
Zirconium Phosphate Heterostructures as Catalyst Support in Hydrodeoxygenation Reactions




Abstract
:1. Introduction
2. Results
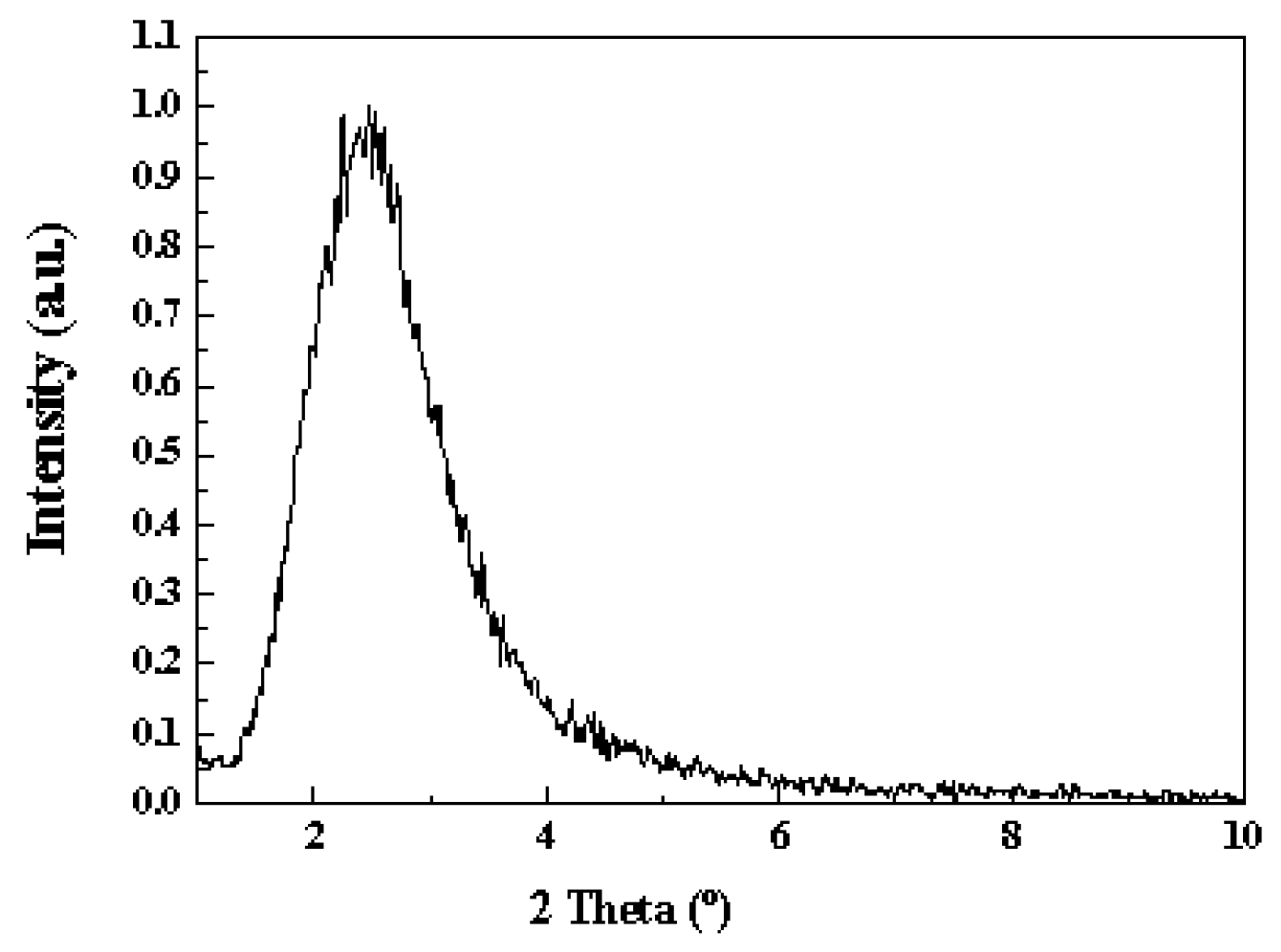
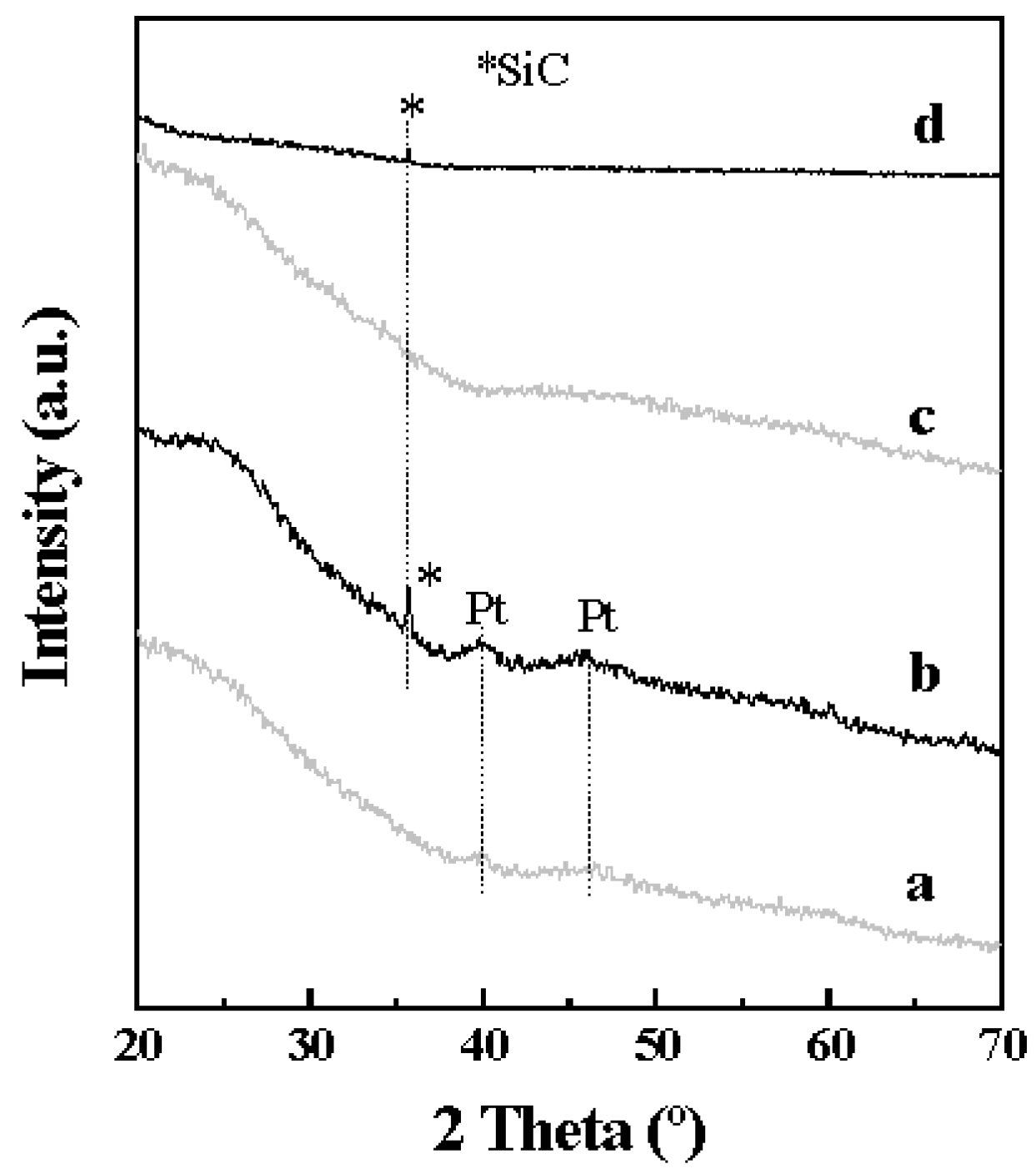
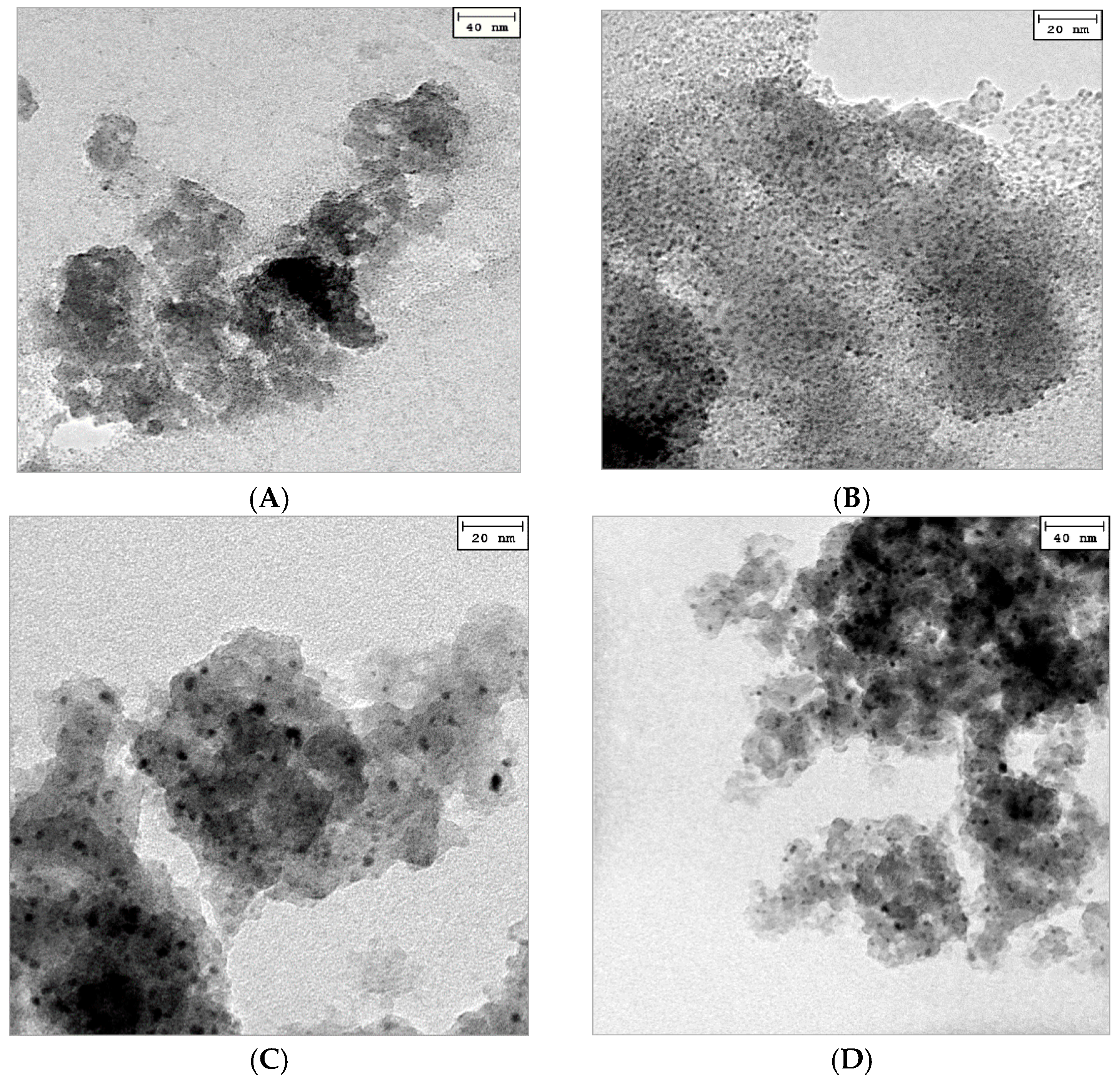
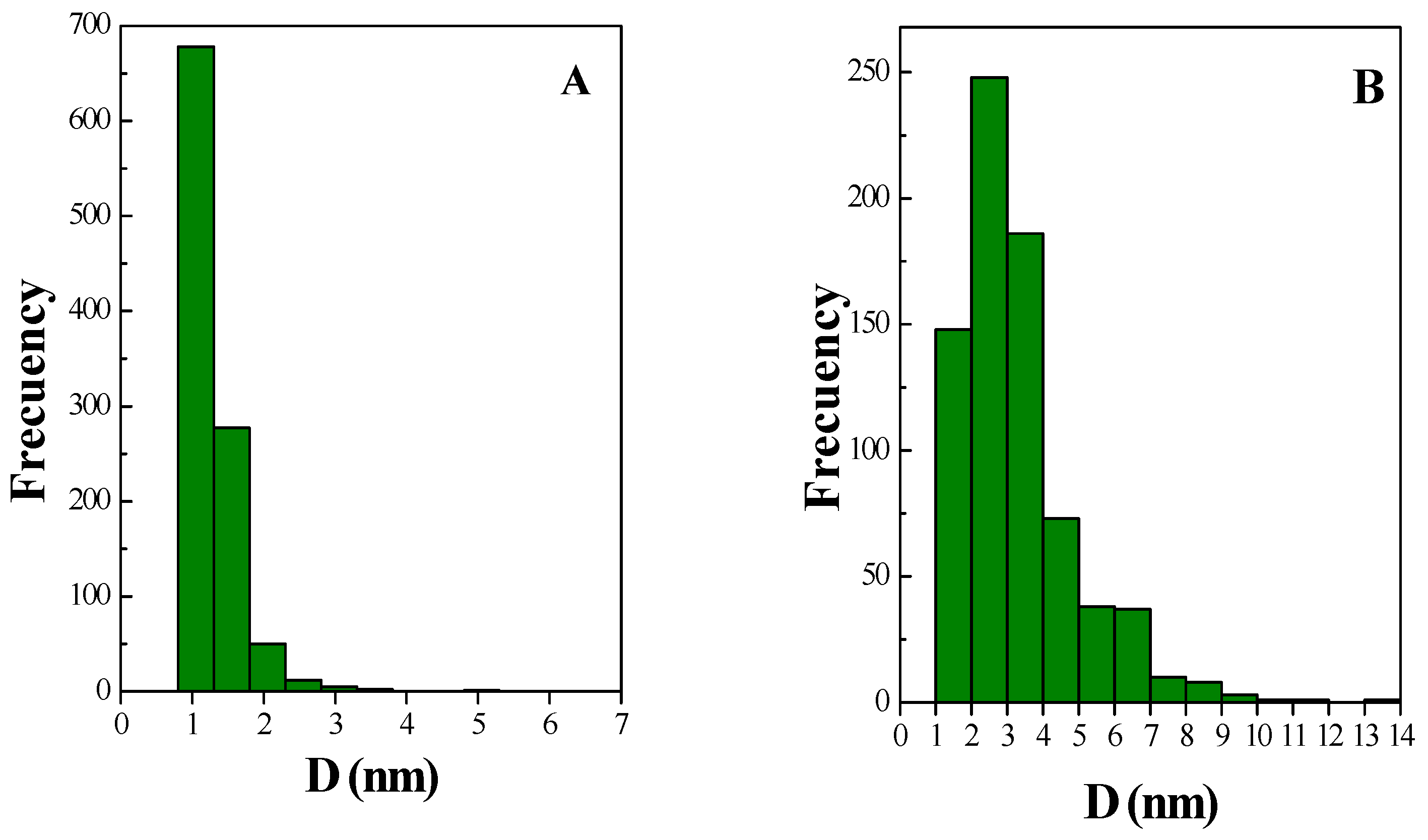
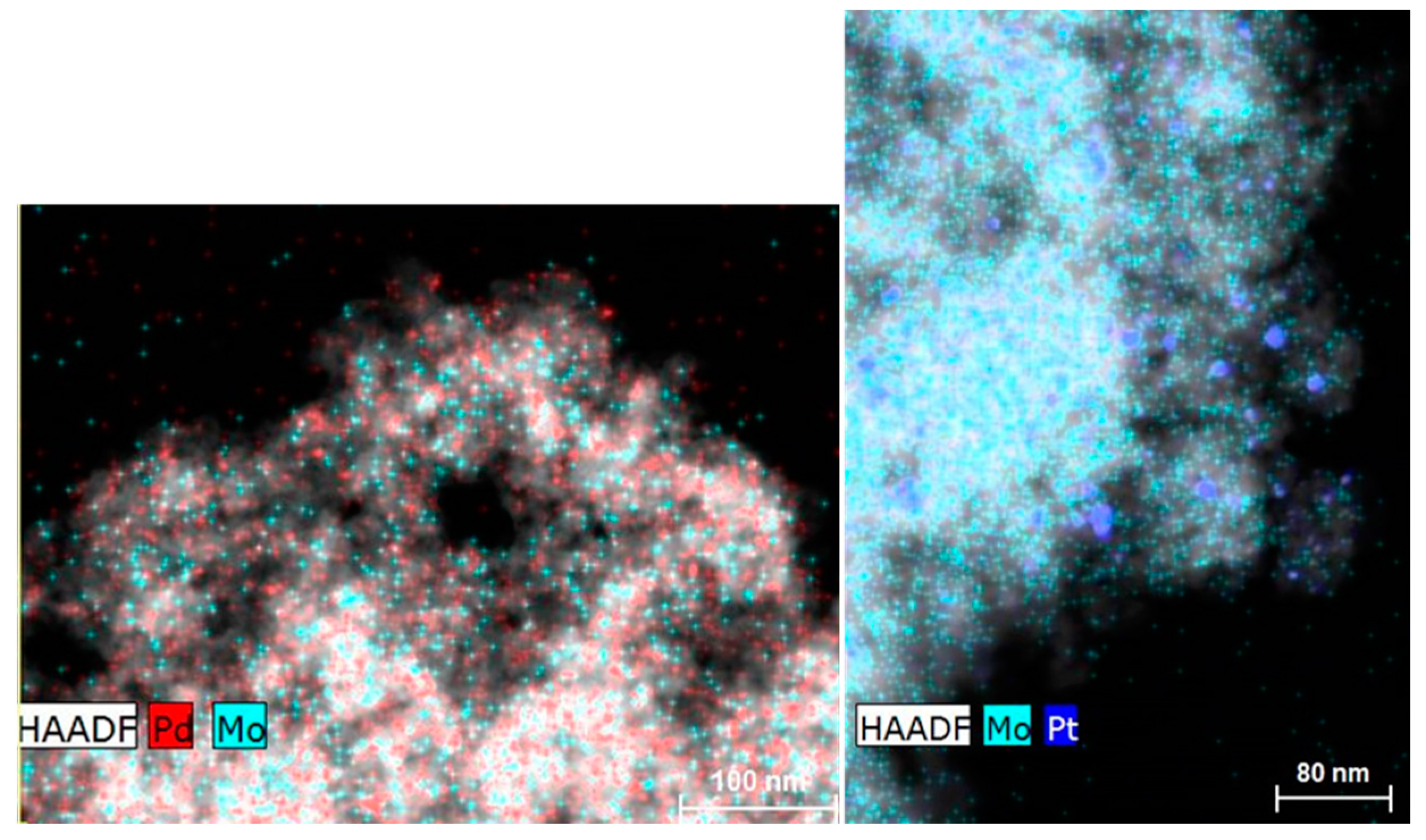
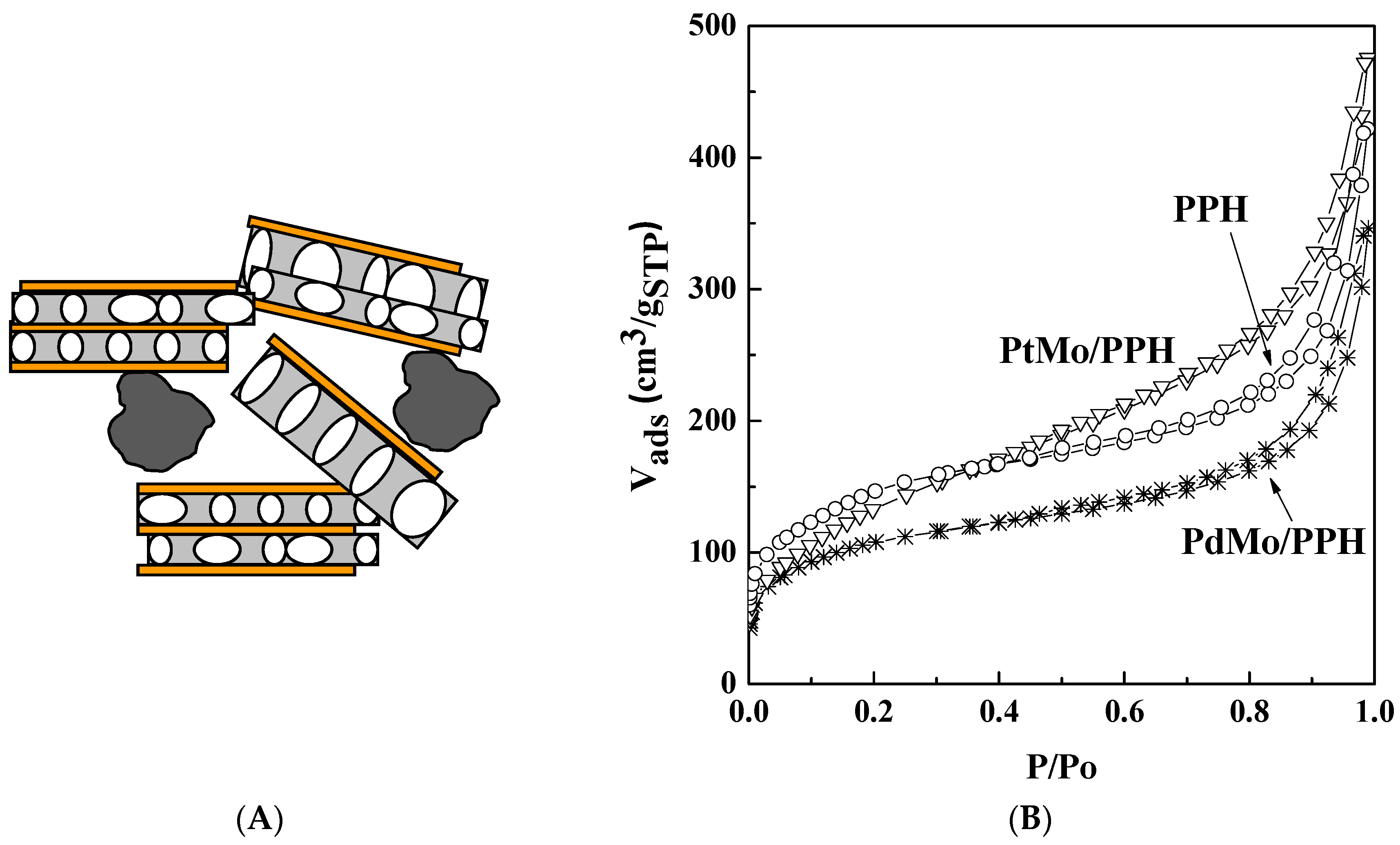
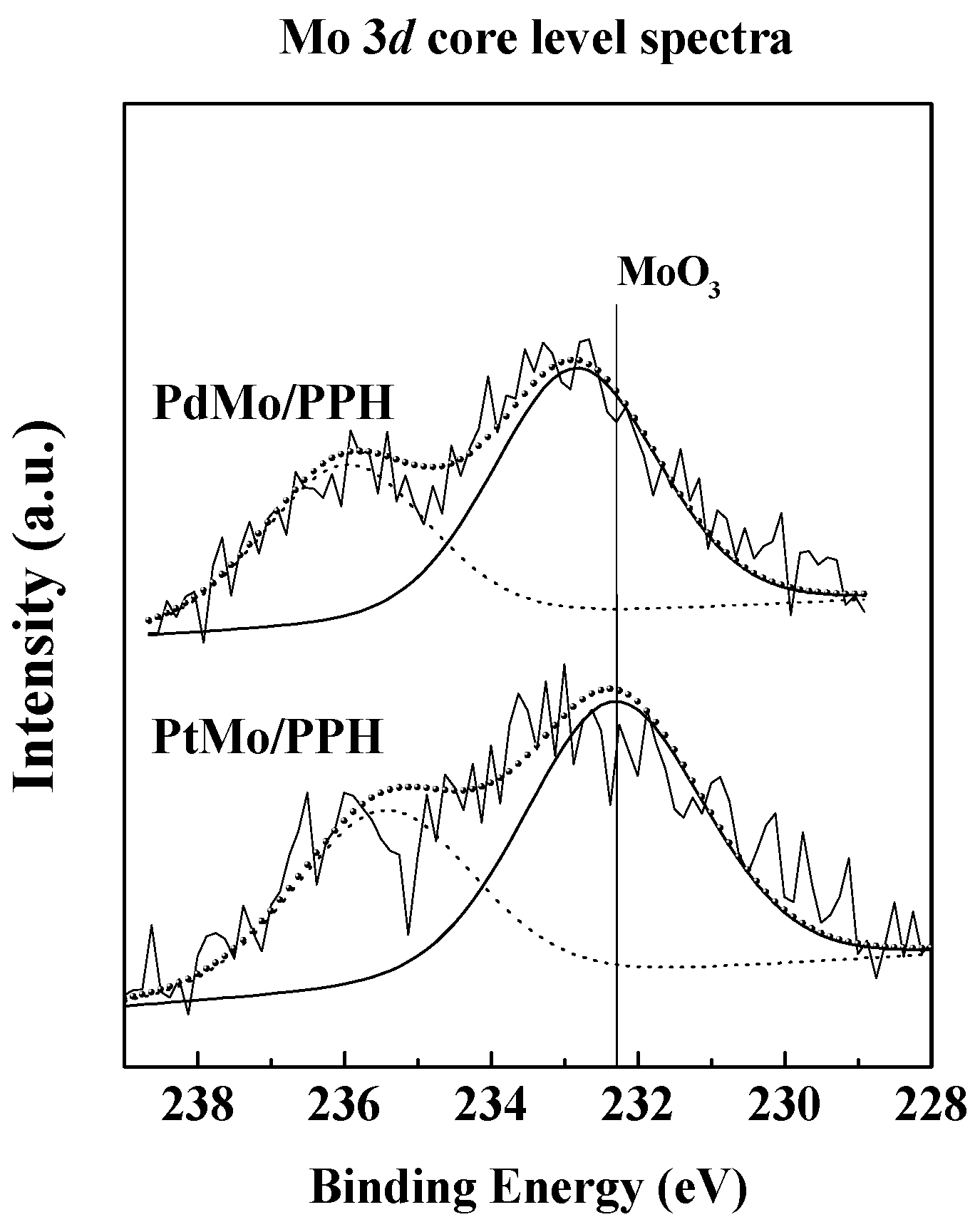
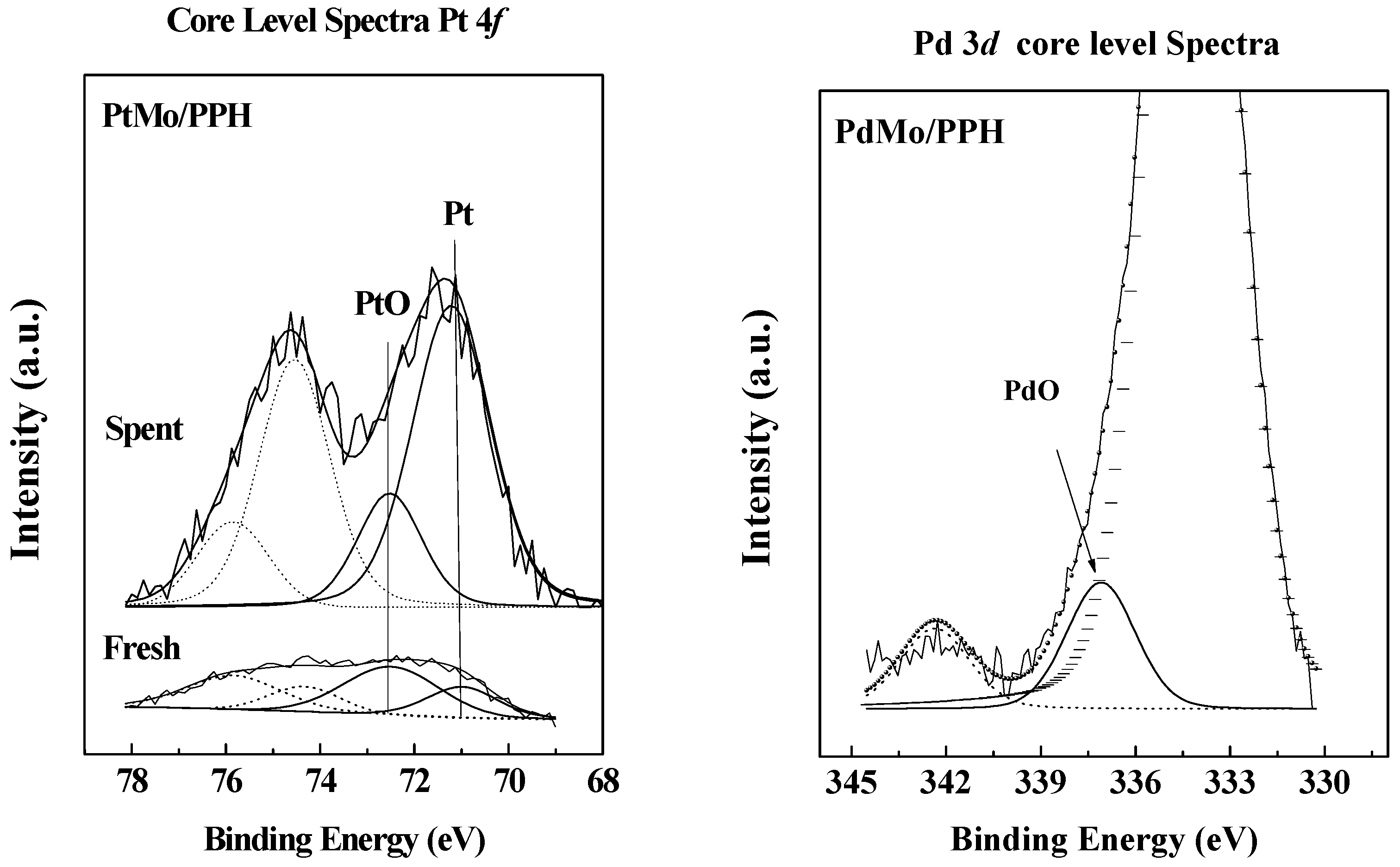
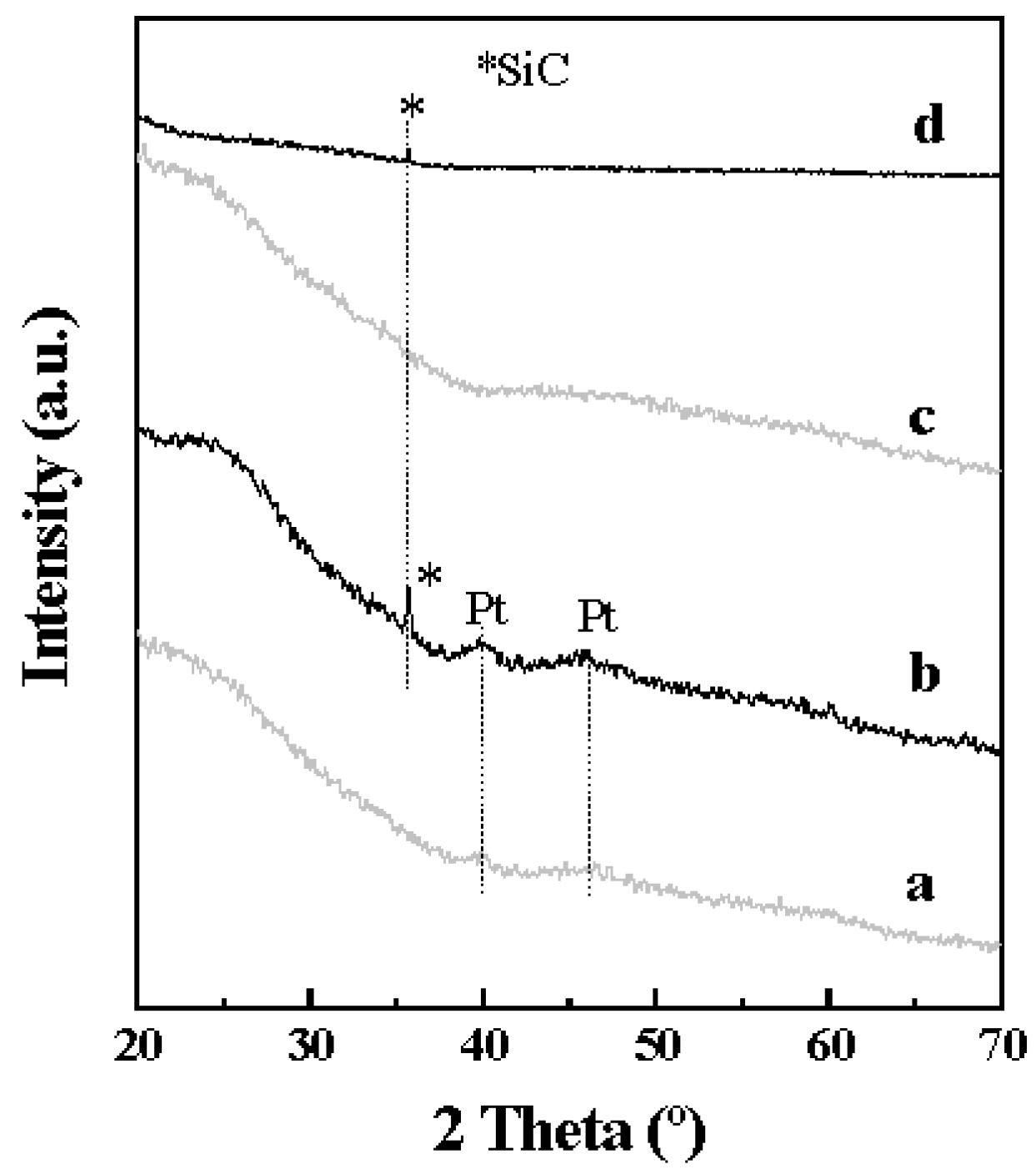
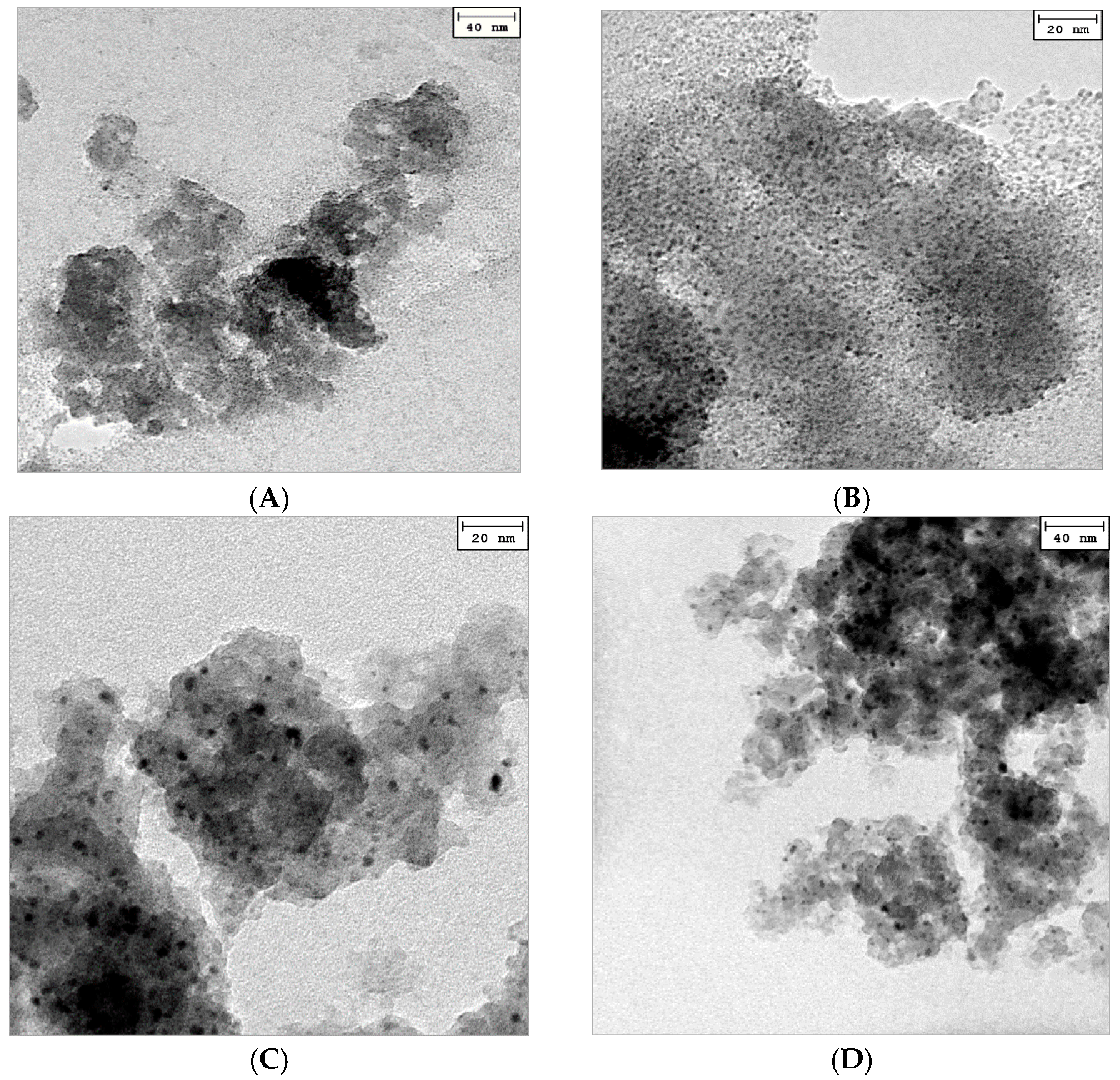
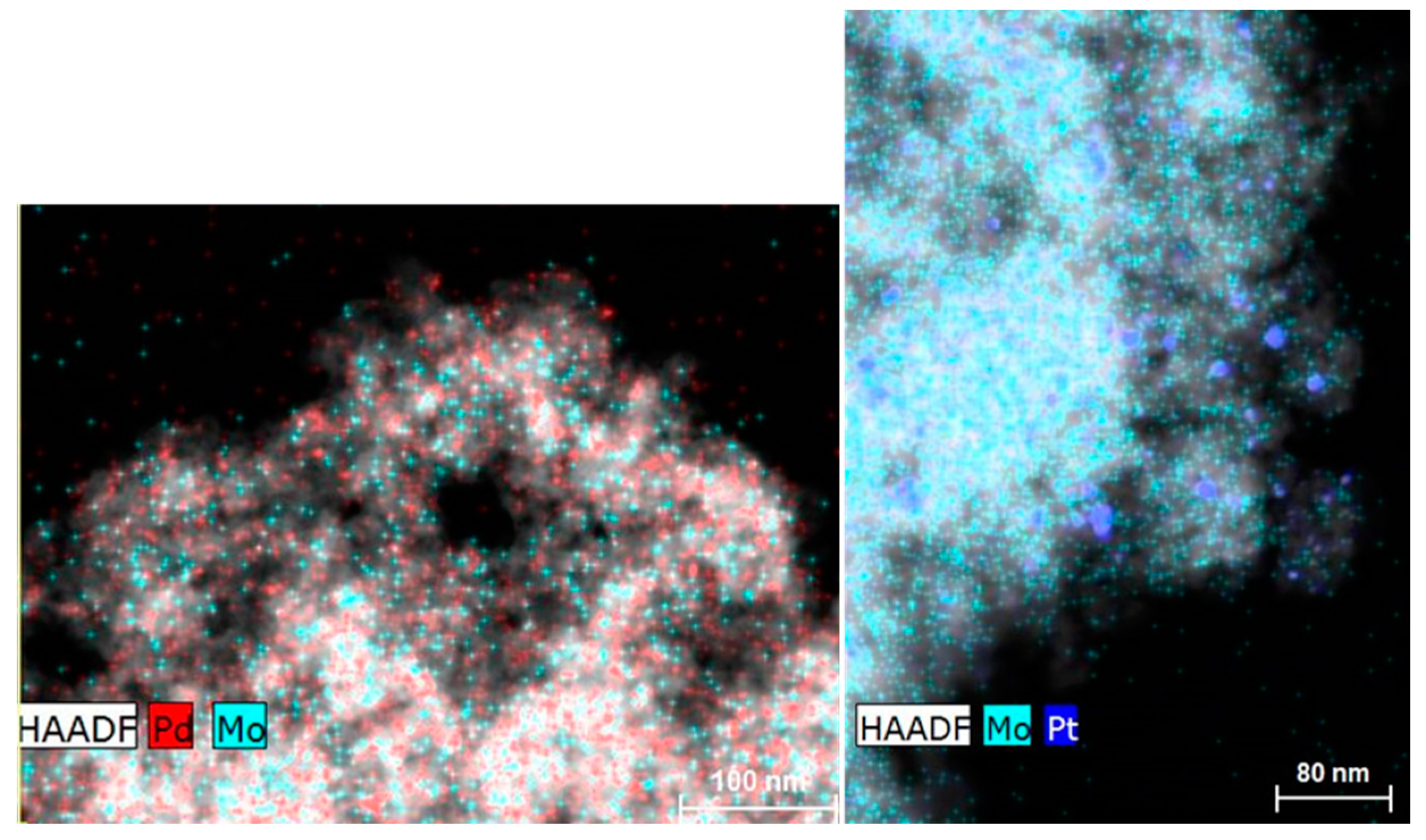
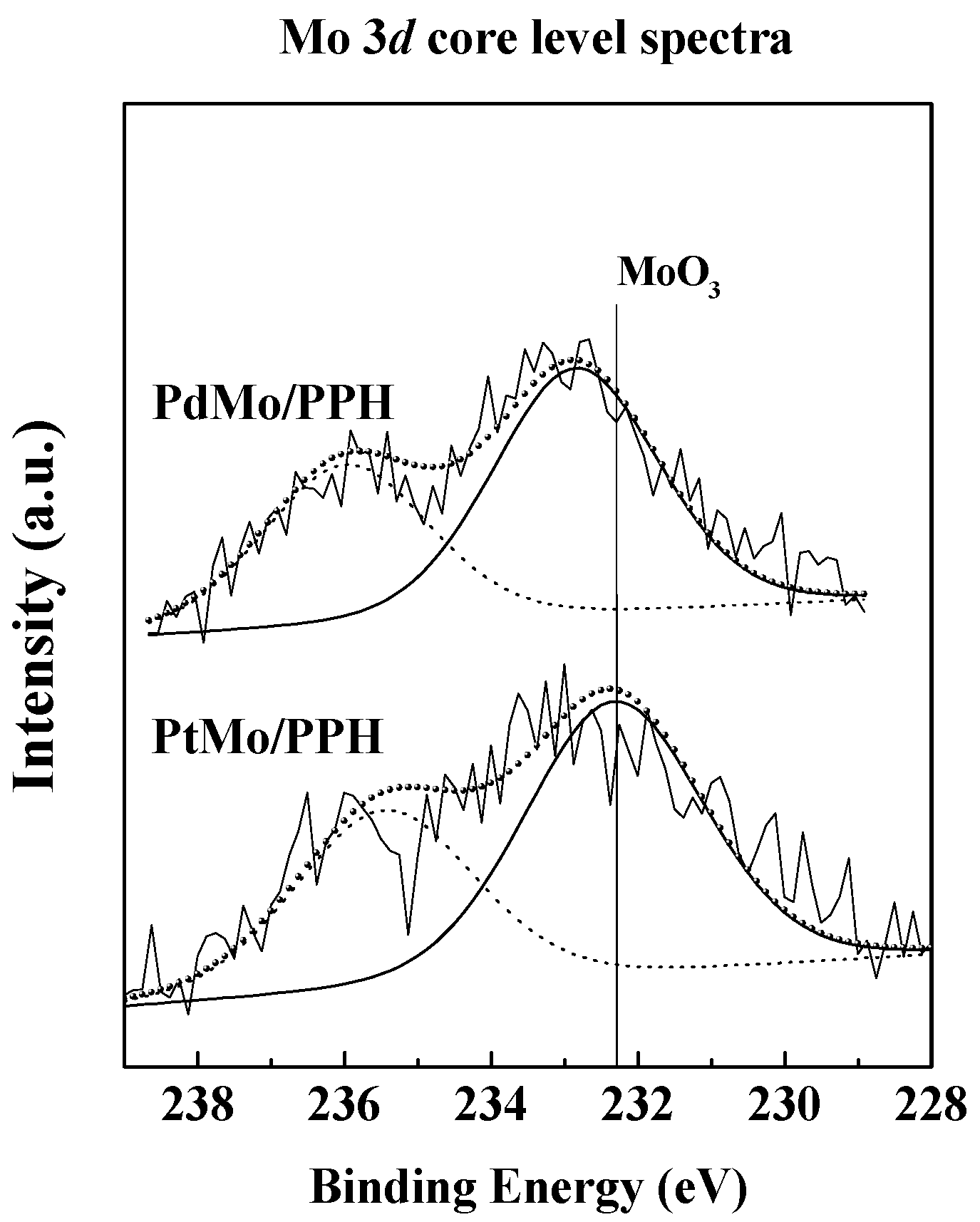
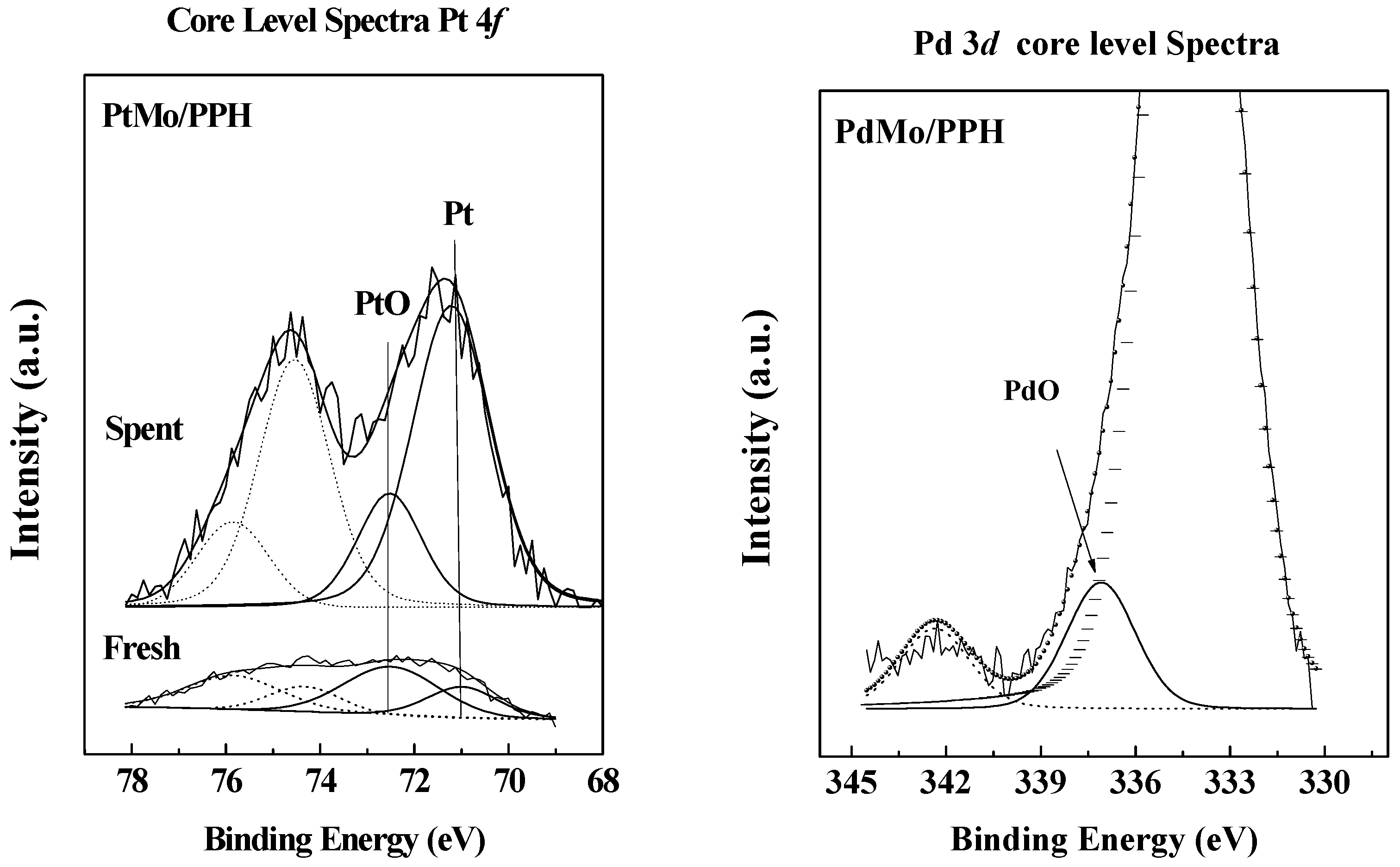
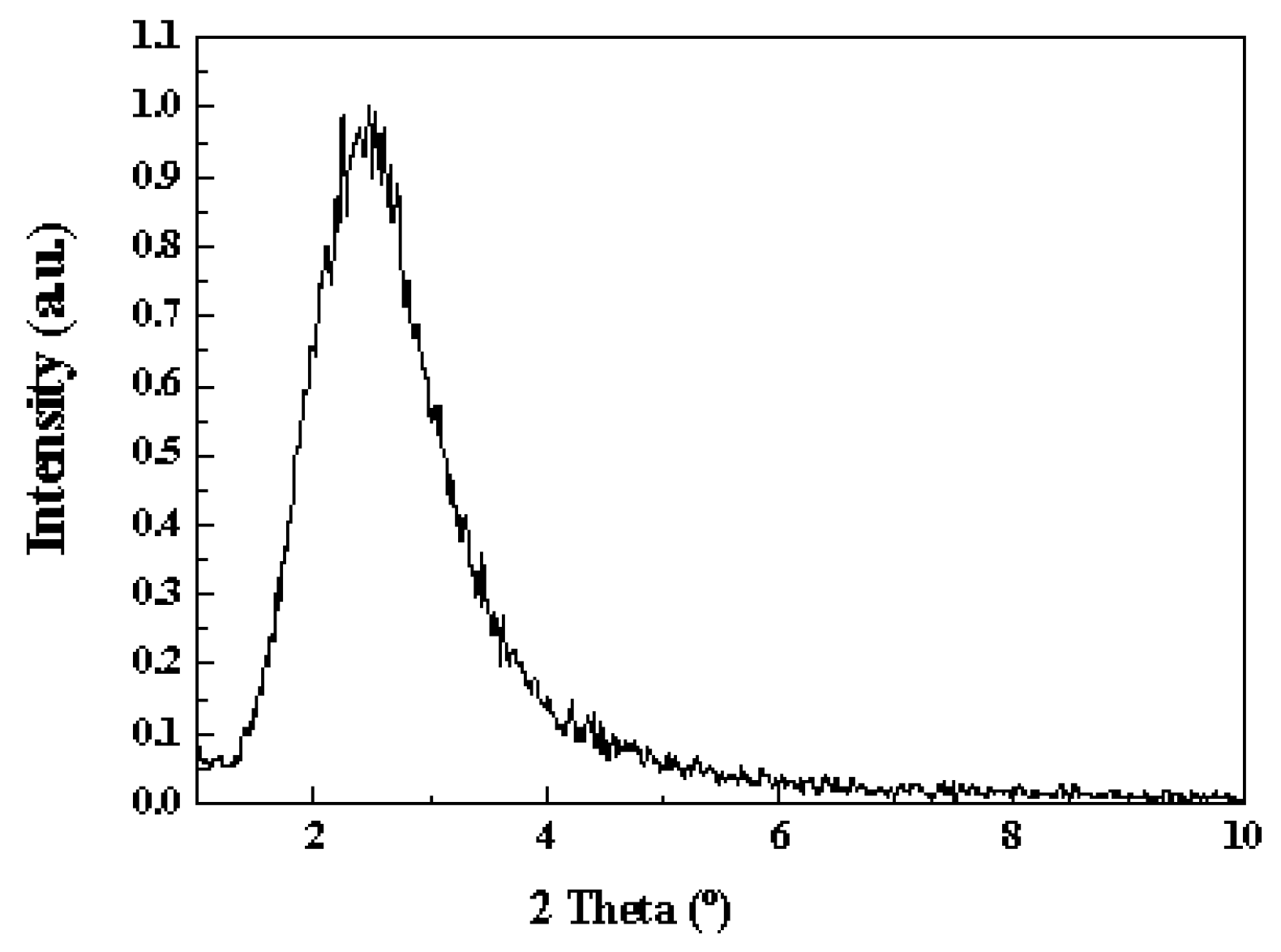
2.1. Catalyst Characterization
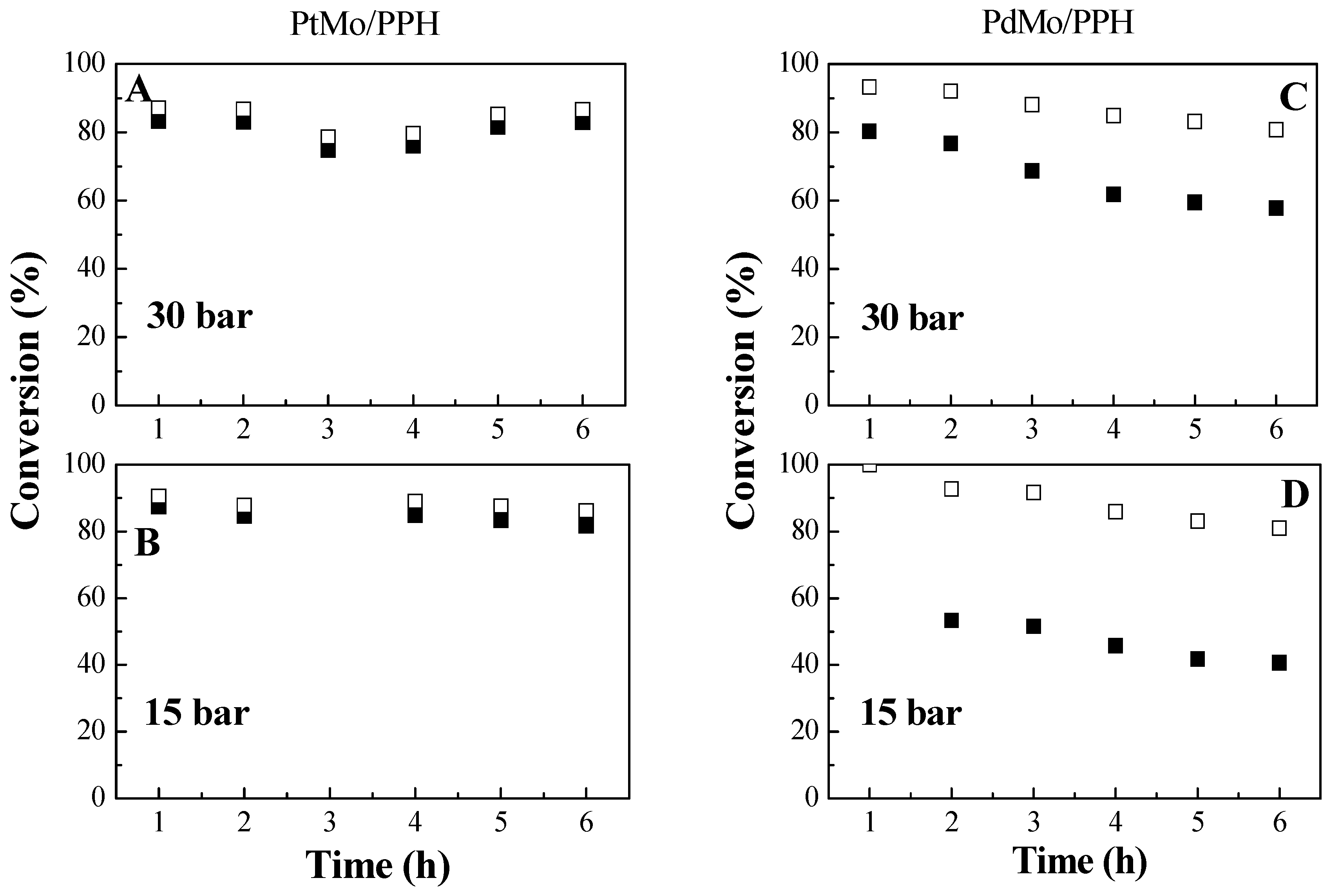
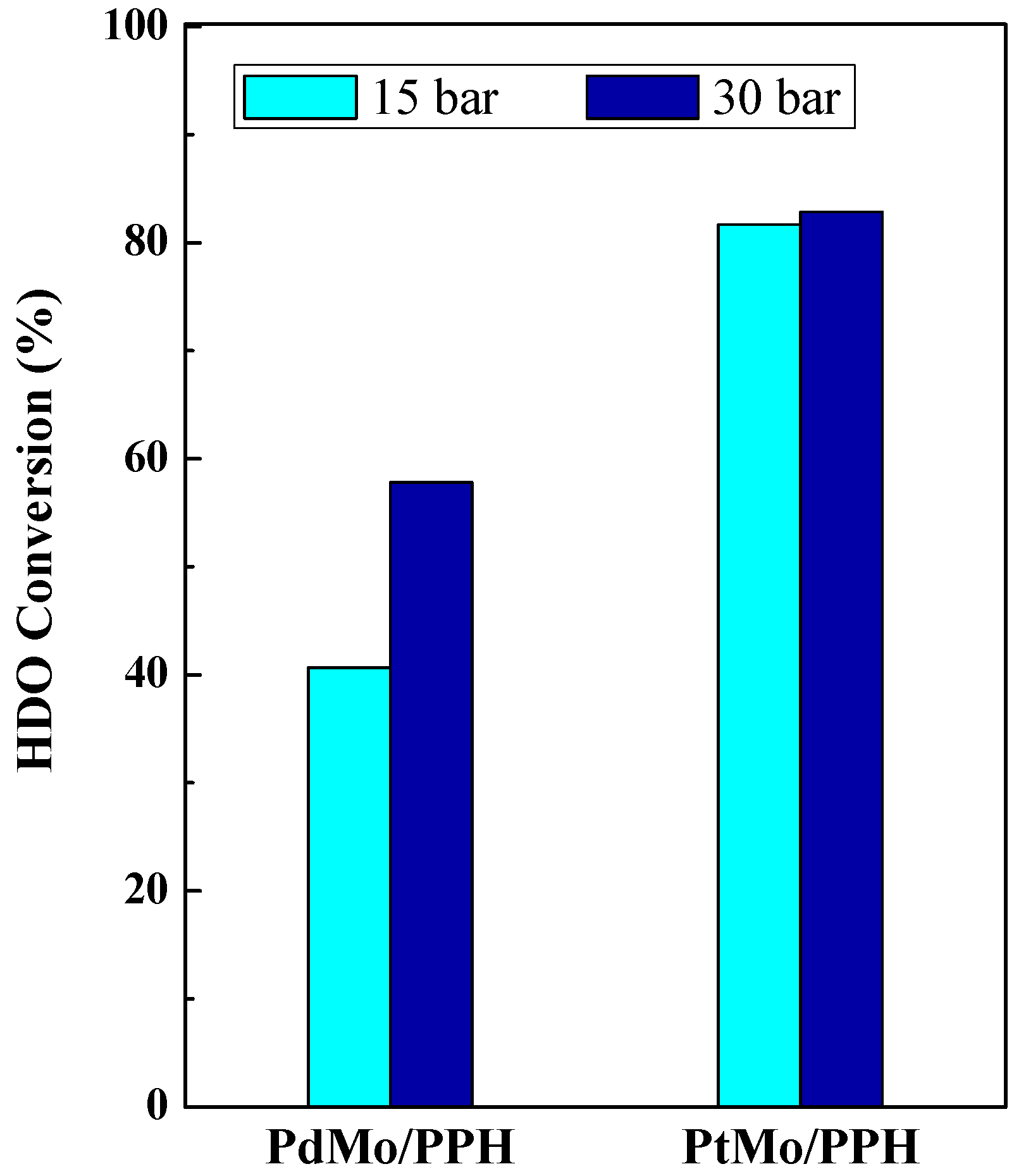
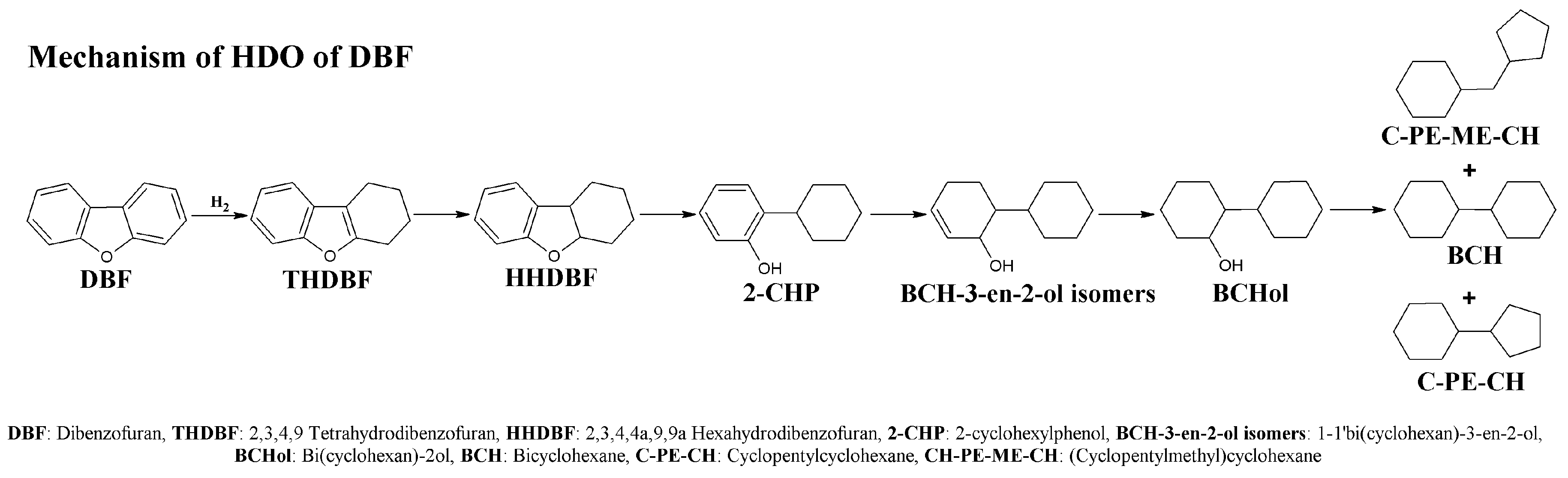
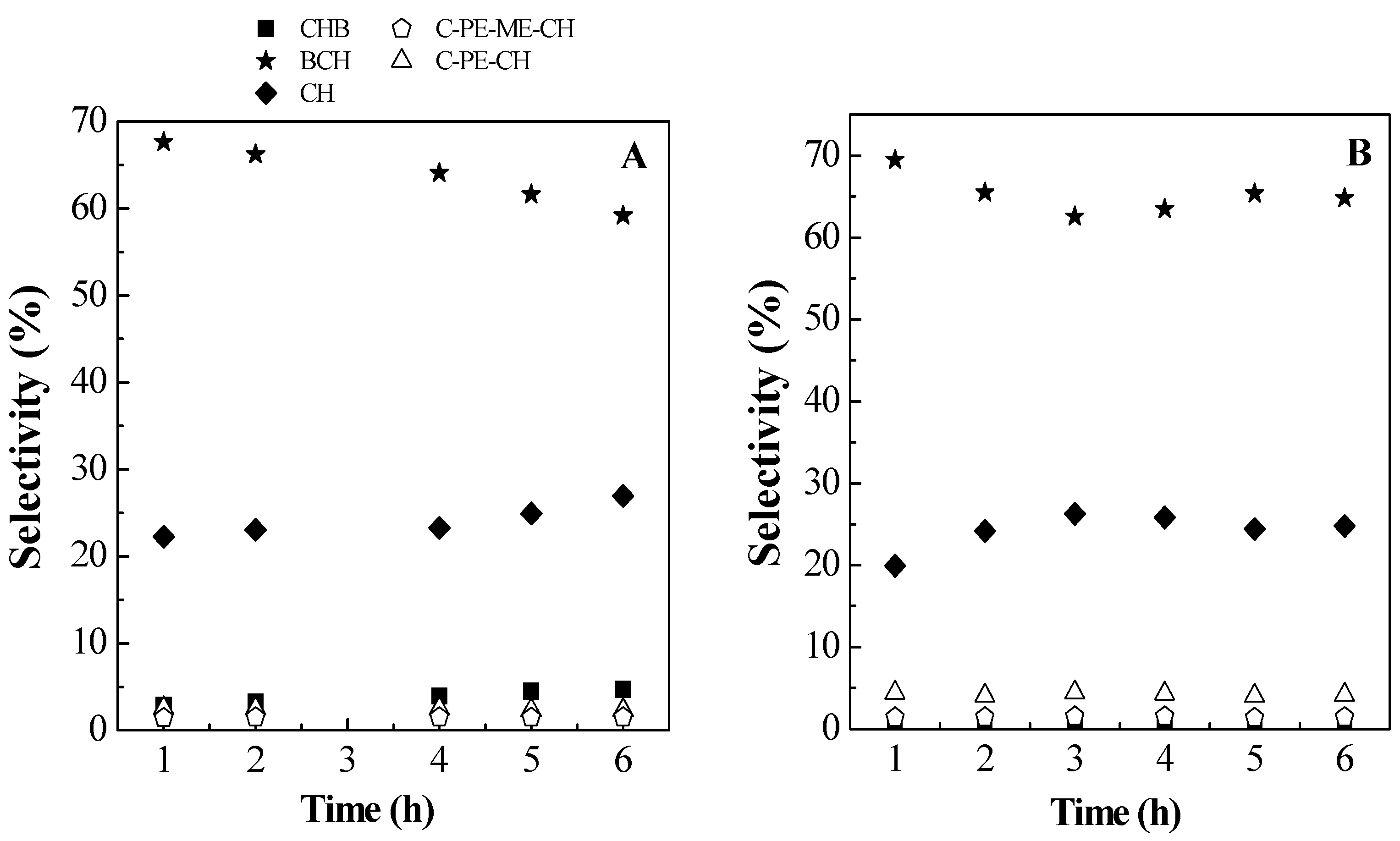
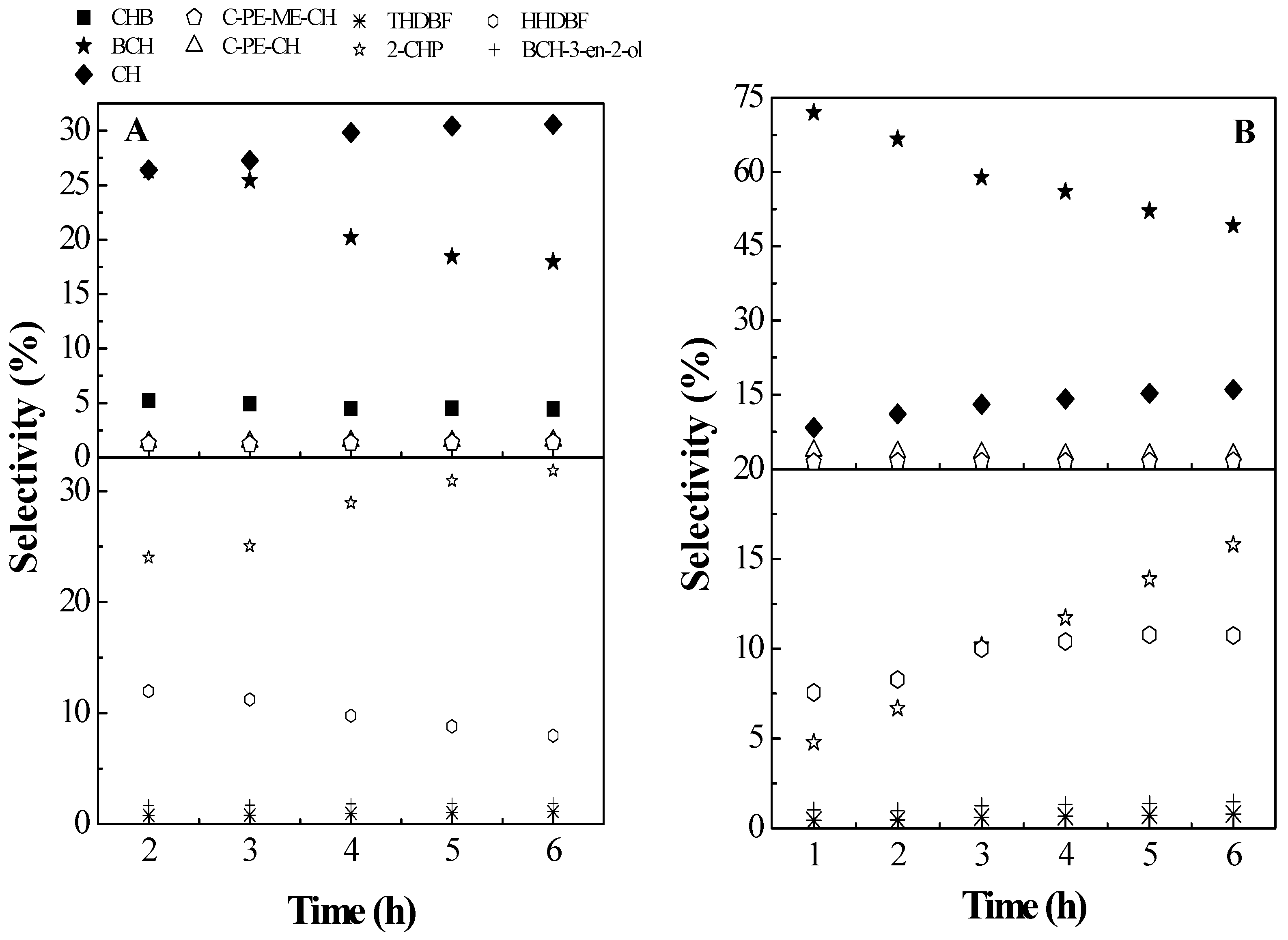
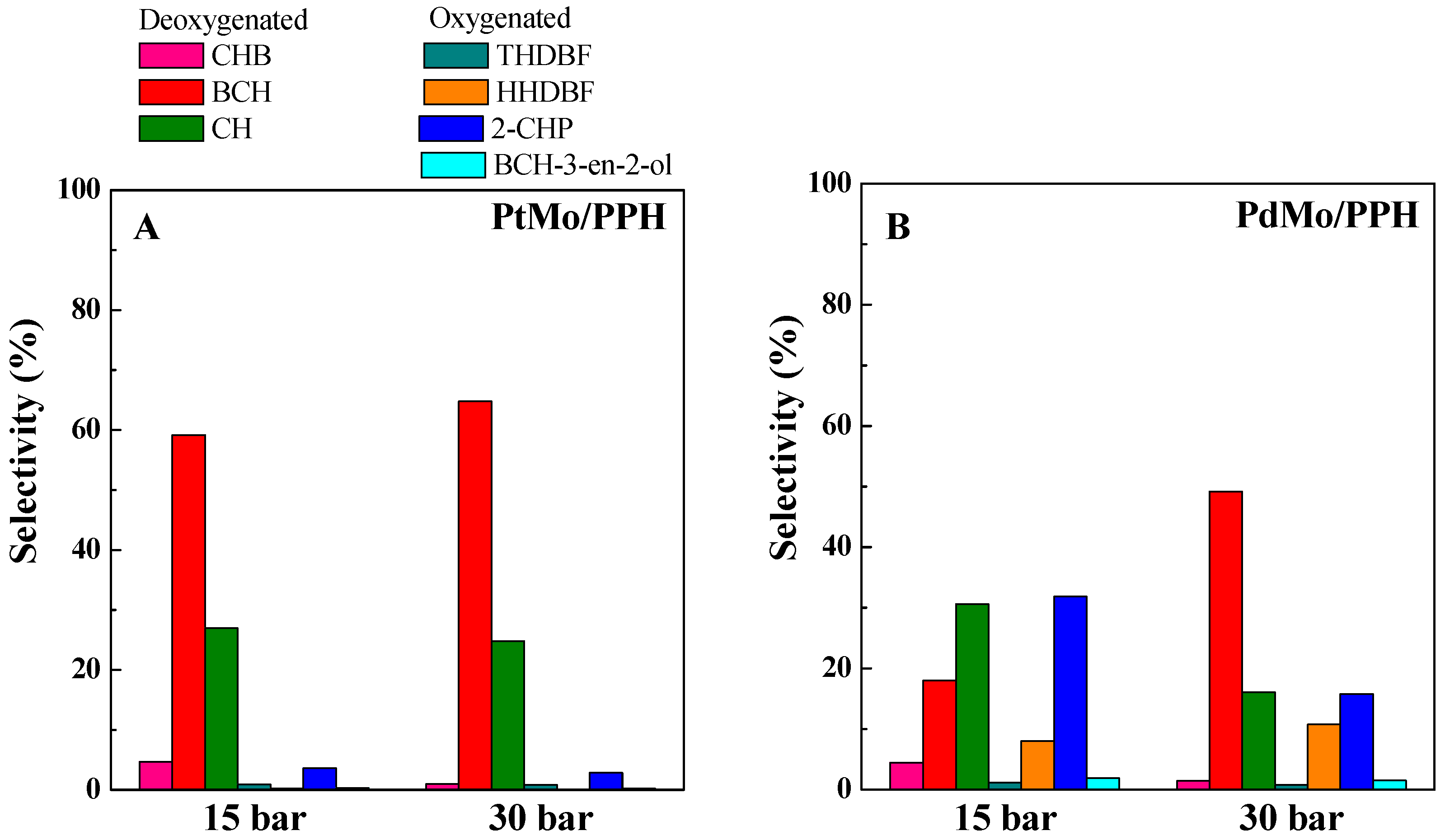
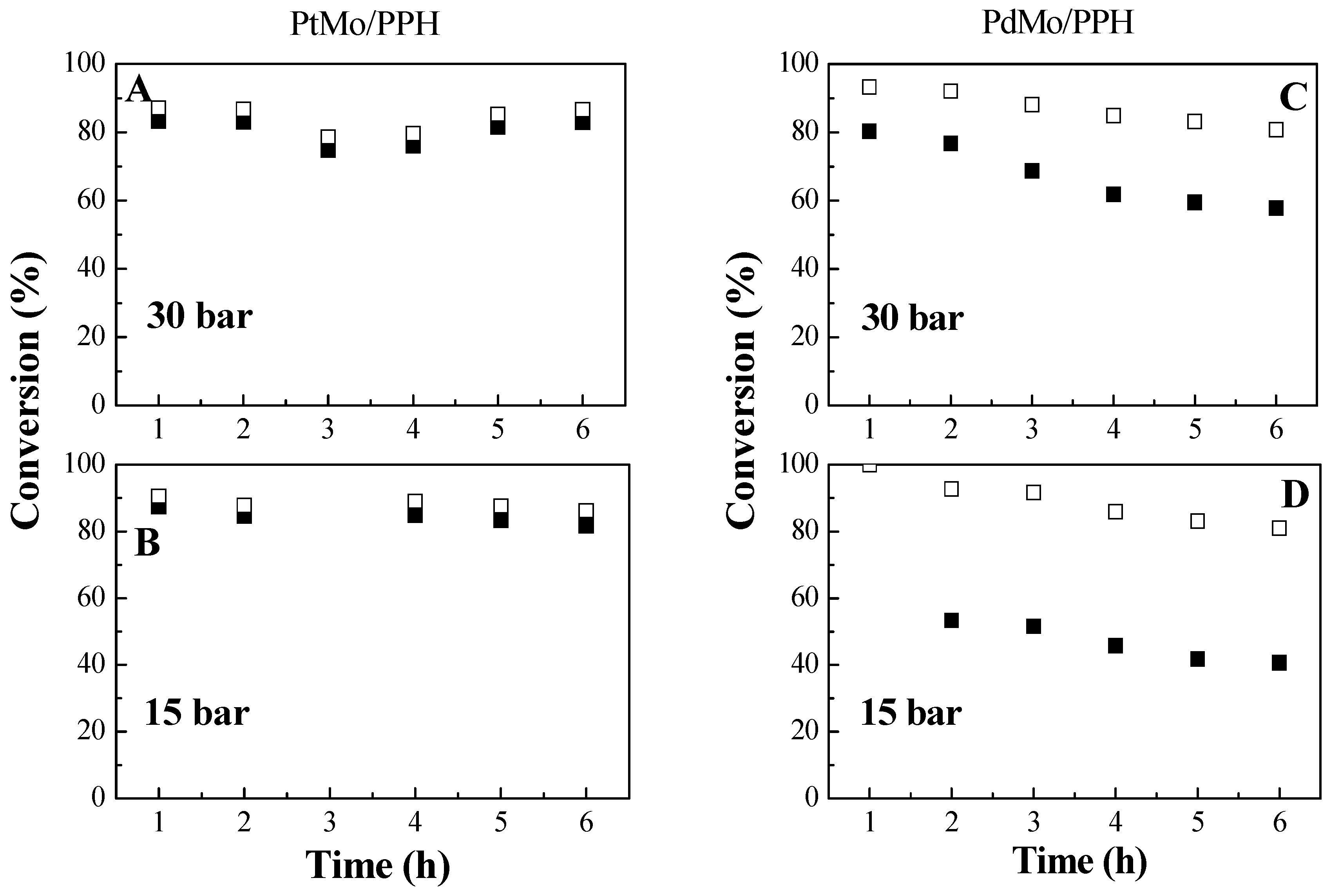
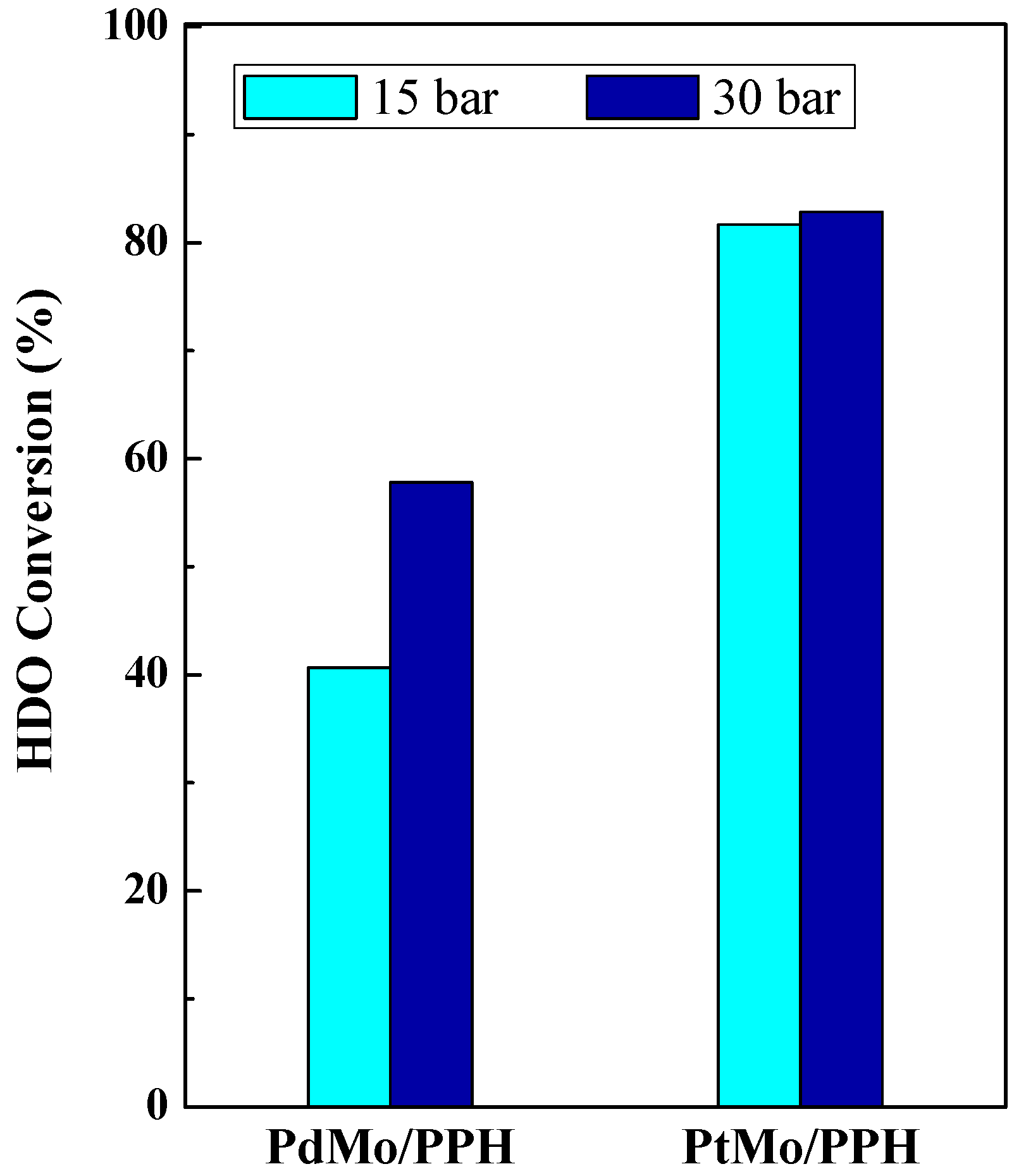
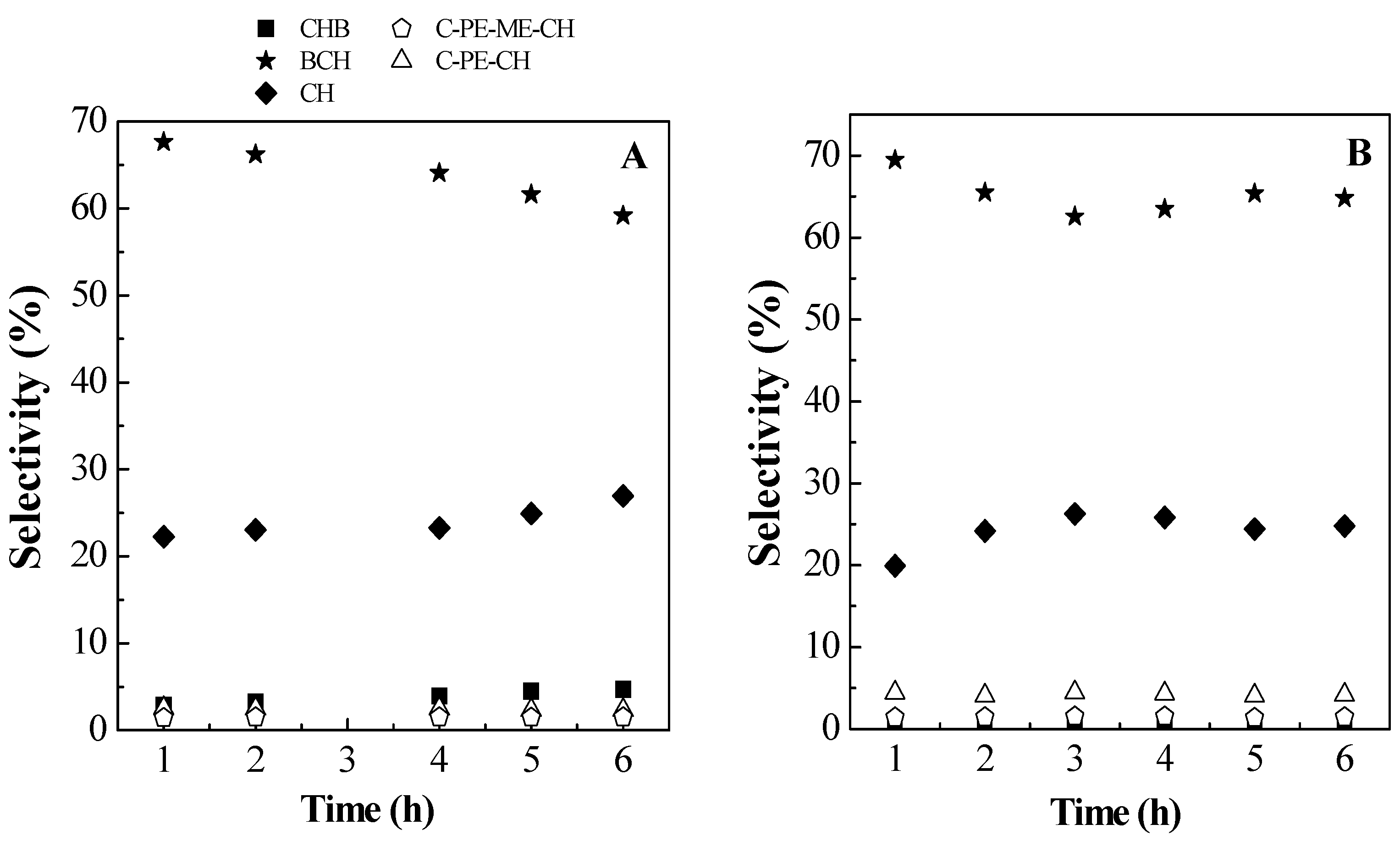
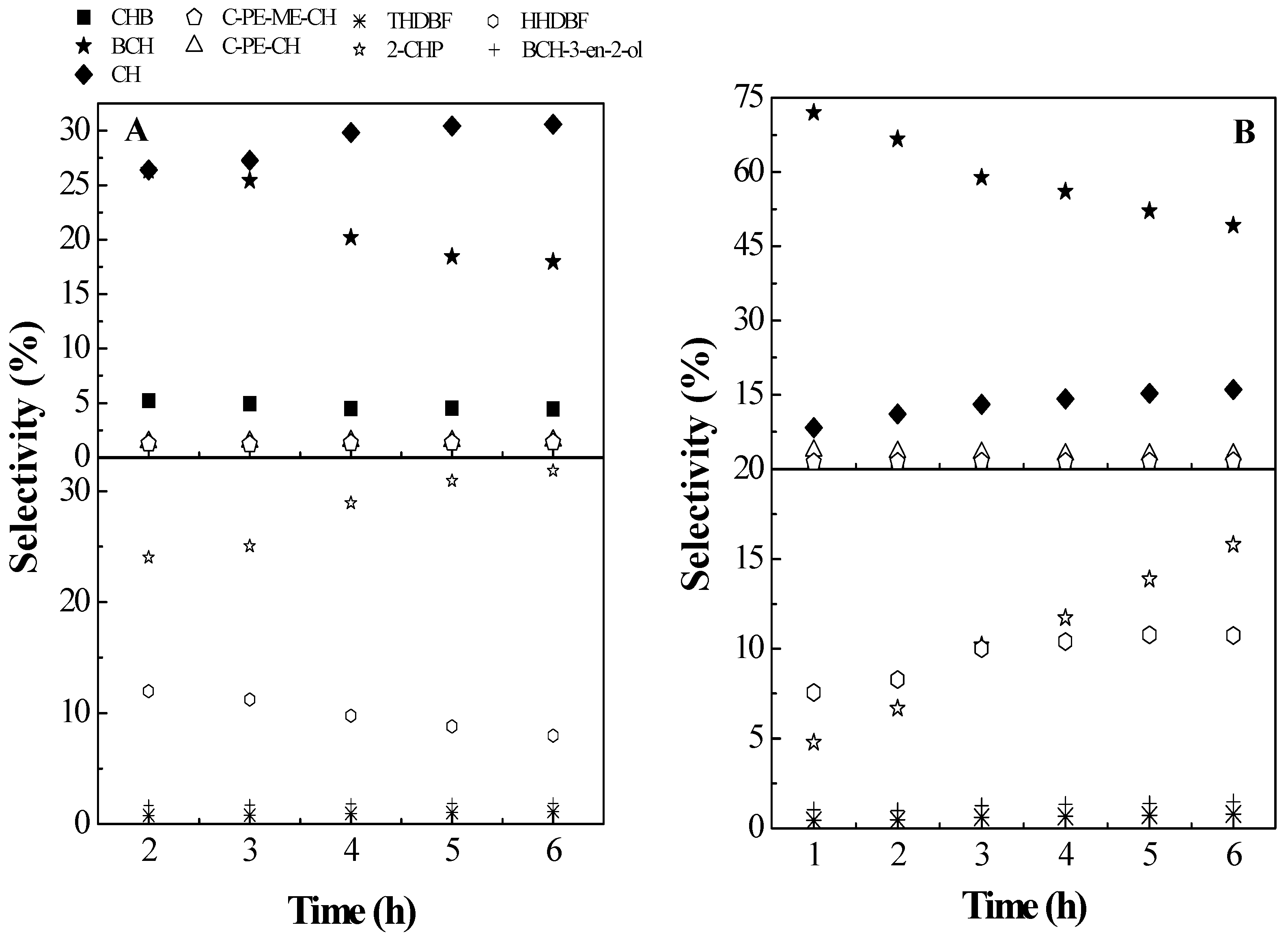
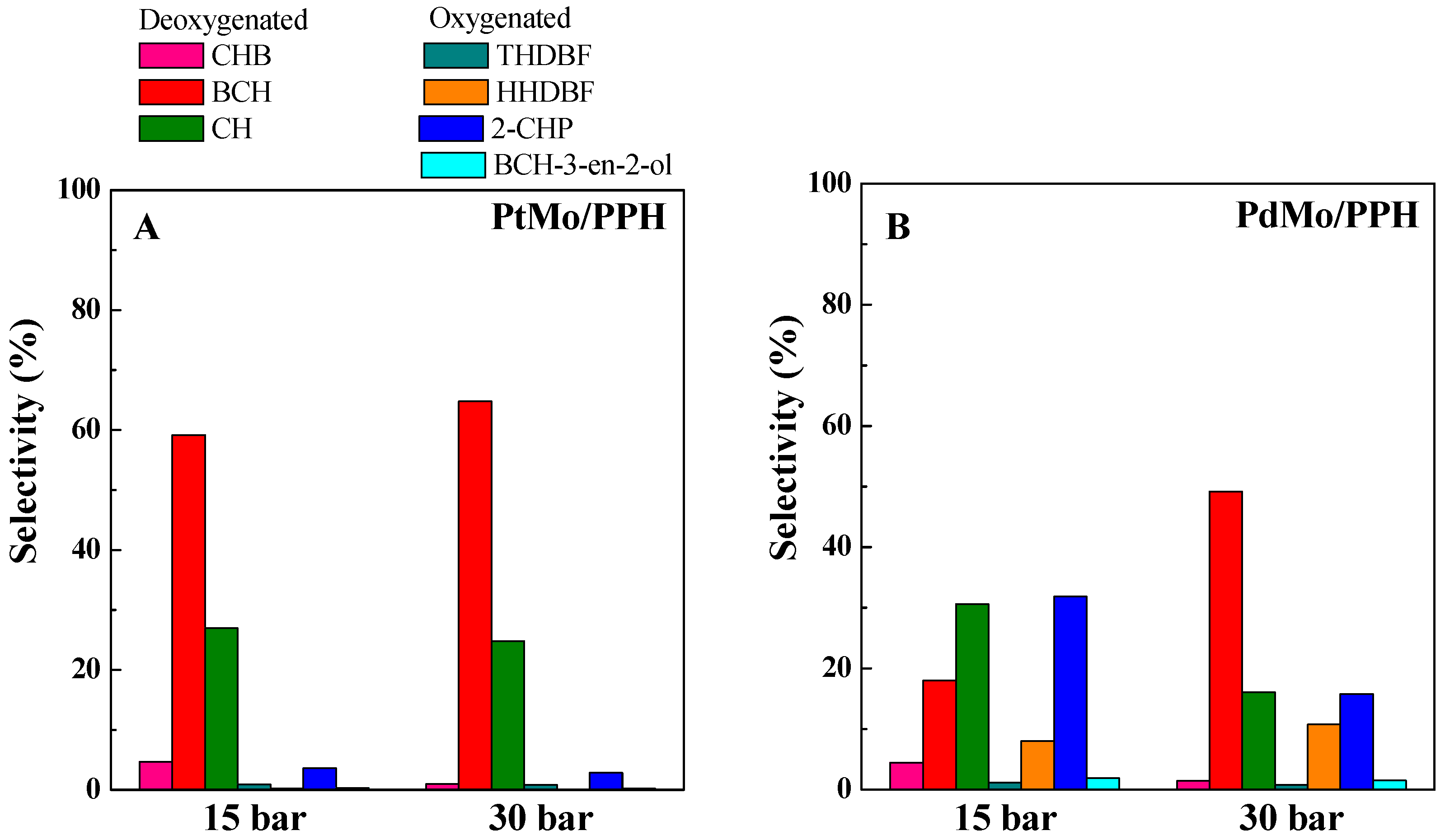
2.2. Catalytic Results
3. Materials and Methods
3.1. Preparation of Catalysts
3.2. Characterization of Catalysts
3.3. Catalytic Activity
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A
References
- Demirbaş, A. Biomass resource facilities and biomass conversion processing for fuels and chemicals. Energy Convers. Manag. 2001, 42, 1357–1378. [Google Scholar] [CrossRef]
- Infantes-Molina, A.; Moretti, E.; Segovia, E.; Lenarda, A.; Rodríguez-Castellón, E. Pd-nb binfunctional catalysts supported on silica and zirconium phosphate heterostructures for o-removal of dibenzofurane. Catal. Today 2016, 277, 143–151. [Google Scholar] [CrossRef]
- Bridgwater, A.V.; Meier, D.; Radlein, D. An overview of fast pyrolysis of biomass. Org. Geochem. 1999, 30, 1479–1493. [Google Scholar] [CrossRef]
- Boullosa-Eiras, S.; Lødeng, R.; Bergem, H.; Stöcker, M.; Hannevold, L.; Blekkan, E.A. Catalytic hydrodeoxygenation (hdo) of phenol over supported molybdenum carbide, nitride, phosphide and oxide catalysts. Catal. Today 2014, 223, 44–53. [Google Scholar] [CrossRef]
- Furimsky, E. Catalytic hydrodeoxygenation. Appl. Catal. A Gen. 2000, 199, 147–190. [Google Scholar] [CrossRef]
- Echeandia, S.; Pawelec, B.; Barrio, V.L.; Arias, P.L.; Cambra, J.F.; Loricera, C.V.; Fierro, J.L.G. Enhancement of phenol hydrodeoxygenation over pd catalysts supported on mixed hy zeolite and Al2O3. An approach to o-removal from bio-oils. Fuel 2014, 117, 1061–1073. [Google Scholar] [CrossRef]
- Han, Y.; McIlroy, D.N.; McDonald, A.G. Hydrodeoxygenation of pyrolysis oil for hydrocarbon production using nanospring based catalysts. J. Anal. Appl. Pyrolysis 2016, 117, 94–105. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Grunwaldt, J.D.; Jensen, P.A.; Knudsen, K.G.; Jensen, A.D. A review of catalytic upgrading of bio-oil to engine fuels. Appl. Catal. A Gen. 2011, 407, 1–19. [Google Scholar] [CrossRef]
- Lee, H.W.; Jun, B.R.; Kim, H.; Kim, D.H.; Jeon, J.-K.; Park, S.H.; Ko, C.H.; Kim, T.-W.; Park, Y.-K. Catalytic hydrodeoxygenation of 2-methoxy phenol and dibenzofuran over pt/mesoporous zeolites. Energy 2015, 81, 33–40. [Google Scholar] [CrossRef]
- Bu, Q.; Lei, H.; Zacher, A.H.; Wang, L.; Ren, S.; Liang, J.; Wei, Y.; Liu, Y.; Tang, J.; Zhang, Q.; et al. A review of catalytic hydrodeoxygenation of lignin-derived phenols from biomass pyrolysis. Bioresour. Technol. 2012, 124, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Kumar, A. Production of renewable diesel through the hydroprocessing of lignocellulosic biomass-derived bio-oil: A review. Renew. Sustain. Energy Rev. 2016, 58, 1293–1307. [Google Scholar] [CrossRef]
- De la Puente, G.; Gil, A.; Pis, J.J.; Grange, P. Effects of support surface chemistry in hydrodeoxygenation reactions over como/activated carbon sulfided catalysts. Langmuir 1999, 15, 5800–5806. [Google Scholar] [CrossRef]
- Dong, P.; Lu, G.-P.; Cai, C. Effective hydrodeoxygenation of dibenzofuran by a bimetallic catalyst in water. New J. Chem. 2016, 40, 1605–1609. [Google Scholar] [CrossRef]
- Yohe, S.L.; Choudhari, H.J.; Mehta, D.D.; Dietrich, P.J.; Detwiler, M.D.; Akatay, C.M.; Stach, E.A.; Miller, J.T.; Delgass, W.N.; Agrawal, R.; et al. High-pressure vapor-phase hydrodeoxygenation of lignin-derived oxygenates to hydrocarbons by a ptmo bimetallic catalyst: Product selectivity, reaction pathway, and structural characterization. J. Catal. 2016, 344, 535–552. [Google Scholar] [CrossRef]
- Ardiyanti, A.R.; Gutierrez, A.; Honkela, M.L.; Krause, A.O.I.; Heeres, H.J. Hydrotreatment of wood-based pyrolysis oil using zirconia-supported mono- and bimetallic (pt, pd, rh) catalysts. Appl. Catal. A Gen. 2011, 407, 56–66. [Google Scholar] [CrossRef]
- Gutierrez, A.; Kaila, R.K.; Honkela, M.L.; Slioor, R.; Krause, A.O.I. Hydrodeoxygenation of guaiacol on noble metal catalysts. Catal. Today 2009, 147, 239–246. [Google Scholar] [CrossRef]
- Wang, Y.; Fang, Y.; He, T.; Hu, H.; Wu, J. Hydrodeoxygenation of dibenzofuran over noble metal supported on mesoporous zeolite. Catal. Commun. 2011, 12, 1201–1205. [Google Scholar] [CrossRef]
- Wang, L.; Li, C.; Jin, S.; Li, W.; Liang, C. Hydrodeoxygenation of dibenzofuran over sba-15 supported pt, pd, and ru catalysts. Catal. Lett. 2014, 144, 809–816. [Google Scholar] [CrossRef]
- Halasz, I.; Brenner, A.; Shelef, M. Catalytic reduction of nitric oxide on pdo—MoO3/γ-Al2O3. Appl. Catal. B Environ. 1993, 2, 131–146. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, M.; Zhang, M.; Sha, G.; Liang, C. Hydrodeoxygenation of dibenzofuran over mesoporous silica cok-12 supported palladium catalysts. Energy Fuels 2013, 27, 2209–2217. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Grunwaldt, J.-D.; Jensen, P.A.; Jensen, A.D. Screening of catalysts for hydrodeoxygenation of phenol as a model compound for bio-oil. ACS Catal. 2013, 3, 1774–1785. [Google Scholar] [CrossRef]
- Shetty, M.; Murugappan, K.; Prasomsri, T.; Green, W.H.; Román-Leshkov, Y. Reactivity and stability investigation of supported molybdenum oxide catalysts for the hydrodeoxygenation (hdo) of m-cresol. J. Catal. 2015, 331, 86–97. [Google Scholar] [CrossRef]
- Nolte, M.W.; Zhang, J.; Shanks, B.H. Ex situ hydrodeoxygenation in biomass pyrolysis using molybdenum oxide and low pressure hydrogen. Green Chem. 2016, 18, 134–138. [Google Scholar] [CrossRef]
- Prasomsri, T.; Nimmanwudipong, T.; Román-Leshkov, Y. Effective hydrodeoxygenation of biomass-derived oxygenates into unsaturated hydrocarbons by MoO3 using low H2 pressures. Energy Environ. Sci. 2013, 6, 1732. [Google Scholar] [CrossRef]
- Prasomsri, T.; Shetty, M.; Murugappan, K.; Román-Leshkov, Y. Insights into the catalytic activity and surface modification of MoO3 during the hydrodeoxygenation of lignin-derived model compounds into aromatic hydrocarbons under low hydrogen pressures. Energy Environ. Sci. 2014, 7, 2660. [Google Scholar] [CrossRef]
- Robinson, A.; Ferguson, G.A.; Gallagher, J.R.; Cheah, S.; Beckham, G.T.; Schaidle, J.A.; Hensley, J.E.; Medlin, J.W. Enhanced hydrodeoxygenation ofm-cresol over bimetallic pt–mo catalysts through an oxophilic metal-induced tautomerization pathway. ACS Catal. 2016, 6, 4356–4368. [Google Scholar] [CrossRef]
- Ardiyanti, A.R.; Khromova, S.A.; Venderbosch, R.H.; Yakovlev, V.A.; Melián-Cabrera, I.V.; Heeres, H.J. Catalytic hydrotreatment of fast pyrolysis oil using bimetallic ni–cu catalysts on various supports. Appl. Catal. A Gen. 2012, 449, 121–130. [Google Scholar] [CrossRef]
- Bui, V.N.; Laurenti, D.; Delichère, P.; Geantet, C. Hydrodeoxygenation of guaiacol. Appl. Catal. B Environ. 2011, 101, 246–255. [Google Scholar] [CrossRef]
- Yan, K.; Liu, Y.; Lu, Y.; Chai, J.; Sun, L. Catalytic application of layered double hydroxide-derived catalysts for the conversion of biomass-derived molecules. Catal. Sci. Technol. 2017, 7, 1622–1645. [Google Scholar] [CrossRef]
- Jiménez-Jiménez, J.; Rubio-Alonso, M.; Quesada, D.E.; Rodríguez-Castellón, E.; Jiménez-López, A. Synthesis and characterisation of acid mesoporous phosphate heterostructure (PPH) materials. J. Mater. Chem. 2005, 15, 3466. [Google Scholar] [CrossRef]
- Eliche-Quesada, D.; Macías-Ortiz, M.I.; Jiménez-Jiménez, J.; Rodríguez-Castellón, E.; Jiménez-López, A. Catalysts based on ru/mesoporous phosphate heterostructures (PPH) for hydrotreating of aromatic hydrocarbons. J. Mol. Catal. A Chem. 2006, 255, 41–48. [Google Scholar] [CrossRef]
- León, M.; Jiménez-Jiménez, J.; Jiménez-López, A.; Rodríguez-Castellón, E.; Soriano, D.; López Nieto, J.M. Vanadium oxide-porous phosphate heterostructure catalysts for the selective oxidation of h2s to sulphur. Solid State Sci. 2010, 12, 996–1001. [Google Scholar] [CrossRef]
- Infantes-Molina, A.; Mérida-Robles, J.; Rodríguez-Castellón, E.; Fierro, J.L.G.; Jiménez-López, A. Effect of molybdenum and tungsten on co/msu as hydrogenation catalysts. J. Catal. 2006, 240, 258–267. [Google Scholar] [CrossRef]
- Kozlova, E.A.; Lyubina, T.P.; Nasalevich, M.A.; Vorontsov, A.V.; Miller, A.V.; Kaichev, V.V.; Parmon, V.N. Influence of the method of platinum deposition on activity and stability of pt/TiO2 photocatalysts in the photocatalytic oxidation of dimethyl methylphosphonate. Catal. Commun. 2011, 12, 597–601. [Google Scholar] [CrossRef]
- Beketov, G.; Heinrichs, B.; Pirard, J.P.; Chenakin, S.; Kruse, N. Xps structural characterization of pd/SiO2 catalysts prepared by cogelation. Appl. Surf. Sci. 2013, 287, 293–298. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SBET (m2 g−1) a | VP (m3 g−1) b | dp (nm) c |
|---|---|---|---|
| PPH | 533 | 0.65 | 4.9 |
| PdMo/PPH | 388 | 0.53 | 5.5 |
| PtMo/PPH | 504 | 0.73 | 5.8 |
| Sample | Acidity (µmol NH3 g−1) | Acidity (µmol NH3 m−2) | |||
|---|---|---|---|---|---|
| Weak a | Average b | Strong c | Total | ||
| PPH | 494 | 285 | 38 | 817 | 1.53 |
| PdMo/PPH | 798 | 496 | 164 | 1459 | 3.76 |
| PtMo/PPH | 1070 | 553 | 135 | 1759 | 3.49 |
| Catalysts | Binding Energy (eV) | Atomic Surface Relations | |||||
|---|---|---|---|---|---|---|---|
| Pd 3d5/2 Pt 4f7/2 | Mo 3d5/2 | Pd(Pt)/Mo | Pd(Pt)/Sup | Mo/Sup | |||
| M° | MOx | Mo(VI) | |||||
| PdMo/PPH | F | - | 337.1 | 232.8 | 0.375 | 0.0020 | 0.0118 |
| U | - | - | - | - | - | - | |
| PtMo/PPH | F | 70.9 | 72.5 | 232.1 | 0.625 | 0.0031 | 0.0131 |
| U | 71.2 | 72.5 | 232.5 | 0.692 | 0.0051 | 0.0074 | |
| Conversion (%) | Selectivity (%) | ||||
|---|---|---|---|---|---|
| Sample | HDO | Total | BCH | CH | CHB |
| PtMo/PPH | 81.66 | 86.21 | 59.2 | 26.9 | 4.7 |
| PtMo/SiO2 | 74.98 | 84.21 | 69.0 | 0.5 | 12.3 |
| PdMo/PPH | 40.6 | 80.9 | 18.0 | 30.6 | 4.4 |
| PdMo/SiO2 | 56.5 | 90.9 | 46.1 | 0.5 | 11.4 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ballesteros-Plata, D.; Infantes-Molina, A.; Rodríguez-Aguado, E.; Braos-García, P.; Jiménez-Jiménez, J.; Rodríguez-Castellón, E. Zirconium Phosphate Heterostructures as Catalyst Support in Hydrodeoxygenation Reactions. Catalysts 2017, 7, 176. https://doi.org/10.3390/catal7060176
Ballesteros-Plata D, Infantes-Molina A, Rodríguez-Aguado E, Braos-García P, Jiménez-Jiménez J, Rodríguez-Castellón E. Zirconium Phosphate Heterostructures as Catalyst Support in Hydrodeoxygenation Reactions. Catalysts. 2017; 7(6):176. https://doi.org/10.3390/catal7060176
Chicago/Turabian StyleBallesteros-Plata, Daniel, Antonia Infantes-Molina, Elena Rodríguez-Aguado, Pilar Braos-García, José Jiménez-Jiménez, and Enrique Rodríguez-Castellón. 2017. "Zirconium Phosphate Heterostructures as Catalyst Support in Hydrodeoxygenation Reactions" Catalysts 7, no. 6: 176. https://doi.org/10.3390/catal7060176
APA StyleBallesteros-Plata, D., Infantes-Molina, A., Rodríguez-Aguado, E., Braos-García, P., Jiménez-Jiménez, J., & Rodríguez-Castellón, E. (2017). Zirconium Phosphate Heterostructures as Catalyst Support in Hydrodeoxygenation Reactions. Catalysts, 7(6), 176. https://doi.org/10.3390/catal7060176