
The Role of Ruthenium in CO2 Capture and Catalytic Conversion to Fuel by Dual Function Materials (DFM)


Abstract
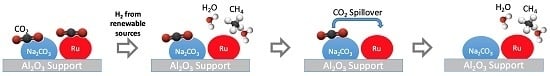
:
1. Introduction
2. Results and Discussion
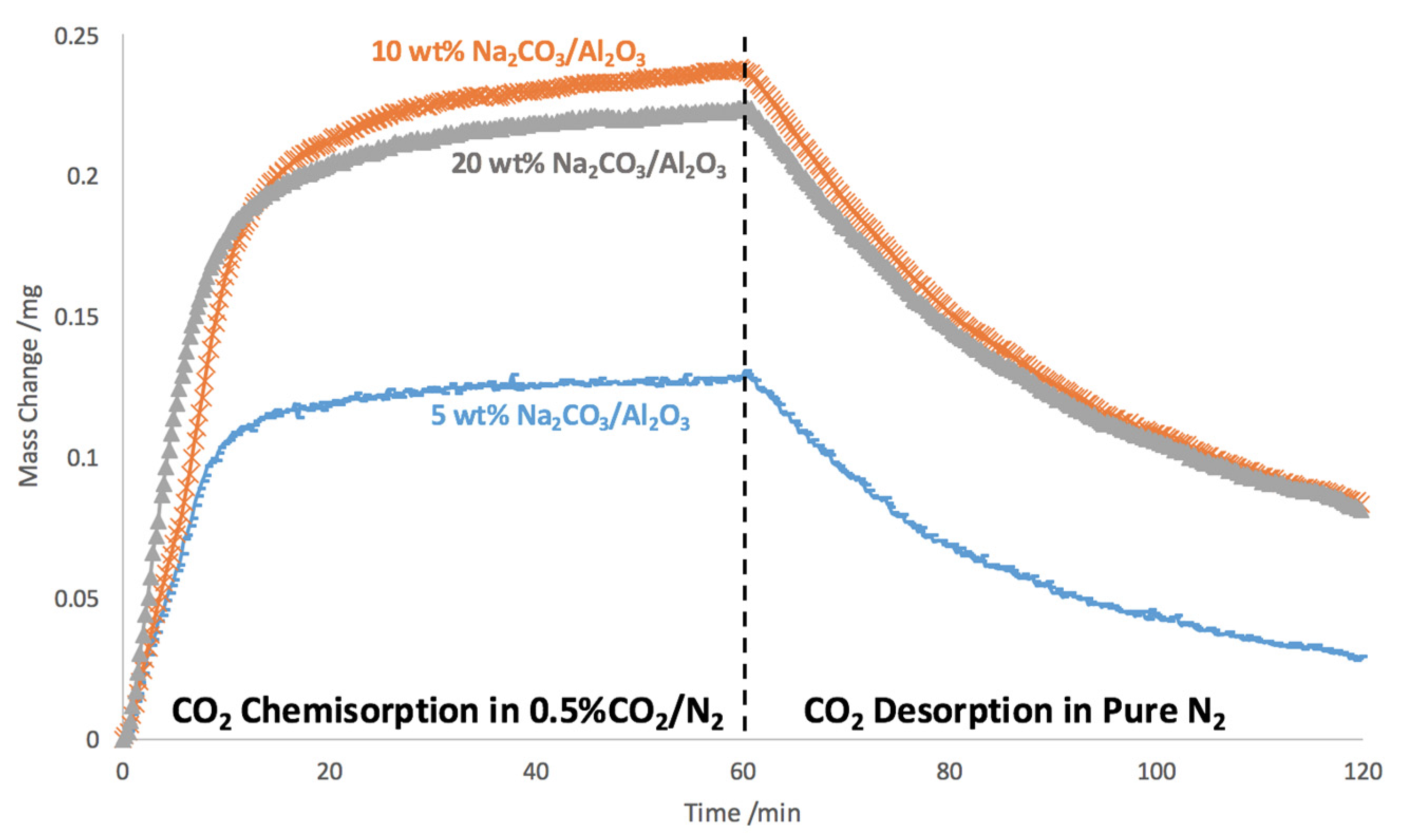
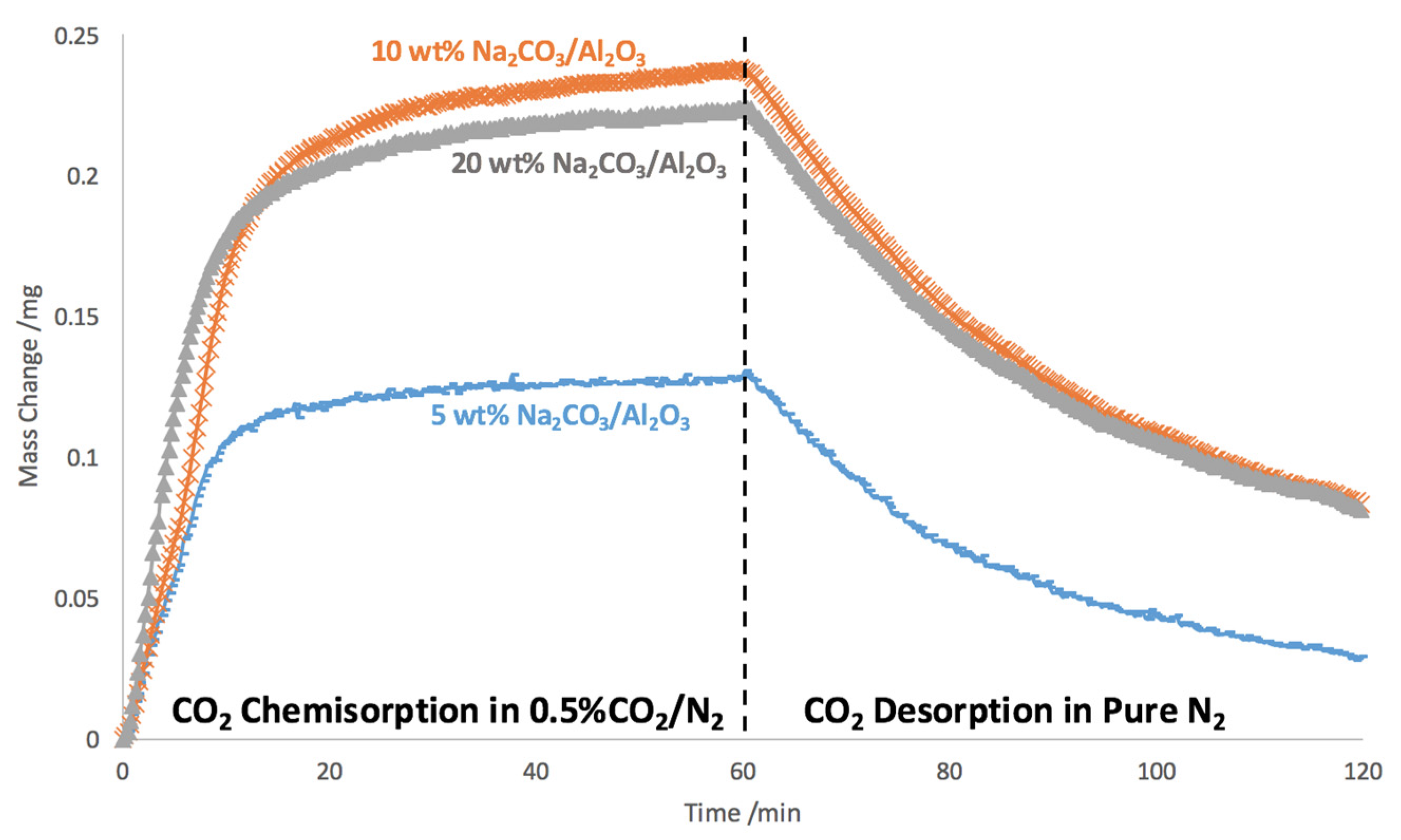
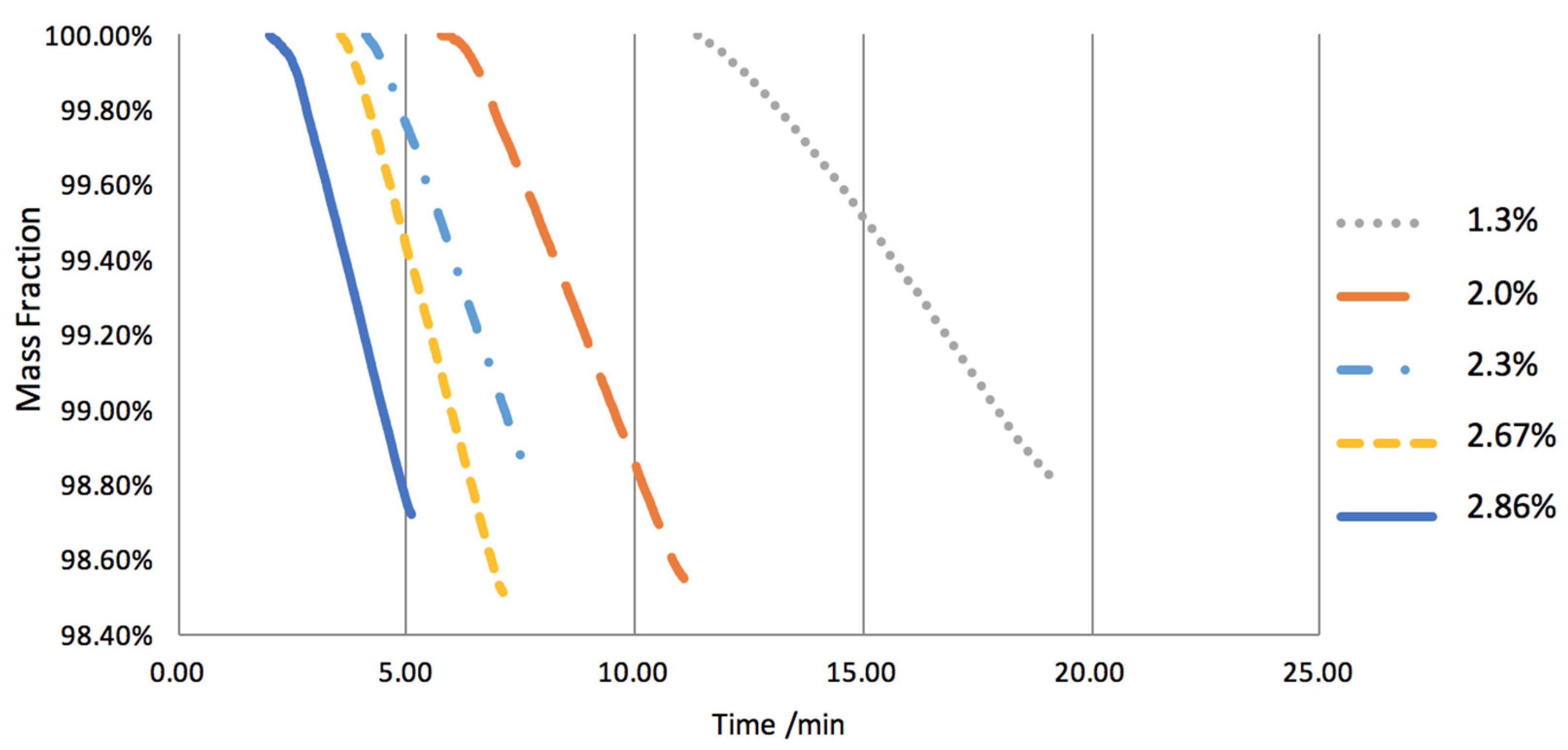
2.1. Na2CO3 Loading Study
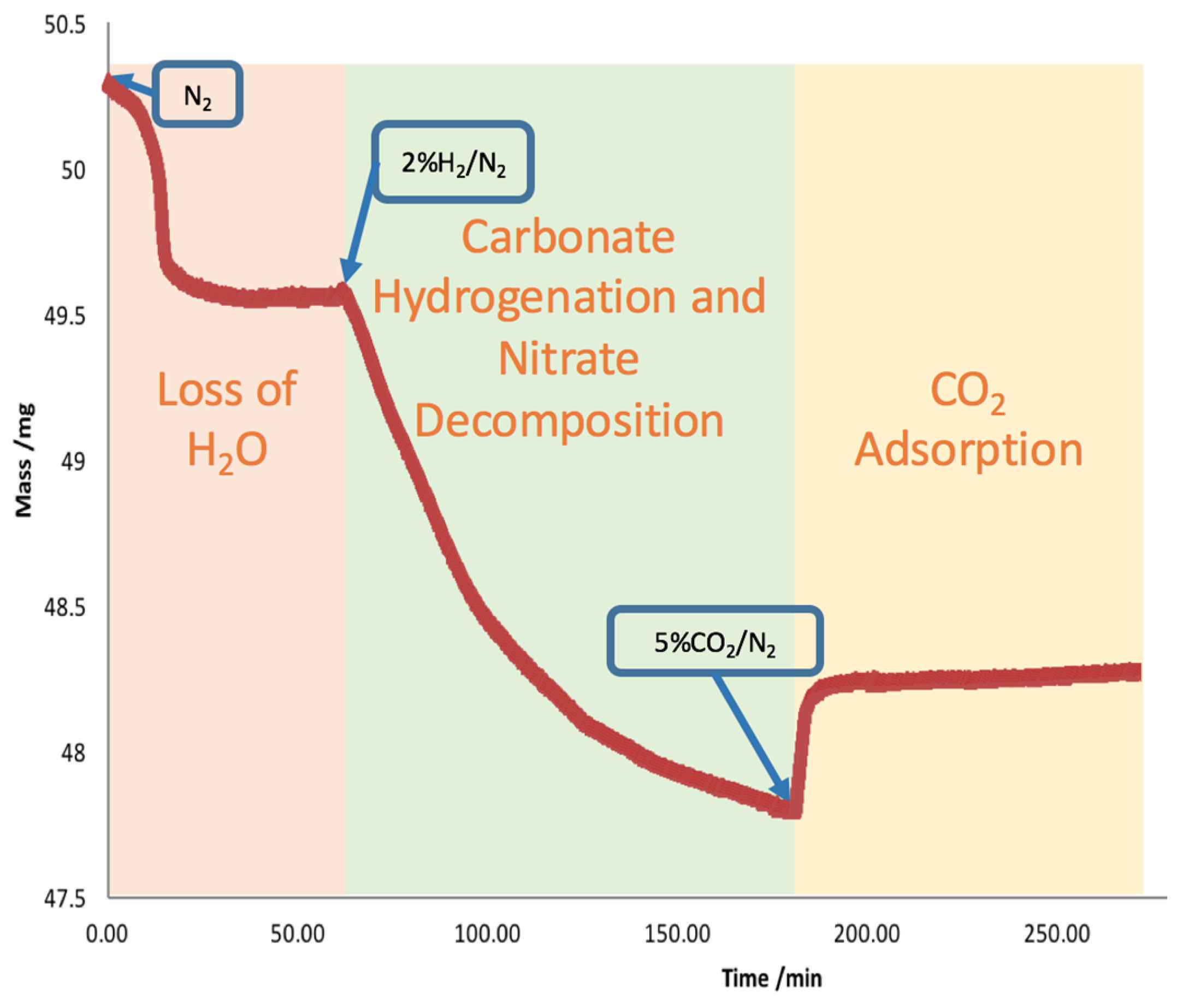
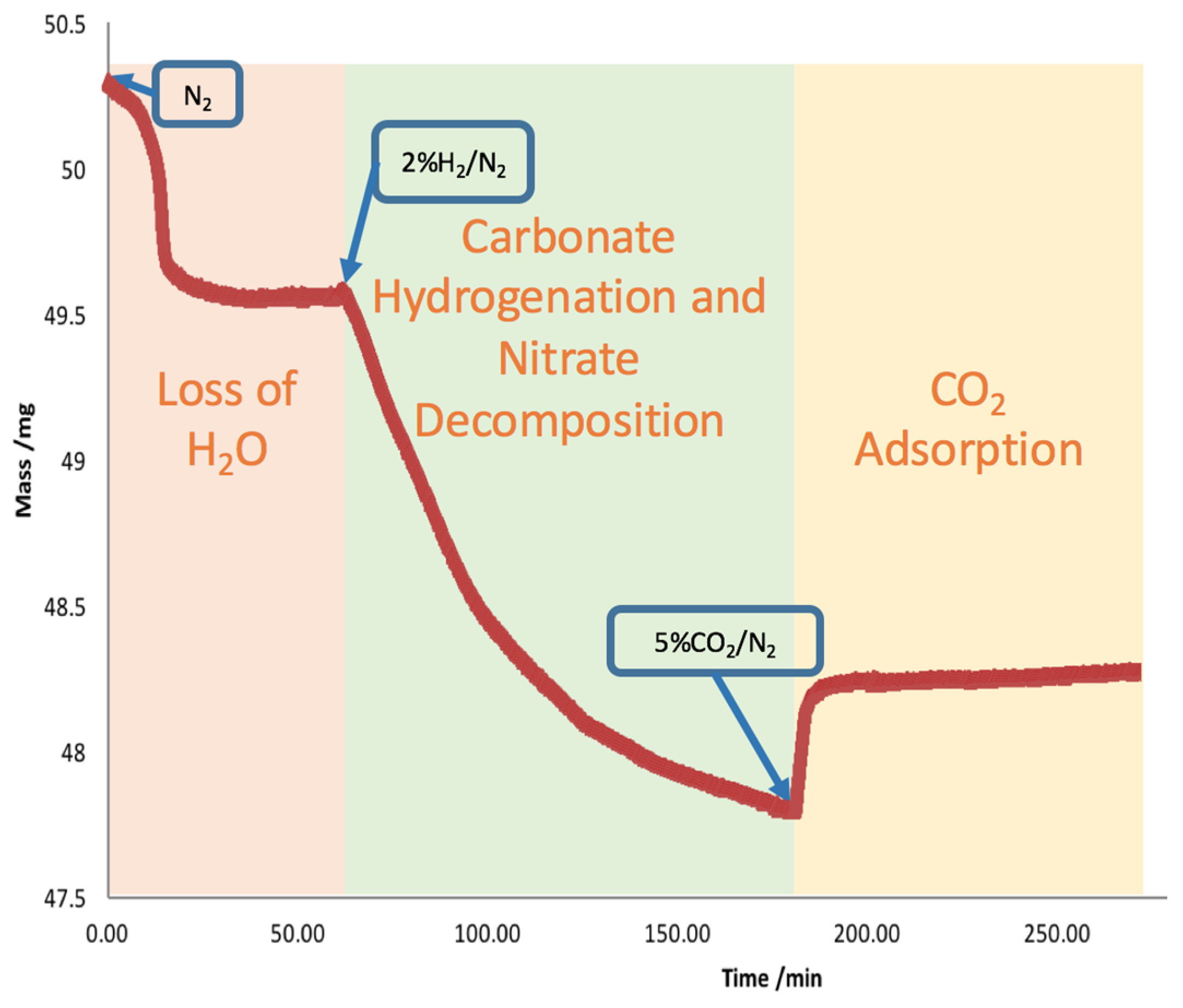
2.2. Carbonate Hydrogenation Study
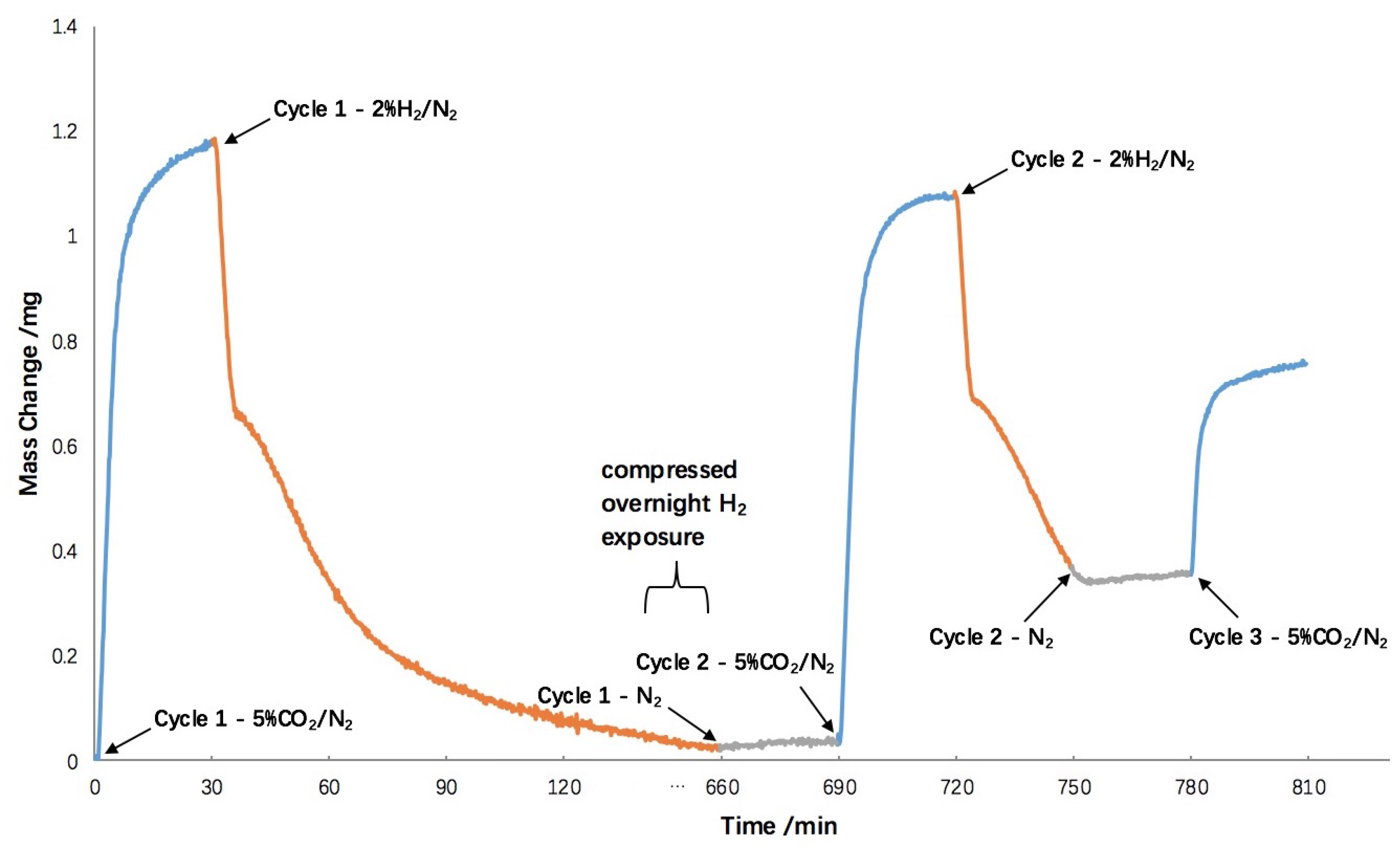
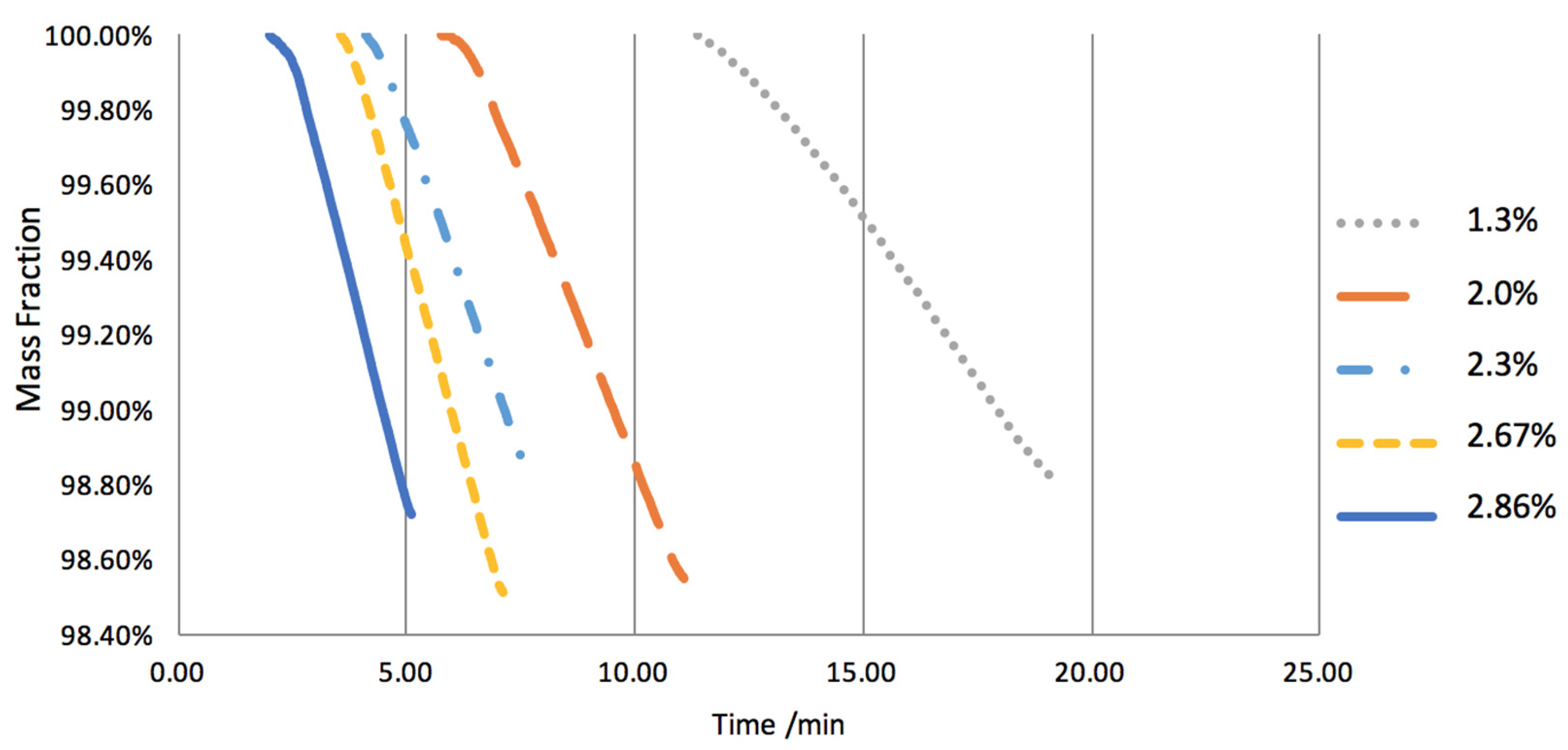
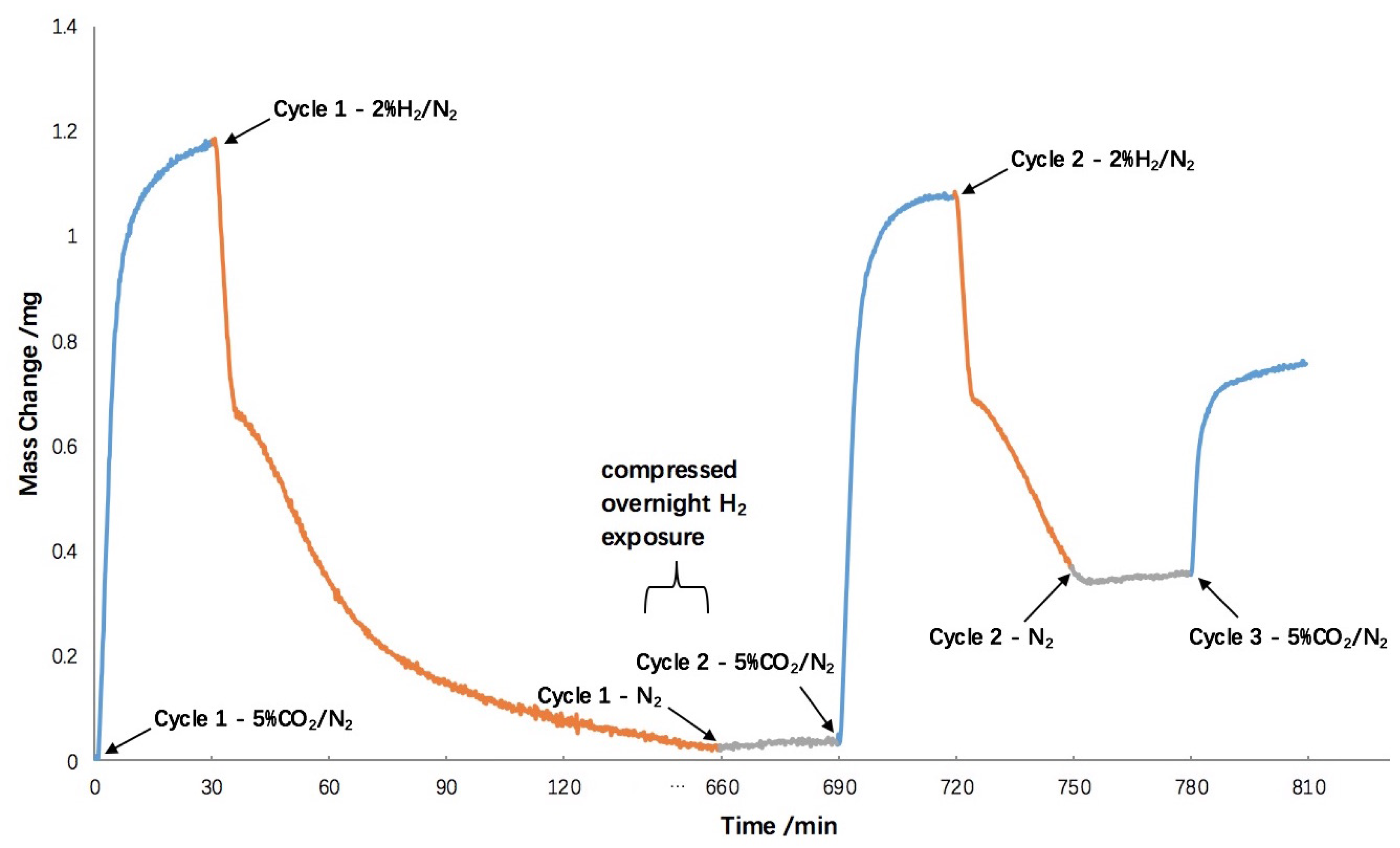
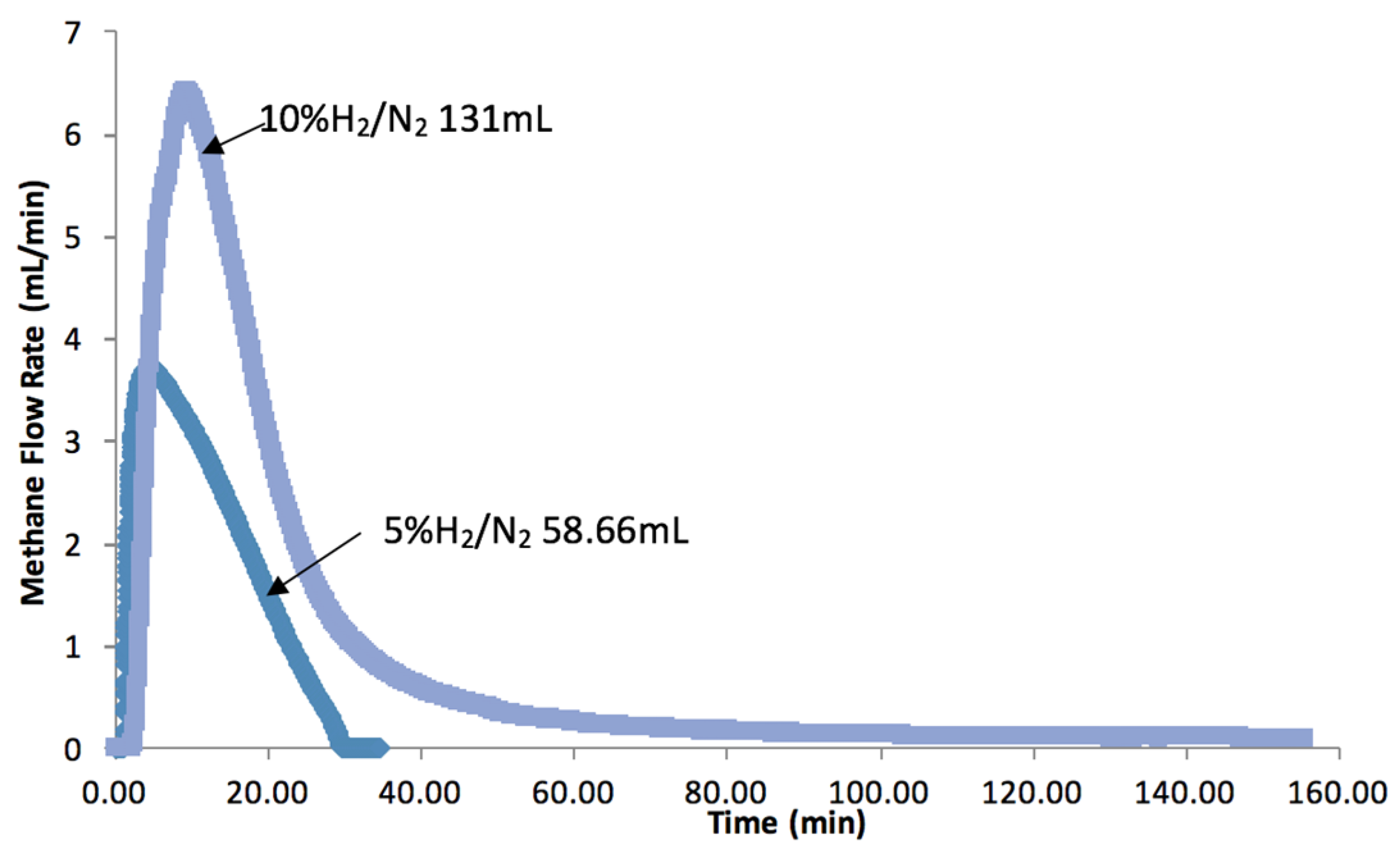
2.3. Hydrogen Exposure Time Study
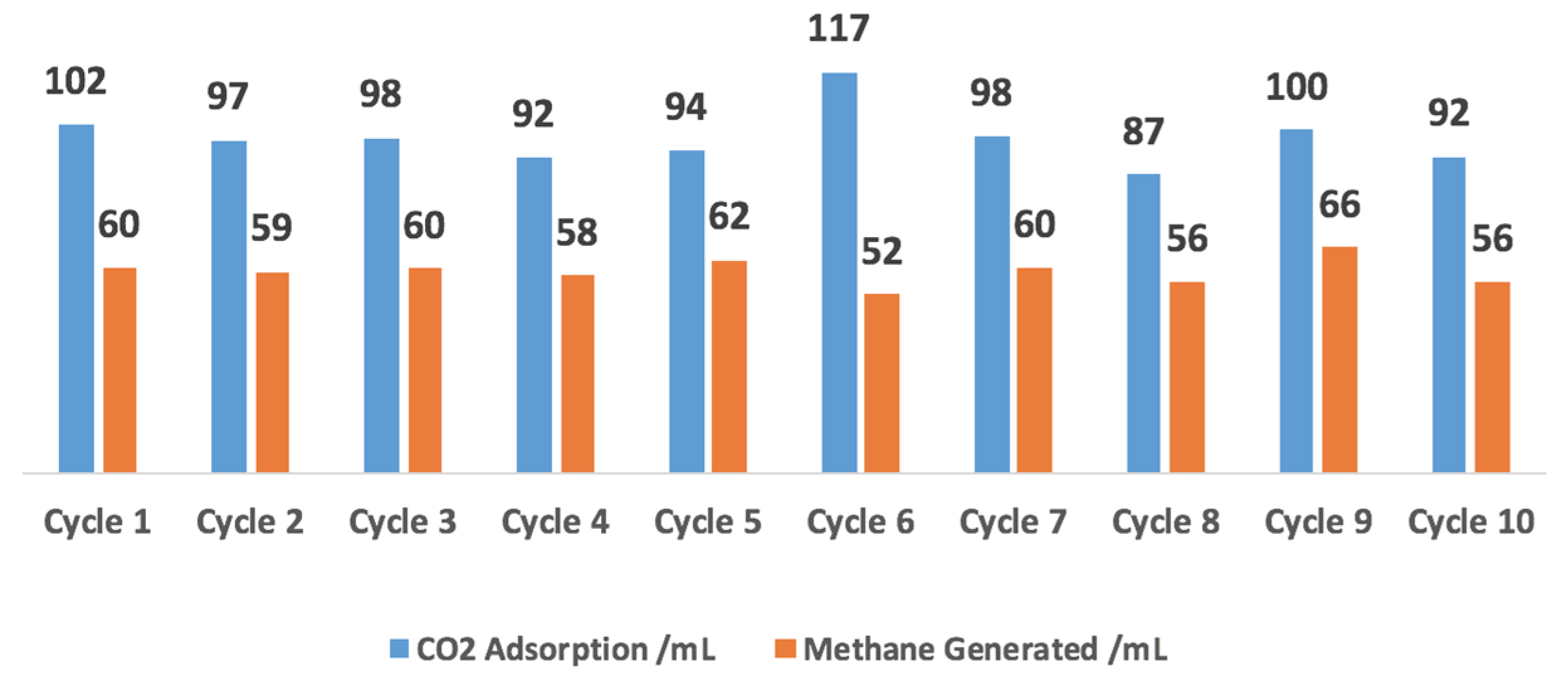
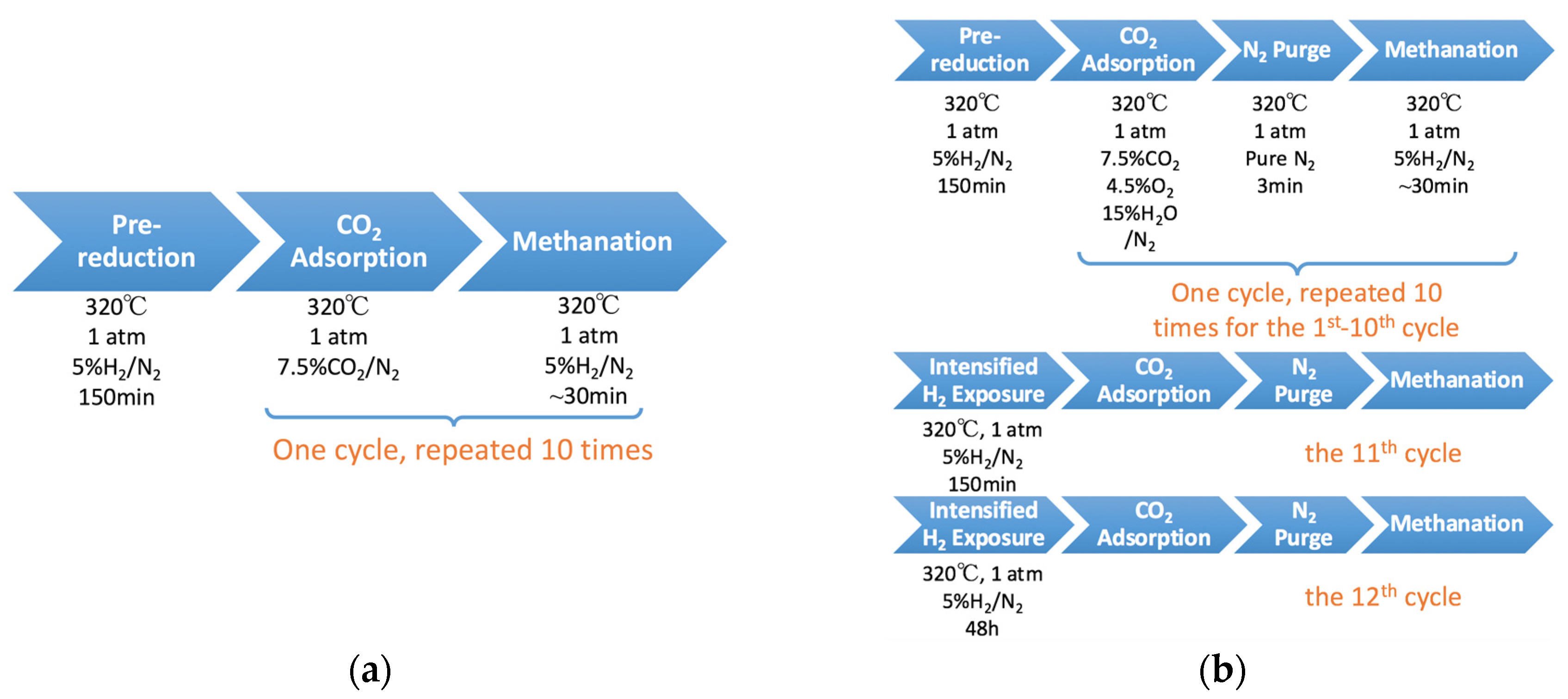
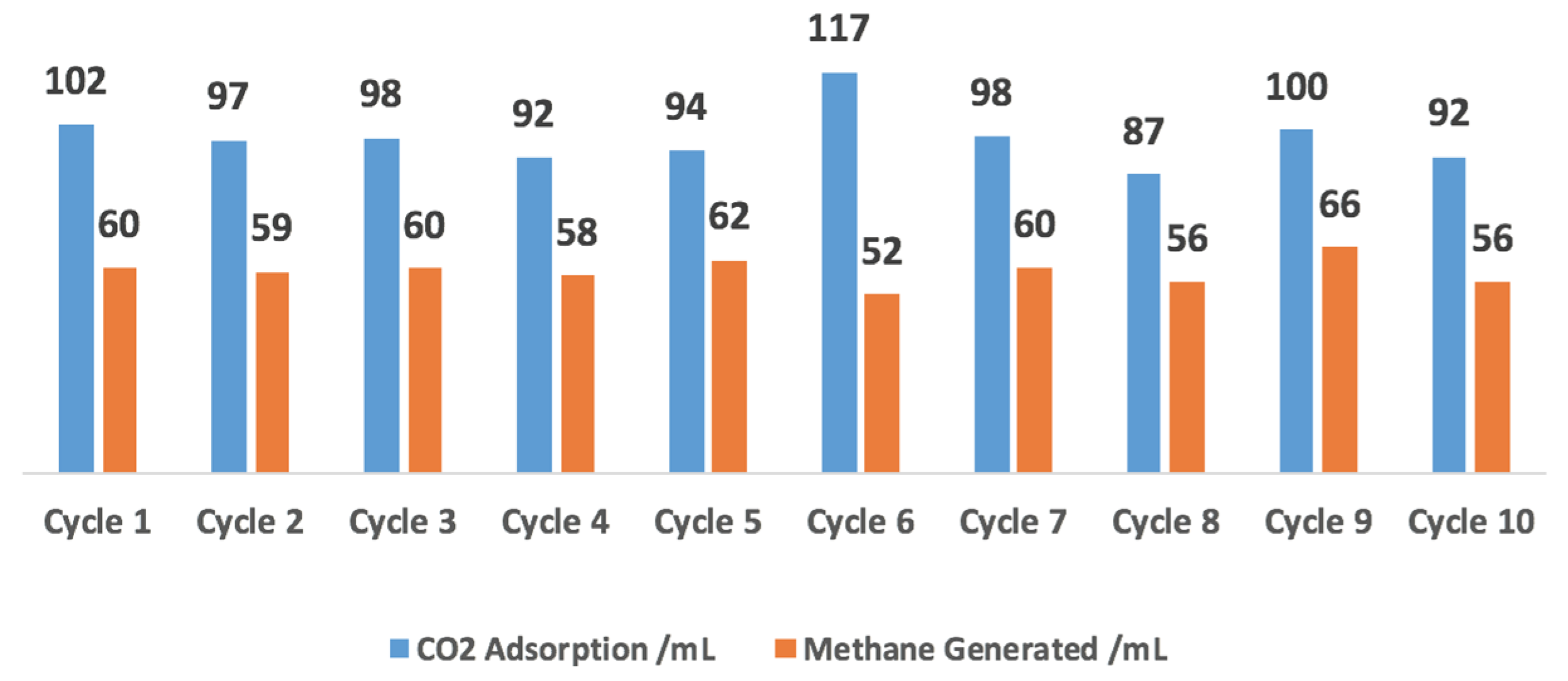
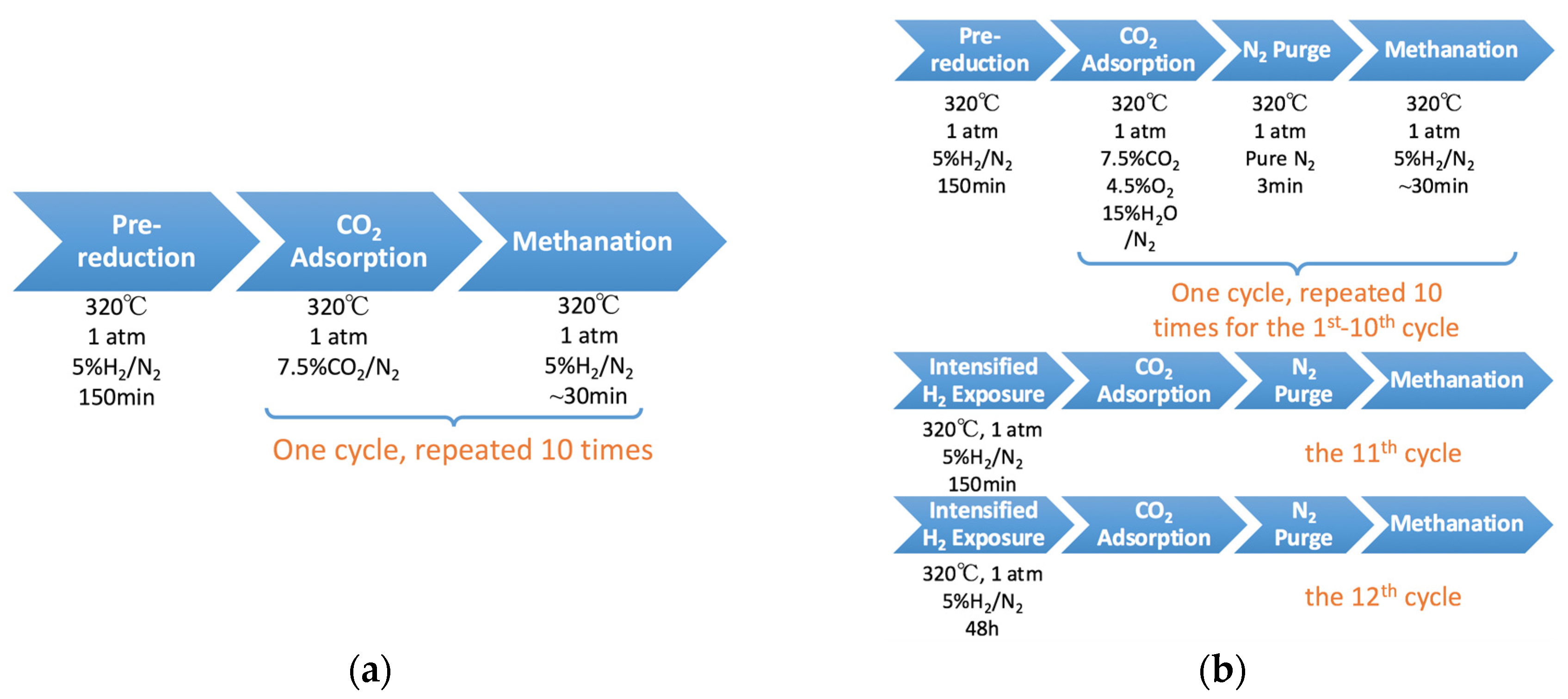
2.4. 10-Cycle Test in CO2 Feed
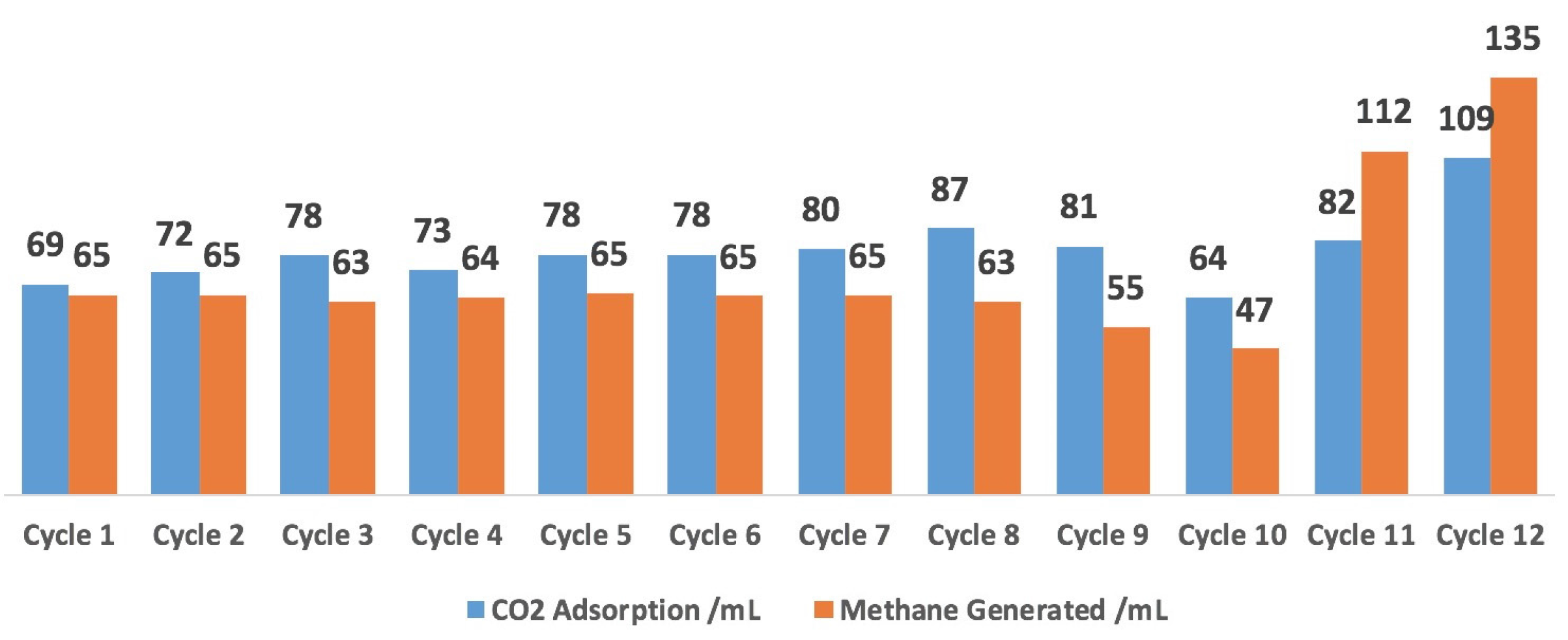
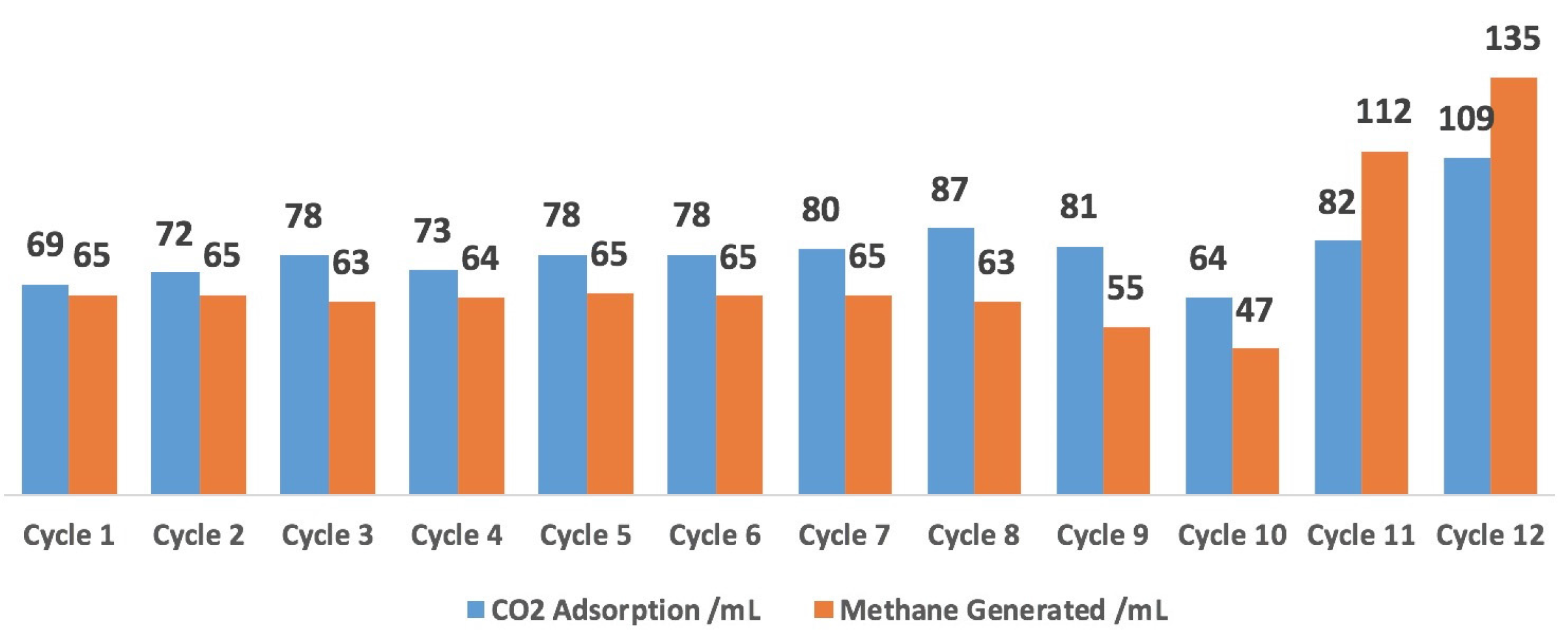
2.5. 12-Cycle Test in Simulated Flue Gas
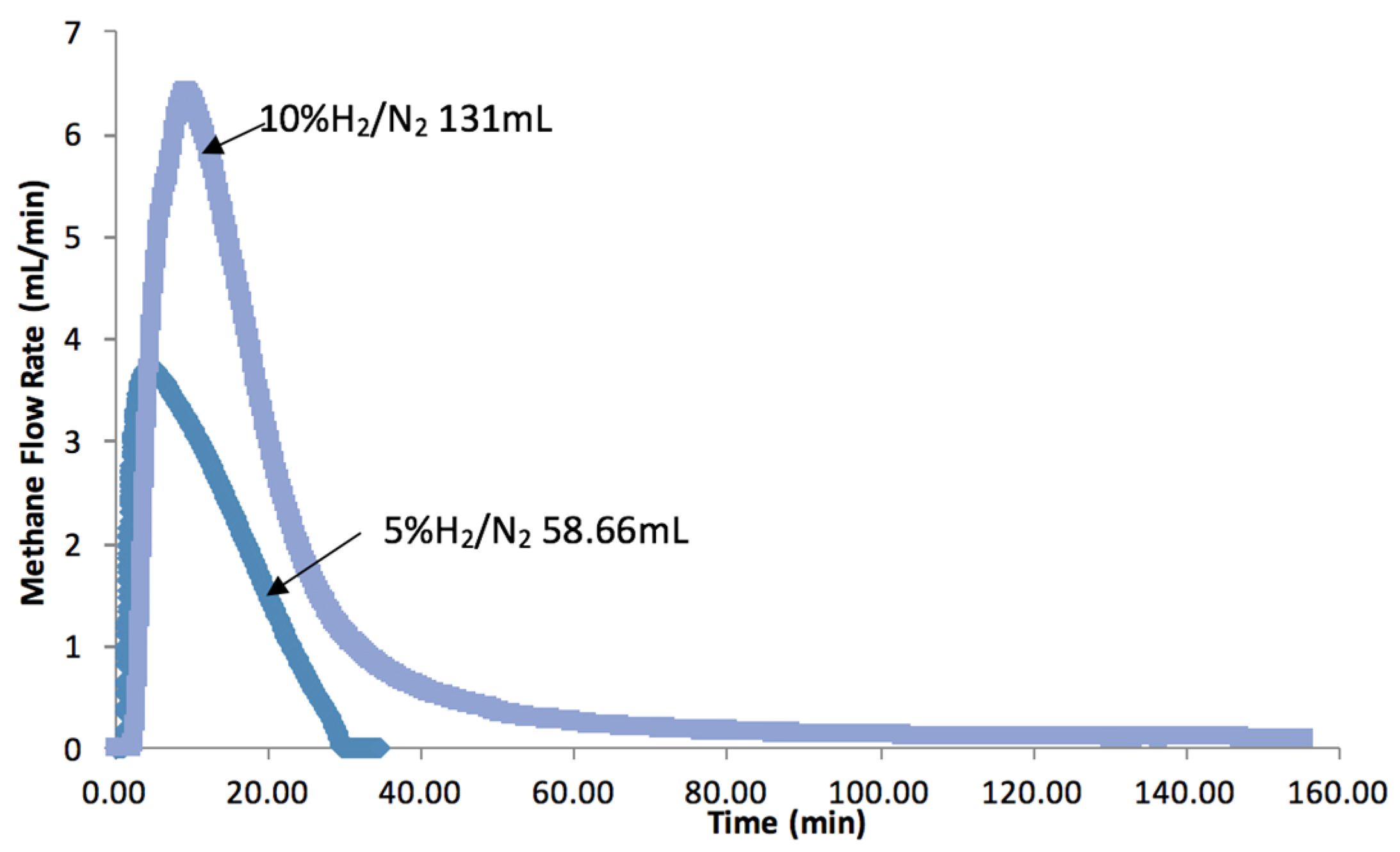
2.6. Effect of H2 Partial Pressure on CO2 Hydrogenation and RuOx Reduction
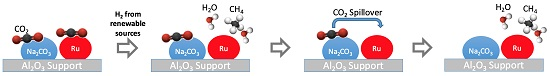
2.7. Speculated Functional Pathway of the Ru-Na2CO3 DFM
3. Experimental
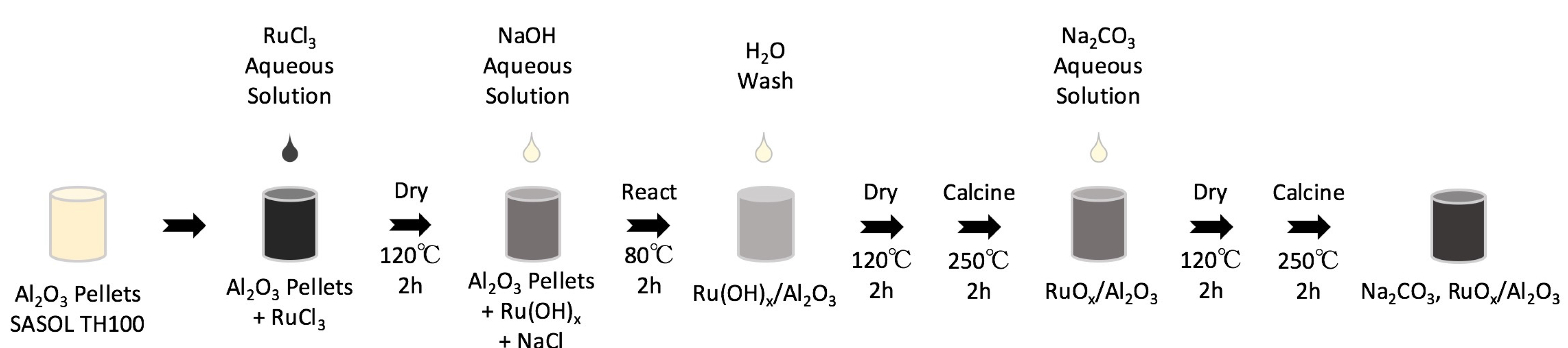
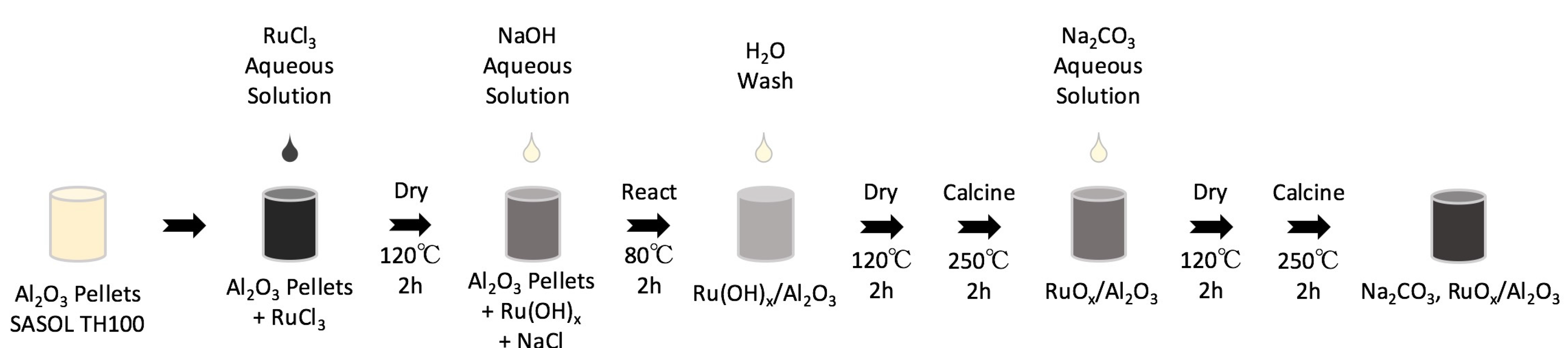
3.1. Material Synthesis
3.2. Thermogravimetric Analysis (TGA)
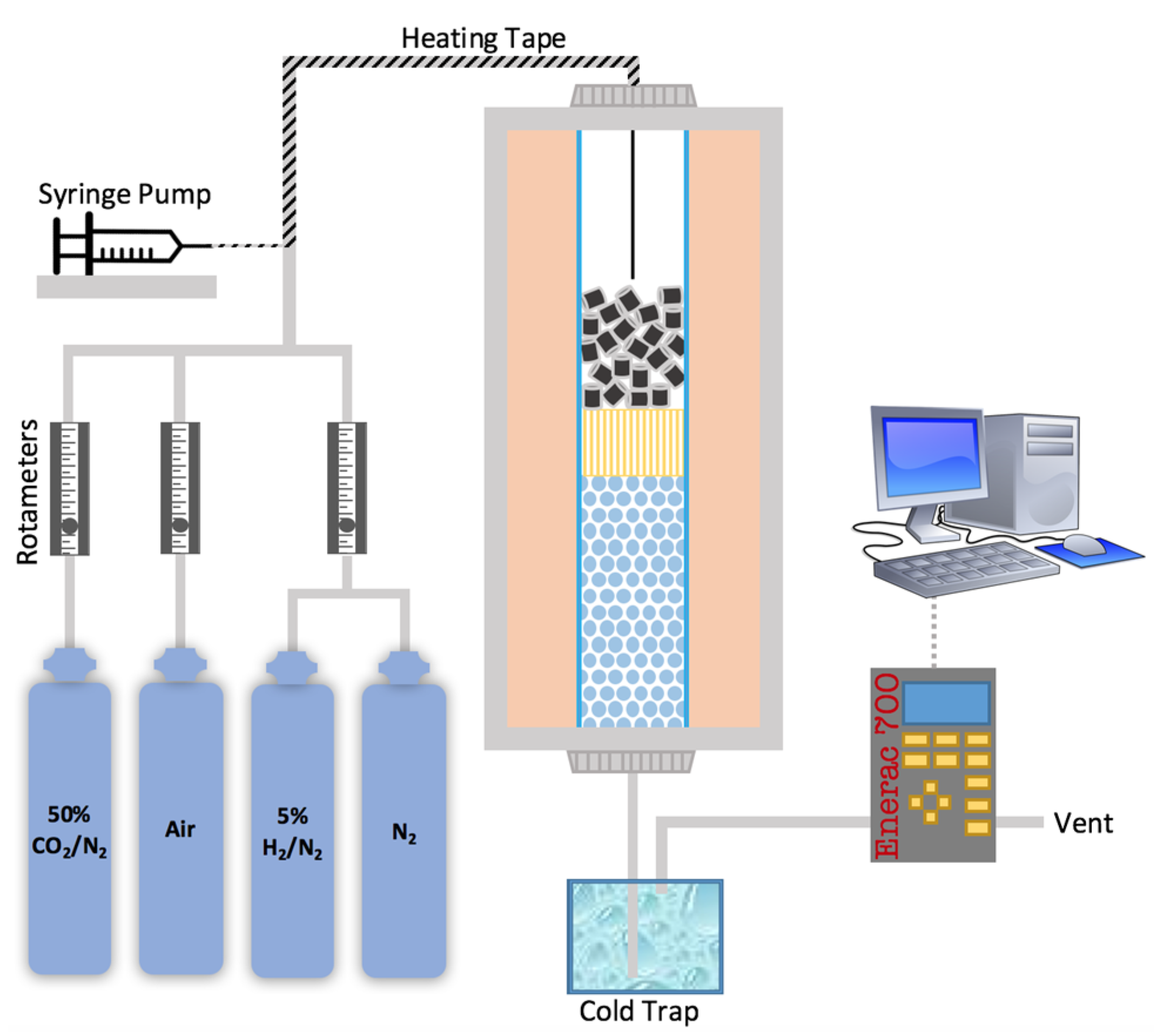
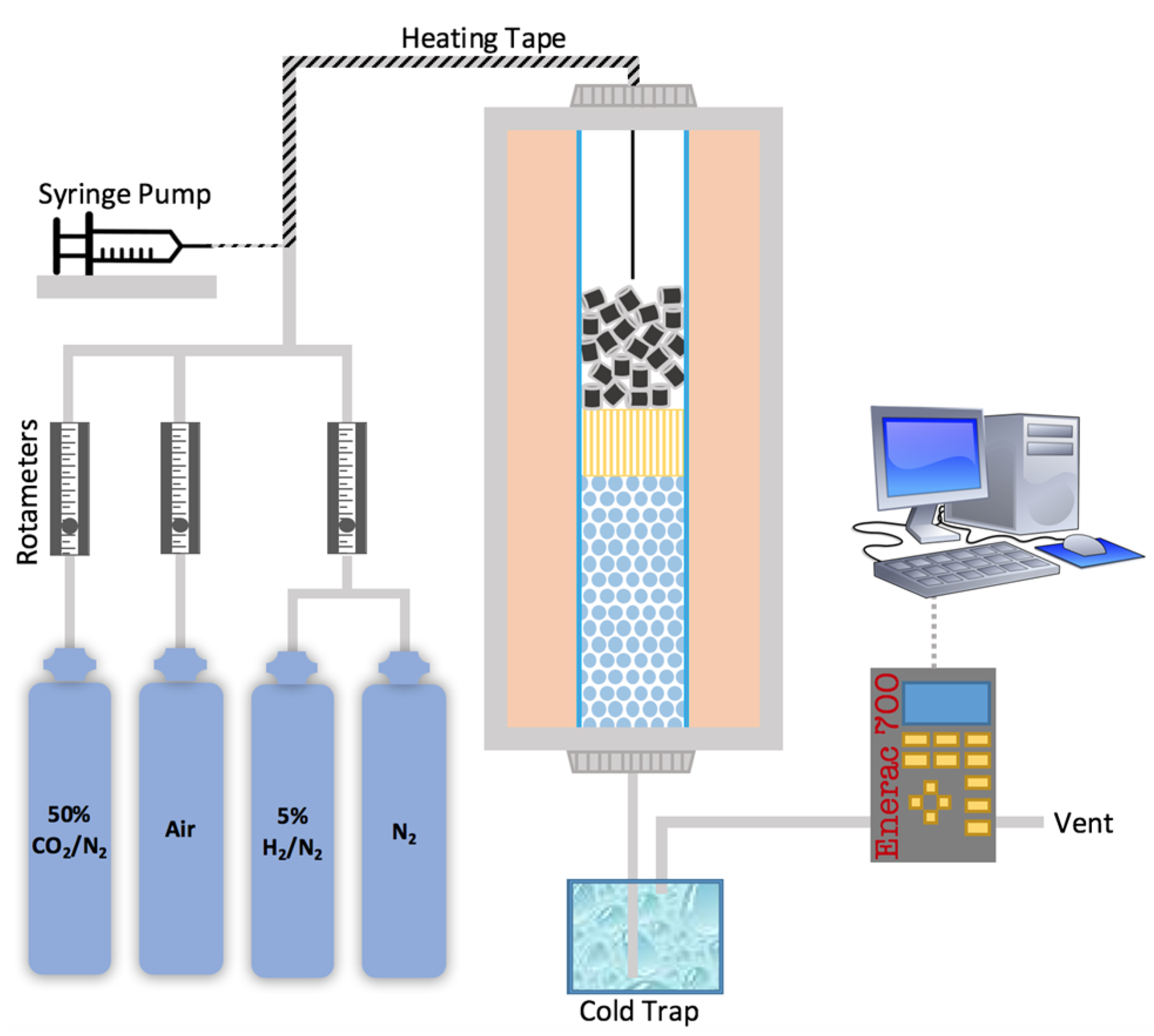
3.3. Scaled-Up Fixed Bed Reactor
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stewart, C.; Hessami, M.-A. A study of methods of carbon dioxide capture and sequestration––The sustainability of a photosynthetic bioreactor approach. Energy Conver. Manag. 2005, 46, 403–420. [Google Scholar] [CrossRef]
- Hasib-ur-Rahman, M.; Siaj, M.; Larachi, F. Chemical Engineering and Processing: Process Intensification. Chem. Eng. Process. Process Intensif. 2010, 49, 313–322. [Google Scholar] [CrossRef]
- Audus, H.; Freund, P.; Smith, A. Global Warming Damage and the Benefits of Mitigation; IEA Greenhouse Gas R&D Programme: Paris, France, December 1995. [Google Scholar]
- Heintz, Y.J.; Sehabiague, L.; Morsi, B.I.; Jones, K.L.; Pennline, H.W. Novel Physical Solvents for Selective CO2 Capture from Fuel Gas Streams at Elevated Pressures and Temperatures. Energy Fuels 2008, 22, 3824–3837. [Google Scholar] [CrossRef]
- Carmo, M.; Fritz, D.L.; Mergel, J.; Stolten, D. A comprehensive review on PEM water electrolysis. Int. J. Hydrog. Energy 2013, 38, 4901–4934. [Google Scholar] [CrossRef]
- Ursua, A.; Gandia, L.M.; Sanchis, P. Hydrogen Production from Water Electrolysis: Current Status and Future Trends. IEEE Proc. 2012, 100, 410–426. [Google Scholar] [CrossRef]
- Lackner, K.S. A guide to CO2 Sequestration. Science 2003, 300, 1677–1678. [Google Scholar] [CrossRef] [PubMed]
- Saeidi, S.; Amin, N.A.S.; Rahimpour, M.R. Hydrogenation of CO2 to value-added products—A review and potential future developments. J. CO2 Util. 2014, 5, 66–81. [Google Scholar] [CrossRef]
- Spigarelli, B.P.; Kawatra, S.K. Opportunities and challenges in carbon dioxide capture. J. CO2 Util. 2013, 1, 69–87. [Google Scholar] [CrossRef]
- Rochelle, G.T. Amine Scrubbing for CO2 Capture. Science 2009, 325, 1652–1654. [Google Scholar] [CrossRef] [PubMed]
- Buelens, L.C.; Buelens, L.C.; Galvita, V.V.; Poelman, H.; Detavernier, C.; Marin, G.B. Super-dry reforming of methane intensifies CO2 utilization via Le Chatelier’s principle. Science 2016, 354, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Bobadilla, L.F.; Riesco-García, J.M.; Penelás-Pérez, G.; Urakawa, A. Enabling continuous capture and catalytic conversion of flue gas CO2 to syngas in one process. J. CO2 Util. 2016, 14, 106–111. [Google Scholar] [CrossRef]
- Bartholomew, C.; Farrauto, R.J. Fundamentals of Industrial Catalytic Processes; Wiley and Sons: New York, NY, USA, 2006; Chapter 7; pp. 490–491. [Google Scholar]
- Bartholomew, C.; Farrauto, R.J. Fundamentals of Industrial Catalytic Processes; Wiley and Sons: New York, NY, USA, 2006; Chapter 6; p. 355. [Google Scholar]
- Bilgrien, C.; Davis, S.; Drago, R. The selective oxidation of primary alcohols to aldehydes by oxygen employing a trinuclear ruthenium carboxylate catalyst. J. Am. Chem. Soc. 1987, 1987, 3786–3787. [Google Scholar] [CrossRef]
- Huang, Y.Y.; Yang, X.; Feng, Y.; Verpoort, F.; Fan, Q.H. Chiral Ru/Ir bimetallic dendronized polymer catalysts constructed through sequential metal coordination and applied in asymmetric hydrogenation of quinaldine. J. Mol. Catal. A Chem. 2014, 393, 150–155. [Google Scholar] [CrossRef]
- Saadatjou, N.; Jafari, A.; Sahebdelfar, S. Ruthenium Nanocatalysts for Ammonia Synthesis: A Review. Chem. Eng. Commun. 2014, 202, 420–448. [Google Scholar] [CrossRef]
- Shen, L.; Zhang, C.; Liu, Y. Meso-macroporous Al2O3 supported Ru catalysts for CO preferential oxidation in hydrogen-rich gases. J. Nat. Gas Chem. 2012, 21, 653–660. [Google Scholar] [CrossRef]
- Steynberg, A.P.; Dry, M.E. Fischer-Tropsch Technology, Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 2004; Chapter 7; pp. 560 and 566. [Google Scholar]
- Menzel, N.; Ortel, E.; Mette, K.; Kraehnert, R.; Strasser, P. Dimensionally stable Ru/Ir/TiO2-anodes with tailored mesoporosity for efficient electrochemical chlorine evolution. ACS Catal. 2013, 3, 1324–1333. [Google Scholar] [CrossRef]
- Garbarino, G.; Bellotti, D.; Riani, P.; Magistri, L.; Busca, G. Methanation of carbon dioxide on Ru/Al2O3 and Ni/Al2O3 catalysts at atmospheric pressure: Catalysts activation, behaviour and stability. Int. J. Hydrog. Energy 2015, 40, 9171–9182. [Google Scholar] [CrossRef]
- Götz, M.; Lefebvre, J.; Mörs, F.; McDaniel Koch, A.; Graf, F.; Bajohr, S.; Reimert, R.; Kolb, T. Renewable Power-to-Gas: A technological and economic review. Renew. Energy 2016, 85, 1371–1390. [Google Scholar] [CrossRef]
- Janke, C.; Duyar, M.S.; Hoskins, M.; Farrauto, R. Catalytic and adsorption studies for the hydrogenation of CO2 to methane. Appl. Catal. B Environ. 2014, 152–153, 184–191. [Google Scholar] [CrossRef]
- Gruene, P.; Belova, A.G.; Yegulalp, T.M.; Farrauto, R.J.; Castaldi, M.J. Dispersed Calcium Oxide as a Reversible and Efficient CO2—Sorbent at Intermediate Temperatures. Ind. Eng. Chem. Res. 2011, 50, 4042–4049. [Google Scholar] [CrossRef]
- Wang, S.; Yan, S.; Ma, X.; Gong, J. Recent advances in capture of carbon dioxide using alkali-metal-based oxides. Energy Environ. Sci. 2011, 4, 3805–3819. [Google Scholar] [CrossRef]
- Tsuneto, A.; Kudo, A.; Saito, N.; Sakata, T. Hydrogenation of solid state carbonates. Chem. Lett. 1992, 21, 831–834. [Google Scholar] [CrossRef]
- Yoshida, N.; Hattori, T.; Komai, E.; Wada, T. Methane formation by metal-catalyzed hydrogenation of solid calcium carbonate. Catal. Lett. 1999, 58, 119–122. [Google Scholar] [CrossRef]
- Proton OnSite Awarded 13 Megawatt Electrolyzers. Available online: https://www.linkedin.com/pulse/proton-onsite-awarded-13-megawatt-electrolyzers-kathleen-mullins (accessed on 20 December 2016).
- Duyar, M.S.; Treviño, M.A.A.; Farrauto, R.J. Dual function materials for CO2 capture and conversion using renewable H2. Appl. Catal. B Environ. 2015, 168, 370–376. [Google Scholar] [CrossRef]
- Duyar, M.S.; Wang, S.; Arellano-Treviño, M.A.; Farrauto, R.J. CO2 utilization with a novel dual function material (DFM) for capture and catalytic conversion to synthetic natural gas: An update. J. CO2 Util. 2016, 15, 65–71. [Google Scholar] [CrossRef]
- Zheng, Q.; Farrauto, R.; Chau Nguyen, A. Adsorption and Methanation of Flue Gas CO2 with Dual Functional Catalytic Materials: A Parametric Study. Ind. Eng. Chem. Res. 2016, 55, 6768–6776. [Google Scholar] [CrossRef]
- Bartholomew, C.H.; Farrauto, R.J. Chemistry of nickel-alumina catalysts. J. Catal. 1976, 45, 41–53. [Google Scholar] [CrossRef]
- Kato, T.; Usami, T.; Tsukada, T.; Shibata, Y.; Kodama, T. Study on volatilization mechanism of ruthenium tetroxide from nitrosyl ruthenium nitrate by using mass spectrometer. J. Nucl. Mater. 2016, 479, 123–129. [Google Scholar] [CrossRef]
- Sawada, K.; Ueda, Y.; Enokida, Y. Ruthenium Release from Thermally Overheated Nitric Acid Solution Containing Ruthenium Nitrosyl Nitrate and Sodium Nitrate to Solidify. Procedia Chem. 2016, 21, 82–86. [Google Scholar] [CrossRef]
- Islam, A.; Ravindra, P. Biodiesel Production with Green Technologies; Springer International Publishing Switzerland: Cham, Switzerland, 2017; Chapter 1. [Google Scholar]
- Batholomew, C.; Farrauto, R.J. Fundamentals of Industrial Catalytic Processes; Wiley and Sons: New York, NY, USA, 2006; Chapter 10. [Google Scholar]
- Batholomew, C.; Farrauto, R.J. Fundamentals of Industrial Catalytic Processes; Wiley and Sons: New York, NY, USA, 2006; Chapter 2; p. 81. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | BET Specific Surface Area (m2/g) | Ru Crystallite Sizes (nm) |
|---|---|---|
| Fresh Ru-Na2CO3 DFM pellet | 103 | 3.98 |
| Flue gas cyclic aged Ru-Na2CO3 DFM pellet | 89 | 5.15 |
| Section | Ru Pathway | Na2CO3 Pathway |
|---|---|---|
| Pretreatment | ||
| O2-free CO2 feed 1 | ||
| O2-containing CO2 feed | ||
| Methanation & hydrogenation |
| Test | Pre-Treatment | Step 1 | Step 2 | Step 3 |
|---|---|---|---|---|
| Na2CO3 Loading Study | 2% H2/N2, 2 h | 0.5% CO2/N2, 1 h | Pure N2, 1 h | - |
| Carbonate Hydrogenation Study | Pure N2, 1 h | 2% H2/N2, 2 h | 5% CO2, 2 h | - |
| Hydrogen Exposure Time Study 1 | 2% H2/N2, 1 h | 5% CO2/N2, 30 m | 2% H2/N2, 10.5 h | Pure N2, 30 m |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Schrunk, E.T.; Mahajan, H.; Farrauto, A.R.J. The Role of Ruthenium in CO2 Capture and Catalytic Conversion to Fuel by Dual Function Materials (DFM). Catalysts 2017, 7, 88. https://doi.org/10.3390/catal7030088
Wang S, Schrunk ET, Mahajan H, Farrauto ARJ. The Role of Ruthenium in CO2 Capture and Catalytic Conversion to Fuel by Dual Function Materials (DFM). Catalysts. 2017; 7(3):88. https://doi.org/10.3390/catal7030088
Chicago/Turabian StyleWang, Shuoxun, Erik T. Schrunk, Harshit Mahajan, and And Robert J. Farrauto. 2017. "The Role of Ruthenium in CO2 Capture and Catalytic Conversion to Fuel by Dual Function Materials (DFM)" Catalysts 7, no. 3: 88. https://doi.org/10.3390/catal7030088
APA StyleWang, S., Schrunk, E. T., Mahajan, H., & Farrauto, A. R. J. (2017). The Role of Ruthenium in CO2 Capture and Catalytic Conversion to Fuel by Dual Function Materials (DFM). Catalysts, 7(3), 88. https://doi.org/10.3390/catal7030088