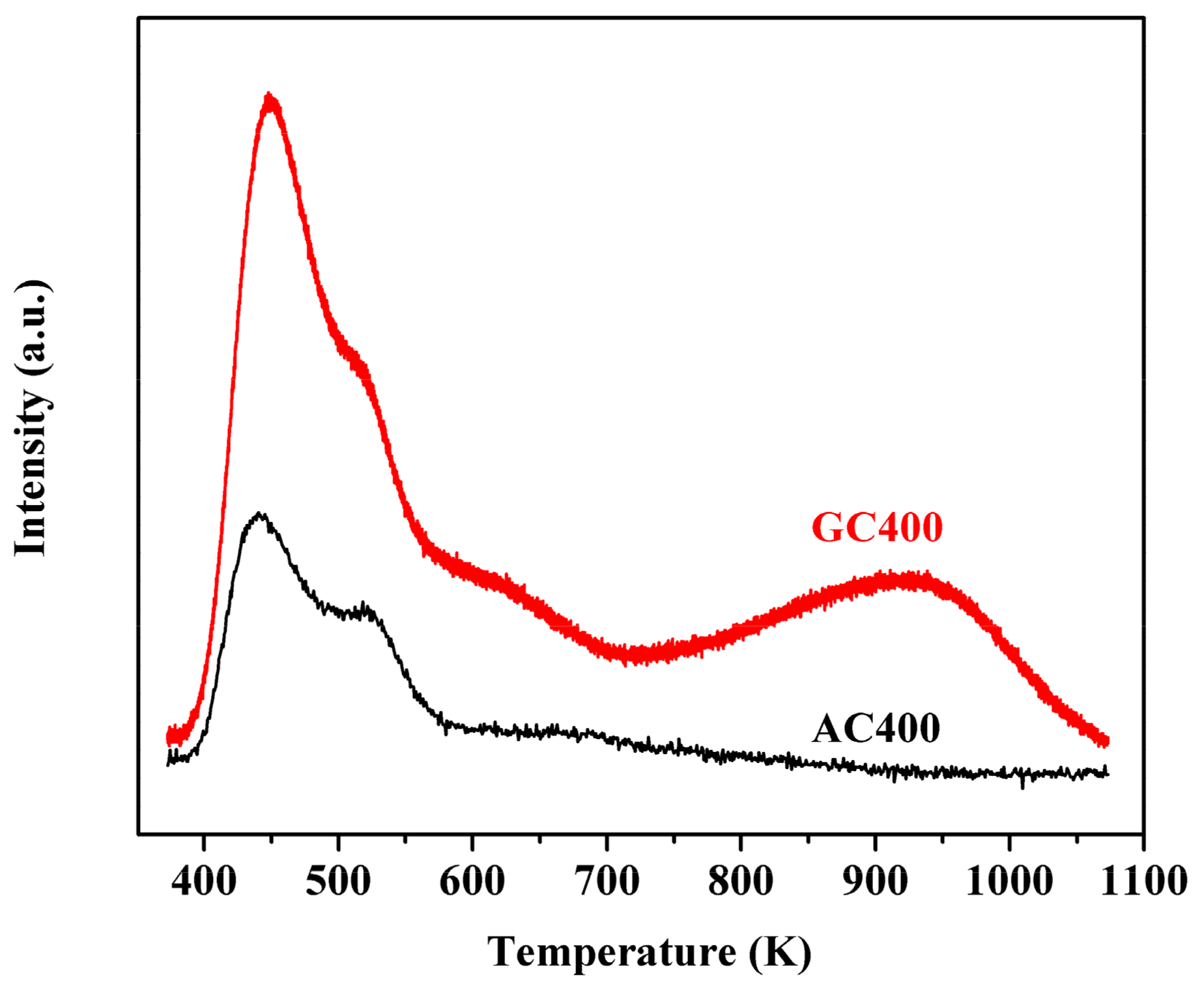
2.1. Catalyst Characterizations
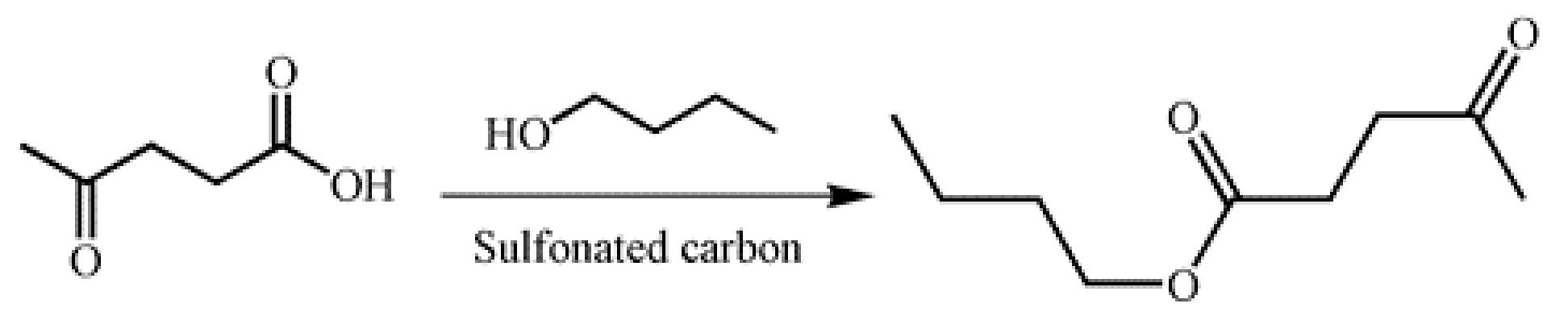
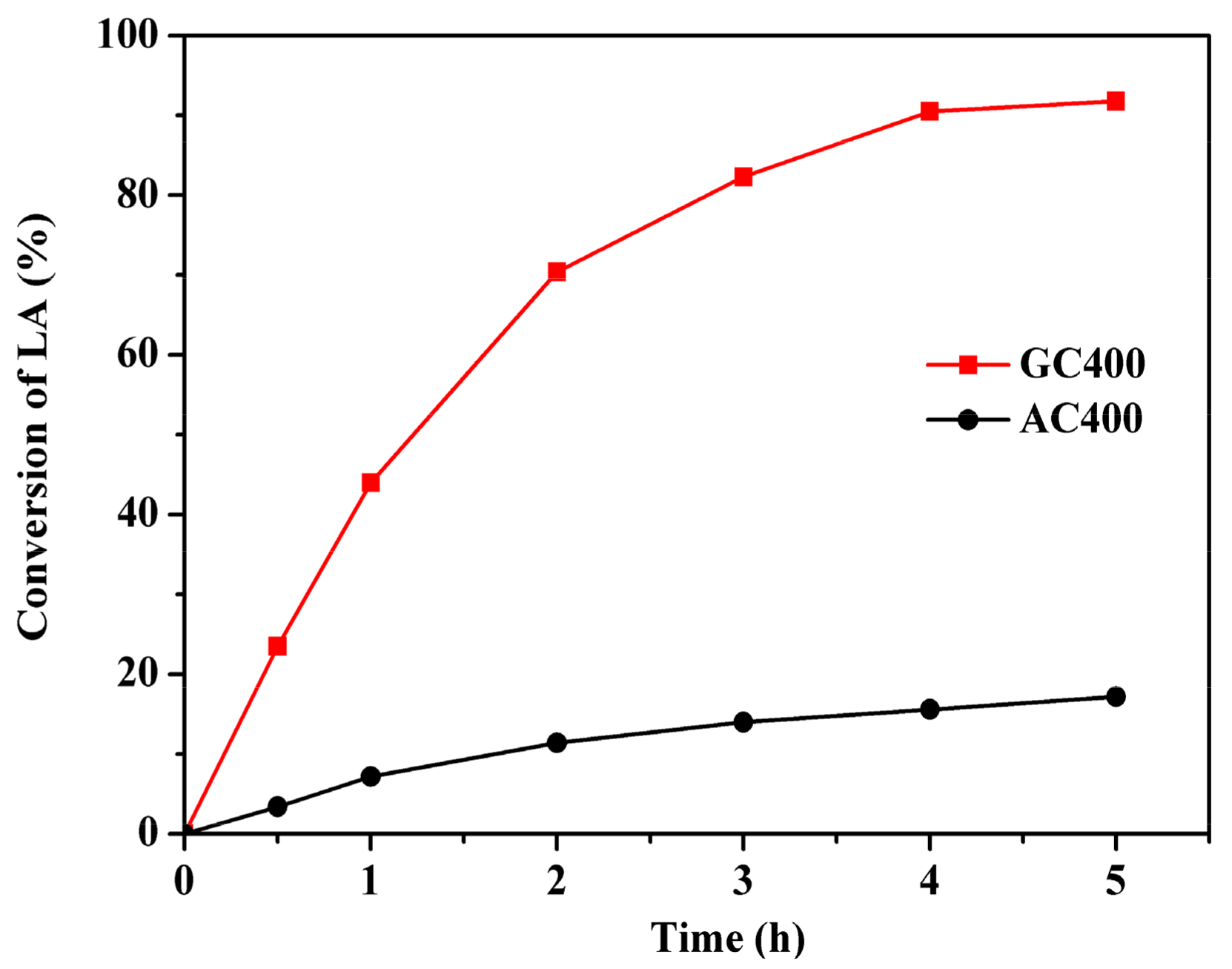
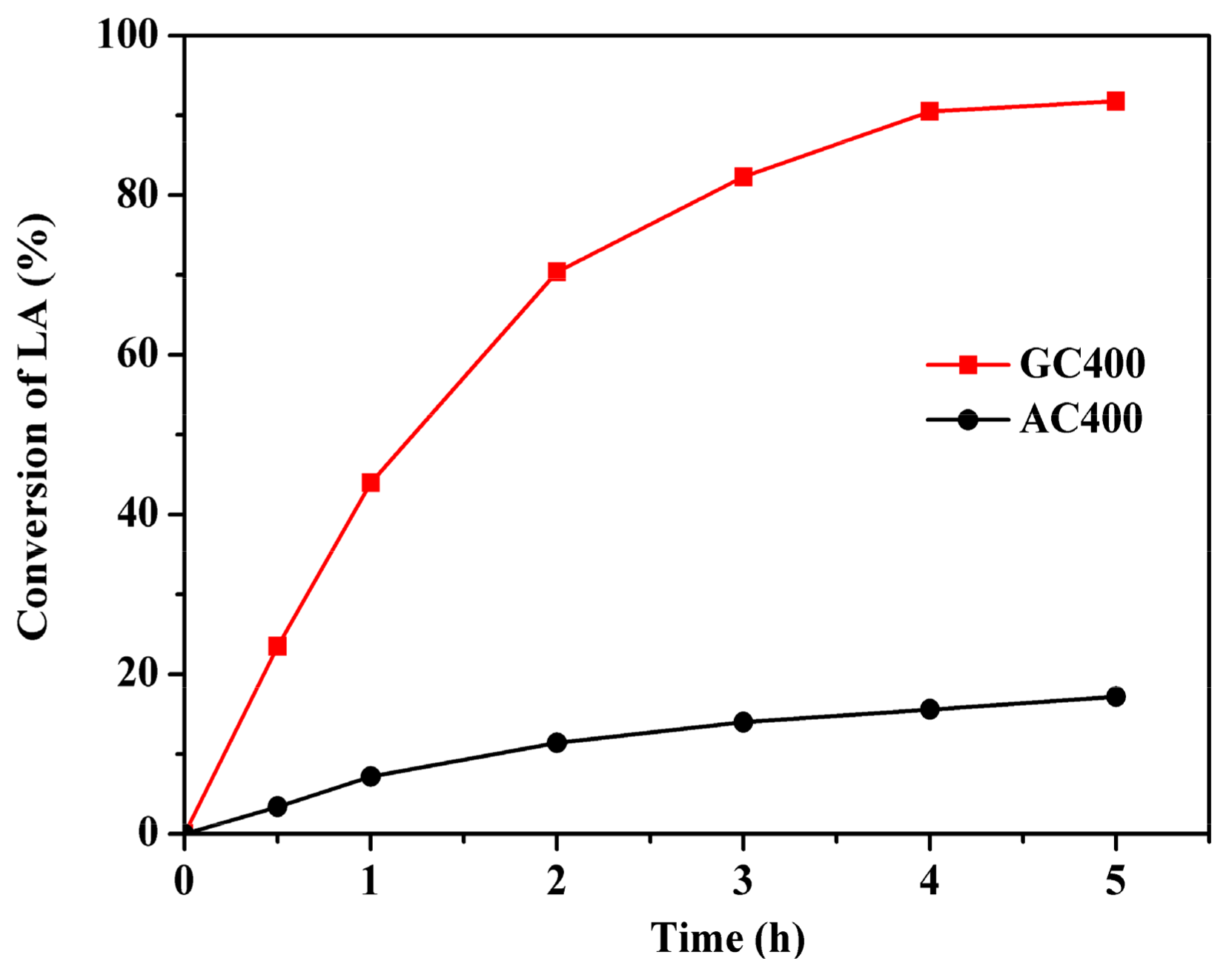
2.2. Esterification Activity over Two Sulfonated Carbon Materials
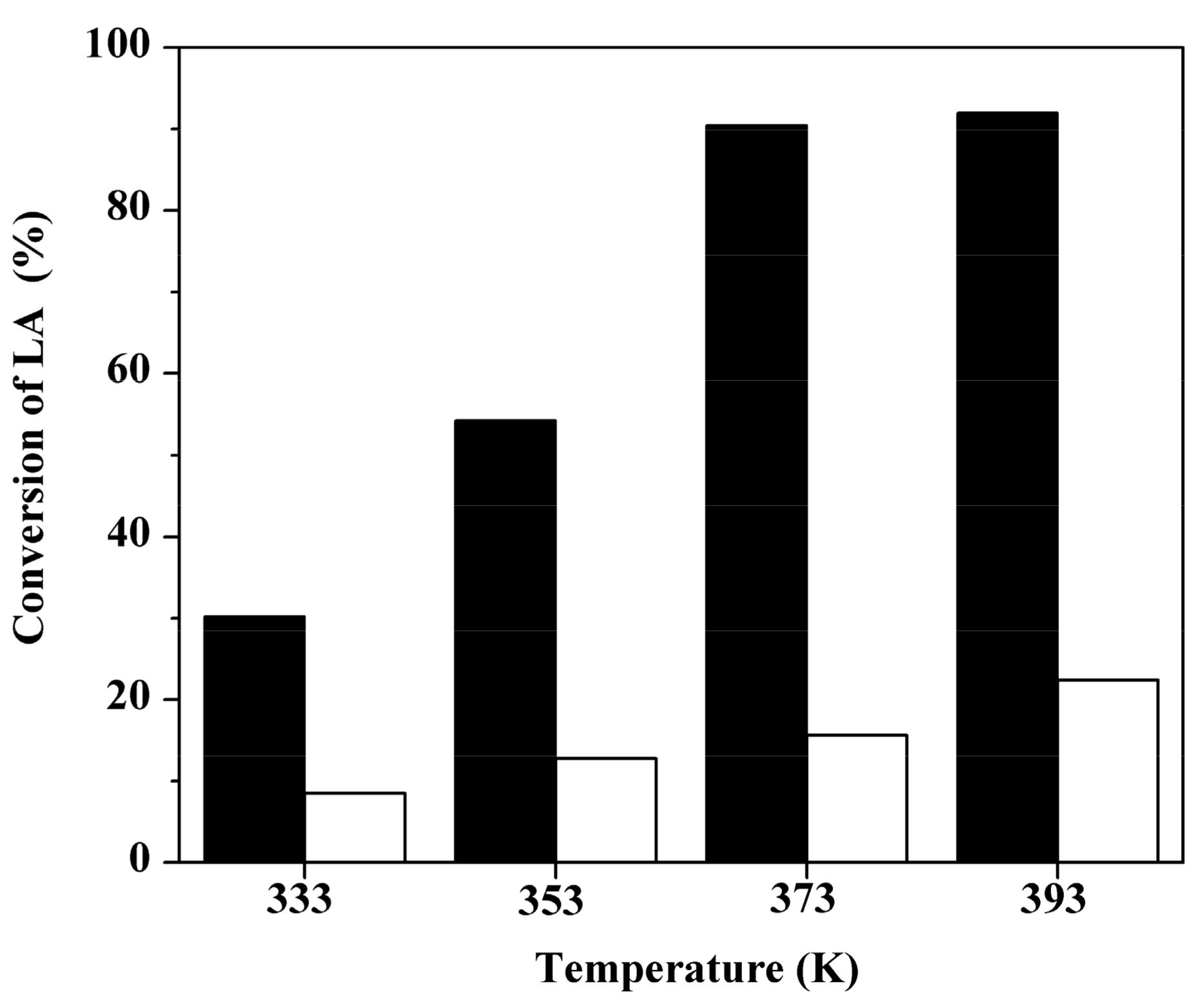
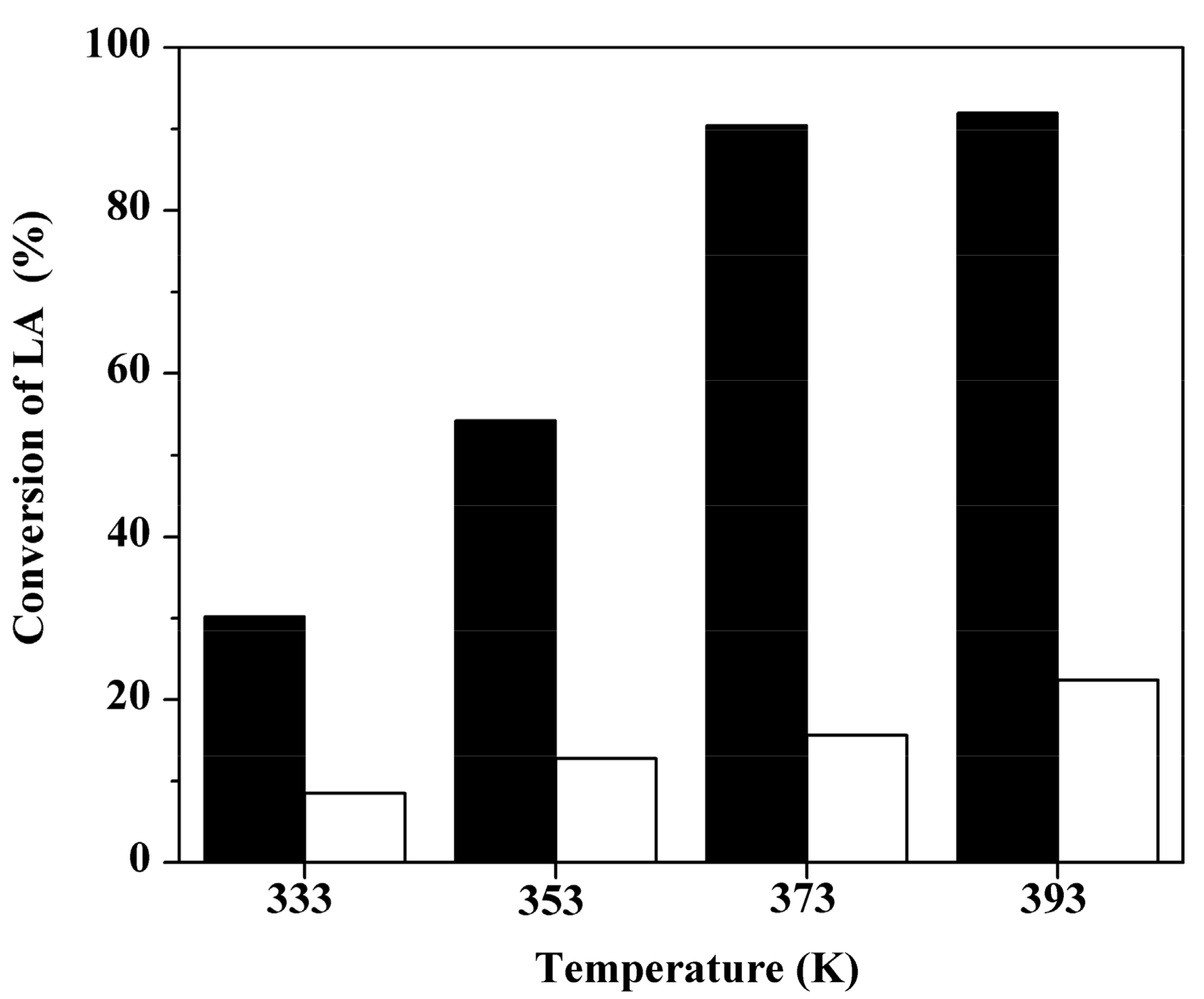
2.3. Effect of Reaction Temperature over Two Sulfonated Carbon Materials
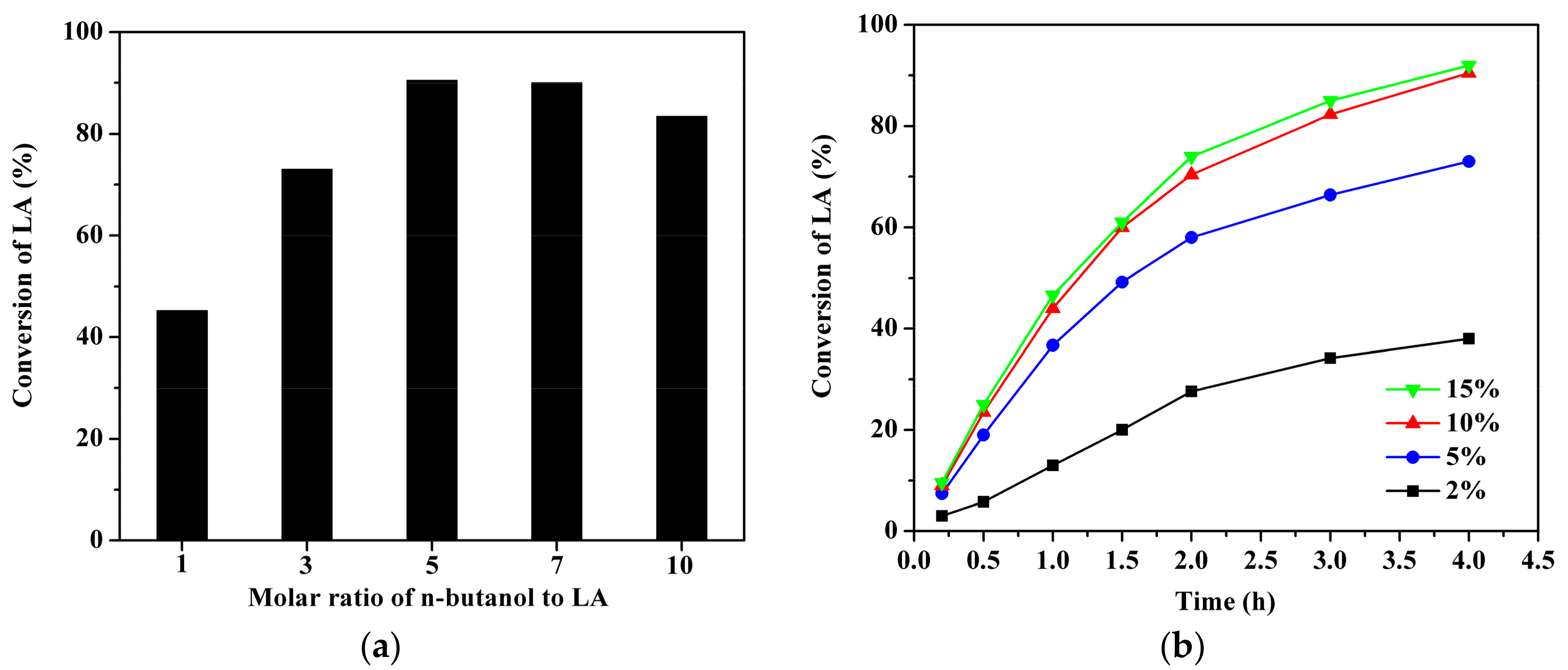
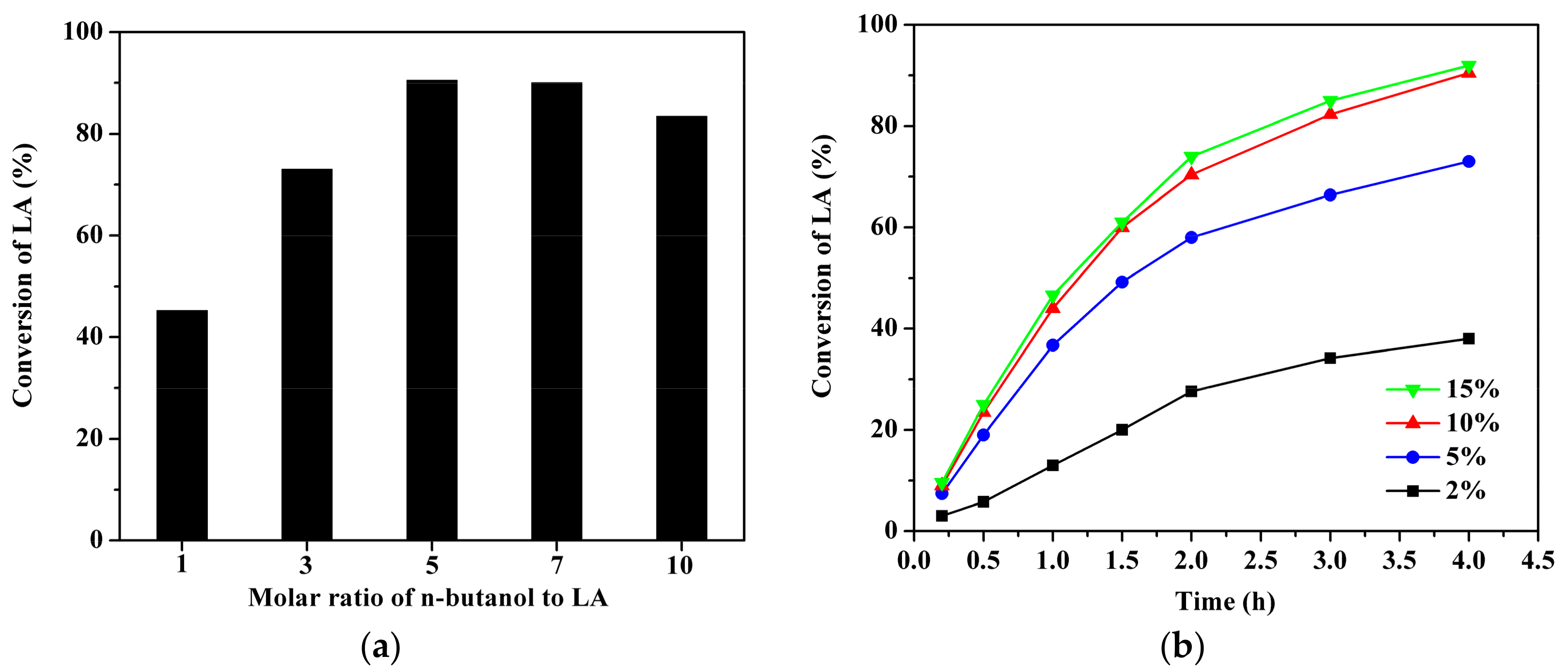
2.4. Effect of Reactant Molar Ratio and Catalyst Loading over GC400
2.5. Comparison with Other Typical Solid Acids
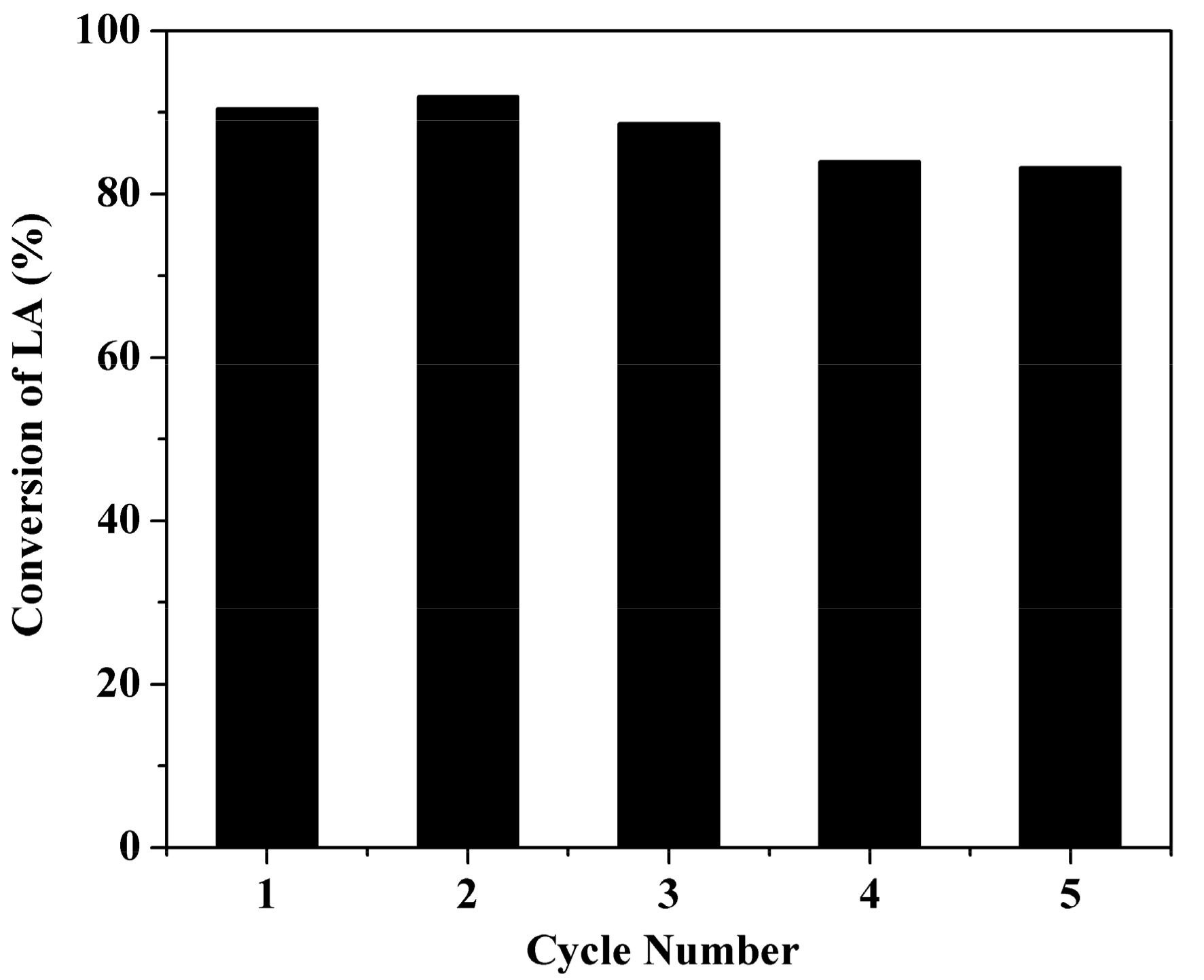
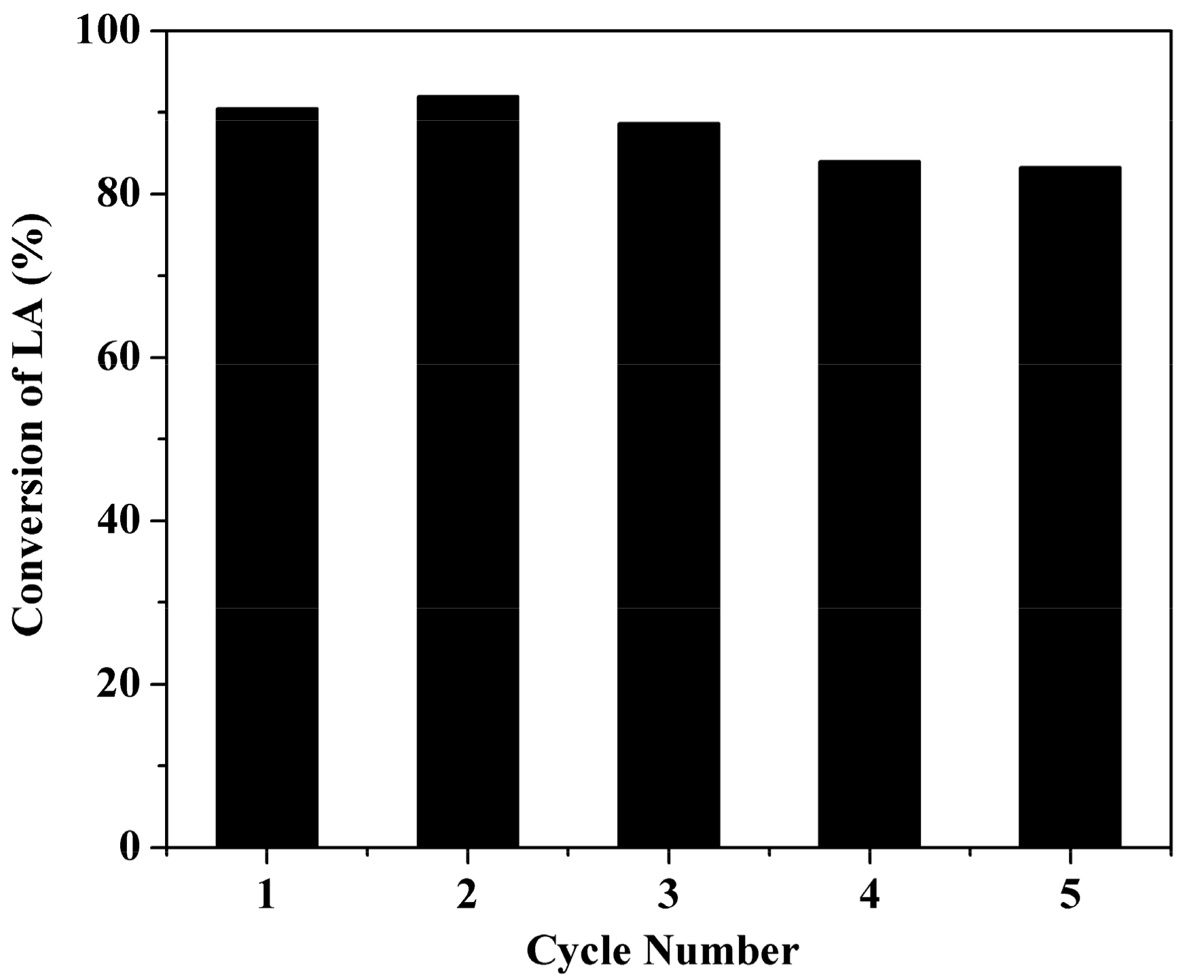
2.6. Stability Test
2.7. Merits of the Present Method
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Catalyst Characterization
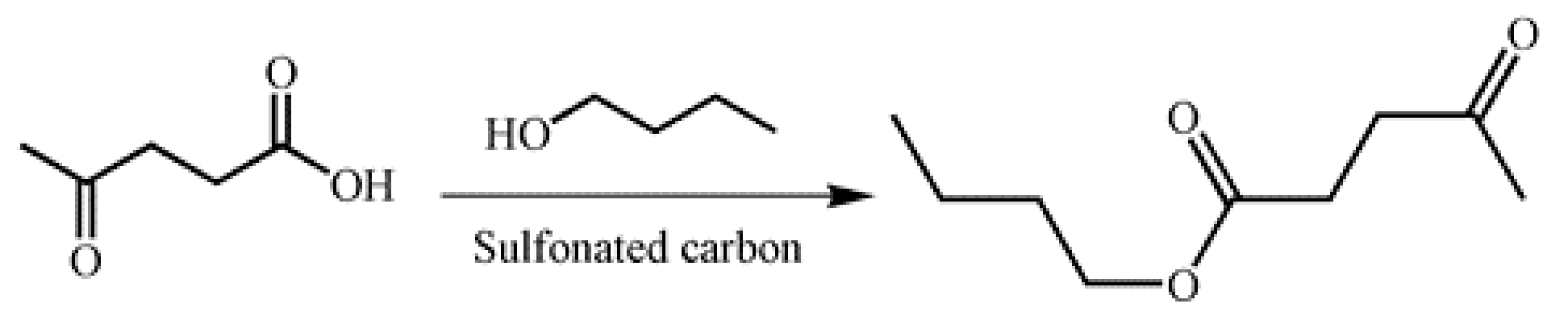
3.3. Esterification of LA and N-Butanol
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [PubMed]
- Alonso, D.M.; Bond, J.Q.; Dumesic, J.A. Catalytic conversion of biomass to biofuels. Green Chem. 2010, 12, 1493–1513. [Google Scholar] [CrossRef]
- Climent, M.J.; Corma, A.; Iborra, S. Conversion of biomass platform molecules into fuel additives and liquid hydrocarbon fuels. Green Chem. 2014, 16, 516–547. [Google Scholar] [CrossRef]
- Corma, A.; Iborra, S.; Velty, A. Chemical routes for the transformation of biomass into chemicals. Chem. Rev. 2007, 107, 2411–2502. [Google Scholar] [CrossRef] [PubMed]
- Mascal, M.; Nikitin, E.B. High-yield conversion of plant biomass into the key value-added feedstocks 5-(hydroxymethyl)furfural, levulinic acid, and levulinic esters via 5-(chloromethyl)furfural. Green Chem. 2010, 12, 370–373. [Google Scholar] [CrossRef]
- Bozell, J.J.; Petersen, G.R. Technology development for the production of biobased products from biorefinery carbohydrates—The US department of energy’s “top 10” revisited. Green Chem. 2010, 12, 539–554. [Google Scholar] [CrossRef]
- Antonetti, C.; Licursi, D.; Fulignati, S.; Valentini, G.; Raspolli Galletti, A. New frontiers in the catalytic synthesis of levulinic acid: From sugars to raw and waste biomass as starting feedstock. Catalysts 2016, 6, 196. [Google Scholar] [CrossRef]
- Hayes, D.J. An examination of biorefining processes, catalysts and challenges. Catal. Lett. 2009, 145, 138–151. [Google Scholar] [CrossRef]
- Nandiwale, K.Y.; Sonar, S.K.; Niphadkar, P.S.; Joshi, P.N.; Deshpande, S.S.; Patil, V.S.; Bokade, V.V. Catalytic upgrading of renewable levulinic acid to ethyl levulinate biodiesel using dodecatungstophosphoric acid supported on desilicated H-ZSM-5 as catalyst. Appl. Catal. A Gen. 2013, 460–461, 90–98. [Google Scholar] [CrossRef]
- Yan, K.; Wu, G.; Wen, J.; Chen, A. One-step synthesis of mesoporous H4SiW12O40-SiO2 catalysts for the production of methyl and ethyl levulinate biodiesel. Catal. Commun. 2013, 34, 58–63. [Google Scholar] [CrossRef]
- Nandiwale, K.Y.; Niphadkar, P.S.; Deshpande, S.S.; Bokade, V.V. Esterification of renewable levulinic acid to ethyl levulinate biodiesel catalyzed by highly active and reusable desilicated H-ZSM-5. J. Chem. Technol. Biotechnol. 2014, 89, 1507–1515. [Google Scholar] [CrossRef]
- Patil, C.R.; Niphadkar, P.S.; Bokade, V.V.; Joshi, P.N. Esterification of levulinic acid to ethyl levulinate over bimodal micro–mesoporous H/BEA zeolite derivatives. Catal. Commun. 2014, 43, 188–191. [Google Scholar] [CrossRef]
- Fernandes, D.R.; Rocha, A.S.; Mai, E.F.; Mota, C.J.A.; Teixeira da Silva, V. Levulinic acid esterification with ethanol to ethyl levulinate production over solid acid catalysts. Appl. Catal. A Gen. 2012, 425–426, 199–204. [Google Scholar] [CrossRef]
- Osatiashtiani, A.; Durndell, L.J.; Manayil, J.C.; Lee, A.F.; Wilson, K. Influence of alkyl chain length on sulfated zirconia catalysed batch and continuous esterification of carboxylic acids by light alcohols. Green Chem. 2016, 18, 5529–5535. [Google Scholar] [CrossRef]
- Oliveira, B.L.; Teixeira da Silva, V. Sulfonated carbon nanotubes as catalysts for the conversion of levulinic acid into ethyl levulinate. Catal. Today 2014, 234, 257–263. [Google Scholar] [CrossRef]
- Budarin, V.L.; Clark, J.H.; Luque, R.; Macquarrie, D.J. Versatile mesoporous carbonaceous materials for acid catalysis. Chem. Commun. 2007, 634–636. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Ma, L.; Song, D.; Zhang, X.; Guo, Y. Design of a highly ordered mesoporous H3PW12O40/ZrO2-Si(Ph)Si hybrid catalyst for methyl levulinate synthesis. Green Chem. 2013, 15, 885–890. [Google Scholar] [CrossRef]
- Christensen, E.; Yanowitz, J.; Ratcliff, M.; McCormick, R.L. Renewable oxygenate blending effects on gasoline properties. Energy Fuels 2011, 25, 4723–4733. [Google Scholar] [CrossRef]
- Shrivastav, G.; Khan, T.S.; Agarwal, M.; Haider, M.A. Reformulation of gasoline to replace aromatics by biomass-derived alkyl levulinates. ACS Sustain. Chem. Eng. 2017, 5, 7118–7127. [Google Scholar] [CrossRef]
- Earl Christensen, A.W.; Paul, S.; Burton, S.; McCormick, A.R.L. Properties and performance of levulinate esters as diesel blend components. Energy Fuels 2011, 25, 5422–5428. [Google Scholar] [CrossRef]
- Dharne, S.; Bokade, V.V. Esterification of levulinic acid to n-butyl levulinate over heteropolyacid supported on acid-treated clay. J. Nat. Gas Chem. 2011, 20, 18–24. [Google Scholar] [CrossRef]
- Maheria, K.C.; Kozinski, J.; Dalai, A. Esterification of levulinic acid to n-butyl levulinate over various acidic zeolites. Catal. Lett. 2013, 143, 1220–1225. [Google Scholar] [CrossRef]
- Nandiwale, K.Y.; Bokade, V.V. Esterification of renewable levulinic acid to n-butyl levulinate over modified h-zsm-5. Chem. Eng. Technol. 2015, 38, 246–252. [Google Scholar] [CrossRef]
- Cirujano, F.G.; Corma, A.; Llabrés i Xamena, F.X. Conversion of levulinic acid into chemicals: Synthesis of biomass derived levulinate esters over Zr-containing MOFs. Chem. Eng. Sci. 2015, 124, 52–60. [Google Scholar] [CrossRef]
- Najafi Chermahini, A.; Nazeri, M. Esterification of the levulinic acid with n-butyl and isobutyl alcohols over aluminum-containing MCM-41. Fuel Process. Technol. 2017, 167, 442–450. [Google Scholar] [CrossRef]
- Tejero, M.A.; Ramírez, E.; Fité, C.; Tejero, J.; Cunill, F. Esterification of levulinic acid with butanol over ion exchange resins. Appl. Catal. A Gen. 2016, 517, 56–66. [Google Scholar] [CrossRef]
- Trombettoni, V.; Bianchi, L.; Zupanic, A.; Porciello, A.; Cuomo, M.; Piermatti, O.; Marrocchi, A.; Vaccaro, L. Efficient catalytic upgrading of levulinic acid into alkyl levulinates by resin-supported acids and flow reactors. Catalysts 2017, 7, 235. [Google Scholar] [CrossRef]
- Nakhate, A.V.; Yadav, G.D. Synthesis and characterization of sulfonated carbon-based graphene oxide monolith by solvothermal carbonization for esterification and unsymmetrical ether formation. ACS Sustain. Chem. Eng. 2016, 4, 1963–1973. [Google Scholar] [CrossRef]
- Badgujar, K.C.; Bhanage, B.M. Thermo-chemical energy assessment for production of energy-rich fuel additive compounds by using levulinic acid and immobilized lipase. Fuel Process. Technol. 2015, 138, 139–146. [Google Scholar] [CrossRef]
- Toda, M.; Takagaki, A.; Okamura, M.; Kondo, J.N.; Hayashi, S.; Domen, K.; Hara, M. Green chemistry: Biodiesel made with sugar catalyst. Nature 2005, 438, 178. [Google Scholar] [CrossRef] [PubMed]
- Okamura, M.; Takagaki, A.; Toda, M.; Kondo, J.N.; Domen, K.; Tatsumi, T.; Hara, M.; Hayashi, S. Acid-catalyzed reactions on flexible polycyclic aromatic carbon in amorphous carbon. Chem. Mater. 2006, 18, 3039–3045. [Google Scholar] [CrossRef]
- Lou, W.; Zong, M.; Duan, Z. Efficient production of biodiesel from high free fatty acid-containing waste oils using various carbohydrate-derived solid acid catalysts. Bioresour. Technol. 2008, 99, 8752–8758. [Google Scholar] [CrossRef] [PubMed]
- Lokman, I.M.; Rashid, U.; Taufiqyap, Y.H.; Yunus, R. Methyl ester production from palm fatty acid distillate using sulfonated glucose-derived acid catalyst. Renew. Energy 2015, 81, 347–354. [Google Scholar] [CrossRef]
- Cao, X.; Sun, S.; Sun, R. Application of biochar-based catalysts in biomass upgrading: A review. RSC Adv. 2017, 7, 48793–48805. [Google Scholar] [CrossRef]
- Dandekar, A.; Baker, R.T.K.; Vannice, M.A. Characterization of activated carbon, graphitized carbon fibers and synthetic diamond powder using TPD and drifts. Carbon 1998, 36, 1821–1831. [Google Scholar] [CrossRef]
- Chen, G.; Fang, B. Preparation of solid acid catalyst from glucose–starch mixture for biodiesel production. Bioresour. Technol. 2011, 102, 2635–2640. [Google Scholar] [CrossRef] [PubMed]
- Dawodu, F.A.; Ayodele, O.; Xin, J.; Zhang, S.; Yan, D. Effective conversion of non-edible oil with high free fatty acid into biodiesel by sulphonated carbon catalyst. Appl. Energy 2014, 114, 819–826. [Google Scholar] [CrossRef]
- Shu, Q.; Gao, J.; Nawaz, Z.; Liao, Y.; Wang, D.; Wang, J. Synthesis of biodiesel from waste vegetable oil with large amounts of free fatty acids using a carbon-based solid acid catalyst. Appl. Energy 2010, 87, 2589–2596. [Google Scholar] [CrossRef]
- Zhou, Y.; Niu, S.; Li, J. Activity of the carbon-based heterogeneous acid catalyst derived from bamboo in esterification of oleic acid with ethanol. Energy Convers. Manag. 2016, 114, 188–196. [Google Scholar] [CrossRef]
- Okuhara, T. Water-tolerant solid acid catalysts. Chem. Rev. 2002, 102, 3641–3666. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, M.; Bringué, R.; Fité, C.; Ramírez, E.; Cunill, F. Liquid-Phase Oligomerization of 1-Hexene Catalyzed by Macroporous Ion-Exchange Resins. Top. Catal. 2011, 54, 998–1008. [Google Scholar] [CrossRef]
- Suganuma, S.; Nakajima, K.; Kitano, M.; Yamaguchi, D.; Kato, H.; Hayashi, S. Hydrolysis of cellulose by amorphous carbon bearing SO3H, COOH, and OH groups. J. Am. Chem. Soc. 2008, 130, 12787–12793. [Google Scholar] [CrossRef] [PubMed]
- Takagaki, A.; Toda, M.; Okamura, M.; Kondo, J.N.; Hayashi, S.; Domen, K.; Hara, M. Esterification of higher fatty acids by a novel strong solid acid. Catal. Today 2006, 116, 157–161. [Google Scholar] [CrossRef]
- Yang, J.; Li, N.; Li, S.; Wang, W.; Li, L.; Wang, A.; Wang, X.; Cong, Y.; Zhang, T. Synthesis of diesel and jet fuel range alkanes with furfural and ketones from lignocellulose under solvent free conditions. Green Chem. 2014, 16, 4879–4884. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Elemental Composition (wt %) | SBET (m2·g−1) | |||
|---|---|---|---|---|---|
| C | H | N | S | ||
| GC400 | 57.76 | 2.33 | 0.13 | 2.13 | 2 |
| AC400 | 73.48 | 0.76 | 0.53 | 0.65 | 708 |
| Catalyst | –H0 | –SO3H Density (mmol·g−1) | Total Acid Density a (mmol·g−1) | LA Conversion b (%) | TOF c (h−1) | Functional Groups |
|---|---|---|---|---|---|---|
| HZSM-5 | 5.6~5.7 [40] | - | 2.0 | 20.8 | 4.3 d | Acidic –OH |
| Hβ | 4.4~5.7 [41] | - | 1.46 | 33.4 | 7.2 d | Acidic –OH |
| Amberlyst-15 | 2.2 [42] | 4.8 | 4.8 | 78.0 | 10.7 | –SO3H |
| Nafion-212 | 11~13 [40] | 0.9 | 0.9 | 93.3 | 59.0 | –SO3H |
| GC400 | 8~11 [43] | 0.66 | 1.1 | 90.5 | 95.8 | –SO3H, –COOH, phenolic –OH |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Li, G.; Zhang, L.; Zhang, S. Efficient Production of N-Butyl Levulinate Fuel Additive from Levulinic Acid Using Amorphous Carbon Enriched with Oxygenated Groups. Catalysts 2018, 8, 14. https://doi.org/10.3390/catal8010014
Yang J, Li G, Zhang L, Zhang S. Efficient Production of N-Butyl Levulinate Fuel Additive from Levulinic Acid Using Amorphous Carbon Enriched with Oxygenated Groups. Catalysts. 2018; 8(1):14. https://doi.org/10.3390/catal8010014
Chicago/Turabian StyleYang, Jinfan, Guangyi Li, Lulu Zhang, and Sufeng Zhang. 2018. "Efficient Production of N-Butyl Levulinate Fuel Additive from Levulinic Acid Using Amorphous Carbon Enriched with Oxygenated Groups" Catalysts 8, no. 1: 14. https://doi.org/10.3390/catal8010014
APA StyleYang, J., Li, G., Zhang, L., & Zhang, S. (2018). Efficient Production of N-Butyl Levulinate Fuel Additive from Levulinic Acid Using Amorphous Carbon Enriched with Oxygenated Groups. Catalysts, 8(1), 14. https://doi.org/10.3390/catal8010014