
Electrocatalysts Prepared by Galvanic Replacement



Abstract
:1. Principle of Galvanic Replacement/Deposition
1.1. Thermodynamic Considerations
1.2. Kinetic Considerations
2. History and Applications of Galvanic Replacement/Deposition
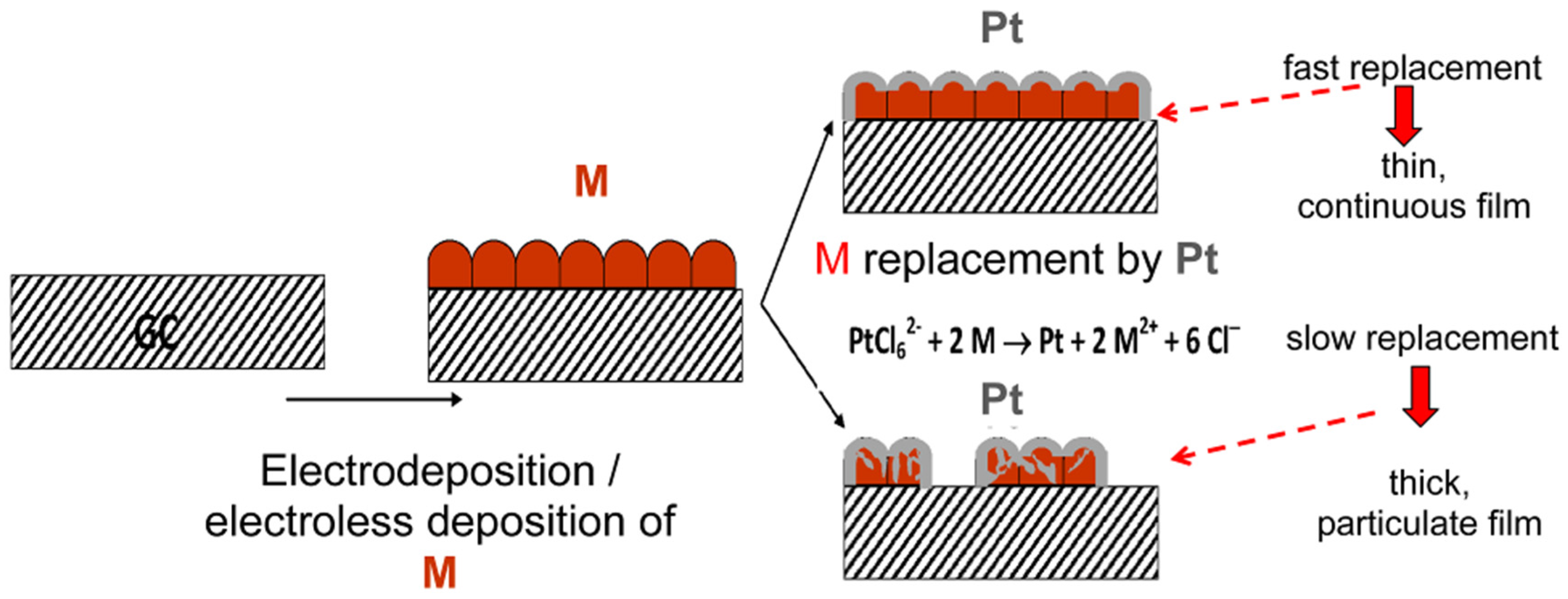
2.1. Metal Finishing, the Electronics Industry, and Metallurgical Applications
2.2. Ultrathin Film Applications
2.3. Nanoparticle and Catalytic Layer Applications
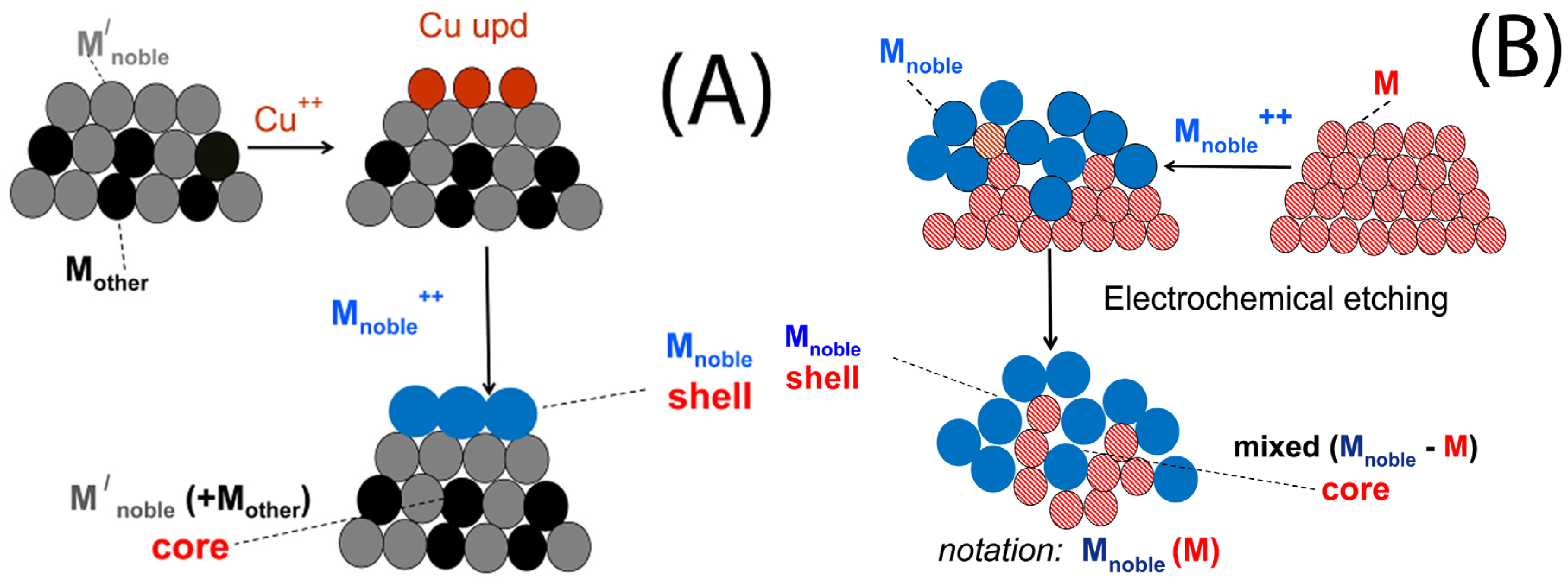
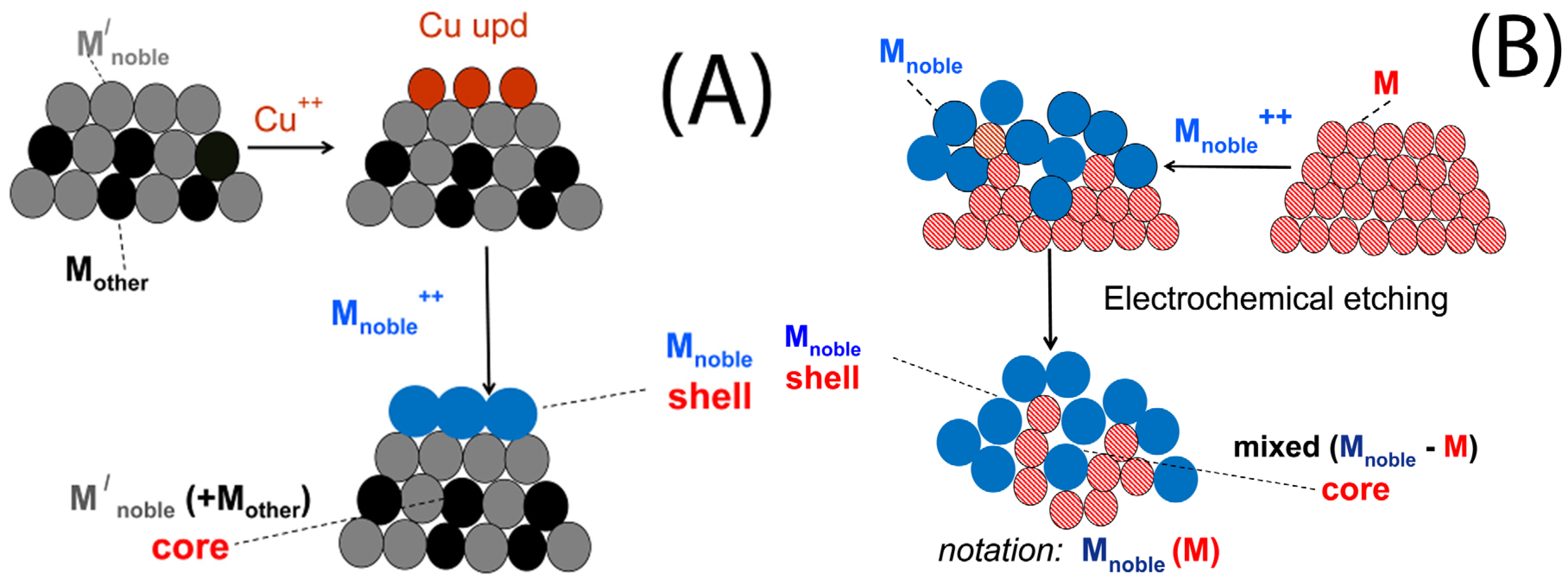
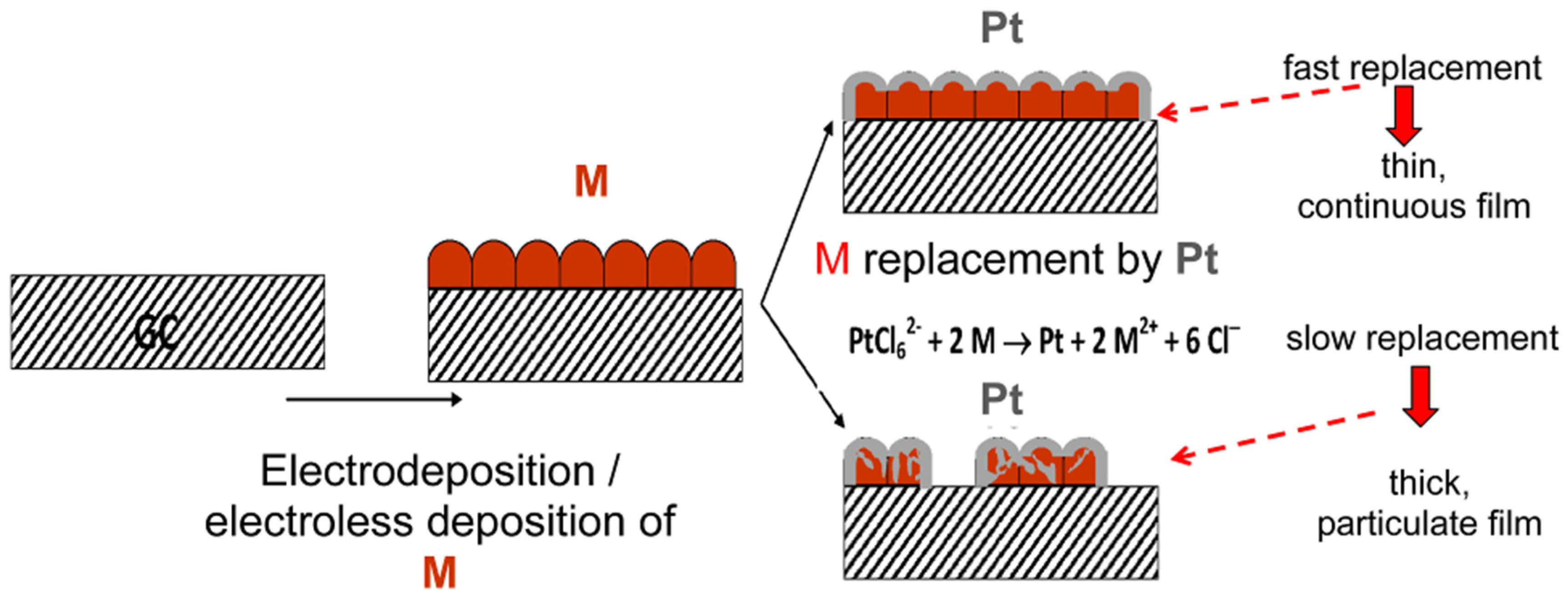
3. Characteristics of the Two Galvanic Replacement Approaches for Electrocatalyst Preparation
4. Types of Support, Methods of Less Noble Metal Preparation/Deposition and Catalyst Characteristics
5. Electrochemical Reactions at Poly-Metallic Catalysts Prepared by Galvanic Reaction
5.1. Oxygen Reduction
5.2. Methanol, Formic Acid, and Ethanol Oxidation
5.3. Hydrogen Evolution and Oxidation
5.4. Oxygen Evolution
5.5. Borohydride Oxidation
5.6. Other Cathodic Reactions
6. Conclusions/Future Avenues
- (i)
- It is a moderate temperature method, resulting in energy savings and most importantly in limited particle aggregation (thus increased surface area).
- (ii)
- It can lead (especially via its Cu, Pb, or H upd replacement variant) to a minimization of noble metal loadings in the polymetallic catalyst, since the noble metal can preferably (or even exclusively) be located at the outer layer(s).
- (iii)
- It is a relatively simple method that involves few steps and chemicals (especially in its variant where a bulk metallic substrate is modified by the more noble metal by simply immersing the former into an ionic solution of the latter).
- (i)
- The galvanic replacement of upd monolayers is perhaps the most mature method for preparing commercial catalysts and has shown remarkable noble metal mass utilization, especially for oxygen reduction and the Pt(Pd) system. The only foreseen hurdle is moving the catalyst preparation industry to use the apparatus needed for the electrodeposition of the sacrificial metal upd onto a fluidized electrode of M or M/C particles, that will be subsequently transformed to Mnoble (M) or Mnoble (M)/C particles. (The alternative, of treating the substrate with hydrogen to create the sacrificial H layers, can only find application for a few metal substrates, e.g., Pd.)
- (ii)
- The direct deposition of Mnoble on M or M/C nanoparticles, despite giving promising results especially for methanol and CO oxidation at Pt(Cu) electrodes, still suffers from rather low noble metal mass utilization. This is due to the penetration of Mnoble into the M core, the need for use of rather large M precursor particles (thus increasing surface area) and, for M/C particles, the simultaneous deposition of Mnoble on C. Ways to move forward with this variant will include the preparation of initially unsupported Mnoble (M) nanoparticles with a thin outer shell and, possibly, a hollow interior too. This will require standardizing existing nanomaterials preparation routes and testing their products as electrocatalysts.
- (iii)
- The direct deposition of Mnoble on M bulk substrates is a straightforward approach that can find applications in alkaline and neutral media such as hydrogen production via water electrolysis and organic electrosynthesis. Future avenues should include the use of inexpensive substrates (e.g., stainless steel) and the screening of many organic reactions.
Conflicts of Interest
References
- Osakada, K. Transmetalation. In Current Methods in Inorganic Chemistry, Fundamentals of Molecular Catalysis; Kurosawa, H., Yamamoto, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2003; Volume 3, pp. 233–291. [Google Scholar]
- Kunces, D. Chemical deposition of metallic films from aqueous solutions. In Electroless Plating: Fundamentals and Applications, 1st ed.; Mallory, G.O., Hajdu, J.B., Eds.; Noyes Publications/William Andrew Publishing, LLC: Norwich, NY, US, 1990; pp. 511–517. [Google Scholar]
- Moon, G.D.; Ko, S.; Min, Y.; Zeng, J.; Xia, Y.; Jeong, U. Chemical transformations of nanostructured materials. Nano Today 2011, 6, 186–203. [Google Scholar] [CrossRef]
- Bard, A.J.; Parsons, R.; Jordan, J. Standard Potentials in Aqueous Solution; Marcel Dekker, Inc.: New York, NY, USA; Basel, Switzerland, 1985. [Google Scholar]
- Beverskog, B.; Puigdomenech, I. Revised pourbaix diagrams for chromium at 25–300 °C. Corros. Sci. 1997, 39, 43–57. [Google Scholar] [CrossRef]
- Brankovic, S.; Zangari, G. Electrochemical surface processes and opportunities for materials synthesis. In Electrochemical Engineering across Scales: From Molecules to Processes; Alkire, R.C., Bartlett, P.N., Lipkowski, J., Eds.; Wiley-VCH Verlag GmbH & Co, KGaA: Weinheim, Germany, 2015; pp. 59–106. [Google Scholar]
- Gokcen, D.; Yuan, Q.; Brankovic, S.R. Nucleation of Pt monolayers deposited via surface limited redox replacement reaction. J. Electrochem. Soc. 2014, 161, D3051–D3056. [Google Scholar] [CrossRef]
- Dimitrov, N.; Vasilic, R.; Vasiljevic, N. A kinetic model for redox replacement of UPD layers. Electrochem. Solid State Lett. 2007, 10, D79–D83. [Google Scholar] [CrossRef]
- Gokcen, D.; Bae, S.-E.; Brankovic, S.R. Kinetics of metal deposition via surface-limited redox replacement reaction. ECS Trans. 2011, 35, 11–22. [Google Scholar]
- Gokcen, D.; Bae, S.-E.; Brankovic, S.R. Reaction kinetics of metal deposition via surface limited red-ox replacement of underpotentially deposited metal monolayers. Electrochim. Acta 2011, 56, 5545–5553. [Google Scholar] [CrossRef]
- Bulut, E.; Wu, D.; Dole, N.; Kilic, H.; Brankovic, S.R. Reaction kinetics of metal deposition via surface limited redox replacement of underpotentially deposited monolayer studied by surface reflectivity and open circuit potential measurements. J. Electrochem. Soc. 2017, 164, D159–D168. [Google Scholar] [CrossRef]
- Mkwizu, T.S.; Cukrowski, I. Physico-chemical modelling of adlayer phase formation via surface-limited reactions of copper in relation to sequential electrodeposition of multilayered platinum on crystalline gold. Electrochim. Acta 2014, 147, 432–441. [Google Scholar] [CrossRef]
- Podlovchenko, B.I.; Gladysheva, T.D.; Filatov, A.Y.; Yashina, L.V. The use of galvanic displacement in synthesizing Pt(Cu) catalysts with the core-shell structure. Russ. J. Electrochem. 2010, 46, 1189–1197. [Google Scholar] [CrossRef]
- Pletcher, D.; Walsh, F. Industrial Electrochemistry, 2nd ed.; Chapman and Hall: London, UK, 1990; pp. 498–502. [Google Scholar]
- Cabrera, N.; Mott, N.F. Theory of the oxidation of metals. Rep. Prog. Phys. 1949, 12, 163–184. [Google Scholar] [CrossRef]
- Werbicki, J.J., Jr. Practical electroless and immersion plating. Plating 1971, 58, 763–767. [Google Scholar]
- Wingenfeld, P. Advanced technology for selective plating of connectors. Met. Finish. 1994, 92, 13–18. [Google Scholar]
- Walsh, D.E.; Milad, G.; Gudeczauskas, D. Final finish: Printed circuit boards. Met. Finish. 2003, 101, 25–26. [Google Scholar] [CrossRef]
- Walsh, D.E.; Milad, G.; Gudeczauskas, D. Printed circuit boards: Final finish options. Prod. Finish. (Cincinnati) 2004, 68, 50–53. [Google Scholar] [CrossRef]
- Couble, E.C.; Dutkewych, O.B.; Florio, S.M.; Marsh, M.V.; Staniunas, R.F. Immersion, non-electrolytic tin/lead plating process. Circuit World 1992, 19, 63–70. [Google Scholar] [CrossRef]
- Milad, G.; Mayes, R. Electroless nickel/immersion gold finishes for application to surface mount technology: A regenerative approach. Met. Finish. 1998, 96, 44–46. [Google Scholar] [CrossRef]
- Gudeczauskas, D.; Hashimoto, S.; Kiso, M. Gold plating. Print. Circuit Fabr. 1999, 22, 30–33. [Google Scholar]
- Blair, A. Silver plating. Plat. Surf. Finish. 2003, 90, 42–47. [Google Scholar]
- Lashmore, D.S. Plating on aluminium: A review. Plat. Surf. Finish. 1985, 72, 36–39. [Google Scholar]
- Pushpavanam, M.; Natarajan, S.R.; Sudha, S.; Vidhya, M. Decorative plating on aluminum. Met. Finish. 1993, 91, 16–20. [Google Scholar]
- Stoyanova, E.; Stoychev, D. Electrochemical aspects of the immersion treatment of aluminium. J. Appl. Electrochem. 1997, 27, 685–690. [Google Scholar] [CrossRef]
- Rossi, S.; Fedrizzi, L.; Deflorian, F. Characterization of commercial metallic coatings for corrosion protection of P/M parts. Int. J. Powder Metall. 2004, 40, 33–40. [Google Scholar]
- Biswas, A.K.; Davenport, W.G. Extractive Metallurgy of Copper, 2nd ed.; Pergamon Press Ltd.: Oxford, UK, 1980; p. 272. [Google Scholar]
- Scott, D.A. Copper and Bronze in Art: Corrosion, Colorants, Conservation; Getty Publications: Los Angeles, CA, USA, 2002; p. 17. [Google Scholar]
- Petala, M.; Tsiridis, V.; Mintsouli, I.; Pliatsikas, N.; Spanos, T.; Rebeyre, P.; Darakas, E.; Patsalas, P.; Vourlias, G.; Kostoglou, M.; et al. Silver deposition on stainless steel container surfaces in contact with disinfectant silver aqueous solutions. Appl. Surf. Sci. 2017, 396, 1067–1075. [Google Scholar] [CrossRef]
- Dimitrov, N. Recent advances in the growth of metals, alloys, and multilayers by surface limited redox replacement (SLRR) based approaches. Electrochim. Acta 2016, 209, 599–622. [Google Scholar] [CrossRef]
- Herrero, E.; Buller, L.J.; Abruña, H.D. Underpotential deposition at single crystal surfaces of Au, Pt, Ag and other materials. Chem. Rev. 2001, 101, 1897–1930. [Google Scholar] [CrossRef] [PubMed]
- Gregory, B.W.; Wayne Suggs, D.; Stickney, J.L. Conditions for the deposition of CdTe by electrochemical atomic layer epitaxy. J. Electrochem. Soc. 1991, 138, 1279–1284. [Google Scholar] [CrossRef]
- Gregory, B.W.; Stickney, J.L. Electrochemical atomic layer epitaxy (ECALE). J. Electroanal. Chem. 1991, 300, 543–561. [Google Scholar] [CrossRef]
- Villegas, I.; Stickney, J.L. Preliminary studies of GaAs deposition on Au(100), (110), and (111) surfaces by electrochemical atomic layer epitaxy. J. Electrochem. Soc. 1992, 139, 686–694. [Google Scholar] [CrossRef]
- Suggs, D.W.; Villegas, I.; Gregory, B.W.; Stickney, J.L. Formation of compound semiconductors by electrochemical atomic layer epitaxy. J. Vac. Sci. Technol. A 1992, 10, 886–891. [Google Scholar] [CrossRef]
- Brankovic, S.R.; Wang, J.X.; Adžić, R.R. Metal monolayer deposition by replacement of metal adlayers on electrode surfaces. Surf. Sci. 2001, 474, L173–L179. [Google Scholar] [CrossRef]
- Mrozek, M.F.; Xie, Y.; Weaver, M.J. Surface-enhanced Raman scattering on uniform platinum-group overlayers: Preparation by redox replacement of underpotential-deposited metals on gold. Anal. Chem. 2001, 73, 5953–5960. [Google Scholar] [CrossRef] [PubMed]
- Vasilic, R.; Dimitrov, N. Epitaxial growth by monolayer-restricted galvanic displacement. Electrochem. Solid State Lett. 2005, 8, C173–C176. [Google Scholar] [CrossRef]
- Vasilic, R.; Viyannalage, L.T.; Dimitrov, N. Epitaxial growth of Ag on Au(111) by galvanic displacement of Pb and Tl monolayers. J. Electrochem. Soc. 2006, 153, C648–C655. [Google Scholar] [CrossRef]
- Viyannalage, L.T.; Vasilic, R.; Dimitrov, N. Epitaxial growth of Cu on Au(111) and Ag(111) by surface limited redox replacement—An electrochemical and STM study. J. Phys. Chem. C 2007, 111, 4036–4041. [Google Scholar] [CrossRef]
- Fayette, M.; Liu, Y.; Bertrand, D.; Nutariya, J.; Vasiljevic, N.; Dimitrov, N. From Au to Pt via surface limited redox replacement of Pb UPD in one-cell configuration. Langmuir 2011, 27, 5650–5658. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, L.; Fayette, M.; Martens, B.; Luo, Z.P.; Wang, Y.; Xu, D.; Zhang, J.; Fang, J.; Dimitrov, N. Catalytic performance comparison of shape-dependent nanocrystals and oriented ultrathin films of pt4cu alloy in the formic acid oxidation process. Electrocatalysis 2013, 4, 24–36. [Google Scholar] [CrossRef]
- Nutariya, J.; Fayette, M.; Dimitrov, N.; Vasiljevic, N. Growth of Pt by surface limited redox replacement of underpotentially deposited hydrogen. Electrochim. Acta 2013, 112, 813–823. [Google Scholar] [CrossRef]
- Ambrozik, S.; Rawlings, B.; Vasiljevic, N.; Dimitrov, N. Metal deposition via electroless surface limited redox replacement. Electrochem. Commun. 2014, 44, 19–22. [Google Scholar] [CrossRef]
- Ambrozik, S.; Dimitrov, N. The deposition of Pt via electroless surface limited redox replacement. Electrochim. Acta 2015, 169, 248–255. [Google Scholar] [CrossRef]
- Kim, Y.-G.; Kim, J.Y.; Vairavapandian, D.; Stickney, J.L. Platinum nanofilm formation by EC-ALE via redox replacement of UPD copper: Studies using in-situ scanning tunneling microscopy. J. Phys. Chem. B 2006, 110, 17998–18006. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Kim, Y.-G.; Stickney, J.L. Studies of Cu atomic layer replacement, formed by underpotential deposits, to form Pt nanofilms using Electrochemical Atomic Layer Epitaxy (EC-ALE). ECS Trans. 2006, 1, 41–48. [Google Scholar]
- Hossain, M.A.; Cummins, K.D.; Park, Y.-S.; Soriaga, M.P.; Stickney, J.L. Layer-by-layer deposition of Pd on Pt(111) electrode: An electron spectroscopy-electrochemistry study. Electrocatalysis 2012, 3, 183–191. [Google Scholar] [CrossRef]
- Jayaraju, N.; Vairavapandian, D.; Kim, Y.G.; Banga, D.; Stickney, J.L. Electrochemical atomic layer deposition (E-ALD) of Pt nanofilms using SLRR cycles. J. Electrochem. Soc. 2012, 159, D616–D622. [Google Scholar] [CrossRef]
- Sheridan, L.B.; Gebregziabiher, D.K.; Stickney, J.L.; Robinson, D.B. Formation of palladium nanofilms using electrochemical atomic layer deposition (E-ALD) with chloride complexation. Langmuir 2013, 29, 1592–1600. [Google Scholar] [CrossRef] [PubMed]
- Jayaraju, N.; Banga, D.; Thambidurai, C.; Liang, X.; Kim, Y.-G.; Stickney, J.L. PtRu nanofilm formation by electrochemical atomic layer deposition (E-ALD). Langmuir 2014, 30, 3254–3263. [Google Scholar] [CrossRef] [PubMed]
- Cappillino, P.J.; Sugar, J.D.; El Gabaly, F.; Cai, T.Y.; Liu, Z.; Stickney, J.L.; Robinson, D.B. Atomic-layer electroless deposition: A scalable approach to surface-modified metal powders. Langmuir 2014, 30, 4820–4829. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Wang, Y.; Ruditskiy, A.; Xia, Y. 25th anniversary article: Galvanic replacement: A simple and versatile route to hollow nanostructures with tunable and well-controlled properties. Adv. Mater. 2013, 25, 6313–6332. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Crooks, R.M. Intradendrimer exchange of metal nanoparticles. Chem. Mater. 1999, 11, 3379–3385. [Google Scholar] [CrossRef]
- Park, J.-I.; Cheon, J. Synthesis of “solid solution” and “core-shell” type cobalt-platinum magnetic nanoparticles via transmetalation reactions. J. Am. Chem. Soc. 2001, 123, 5743–5746. [Google Scholar] [CrossRef] [PubMed]
- Shon, Y.-S.; Dawson, G.B.; Porter, M.; Murray, R.W. Monolayer-protected bimetal cluster synthesis by core metal galvanic exchange reaction. Langmuir 2002, 18, 3880–3885. [Google Scholar] [CrossRef]
- Sun, Y.; Mayers, B.T.; Xia, Y. Template-engaged replacement reaction: A one-step approach to the large-scale synthesis of metal nanostructures with hollow interiors. Nano Lett. 2002, 2, 481–485. [Google Scholar] [CrossRef]
- Chen, J.; Glaus, C.; Laforest, R.; Zhang, Q.; Yang, M.; Gidding, M.; Welch, M.J.; Xia, Y. Gold nanocages as photothermal transducers for cancer treatment. Small 2010, 6, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.; Luehmann, H.; Xia, X.; Wan, D.; Cutler, C.; Xia, Y. Radioluminescent gold nanocages with controlled radioactivity for real-time in vivo imaging. Nano Lett. 2013, 13, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Gan, Y.; Du, J.; Tian, D.; Zhang, R.; Yang, C.; Dai, Z. A review of hollow Pt-based nanocatalysts applied in proton exchange membrane fuel cells. J. Power Sources 2013, 232, 310–322. [Google Scholar] [CrossRef]
- Liu, B.; Liao, S.; Liang, Z. Core-shell structure: The best way to achieve low-Pt fuel cell electrocatalysts. Prog. Chem. 2011, 23, 852–859. [Google Scholar]
- Lai, X.; Halpert, J.E.; Wang, D. Recent advances in micro-/nano-structured hollow spheres for energy applications: From simple to complex systems. Energy Environ. Sci. 2012, 5, 5604–5618. [Google Scholar] [CrossRef]
- Adzic, R.R.; Zhang, J.; Sasaki, K.; Vukmirovic, M.B.; Shao, M.; Wang, J.X.; Nilekar, A.U.; Mavrikakis, M.; Valerio, J.A.; Uribe, F. Platinum monolayer fuel cell electrocatalysts. Top. Catal. 2007, 46, 249–262. [Google Scholar] [CrossRef]
- Sasaki, K.; Vukmirovic, M.B.; Wang, J.X.; Adzic, R.R. Platinum monolayer electrocatalysts: Improving structure and activity. In Fuel Cell Science: Theory, Fundamentals, and Biocatalysis; Wieckowski, A., Nørskov, J.K., Eds.; Wiley: Hoboken, NJ, USA, 2010; pp. 215–236. [Google Scholar]
- Bliznakov, S.; Vukmirovic, M.; Adzic, R. Electrochemical atomic-level controlled syntheses of electrocatalysts for the oxygen reduction reaction. RSC Catal. Ser. 2015, 22, 144–166. [Google Scholar]
- Papadimitriou, S.; Armyanov, S.; Valova, E.; Hubin, A.; Steenhaut, O.; Pavlidou, E.; Kokkinidis, G.; Sotiropoulos, S. Methanol oxidation at Pt-Cu, Pt-Ni, and Pt-Co electrode coatings prepared by a galvanic replacement process. J. Phys. Chem. C 2010, 114, 5217–5223. [Google Scholar] [CrossRef]
- Geboes, B.; Mintsouli, I.; Wouters, B.; Georgieva, J.; Kakaroglou, A.; Sotiropoulos, S.; Valova, E.; Armyanov, S.; Hubin, A.; Breugelmans, T. Surface and electrochemical characterisation of a Pt-Cu/C nano-structured electrocatalyst, prepared by galvanic displacement. Appl. Catal. B 2014, 150–151, 249–256. [Google Scholar] [CrossRef]
- Kuznetsov, V.V.; Podlovchenko, B.I.; Kavyrshina, K.V.; Maksimov, Yu.M. Oxidation of methanol on Pt(Mo) electrodes obtained using galvanic displacement method. Russ. J. Electrochem. 2010, 46, 1353–1359. [Google Scholar] [CrossRef]
- Podlovchenko, B.I.; Krivchenko, V.A.; Maksimov, Y.M.; Gladysheva, T.D.; Yashina, L.V.; Evlashin, S.A.; Pilevsky, A.A. Specific features of the formation of Pt(Cu) catalysts by galvanic displacement with carbon nanowalls used as support. Electrochim. Acta 2012, 76, 137–144. [Google Scholar] [CrossRef]
- Vázquez-Gómez, L.; Cattarin, S.; Guerriero, P.; Musiani, M. Hydrogen evolution on porous Ni cathodes modified by spontaneous deposition of Ru or Ir. Electrochim. Acta 2008, 53, 8310–8318. [Google Scholar] [CrossRef]
- Verlato, E.; Cattarin, S.; Comisso, N.; Gambirasi, A.; Musiani, M.; Vázquez-Gómez, L. Preparation of Pd-modified Ni foam electrodes and their use as anodes for the oxidation of alcohols in basic media. Electrocatalysis 2012, 3, 48–58. [Google Scholar] [CrossRef]
- Kokkinidis, G.; Papoutsis, A.; Stoychev, D.; Milchev, A. Electroless deposition of Pt on Ti—Catalytic activity for the hydrogen evolution reaction. J. Electroanal. Chem. 2000, 486, 48–55. [Google Scholar] [CrossRef]
- Kokkinidis, G.; Stoychev, D.; Lazarov, V.; Papoutsis, A.; Milchev, A. Electroless deposition of Pt on Ti: Part II. Catalytic activity for oxygen reduction. J. Electroanal. Chem. 2001, 511, 20–30. [Google Scholar] [CrossRef]
- Van Brussel, M.; Kokkinidis, G.; Vandendael, I.; Buess-Herman, C. High performance gold-supported platinum electrocatalyst for oxygen reduction. Electrochem. Commun. 2002, 4, 808–813. [Google Scholar] [CrossRef]
- Van Brussel, M.; Kokkinidis, G.; Hubin, A.; Buess-Herman, C. Oxygen reduction at platinum modified gold electrodes. Electrochim. Acta 2003, 48, 3909–3919. [Google Scholar] [CrossRef]
- Brankovic, S.R.; McBreen, J.; Adžić, R.R. Spontaneous deposition of Pt on the Ru(0001) surface. J. Electroanal. Chem. 2001, 503, 99–104. [Google Scholar] [CrossRef]
- Brankovic, S.R.; Wang, J.X.; Adžić, R.R. Pt submonolayers on Ru nanoparticles. A novel low Pt loading, high CO tolerance fuel cell electrocatalyst. Electrochem. Solid State Lett. 2001, 4, A217–A220. [Google Scholar] [CrossRef]
- Sasaki, K.; Mo, Y.; Wang, J.X.; Balasubramanian, M.; Uribe, F.; McBreen, J.; Adzic, R.R. Pt submonolayers on metal nanoparticles—Novel electrocatalysts for H2 oxidation and O2 reduction. Electrochim. Acta 2003, 48, 3841–3849. [Google Scholar] [CrossRef]
- Sasaki, K.; Wang, J.X.; Balasubramanian, M.; McBreen, J.; Uribe, F.; Adzic, R.R. Ultra-low platinum content fuel cell anode electrocatalyst with a long-term performance stability. Electrochim. Acta 2004, 49, 3873–3877. [Google Scholar] [CrossRef]
- Zhang, J.; Vukmirovic, M.B.; Xu, Y.; Mavrikakis, M.; Adzic, R.R. Controlling the catalytic activity of platinum-monolayer electrocatalysts for oxygen reduction with different substrates. Angew. Chem. Int. Ed. 2005, 44, 2132–2135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Vukmirovic, M.B.; Sasaki, K.; Nilekar, A.U.; Mavrikakis, M.; Adzic, R.R. Mixed-metal Pt monolayer electrocatalysts for enhanced oxygen reduction kinetics. J. Am. Chem. Soc. 2005, 127, 12480–12481. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lima, F.H.B.; Shao, M.H.; Sasaki, K.; Wang, J.X.; Hanson, J.; Adzic, R.R. Platinum monolayer on nonnoble metal-noble metal core-shell nanoparticle electrocatalysts for O2 reduction. J. Phys. Chem. B 2005, 109, 22701–22704. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Zhang, J.; Wang, J.; Uribe, F.; Adzic, R. Platinum submonolayer-monolayer electrocatalysts: An electrochemical and X-ray absorption spectroscopy study. Res. Chem. Intermed. 2006, 32, 543–559. [Google Scholar] [CrossRef]
- Zhang, J.; Shao, M.H.; Sasaki, K.; Vukmirovic, M.B.; Uribe, F.; Adzic, R.R. Platinum and mixed platinum-metal monolayer fuel cell electrocatalysts: Design, activity and long-term performance stability. ECS Trans. 2006, 3, 31–36. [Google Scholar]
- Vukmirovic, M.B.; Zhang, J.; Sasaki, K.; Nilekar, A.U.; Uribe, F.; Mavrikakis, M.; Adzic, R.R. Platinum monolayer electrocatalysts for oxygen reduction. Electrochim. Acta 2007, 52, 2257–2263. [Google Scholar] [CrossRef]
- Nilekar, A.U.; Xu, Y.; Zhang, J.; Vukmirovic, M.B.; Sasaki, K.; Adzic, R.R.; Mavrikakis, M. Bimetallic and ternary alloys for improved oxygen reduction catalysis. Top. Catal. 2007, 46, 276–284. [Google Scholar] [CrossRef]
- Xu, Y.; Shao, M.; Mavrikakis, M.; Adzic, R.R. Recent Developments in the electrocatalysis of the O2 reduction reaction. In Fuel Cell Catalysis: A Surface Science Approach; Koper, M.T.M., Ed.; Wiley: Hoboken, NJ, USA, 2009; pp. 271–315. [Google Scholar]
- Ball, S.; Burton, S.L.; Fisher, J.; O’Malley, R.; Tessier, B.; Theobald, B.R.C.; Thompsett, D.; Zhou, W.P.; Su, D.; Zhu, Y.; et al. Structure and activity of novel Pt core-shell catalysts for the oxygen reduction reaction. ECS Trans. 2009, 25, 1023–1036. [Google Scholar]
- Sasaki, K.; Wang, J.X.; Naohara, H.; Marinkovic, N.; More, K.; Inada, H.; Adzic, R.R. Recent advances in platinum monolayer electrocatalysts for oxygen reduction reaction: Scale-up synthesis, structure and activity of Pt shells on Pd cores. Electrochim. Acta 2010, 55, 2645–2652. [Google Scholar] [CrossRef]
- Sasaki, K.; Naohara, H.; Cai, Y.; Choi, Y.M.; Liu, P.; Vukmirovic, M.B.; Wang, J.X.; Adzic, R.R. Core-protected platinum monolayer shell high-stability electrocatalysts for fuel-cell cathodes. Angew. Chem. Int. Ed. 2010, 49, 8602–8607. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Adzic, R.R. Platinum monolayer electrocatalysts for the oxygen reduction reaction: Improvements induced by surface and subsurface modifications of cores. Adv. Phys. Chem. 2011, 2011, 530397. [Google Scholar] [CrossRef]
- Adzic, R.R. Platinum monolayer electrocatalysts: Tunable activity, stability, and self-healing properties. Electrocatalysis 2012, 3, 163–169. [Google Scholar] [CrossRef]
- Li, M.; Liu, P.; Adzic, R.R. Platinum monolayer electrocatalysts for anodic oxidation of alcohols. J. Phys. Chem. Lett. 2012, 3, 3480–3485. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Adzic, R.R. Low-platinum-content electrocatalysts for methanol and ethanol electrooxidation. In Electrocatalysis in Fuel Cells; Lecture Notes in Energy; Springer: London, UK, 2013. [Google Scholar]
- Kuttiyiel, K.A.; Choi, Y.M.; Sasaki, K.; Su, D.; Hwang, S.-M.; Yim, S.-D.; Yang, T.-H.; Park, G.-G.; Adzic, R.R. Tuning electrocatalytic activity of Pt monolayer shell by bimetallic Ir-M (M = Fe, Co, Ni or Cu) cores for the oxygen reduction reaction. Nano Energy 2016, 29, 261–267. [Google Scholar] [CrossRef]
- Sasaki, K.; Marinkovic, N.; Isaacs, H.S.; Adzic, R.R. Synchrotron-based in situ characterization of carbon-supported platinum and platinum monolayer electrocatalysts. ACS Catal. 2016, 6, 69–76. [Google Scholar] [CrossRef]
- Khatee, S.; Guerreo, S.; Su, D.; Darling, R.M.; Protsailo, L.V.; Shao, M. Fuel cell performance of palladium-platinum core-shell electrocatalysts synthesized in gram-scale batches. J. Electrochem. Soc. 2016, 163, F708–F713. [Google Scholar] [CrossRef]
- Zhu, S.; Yue, J.; Qin, X.; Wei, Z.; Liang, Z.; Adzic, R.R.; Brankovic, S.R.; Du, Z.; Shao, M. The role of citric acid in perfecting platinum monolayer on palladium nanoparticles during the surface limited redox replacement reaction. J. Electrochem. Soc. 2016, 163, D3040–D3046. [Google Scholar] [CrossRef]
- Papadimitriou, S.; Tegou, A.; Pavlidou, E.; Kokkinidis, G.; Sotiropoulos, S. Methanol oxidation at platinized lead coatings prepared by a two-step electrodeposition-electroless deposition process on glassy carbon and platinum substrates. Electrochim. Acta 2007, 52, 6254–6260. [Google Scholar] [CrossRef]
- Tegou, A.; Papadimitriou, S.; Pavlidou, E.; Kokkinidis, G.; Sotiropoulos, S. Oxygen reduction at platinum- and gold-coated copper deposits on glassy carbon substrates. J. Electroanal. Chem. 2007, 608, 67–77. [Google Scholar] [CrossRef]
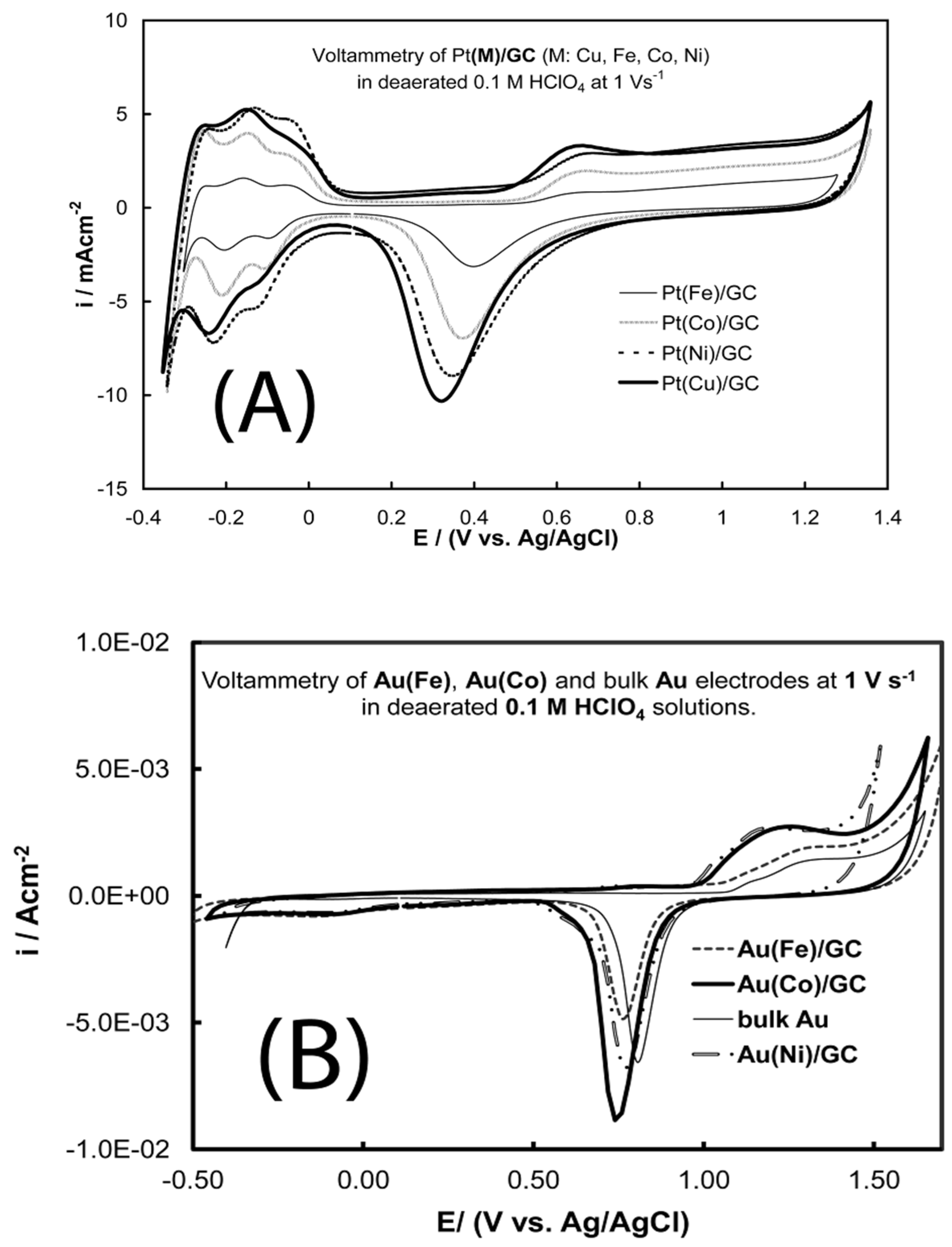
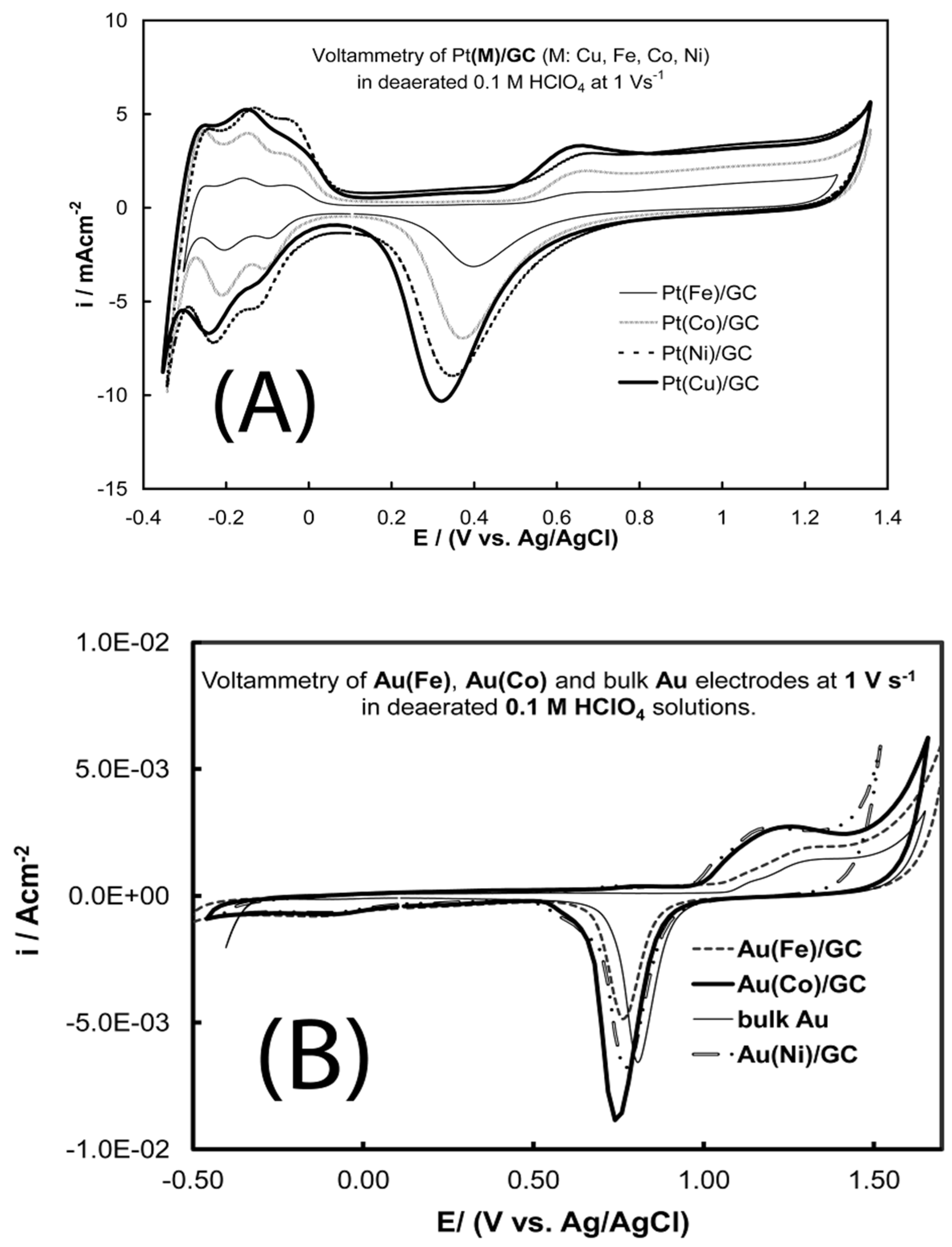
- Papadimitriou, S.; Tegou, A.; Pavlidou, E.; Armyanov, S.; Valova, E.; Kokkinidis, G.; Sotiropoulos, S. Preparation and characterisation of platinum- and gold-coated copper, iron, cobalt and nickel deposits on glassy carbon substrates. Electrochim. Acta 2008, 53, 6559–6567. [Google Scholar] [CrossRef]
- Tegou, A.; Papadimitriou, S.; Armyanov, S.; Valova, E.; Kokkinidis, G.; Sotiropoulos, S. Oxygen reduction at platinum- and gold-coated iron, cobalt, nickel and lead deposits on glassy carbon substrates. J. Electroanal. Chem. 2008, 623, 187–196. [Google Scholar] [CrossRef]
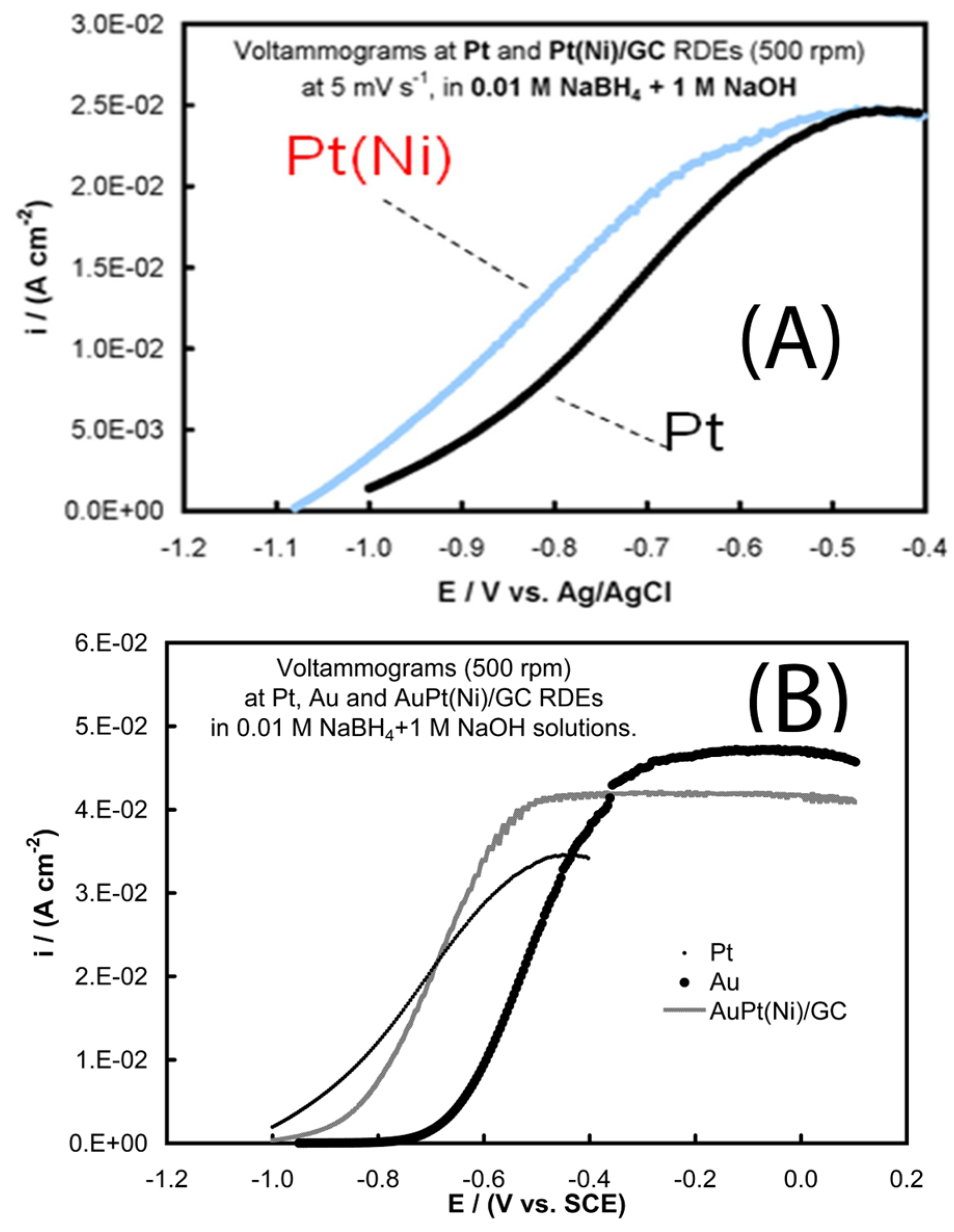
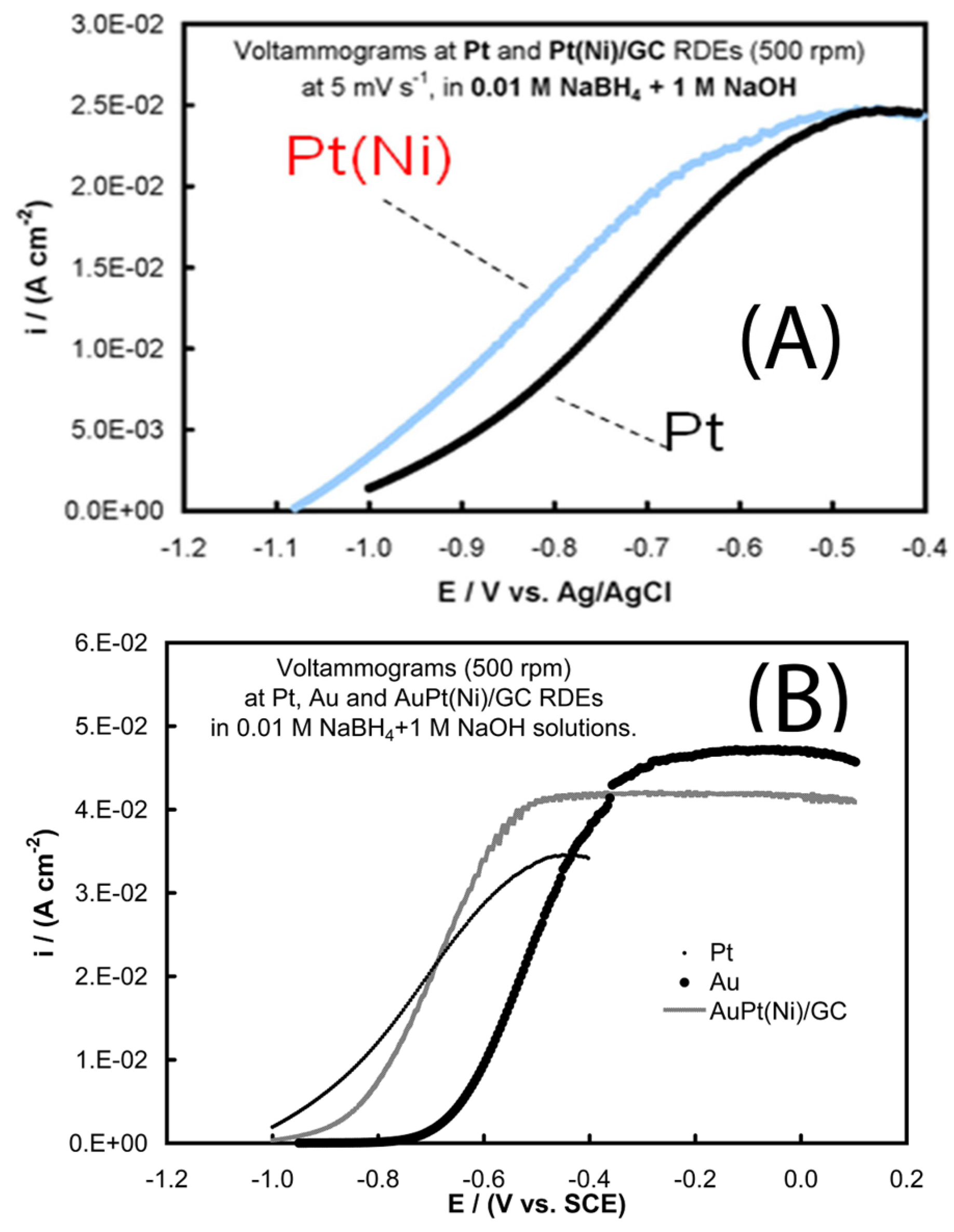
- Tegou, A.; Armyanov, S.; Valova, E.; Steenhaut, O.; Hubin, A.; Kokkinidis, G.; Sotiropoulos, S. Mixed platinum-gold electrocatalysts for borohydride oxidation prepared by the galvanic replacement of nickel deposits. J. Electroanal. Chem. 2009, 634, 104–110. [Google Scholar] [CrossRef]
- Tegou, A.; Papadimitriou, S.; Kokkinidis, G.; Sotiropoulos, S. A rotating disc electrode study of oxygen reduction at platinised nickel and cobalt coatings. J. Solid State Electrochem. 2010, 14, 175–184. [Google Scholar] [CrossRef]
- Tegou, A.; Papadimitriou, S.; Mintsouli, I.; Armyanov, S.; Valova, E.; Kokkinidis, G.; Sotiropoulos, S. Rotating disc electrode studies of borohydride oxidation at Pt and bimetallic Pt-Ni and Pt-Co electrodes. Catal. Today 2011, 170, 126–133. [Google Scholar] [CrossRef]
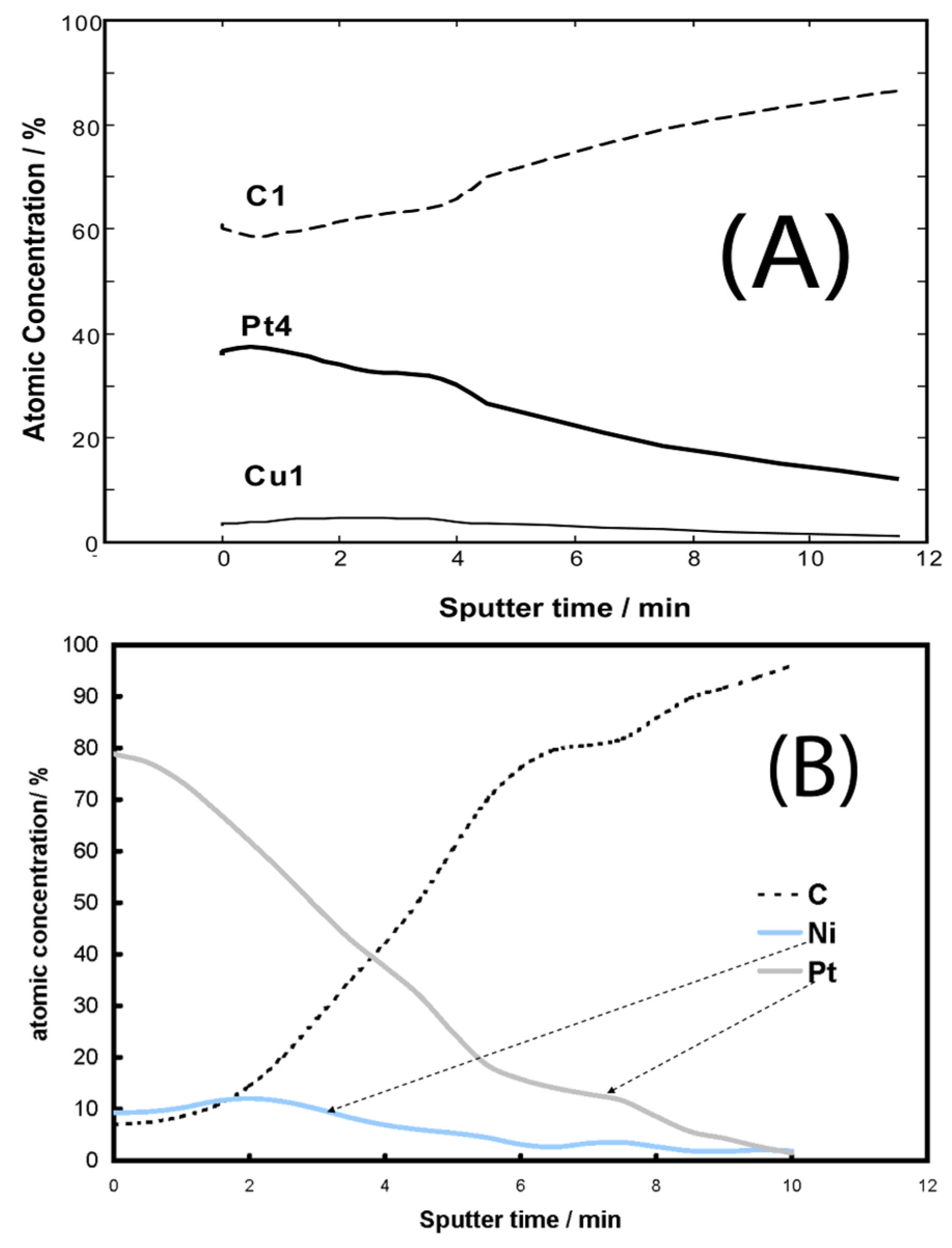
- Mintsouli, I.; Georgieva, J.; Valova, E.; Armyanov, S.; Kakaroglou, A.; Hubin, A.; Steenhaut, O.; Dille, J.; Papaderakis, A.; Kokkinidis, G.; et al. Pt-Ni carbon-supported catalysts for methanol oxidation prepared by Ni electroless deposition and its galvanic replacement by Pt. J. Solid State Electrochem. 2013, 17, 435–443. [Google Scholar] [CrossRef]
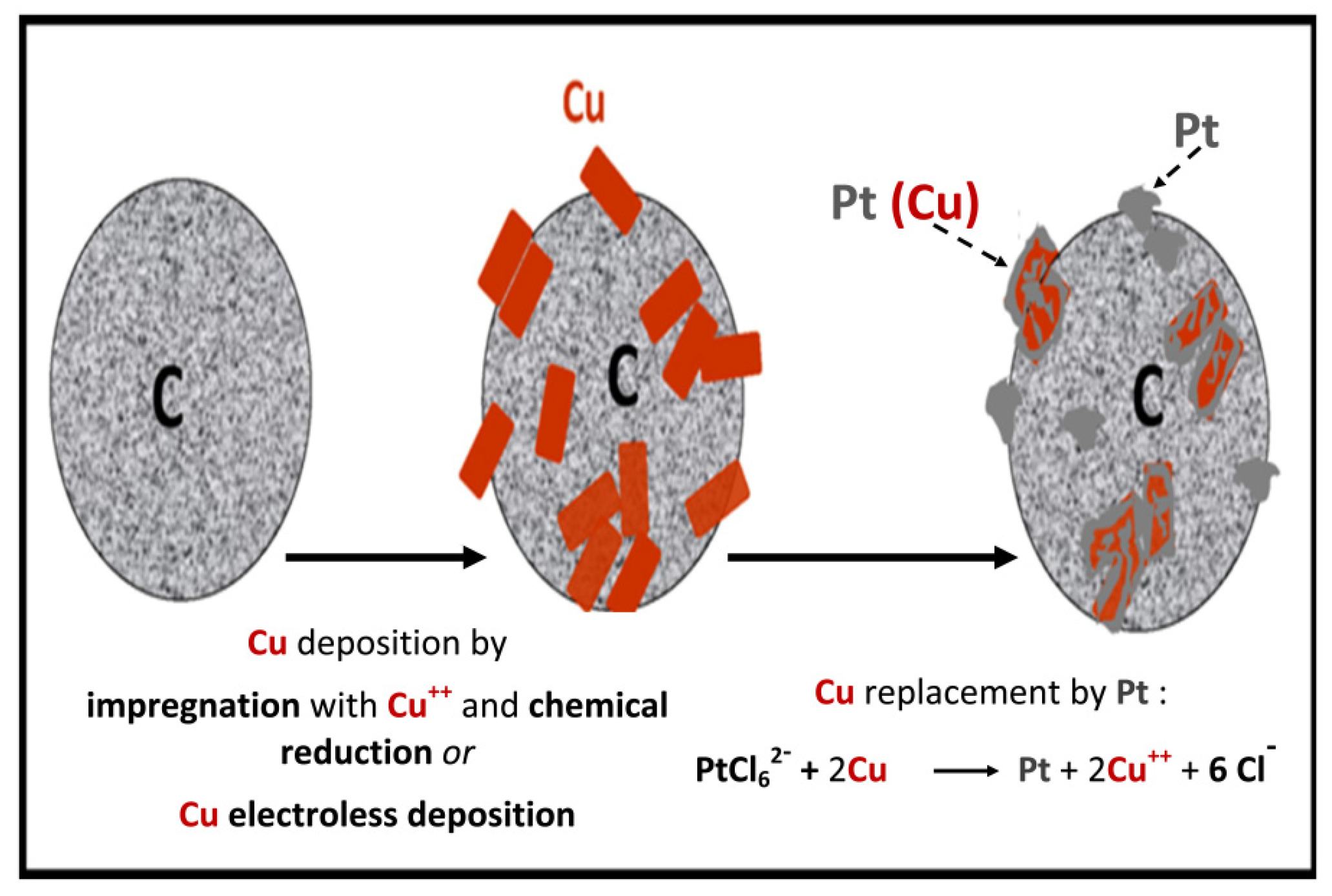
- Mintsouli, I.; Georgieva, J.; Armyanov, S.; Valova, E.; Avdeev, G.; Hubin, A.; Steenhaut, O.; Dille, J.; Tsiplakides, D.; Balomenou, S.; et al. Pt-Cu electrocatalysts for methanol oxidation prepared by partial galvanic replacement of Cu/carbon powder precursors. Appl. Catal. B 2013, 136–137, 160–167. [Google Scholar] [CrossRef]
- Papaderakis, A.; Pliatsikas, N.; Prochaska, C.; Papazisi, K.M.; Balomenou, S.P.; Tsiplakides, D.; Patsalas, P.; Sotiropoulos, S. Ternary Pt-Ru-Ni catalytic layers for methanol electrooxidation prepared by electrodeposition and galvanic replacement. Front. Chem. 2014, 2, 29. [Google Scholar] [CrossRef] [PubMed]
- Georgieva, J.; Valova, E.; Mintsouli, I.; Sotiropoulos, S.; Armyanov, S.; Kakaroglou, A.; Hubin, A.; Steenhaut, O.; Dille, J. Carbon-supported Pt(Cu) electrocatalysts for methanol oxidation prepared by Cu electroless deposition and its galvanic replacement by Pt. J. Appl. Electrochem. 2014, 44, 215–224. [Google Scholar] [CrossRef]
- Georgieva, J.; Sotiropoulos, S.; Valova, E.; Armyanov, S.; Karanasios, N. Methanol oxidation and photo-oxidation at Pt/WO3 electrocatalysts on graphite substrates. J. Electroanal. Chem. 2014, 727, 135–140. [Google Scholar] [CrossRef]
- Georgieva, J.; Valova, E.; Mintsouli, I.; Sotiropoulos, S.; Tatchev, D.; Armyanov, S.; Hubin, A.; Dille, J.; Hoell, A.; Raghuwanshi, V.; et al. Pt(Ni) electrocatalysts for methanol oxidation prepared by galvanic replacement on TiO2 and TiO2/C powder supports. J. Electroanal. Chem. 2015, 754, 65–74. [Google Scholar] [CrossRef]
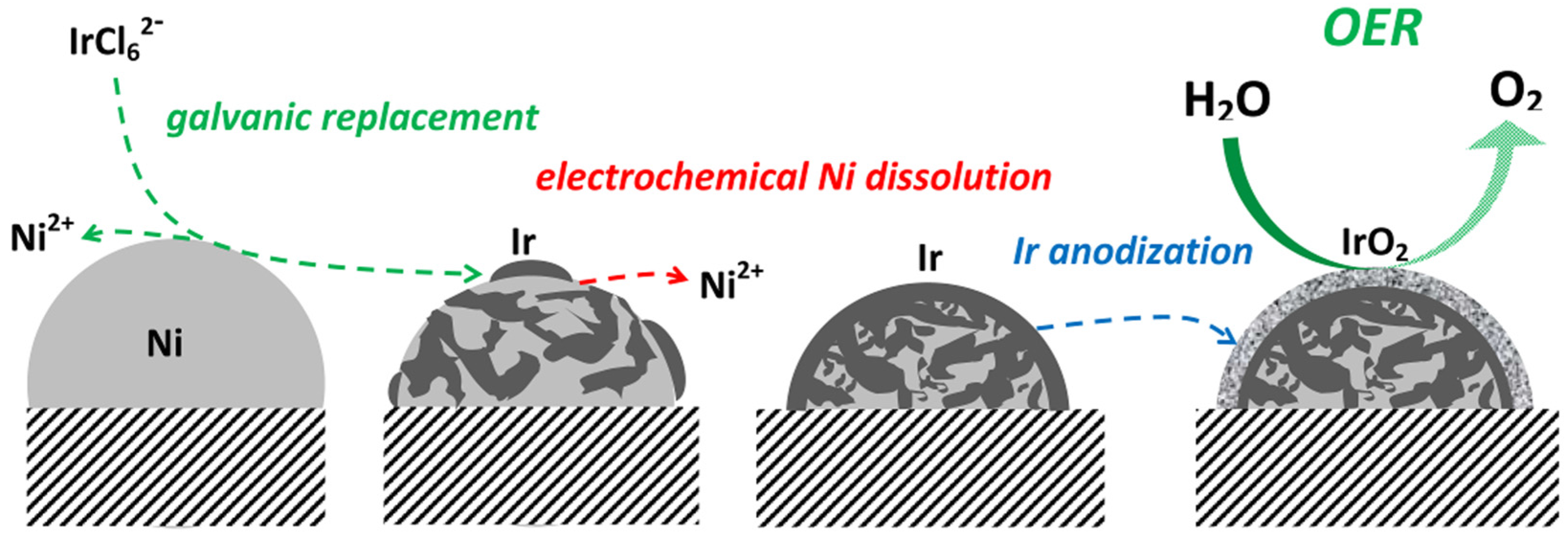
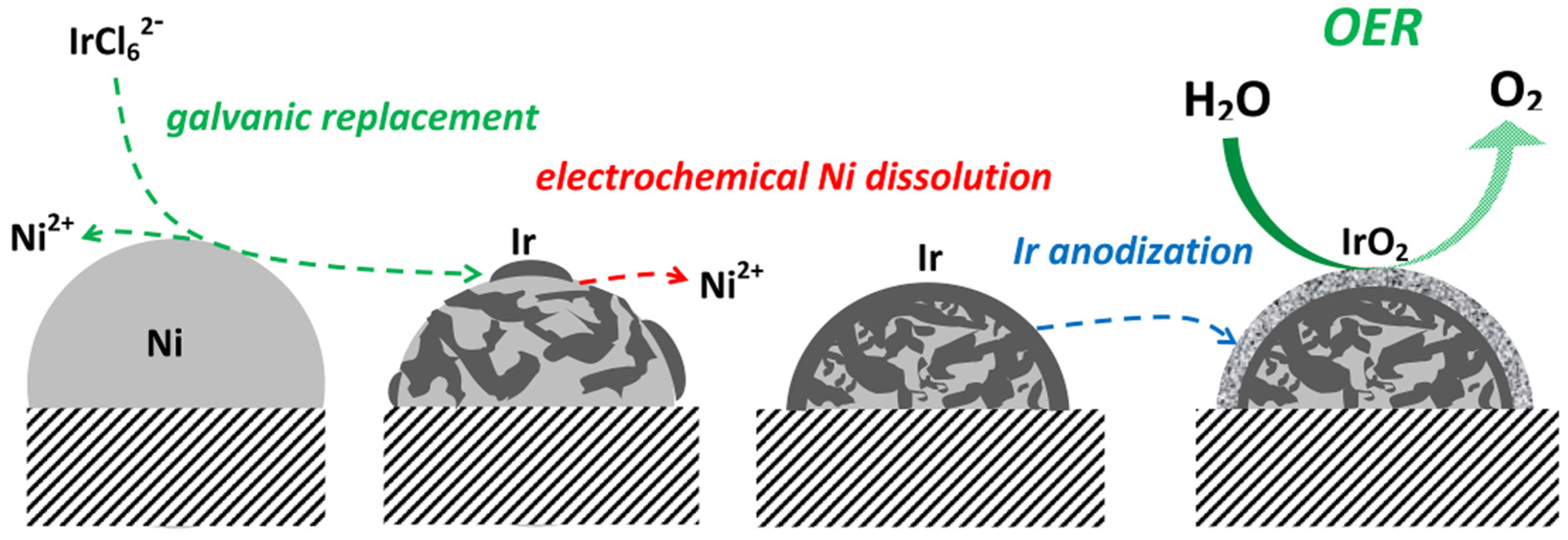
- Papaderakis, A.; Pliatsikas, N.; Prochaska, C.; Vourlias, G.; Patsalas, P.; Tsiplakides, D.; Balomenou, S.; Sotiropoulos, S. Oxygen evolution at IrO2 shell-Ir-Ni core electrodes prepared by galvanic replacement. J. Phys. Chem. C 2016, 120, 19995–20005. [Google Scholar] [CrossRef]
- Tveritinova, E.A.; Maksimov, Y.M.; Zhitnev, Y.N.; Podlovchenko, B.I.; Lunin, V.V. Use of galvanic displacement in the synthesis of a Pd(Cu) hydrodechlorination catalyst. Mendeleev Commun. 2010, 20, 10–11. [Google Scholar] [CrossRef]
- Zhumaev, U.E.; Maksimov, Y.M.; Podlovchenko, B.I. Galvanic replacement of copper adatoms from a Pt/Pt electrode surface in H2PtCl6 solutions. Mendeleev Commun. 2011, 21, 29–30. [Google Scholar] [CrossRef]
- Podlovchenko, B.I.; Zhumaev, U.E.; Maksimov, Y.M. Galvanic displacement of copper adatoms on platinum in PtCl42− solutions. J. Electroanal. Chem. 2011, 651, 30–37. [Google Scholar] [CrossRef]
- Podlovchenko, B.I.; Gladysheva, T.D.; Filatov, A.Y.; Yashina, L.V. Peculiarities of the Pt(Cu)/C catalyst formation by galvanic displacement of copper in H2PtCl4 solutions. Russ. J. Electrochem. 2012, 48, 173–180. [Google Scholar] [CrossRef]
- Kuznetsov, V.V.; Kavyrshina, K.V.; Podlovchenko, B.I. Formation and electrocatalytic properties of Pd deposits on Mo obtained by galvanic displacement. Russ. J. Electrochem. 2012, 48, 467–473. [Google Scholar] [CrossRef]
- Podlovchenko, B.I.; Gladysheva, T.D.; Krivchenko, V.A.; Maksimov, Y.M.; Filatov, A.Y.; Yashina, L.V. Effect of copper deposit morphology on the characteristics of a Pt(Cu)/C-catalyst obtained by galvanic displacement. Mendeleev Commun. 2012, 22, 203–205. [Google Scholar] [CrossRef]
- Kuznetsov, V.V.; Kavyrshina, K.V.; Podlovchenko, B.I. Carbon monoxide adsorption and electrooxidation at a Pd(Mo) electrode prepared by galvanic displacement. Mendeleev Commun. 2012, 22, 206–207. [Google Scholar] [CrossRef]
- Podlovchenko, B.I.; Maksimov, Y.M. Open-circuit potentials established on platinum and gold electrodes in PtCl2−4 solutions after the displacement of copper adatoms. Mendeleev Commun. 2013, 23, 157–159. [Google Scholar] [CrossRef]
- Podlovchenko, B.I.; Maksimov, Y.M.; Maslakov, K.I. Electrocatalytic properties of Au electrodes decorated with Pt submonolayers by galvanic displacement of copper adatoms. Electrochim. Acta 2014, 130, 351–360. [Google Scholar] [CrossRef]
- Gladysheva, T.D.; Filatov, A.Y.; Podlovchenko, B.I. Modification of platinum electrodeposits with an ultralow amount of palladium through the galvanic displacement of hydrogen and copper adatoms. Mendeleev Commun. 2015, 25, 56–58. [Google Scholar] [CrossRef]
- Podlovchenko, B.I.; Maksimov, Y.M.; Evlashin, S.A.; Gladysheva, T.D.; Maslakov, K.I.; Krivchenko, V.A. The use of galvanic displacement in synthesizing Pt0(Bi)/CNW catalysts highly active in electrooxidation of formic acid. J. Electroanal. Chem. 2015, 743, 93–98. [Google Scholar] [CrossRef]
- Podlovchenko, B.I.; Gladysheva, T.D.; Filatov, A.Y. Galvanic-displacement modification of Pd deposits with ultralow amounts of platinum and the electrocatalytic properties of the mixed catalyst. Mendeleev Commun. 2015, 25, 293–295. [Google Scholar] [CrossRef]
- Podlovchenko, B.I.; Maksimov, Y.M. Peculiarities in the electrocatalytic behavior of ultralow platinum deposits on gold synthesized by galvanic displacement. J. Electroanal. Chem. 2015, 756, 140–146. [Google Scholar] [CrossRef]
- Podlovchenko, B.I.; Kuznetsov, V.V.; Batalov, R.S. Palladium catalyst modified with molybdenum bronze as a possible alternative to platinum in the methanol oxidation reaction. J. Solid State Electr. 2016, 20, 589–595. [Google Scholar] [CrossRef]
- Kuznetsov, V.V.; Batalov, R.S.; Podlovchenko, B.I. nPd0·(Hx−2nMoO3) composites as catalysts of methanol and formic acid electrooxidation. Russ. J. Electrochem. 2016, 52, 408–419. [Google Scholar] [CrossRef]
- Vázquez-Gómez, L.; Cattarin, S.; Gerbasi, R.; Guerriero, P.; Musiani, M. Activation of porous Ni cathodes towards hydrogen evolution by electrodeposition of Ir nuclei. J. Appl. Electrochem. 2009, 39, 2165–2172. [Google Scholar] [CrossRef]
- Verlato, E.; Cattarin, S.; Comisso, N.; Guerriero, P.; Musiani, M.; Vázquez-Gómez, L. Preparation of catalytic anodes for methanol oxidation by spontaneous deposition of Pd onto porous Ni or porous Co. Electrochem. Commun. 2010, 12, 1120–1123. [Google Scholar] [CrossRef]
- Vázquez-Gómez, L.; Cattarin, S.; Comisso, N.; Guerriero, P.; Musiani, M.; Verlato, E. Spontaneous deposition of Pd onto Fe-Cr-Al alloys. Electrochim. Acta 2012, 68, 114–122. [Google Scholar] [CrossRef]
- Fiameni, S.; Herraiz-Cardona, I.; Musiani, M.; Pérez-Herranz, V.; Vázquez-Gómez, L.; Verlato, E. The HER in alkaline media on Pt-modified three-dimensional Ni cathodes. Int. J. Hydrogen Energy 2012, 37, 10507–10516. [Google Scholar] [CrossRef]
- Cimino, S.; Gerbasi, R.; Lisi, L.; Mancino, G.; Musiani, M.; Vázquez-Gómez, L.; Verlato, E. Oxidation of CO and CH4 on Pd-FeCr alloy foam catalysts prepared by spontaneous deposition. Chem. Eng. J. 2013, 230, 422–431. [Google Scholar] [CrossRef]
- Verlato, E.; Cattarin, S.; Comisso, N.; Mattarozzi, L.; Musiani, M.; Vázquez-Gómez, L. Reduction of nitrate ions at Rh-modified Ni foam electrodes. Electrocatalysis 2013, 4, 203–211. [Google Scholar] [CrossRef]
- Verlato, E.; Cattarin, S.; Comisso, N.; Mattarozzi, L.; Musiani, M.; Vázquez-Gómez, L. Electrochemical Impedance Spectroscopy study of the preparation of electrocatalysts through galvanic displacement reactions. J. Electroanal. Chem. 2015, 737, 100–107. [Google Scholar] [CrossRef]
- Musiani, M.; Cattarin, S.; Cimino, S.; Comisso, N.; Mattarozzi, L.; Vázquez-Gómez, L.; Verlato, E. Preparation of 3D electrocatalysts and catalysts for gas-phase reactions, through electrodeposition or galvanic displacement. J. Appl. Electrochem. 2015, 45, 715–725. [Google Scholar] [CrossRef]
- Verlato, E.; He, W.; Amrane, A.; Barison, S.; Floner, D.; Fourcade, F.; Geneste, F.; Musiani, M.; Seraglia, R. Preparation of silver-modified nickel foams by galvanic displacement and their use as cathodes for the reductive dechlorination of herbicides. ChemElectroChem 2016. [Google Scholar] [CrossRef]
- Comisso, N.; Cattarin, S.; Guerriero, P.; Mattarozzi, L.; Musiani, M.; Vázquez-Gómez, L.; Verlato, E. Study of Cu, Cu-Ni and Rh-modified Cu porous layers as electrode materials for the electroanalysis of nitrate and nitrite ions. J. Solid State Electr. 2016, 20, 1139–1148. [Google Scholar] [CrossRef]
- Bianchi, I.; Guerrini, E.; Trasatti, S. Electrocatalytic activation of Ni for H2 evolution by spontaneous deposition of Ru. Chem. Phys. 2005, 319, 192–199. [Google Scholar] [CrossRef]
- Duca, M.; Guerrini, E.; Colombo, A.; Trasatti, S. Activation of nickel for hydrogen evolution by spontaneous deposition of iridium. Electrocatalysis 2013, 4, 338–345. [Google Scholar] [CrossRef]
- Oh, M.H.; Yu, T.; Yu, S.-H.; Lim, B.; Ko, K.-T.; Willinger, M.-G.; Seo, D.-H.; Kim, B.H.; Cho, M.G.; Park, J.-H.; et al. Galvanic replacement reactions in metal oxide nanocrystals. Science 2013, 340, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Comisso, N.; Cattarin, S.; Guerriero, P.; Mattarozzi, L.; Musiani, M.; Verlato, E. Conversion of porous PbO2 layers through galvanic displacement reaction with Mn2+ ions. Electrochem. Commun. 2016, 73, 59–62. [Google Scholar] [CrossRef]
- Ruban, A.; Hammer, B.; Stoltze, P.; Skriver, H.L.; Nørskov, J.K. Surface electronic structure and reactivity of transition and noble metals. J. Mol. Catal. A 1997, 115, 421–429. [Google Scholar] [CrossRef]
- Kitchin, J.R.; Nørskov, J.K.; Barteau, M.A.; Chen, J.G. Modification of the surface electronic and chemical properties of Pt(111) by subsurface 3d transition metals. J. Chem. Phys. 2004, 120, 10240–10246. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.-E.; Gokcen, D.; Liu, P.; Mohammadi, P.; Brankovic, S.R. Size effects in monolayer catalysis-model study: Pt submonolayers on Au(111). Electrocatalysis 2012, 3, 203–210. [Google Scholar] [CrossRef]
- Loukrakpam, R.; Yuan, Q.; Petkov, V.; Gan, L.; Rudi, S.; Yang, R.; Huang, Y.; Brankovic, S.R.; Strasser, P. Efficient C-C bond splitting on Pt monolayer and sub-monolayer catalysts during ethanol electro-oxidation: Pt layer strain and morphology effects. Phys. Chem. Chem. Phys. 2014, 16, 18866–18876. [Google Scholar] [CrossRef] [PubMed]
- Grabow, L.C.; Yuan, Q.; Doan, H.A.; Brankovic, S.R. Novel 2D RuPt core-edge nanocluster catalyst for CO electro-oxidation. Surf. Sci. 2015, 640, 50–58. [Google Scholar] [CrossRef]
- Kühl, S.; Strasser, P. Oxygen electrocatalysis on dealloyed Pt nanocatalysts. Top. Catal. 2016, 59, 1628–1637. [Google Scholar] [CrossRef]
- Kibler, L.A. Dependence of electrocatalytic activity on film thickness for the hydrogen evolution reaction of Pd overlayers on Au(111). Electrochim. Acta 2008, 53, 6824–6828. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.; Wang, R.; Wang, Q.; Lei, Z. Carbon-supported platinum-decorated nickel nanoparticles for enhanced methanol oxidation in acid media. J. Solid State Electr. 2012, 16, 1049–1054. [Google Scholar] [CrossRef]
- Ivanov, S.; Mintsouli, I.; Georgieva, J.; Armyanov, S.; Valova, E.; Kokkinidis, G; Sotiropoulos, S. Platinized TiO2 electrode coatings prepared by a two-step electrodeposition-galvanic replacement process for methanol oxidation in the dark and under UV light illumination. J. Electrochem. Sci. Eng. 2012, 2, 155–169. [Google Scholar]
- Mintsouli, I.; Georgieva, J.; Papaderakis, A.; Tsiplakides, D.; Balomenou, S.; Sotiropoulos, S. Methanol oxidation at platinized copper particles prepared by galvanic replacement. J. Electrochem. Sci. Eng. 2016, 6, 17–28. [Google Scholar] [CrossRef]
- Teng, X.; Du, W.; Wang, Q. Synthesis of Pt-containing metals alloy and hybrid nanowires and investigation of electronic structure using synchrotron-based X-ray absorption techniques. In Nanowires Fundamental Research; Hashim, A., Ed.; InTech: Shanghai, China, 2010; pp. 205–224. [Google Scholar]
- Westsson, E.; Koper, G.J.M. How to determine the core-shell nature in bimetallic catalyst particles? Catalysts 2014, 4, 375–396. [Google Scholar] [CrossRef]
- Sutter, E.; Jungjohann, K.; Bliznakov, S.; Courty, A.; Maisonhaute, E.; Tenney, S.; Sutter, P. In situ liquid-cell electron microscopy of silver-palladium galvanic replacement reactions on silver nanoparticles. Nat. Commun. 2014, 5, 4946. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Dong, S.; Wang, E. A general method for the rapid synthesis of hollow metallic or bimetallic nanoelectrocatalysts with urchinlike morphology. Chem. Eur. J. 2008, 14, 4689–4695. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.-W.; Zhang, R.-H.; Zhou, Z.-Y.; Sun, S.-G. Preparation of PtNi hollow nanospheres for the electrocatalytic oxidation of methanol. J. Power Sources 2011, 196, 5844–5848. [Google Scholar] [CrossRef]
- Song, C.; Zhang, J. Electrocatalytic oxygen reduction reaction. In PEM Fuel Cell Electrocatalysts and Catalyst Layers Fundamentals and Applications; Zhang, J., Ed.; Springer: London, UK, 2008; pp. 89–134. [Google Scholar]
- Kinoshita, K. Electrochemical Oxygen Technology; Wiley-Interscience: Ney York, NY, USA, 1992. [Google Scholar]
- Thompsett, D. Pt alloys as oxygen reduction catalysts. In Handbook of Fuel Cells: Fundamentals, Technology, Applications; Vielstich, W., Lamm, A., Gasteiger, H., Eds.; John Wiley and Sons Ltd.: London, UK, 2003; Volume 3, pp. 467–481. [Google Scholar]
- Antolini, E.; Salgado, J.R.C.; Giz, M.J.; Gonzalez, E.R. The stability of Pt-M (M = first row transition metal) alloy catalysts and its effect on the activity in low temperature fuel cells. A literature review and tests on a Pt-Co catalyst. J. Power Sources 2006, 160, 957–968. [Google Scholar] [CrossRef]
- Antolini, E. Formation of carbon-supported PtM alloys for low temperature fuel cells: A review. Mater. Chem. Phys. 2003, 78, 563–573. [Google Scholar] [CrossRef]
- Shao, M.H.; Sasaki, K.; Liu, P.; Adzic, R.R. Pd3Fe and Pt monolayer-modified Pd3Fe electrocatalysts for oxygen reduction. Z. Phys. Chem. 2007, 221, 1175–1190. [Google Scholar] [CrossRef]
- Shao, M.; Sasaki, K.; Marinkovic, N.S.; Zhang, L.; Adzic, R.R. Synthesis and characterization of platinum monolayer oxygen-reduction electrocatalysts with Co-Pd core-shell nanoparticle supports. Electrochem. Commun. 2007, 9, 2848–2853. [Google Scholar] [CrossRef]
- Zhou, W.-P.; Sasaki, K.; Su, D.; Zhu, Y.; Wang, J.X.; Adzic, R.R. Gram-scale-synthesized Pd2Co-supported Pt monolayer electrocatalysts for oxygen reduction reaction. J. Phys. Chem. C 2010, 114, 8950–8957. [Google Scholar] [CrossRef]
- Gong, K.; Chen, W.-F.; Sasaki, K.; Su, D.; Vukmirovic, M.B.; Zhou, W.; Izzo, E.L.; Perez-Acosta, C.; Hirunsit, P.; Balbuena, P.B.; et al. Platinum-monolayer electrocatalysts: Palladium interlayer on IrCo alloy core improves activity in oxygen-reduction reaction. J. Electroanal. Chem. 2010, 649, 232–237. [Google Scholar] [CrossRef]
- Sasaki, K.; Kuttiyiel, K.A.; Su, D.; Adzic, R.R. Platinum monolayer on IrFe core-shell nanoparticle electrocatalysts for the oxygen reduction reaction. Electrocatalysis 2011, 2, 134–140. [Google Scholar] [CrossRef]
- Kuttiyiel, K.A.; Sasaki, K.; Choi, Y.; Su, D.; Liu, P.; Adzic, R.R. Bimetallic IrNi core platinum monolayer shell electrocatalysts for the oxygen reduction reaction. Energy Environ. Sci. 2012, 5, 5297–5304. [Google Scholar] [CrossRef]
- Karan, H.I.; Sasaki, K.; Kuttiyiel, K.; Farberow, C.A.; Mavrikakis, M.; Adzic, R.R. Catalytic activity of platinum monolayer on iridium and rhenium alloy nanoparticles for the oxygen reduction reaction. ACS Catal. 2012, 2, 817–824. [Google Scholar] [CrossRef]
- Choi, Y.; Kuttiyiel, K.A.; Labis, J.P.; Sasaki, K.; Park, G.-G.; Yang, T.-H.; Adzic, R.R. Enhanced oxygen reduction activity of IrCu core platinum monolayer shell nano-electrocatalysts. Top. Catal. 2013, 56, 1059–1064. [Google Scholar] [CrossRef]
- Shao, M.-H.; Sasaki, K.; Adzic, R.R. Pd-Fe nanoparticles as electrocatalysts for oxygen reduction. J. Am. Chem. Soc. 2006, 128, 3526–3527. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.H.; Huang, T.; Liu, P.; Zhang, J.; Sasaki, K.; Vukmirovic, M.B.; Adzic, R.R. Palladium monolayer and palladium alloy electrocatalysts for oxygen reduction. Langmuir 2006, 22, 10409–10415. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Liu, P.; Zhang, J.; Adzic, R.R. Origin of enhanced activity in palladium alloy electrocatalysts for oxygen reduction reaction. J. Phys. Chem. B 2007, 111, 6772–6775. [Google Scholar] [CrossRef] [PubMed]
- Gong, K.; Su, D.; Adzic, R.R. Platinum-monolayer shell on AuNi0.5Fe nanoparticle core electrocatalyst with high activity and stability for the oxygen reduction reaction. J. Am. Chem. Soc. 2010, 132, 14364–14366. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Cai, Y.; Vukmirovic, M.B.; Zhou, W.-P.; Karan, H.; Wang, J.X.; Adzic, R.R. Enhancing oxygen reduction reaction activity via Pd-Au alloy sublayer mediation of Pt monolayer electrocatalysts. J. Phys. Chem. Lett. 2010, 1, 3238–3242. [Google Scholar] [CrossRef]
- Kuttiyiel, K.A.; Sasaki, K.; Su, D.; Vukmirovic, M.B.; Marinkovic, N.S.; Adzic, R.R. Pt monolayer on Au-stabilized PdNi core-shell nanoparticles for oxygen reduction reaction. Electrochim. Acta 2013, 110, 267–272. [Google Scholar] [CrossRef]
- Chen, G.; Kuttiyiel, K.A.; Su, D.; Li, M.; Wang, C.-H.; Buceta, D.; Du, C.; Cao, Y.; Yin, G.; Sasaki, K.; et al. Oxygen reduction kinetics on Pt monolayer shell highly affected by the structure of bimetallic AuNi cores. Chem. Mater. 2016, 28, 5274–5281. [Google Scholar] [CrossRef]
- Kuttiyiel, K.A.; Choi, Y.; Hwang, S.-M.; Park, G.-G.; Yang, T.-H.; Su, D.; Sasaki, K.; Liu, P.; Adzic, R.R. Enhancement of the oxygen reduction on nitride stabilized Pt-M (M = Fe, Co, and Ni) core-shell nanoparticle electrocatalysts. Nano Energy 2015, 13, 442–449. [Google Scholar] [CrossRef]
- Hu, J.; Kuttiyiel, K.A.; Sasaki, K.; Su, D.; Yang, T.H.; Park, G.G.; Zhang, C.; Chen, G.; Adzic, R.R. Pt monolayer shell on nitrided alloy core—A path to highly stable oxygen reduction catalyst. Catalysts 2015, 5, 1321–1332. [Google Scholar] [CrossRef]
- Knupp, S.L.; Vukmirovic, M.B.; Haldar, P.; Herron, J.A.; Mavrikakis, M.; Adzic, R.R. Platinum Monolayer Electrocatalysts for O2 Reduction: Pt Monolayer on Carbon-Supported PdIr Nanoparticles. Electrocatalysis 2010, 1, 213–223. [Google Scholar] [CrossRef]
- Sun, Z.; Masa, J.; Xia, W.; König, D.; Ludwig, A.; Li, Z.-A.; Farle, M.; Schuhmann, W.; Muhler, M. Rapid and surfactant-free synthesis of bimetallic Pt-Cu nanoparticles simply via ultrasound-assisted redox replacement. ACS Catal. 2012, 2, 1647–1653. [Google Scholar] [CrossRef]
- Jung, N.; Sohn, Y.; Park, J.H.; Nahm, K.S.; Kim, P.; Yoo, S.J. High-performance PtCux at Pt core-shell nanoparticles decorated with nanoporous Pt surfaces for oxygen reduction reaction. Appl. Catal. B 2016, 196, 199–206. [Google Scholar] [CrossRef]
- Chen, D.; Ye, F.; Liu, H.; Yang, J. Cage-bell Pt-Pd nanostructures with enhanced catalytic properties and superior methanol tolerance for oxygen reduction reaction. Sci. Rep. 2016, 6, 24600. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Liu, X.; Bai, C.; Chen, Y.; Wang, L.; Zheng, M.; Dong, Q.; Peng, D.-L. Effect of component distribution and nanoporosity in CuPt nanotubes on electrocatalysis of the oxygen reduction reaction. ChemSusChem 2015, 8, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Peng, Z.; Yang, H. Supportless oxygen reduction electrocatalysts of CoCuPt hollow nanoparticles. Philos. Trans. A 2010, 368, 4261–4274. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.J.; Yoo, S.J.; Lim, Y.; Kim, S.; Lim, Y.; Choi, J.; Nahm, K.S.; Hwang, S.J.; Lim, T.-H.; Kim, S.-K.; et al. Facile preparation of carbon-supported PtNi hollow nanoparticles with high electrochemical performance. J. Mater. Chem. 2012, 22, 8820–8825. [Google Scholar] [CrossRef]
- Dubau, L.; Asset, T.; Chattot, R.; Bonnaud, C.; Vanpeene, V.; Nelayah, J.; Maillard, F. Tuning the performance and the stability of porous hollow PtNi/C nanostructures for the oxygen reduction reaction. ACS Catal. 2015, 5, 5333–5341. [Google Scholar] [CrossRef]
- Nagai, H.; Aso, H.; Kawabuchi, M.; Kondo, T. Electrochemical construction of Ni and co core—Pt shell nanoparticles as catalysts for oxygen reduction reaction. ECS Trans. 2013, 58, 27–31. [Google Scholar] [CrossRef]
- Liu, M.; Chi, F.; Liu, J.; Song, Y.; Wang, F. A novel strategy to synthesize bimetallic Pt-Ag particles with tunable nanostructures and their superior electrocatalytic activities toward the oxygen reduction reaction. RSC Adv. 2016, 6, 62327–62335. [Google Scholar] [CrossRef]
- Fu, T.; Fang, J.; Wang, C.; Zhao, J. Hollow porous nanoparticles with Pt skin on a Ag-Pt alloy structure as a highly active electrocatalyst for the oxygen reduction reaction. J. Mater. Chem. A 2016, 4, 8803–8811. [Google Scholar] [CrossRef]
- Chen, D.; Li, C.; Liu, H.; Ye, F.; Yang, J. Core-shell Au@Pd nanoparticles with enhanced catalytic activity for oxygen reduction reaction via core-shell Au@Ag/Pd constructions. Sci. Rep. 2015, 5, 11949. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, W.; Wang, J.; Wexler, D.; Poynton, S.D.; Slade, R.C.T.; Liu, H.; Winther-Jensen, B.; Kerr, R.; Shi, D.; et al. PdNi hollow nanoparticles for improved electrocatalytic oxygen reduction in alkaline environments. ACS Appl. Mater. Interfaces 2013, 5, 12708–12715. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.; Huang, C.; Hao, Y.; Liu, F. Synthesis of highly active and stable Au-PtCu core-shell nanoparticles for oxygen reduction reaction. Phys. Chem. Chem. Phys. 2012, 14, 14696–14701. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Liu, H.; Hu, W.; Zhong, J.; Chen, Y.; Cao, H.; Yang, J. Heterogeneous Au-Pt nanostructures with enhanced catalytic activity toward oxygen reduction. Dalton Trans. 2012, 41, 2898–2903. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jin, M.; Wang, J.; Li, W.; Camargo, P.H.C.; Kim, M.J.; Yang, D.; Xie, Z.; Xia, Y. Synthesis of Pd-Pt bimetallic nanocrystals with a concave structure through a bromide-induced galvanic replacement reaction. J. Am. Chem. Soc. 2011, 133, 6078–6089. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Cheng, C.H.; Zhou, W.; Lee, J.Y.; Liu, Z. Methanol-tolerant heterogeneous PdCo@PdPt/C electrocatalyst for the oxygen reduction reaction. Fuel Cells 2010, 10, 907–913. [Google Scholar] [CrossRef]
- Yaldagard, M.; Seghatoleslami, N.; Jahanshahi, M. Oxygen reduction reaction activity improvement in Cu/PtPd nanocatalyst based on core-shell structured through electrochemical synthesis on porous gas diffusion electrodes in polymer electrolyte membrane fuel cells. J. Nano Res. 2015, 31, 62–80. [Google Scholar] [CrossRef]
- Caixia, X.; Yan, Z.; Liqiang, X.; Xiufang, B.; Houyi, M.; Yi, D. Nanotubular mesoporous PdCu bimetallic electrocatalysts toward oxygen reduction reaction. Chem. Mater. 2009, 21, 3110–3116. [Google Scholar]
- Stamenkovic, V.R.; Mun, B.S.; Mayrhofer, K.J.J.; Ross, P.N.; Markovic, N.M. Effect of surface composition on electronic structure, stability, and electrocatalytic properties of Pt-transition metal alloys: Pt-skin versus Pt-skeleton surfaces. J. Am. Chem. Soc. 2006, 128, 8813–8819. [Google Scholar] [CrossRef] [PubMed]
- Stamenkovic, V.R.; Mun, B.S.; Arenz, M.; Mayrhofer, K.J.J.; Lucas, C.A.; Wang, G.; Ross, P.N.; Markovic, N.M. Trends in electrocatalysis on extended and nanoscale Pt-bimetallic alloy surfaces. Nat. Mater. 2007, 6, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Wakisaka, M.; Mitsui, S.; Hirose, Y.; Kawashima, K.; Uchida, H.; Watanabe, M. Electronic structures of Pt-Co and Pt-Ru alloys for CO-tolerant anode catalysts in polymer electrolyte fuel cells studied by EC-XPS. J. Phys. Chem. B 2006, 110, 23489–23496. [Google Scholar] [CrossRef] [PubMed]
- Paulus, U.A.; Wokaun, A.; Scherer, G.G.; Schmidt, T.J.; Stamenkovic, V.; Radmilovic, V.; Markovic, N.M.; Ross, P.N. Oxygen reduction on carbon-supported Pt-Ni and Pt-Co alloy catalysts. J. Phys. Chem. B 2002, 106, 4181–4191. [Google Scholar] [CrossRef]
- Paulus, U.A.; Wokaun, A.; Scherer, G.G.; Schmidt, T.J.; Stamenkovic, V.; Markovic, N.M.; Ross, P.N. Oxygen reduction on high surface area Pt-based alloy catalysts in comparison to well defined smooth bulk alloy electrodes. Electrochim. Acta 2002, 47, 3787–3798. [Google Scholar] [CrossRef]
- Stamenkovic, V.R.; Schmidt, T.J.; Ross, P.N.; Markovic, N.M. Surface composition effects in electrocatalysis: Kinetics of oxygen reduction on well-defined Pt3Ni and Pt3Co alloy surfaces. J. Phys. Chem. B 2002, 106, 11970–11979. [Google Scholar] [CrossRef]
- Stamenkovic, V.R.; Mun, B.S.; Mayrhofer, K.J.J.; Ross, P.N.; Markovic, N.M.; Rossmeil, J.; Greeley, J.; Nørskov, J.K. Changing the activity of electrocatalysts for oxygen reduction by tuning the surface electronic structure. Angew. Chem. Int. Ed. 2006, 45, 2897–2901. [Google Scholar] [CrossRef] [PubMed]
- Stamenkovic, V.R.; Fowler, B.; Mun, B.S.; Wang, G.; Ross, P.N.; Lucas, C.A.; Markovic, N.M. Improved oxygen reduction activity on Pt3Ni(111) via increased surface site availability. Science 2007, 315, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Wakisaka, M.; Suzuki, H.; Mitsui, S.; Uchida, H.; Watanabe, M. Increased oxygen coverage at Pt-Fe alloy cathode for the enhanced oxygen reduction reaction studied by EC-XPS. J. Phys. Chem. C 2008, 112, 2750–2755. [Google Scholar] [CrossRef]
- Lamy, C.; Léger, J.-M.; Srinivasan, S. Direct methanol fuel cells: From a twentieth century electrochemist’s dream to a twenty-first century emerging technology. In Modern Aspects of Electrochemistry; Bockris, J.O.M., Conway, B.E., Eds.; Plenum Press: Plattsburgh, NY, USA, 2000; Volume 34, pp. 53–118. [Google Scholar]
- Hamnett, A. Direct methanol fuel cells (DMFC). In Handbook of Fuel Cells: Fundamentals Technology and Applications; Vielstich, W., Lamm, A., Gasteiger, H.A., Eds.; Wiley: Chichester, UK, 2003; Volume 1, pp. 305–316. [Google Scholar]
- Beden, B.; Kadirgan, F.; Lamy, C.; Léger, J.-M. Electrocatalytic oxidation of methanol on platinum-based binary electrodes. J. Electroanal. Chem. 1981, 127, 75–85. [Google Scholar] [CrossRef]
- Chrzanowski, W.; Kim, H.; Wieckowski, A. Enhancement in methanol oxidation by spontaneously deposited ruthenium on low-index platinum electrodes. Catal. Lett. 1998, 50, 69–75. [Google Scholar] [CrossRef]
- Lamy, C.; Lima, A.; LeRhun, V.; Delime, F.; Coutencau, C.; Léger, J.-M. Recent advances in the development of direct alcohol fuel cells (DAFC). J. Power Sources 2002, 105, 283–296. [Google Scholar] [CrossRef]
- Antolini, E.; Lopes, T.; Gonzalez, E.R. An overview of platinum-based catalysts as methanol-resistant oxygen reduction materials for direct methanol fuel cells. J. Alloy. Compd. 2008, 461, 253–262. [Google Scholar] [CrossRef]
- Antolini, E.; Salgado, J.R.C.; dos Santos, A.M.; Gonzalez, E.R. The methanol oxidation reaction on platinum alloys with the first row transition metals: The case of Pt-Co and -Ni alloy electrocatalysts for DMFCs: A short review. Appl. Catal. B 2006, 63, 137–149. [Google Scholar] [CrossRef]
- Antolini, E. Platinum-based ternary catalysts for low temperature fuel cells. Part I. Preparation methods and structural characteristics. Appl. Catal. B 2007, 74, 324–336. [Google Scholar] [CrossRef]
- Sasaki, K.; Adzic, R.R. Monolayer-level Ru- and NbO2-supported platinum electrocatalysts for methanol oxidation. J. Electrochem. Soc. 2008, 155, B180–B186. [Google Scholar] [CrossRef]
- Liu, H.; Adzic, R.R.; Wong, S.S. Multifunctional ultrathin PdxCu1−x and Pt∼PdxCu1−x one-dimensional nanowire motifs for various small molecule oxidation reactions. ACS Appl. Mater. Interfaces 2015, 7, 26145–26157. [Google Scholar] [CrossRef] [PubMed]
- Ando, Y.; Sasaki, K.; Adzic, R. Electrocatalysts for methanol oxidation with ultra low content of Pt and Ru. Electrochem. Commun. 2009, 11, 1135–1138. [Google Scholar] [CrossRef]
- Kusnetsov, V.V.; Podlovchenko, B.I.; Shakurov, R.I.; Kavyrshina, K.V.; Lyahenko, S.E. NPt0 (Hx−2nMoO3) as a promising catalyst for the oxidation of methanol. Synthesis and electrocatalytic properties. Int. J. Hydrogen Energy 2014, 39, 829–836. [Google Scholar] [CrossRef]
- Zhou, X.-W.; Gan, Y.-L.; Sun, S.-G. Studies of oxidation processes of methanol on hollow CoPt nanospheres and in situ electrochemical Fourier transform infrared spectroscopy. Acta Phys. Chim. Sin. 2012, 28, 2071–2076. [Google Scholar]
- Zhou, X.-W.; Chen, Q.-S.; Zhou, Z.-Y.; Sun, S.-G. Synthesis, electrocatalytic and anomalous IR properties of hollow CoPt chainlike nanomaterials. J. Nanosci. Nanotechnol. 2009, 9, 2392–2397. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.-S.; Sun, S.-G.; Zhou, Z.-Y.; Chen, Y.-X.; Deng, S.-B. CoPt nanoparticles and their catalytic properties in electrooxidation of CO and CH3OH studied by in situ FTIRS. Phys. Chem. Chem. Phys. 2008, 10, 3645–3654. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xia, T.; Wang, S.; Yang, G.; Dong, B.; Wang, C.; Ma, Q.; Sun, Y.; Wang, R. Oriented-assembly of hollow FePt nanochains with tunable catalytic and magnetic properties. Nanoscale 2016, 8, 11432–11440. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Fang, Y.; Dong, S.; Wang, E. High-efficiency and low-cost hybrid nanomaterial as enhancing electrocatalyst: Spongelike Au/Pt core/shell nanomaterial with hollow cavity. J. Phys. Chem. C 2007, 111, 17104–17109. [Google Scholar] [CrossRef]
- Sieben, J.M.; Alvarez, A.E.; Comignani, V.; Duarte, M.M.E. Methanol and ethanol oxidation on carbon supported nanostructured Cu core Pt-Pd shell electrocatalysts synthesized via redox displacement. Int. J. Hydrogen Energy 2014, 39, 11547–11556. [Google Scholar] [CrossRef]
- Li, Q.; Xu, P.; Zhang, B.; Wu, G.; Zhao, H.; Fu, E.; Wang, H.-L. Self-supported Pt nanoclusters via galvanic replacement from Cu2O nanocubes as efficient electrocatalysts. Nanoscale 2013, 5, 7397–7402. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, M.G.; Abdolmaleki, M.; Ashrafpoor, S. Methanol electro-oxidation on a porous nanostructured Ni/Pd-Ni electrode in alkaline media. Chin. J. Catal. 2013, 34, 1712–1719. [Google Scholar] [CrossRef]
- Zhao, D.; Yan, B.; Xu, B.-Q. Proper alloying of Pt with underlying Ag nanoparticles leads to dramatic activity enhancement of Pt electrocatalyst. Electrochem. Commun. 2008, 10, 884–887. [Google Scholar] [CrossRef]
- Zhang, W.; Yang, J.; Lu, X. Tailoring galvanic replacement reaction for the preparation of Pt/Ag bimetallic hollow nanostructures with controlled number of voids. ACS Nano 2012, 6, 7397–7405. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Luk, S.-Y.; Kwong, T.-L.; Yung, K.-F. Synthesis of hollow PtAg alloy nanospheres with excellent electrocatalytic performances towards methanol and formic acid oxidations. RSC Adv. 2016, 6, 44902–44907. [Google Scholar] [CrossRef]
- Rashid, M.; Jun, T.-S.; Jung, Y.; Kim, Y.S. Bimetallic core-shell Ag@Pt nanoparticle-decorated MWNT electrodes for amperometric H2 sensors and direct methanol fuel cells. Sens. Actuators B 2014, 208, 7–13. [Google Scholar] [CrossRef]
- Zhao, D.; Wang, Y.-H.; Yan, B.; Xu, B.-Q. Manipulation of Pt Ag nanostructures for advanced electrocatalyst. J. Phys. Chem. C 2009, 113, 1242–1250. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Liao, M.-S.; Cabrera, C.R. A theory-guided design of bimetallic nanoparticle catalysts for fuel cell applications. In Computational Materials Science, Theoretical and Computational Chemistry Series; Leszzynski, J., Ed.; Elsevier Science: Amsterdam, The Netherlands, 2004; Volume 15, pp. 125–154. [Google Scholar]
- Yu, X.; Pickup, P.G. Recent advances in direct formic acid fuel cells (DFAFC). J. Power Sources 2008, 182, 124–132. [Google Scholar] [CrossRef]
- Jiang, K.; Zhang, H.-X.; Zou, S.; Cai, W.-B. Electrocatalysis of formic acid on palladium and platinum surfaces: From fundamental mechanisms to fuel cell applications. Phys. Chem. Chem. Phys. 2014, 16, 20360–20376. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Qiu, C.; Ma, H.; Liu, X. Facile fabrication and unexpected electrocatalytic activity of palladium thin films with hierarchical architectures. J. Phys. Chem. C 2008, 112, 13970–13975. [Google Scholar] [CrossRef]
- Liu, Z.; Zhao, B.; Guo, C.; Sun, Y.; Xu, F.; Yang, H.; Li, Z. Novel hybrid electrocatalyst with enhanced performance in alkaline media: Hollow Au/Pd core/shell nanostructures with a raspberry surface. J. Phys. Chem. C 2009, 113, 16766–16771. [Google Scholar] [CrossRef]
- Gu, X.; Cong, X.; Ding, Y. Platinum-decorated Au porous nanotubes as highly efficient catalysts for formic acid electro-oxidation. ChemPhysChem 2010, 11, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Hao, H.; Cai, W.-B.; Huang, T.; Yu, A. Preparation of carbon supported Pd-Pb hollow nanospheres and their electrocatalytic activities for formic acid oxidation. Electrochem. Commun. 2010, 12, 901–904. [Google Scholar] [CrossRef]
- Lu, X.; McKiernan, M.; Peng, Z.; Lee, E.P.; Yang, H.; Xia, Y. Noble-metal nanotubes prepared via a galvanic replacement reaction between Cu nanowires and aqueous HAuCl4, H2PtCl6, or Na2PdCl4. Sci. Adv. Mater. 2010, 2, 413–420. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, W. Nanoneedle-covered Pd-Ag nanotubes: High electrocatalytic activity for formic acid oxidation. J. Phys. Chem. C 2010, 114, 21190–21200. [Google Scholar] [CrossRef]
- Qiu, C.; Guo, Y.; Zhang, J.; Ma, H.; Cai, Y. Bimetallic Pt-Au thin film electrocatalysts with hierarchical structures for the oxidation of formic acid. Mater. Chem. Phys. 2011, 127, 484–488. [Google Scholar] [CrossRef]
- Dai, L.; Zou, S. Enhanced formic acid oxidation on Cu-Pd nanoparticles. J. Power Sources 2011, 196, 9369–9372. [Google Scholar] [CrossRef]
- Jiang, Y.; Lu, Y.; Han, D.; Zhang, Q.; Niu, L. Hollow Ag@Pd core-shell nanotubes as highly active catalysts for the electro-oxidation of formic acid. Nanotechnology 2012, 23, 105609. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, S.; Cui, X.; Wang, L.; Shi, X. Ultralow platinum-loading bimetallic nanoflowers: Fabrication and high-performance electrocatalytic activity towards the oxidation of formic acid. Electrochem. Commun. 2012, 25, 19–22. [Google Scholar] [CrossRef]
- Lee, D.; Jang, H.Y.; Hong, S.; Park, S. Synthesis of hollow and nanoporous gold/platinum alloy nanoparticles and their electrocatalytic activity for formic acid oxidation. J. Colloid Interface Sci. 2012, 388, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhao, T.S. Highly active carbon nanotube-supported Pd electrocatalyst for oxidation of formic acid prepared by etching copper template method. Int. J. Hydrogen Energy 2013, 38, 1391–1396. [Google Scholar] [CrossRef]
- Ren, M.; Zhou, Y.; Tao, F.; Zou, Z.; Akins, D.L.; Yang, H. Controllable modification of the electronic structure of carbon-supported core-shell Cu@Pd catalysts for formic acid oxidation. J. Phys. Chem. C 2014, 118, 12669–12675. [Google Scholar] [CrossRef]
- Rasouli, H.; Tabaian, S.H.; Rezaei, M. Galvanic replacement of electrodeposited nickel by palladium and investigation of the electrocatalytic activity of synthesized Pd/(Ni) for hydrogen evolution and formic acid oxidation. RSC Adv. 2016, 6, 22500–22510. [Google Scholar] [CrossRef]
- Yu, B.; Wen, W.; Li, W.; Yang, Y.; Hou, D.; Liu, C. Fabrication of high performance carbon-supported ternary Pd-Cu-Fe electrocatalysts for formic acid electrooxidation via partly galvanic sacrifice of tunable binary Cu-Fe alloy templates. Electrochim. Acta 2016, 196, 223–230. [Google Scholar] [CrossRef]
- Hu, J.; Li, H.; Gan, Q.-M.; Li, Y.-J. Three-dimensional porous Au nanocoral structure decorated with Pt submonolayer via galvanic displacement of copper adatoms for electrooxidation of formic acid. Russ. J. Electrochem. 2016, 52, 355–361. [Google Scholar] [CrossRef]
- Mkwizu, T.S.; Mathe, M.K.; Cukrowski, I. Multilayered nanoclusters of platinum and gold: Insights on electrodeposition pathways, electrocatalysis, surface and bulk compositional properties. J. Electrochem. Soc. 2013, 160, H529–H546. [Google Scholar] [CrossRef]
- Zhou, Y.; Du, C.; Han, G.; Gao, Y.; Yin, G. Ultra-low Pt decorated PdFe alloy nanoparticles for formic acid electro-oxidation. Electrochim. Acta 2016, 217, 203–209. [Google Scholar] [CrossRef]
- Wang, Y.; Zou, S.; Cai, W.-B. Recent advances on electro-oxidation of ethanol on Pt- and Pd-based catalysts: From reaction mechanisms to catalytic materials. Catalysts 2015, 5, 1507–1534. [Google Scholar] [CrossRef]
- Brouzgou, A.; Podias, A.; Tsiakaras, P. PEMFCs and AEMFCs directly fed with ethanol: A current status comparative review. J. Appl. Electrochem. 2013, 43, 119–136. [Google Scholar] [CrossRef]
- Prieto, M.J.; Rodrigues Filho, U.P.; Landers, R.; Tremiliosi-Filho, G. The ethanol electrooxidation at Pt layers deposited on polycrystalline Au. Phys. Chem. Chem. Phys. 2012, 14, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Su, Y.; Lv, X.; Shi, H.; Yang, X.; Wang, Y. Enhanced ethanol electrooxidation of hollow Pd nanospheres prepared by galvanic exchange reactions. Mater. Lett. 2012, 69, 92–95. [Google Scholar] [CrossRef]
- Tamasauskait-Tamasuinaite, L.; Balčiunaite, A.; Vaiciukevičiene, A.; Selskis, A.; Pakštas, V. Investigation of nanostructured platinum-nickel supported on the titanium surface as electrocatalysts for alkaline fuel cells. J. Power Sources 2012, 208, 242–247. [Google Scholar] [CrossRef]
- Song, H.M.; Anjum, D.H.; Sougrat, R.; Hedhili, M.N.; Khashab, N.M. Hollow Au@Pd and Au@Pt core-shell nanoparticles as electrocatalysts for ethanol oxidation reactions. J. Mater. Chem. 2012, 22, 25003–25010. [Google Scholar] [CrossRef]
- Huang, S.-J.; Chen, P.-Y. Fabrication of macroporous Pt and PtAu electrodes for electrochemical application through galvanic replacement at macroporous Cu electrode electrodeposited at polystyrene template from room temperature ionic liquid. Electrochim. Acta 2013, 89, 180–190. [Google Scholar] [CrossRef]
- Abbasi, N.; Shahbazi, P.; Kiani, A. Electrocatalytic oxidation of ethanol at Pd/Ag nanodendrites prepared via low support electrodeposition and galvanic replacement. J. Mater. Chem. A 2013, 1, 9966–9972. [Google Scholar] [CrossRef]
- Hong, W.; Liu, Y.; Wang, J.; Wang, E. A new kind of highly active hollow flower-like NiPdPt nanoparticles supported by multiwalled-carbon nanotubes toward ethanol electrooxidation. J. Power Sources 2013, 241, 751–755. [Google Scholar] [CrossRef]
- Chen, D.; Zhao, Y.; Fan, Y.; Peng, X.; Wang, X.; Tian, J. Synthesis of Ni@PbPt supported on graphene by galvanic displacement reaction for improving ethanol electro-oxidation. J. Mater. Chem. A 2013, 1, 13227–13232. [Google Scholar] [CrossRef]
- Sieben, J.M.; Comignani, V.; Alvarez, A.E.; Duarte, M.M.E. Synthesis and characterization of Cu core Pt-Ru shell nanoparticles for the electro-oxidation of alcohols. Int. J. Hydrogen Energy 2014, 39, 8667–8674. [Google Scholar] [CrossRef]
- Cai, J.; Zeng, Y.; Guo, Y. Copper@palladium-copper core-shell nanospheres as a highly effective electrocatalyst for ethanol electro-oxidation in alkaline media. J. Power Sources 2014, 270, 257–261. [Google Scholar] [CrossRef]
- Peng, C.; Hu, Y.; Liu, M.; Zheng, Y. Hollow raspberry-like PdAg alloy nanospheres: High electrocatalytic activity for ethanol oxidation in alkaline media. J. Power Sources 2015, 278, 69–75. [Google Scholar] [CrossRef]
- Chen, Y.; Lai, S.; Jiang, S.; Liu, Y.; Fu, C.; Li, A.; Chen, Y.; Lai, X.; Hu, J. Synthesis and enhanced electrocatalytic properties of Au/Pd/Pt nanohollows. Mater. Lett. 2015, 157, 15–18. [Google Scholar] [CrossRef]
- Shi, Q.; Zhang, P.; Li, Y.; Xia, H.; Wang, D.; Tao, X. Synthesis of open-mouthed, yolk-shell Au@AgPd nanoparticles with access to interior surfaces for enhanced electrocatalysis. Chem. Sci. 2015, 6, 4350–4357. [Google Scholar] [CrossRef]
- Gnanaprakasam, P.; Jeena, S.E.; Selvaraju, T. Hierarchical electroless Pt deposition at Au decorated reduced graphene oxide via a galvanic exchanged process: An electrocatalytic nanocomposite with enhanced mass activity for methanol and ethanol oxidation. J. Mater. Chem. A 2015, 3, 18010–18018. [Google Scholar] [CrossRef]
- Zhao, T.-T.; Wang, H.; Han, X.; Jiang, K.; Lin, H.; Xie, Z.; Cai, W.-B. A comparative investigation of electrocatalysis at Pt monolayers on shape-controlled Au nanocrystals: Facet effect versus strain effect. J. Mater. Chem. A 2016, 4, 15845–15850. [Google Scholar] [CrossRef]
- Liu, P.; Cheng, Z.; Ma, L.; Zhang, M.; Qiu, Y.; Chen, M.; Cheng, F. Cuprous oxide template synthesis of hollow-cubic Cu2O@PdxRuy nanoparticles for ethanol electrooxidation in alkaline media. RSC Adv. 2016, 6, 76684–76690. [Google Scholar] [CrossRef]
- Bin, D.; Yang, B.; Zhang, K.; Wang, C.; Wang, J.; Zhong, J.; Feng, Y.; Guo, J.; Du, Y. Design of PdAg hollow nanoflowers through galvanic replacement and their application for ethanol electrooxidation. Chem. Eur. J. 2016, 22, 16642–16647. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.-G.; Kim, S.-M.; Kim, J.-W.; Lee, S.-Y. Composition-tuned porous Pd-Ag bimetallic dendrites for the enhancement of ethanol oxidation reactions. J. Alloys Compd. 2016, 688, 447–453. [Google Scholar] [CrossRef]
- Peng, C.; Yang, W.; Wu, E.; Ma, Y.; Zheng, Y.; Nie, Y.; Zhang, H.; Xu, J. PdAg alloy nanotubes with porous walls for enhanced electrocatalytic activity towards ethanol electrooxidation in alkaline media. J. Alloys Compd. 2017, 698, 250–258. [Google Scholar] [CrossRef]
- Guerrini, E.; Trasatti, S. Electrocatalysis in water electrolysis. In Catalysis for Sustainable Energy Production; Barbaro, P., Bianchini, C., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; pp. 235–269. [Google Scholar]
- Lamy, C. From hydrogen production by water electrolysis to its utilization in a PEM fuel cell or in a SO fuel cell: Some considerations on the energy efficiencies. Int. J. Hydrogen Energy 2016, 41, 15415–15425. [Google Scholar] [CrossRef]
- Bansal, V.; O’Mullane, A.P.; Bhargava, S.K. Galvanic replacement mediated synthesis of hollow Pt nanocatalysts: Significance of residual Ag for the H2 evolution reaction. Electrochem. Commun. 2009, 11, 1639–1642. [Google Scholar] [CrossRef]
- Raoof, J.-B.; Ojani, R.; Kiani, A.; Rashid-Nadimi, S. Fabrication of highly porous Pt coated nanostructured Cu-foam modified copper electrode and its enhanced catalytic ability for hydrogen evolution reaction. Int. J. Hydrogen Energy 2010, 35, 452–458. [Google Scholar] [CrossRef]
- Raoof, J.B.; Ojani, R.; Esfeden, S.A.; Nadimi, S.R. Fabrication of bimetallic Cu/Pt nanoparticles modified glassy carbon electrode and its catalytic activity toward hydrogen evolution reaction. Int. J. Hydrogen Energy 2010, 35, 3937–3944. [Google Scholar] [CrossRef]
- Ojani, R.; Raoof, J.B.; Hasheminejad, E. One-step electroless deposition of Pd/Pt bimetallic microstructures by galvanic replacement on copper substrate and investigation of its performance for the hydrogen evolution reaction. Int. J. Hydrogen Energy 2013, 38, 92–99. [Google Scholar] [CrossRef]
- Rezaei, B.; Mokhtarianpour, M.; Ensafi, A.A. Fabricated of bimetallic Pd/Pt nanostructure deposited on copper nanofoam substrate by galvanic replacement as an effective electrocatalyst for hydrogen evolution reaction. Int. J. Hydrogen Energy 2015, 40, 6754–6762. [Google Scholar] [CrossRef]
- Shen, Y.; Lua, A.C.; Xi, J.; Qiu, X. Ternary platinum-copper-nickel nanoparticles anchored to hierarchical carbon supports as free-standing hydrogen evolution electrodes. ACS Appl. Mater. Interfaces 2016, 8, 3464–3472. [Google Scholar] [CrossRef] [PubMed]
- Tölle, R.; Otto, A. Hydrogen-evolution and -oxidation on Ag(111) covered by Pt. Surf. Sci. 2005, 597, 110–118. [Google Scholar] [CrossRef]
- Mitchell, P.C.H.; Wolohan, P.; Thompsett, D.; Cooper, S.J. Experimental and theoretical studies of fuel cell catalysts: Density functional theory calculations of H2 dissociation and CO chemisorption on fuel cell metal dimers. J. Mol. Catal. A 1997, 119, 223–233. [Google Scholar] [CrossRef]
- Najdovski, I.; O’Mullane, A.P.; Bhargava, S.K. Electrochemical properties of galvanically replaced iron nanocubes with gold and palladium. Electrochem. Commun. 2010, 12, 1535–1538. [Google Scholar] [CrossRef]
- Trasatti, S. Work function, electronegativity, and electrochemical behaviour of metals. III. Electrolytic hydrogen evolution in acid solutions. J. Electroanal. Chem. 1972, 39, 163–184. [Google Scholar] [CrossRef]
- Greeley, J.; Jaramillo, T.F.; Bonde, J.; Chorkendorff, I.B.; Nørskov, J.K. Computational high-throughput screening of electrocatalytic materials for hydrogen evolution. Nat. Mater. 2006, 5, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Greeley, J.; Nørskov, J.K. Large-scale, density functional theory-based screening of alloys for hydrogen evolution. Surf. Sci. 2007, 601, 1590–1598. [Google Scholar] [CrossRef]
- Greeley, J.; Mavrikakis, M. Alloy catalysts designed from first principles. Nat. Mater. 2004, 3, 810. [Google Scholar] [CrossRef] [PubMed]
- Greeley, J.; Mavrikakis, M. Near-surface alloys for hydrogen fuel cell applications. Catal. Today 2006, 111, 52–58. [Google Scholar] [CrossRef]
- Elbert, K.; Hu, J.; Ma, Z.; Zhang, Y.; Chen, G.; An, W.; Liu, P.; Isaacs, H.S.; Adzic, R.R.; Wang, J.X. Elucidating hydrogen oxidation/evolution kinetics in base and acid by enhanced activities at the optimized Pt shell thickness on the Ru core. ACS Catal. 2015, 5, 6764–6772. [Google Scholar] [CrossRef]
- Shao, Y.; Liu, J.; Wang, Y. Oxygen electrocatalysts for water electrolyzers and reversible fuel cells: Status and perspective. Energy Environ. Sci. 2012, 5, 9331–9344. [Google Scholar]
- Hu, S.; Goenaga, G.; Melton, C.; Zawodzinski, T.A.; Mukherjee, D. PtCo/CoOx nanocomposites: Bifunctional electrocatalysts for oxygen reduction and evolution reactions synthesized via tandem laser ablation synthesis in solution-galvanic replacement reactions. Appl. Catal. B 2016, 182, 286–296. [Google Scholar] [CrossRef]
- Pei, J.; Mao, J.; Liang, X.; Chen, C.; Peng, Q.; Wang, D.; Li, Y. Ir-Cu nanoframes: One-pot synthesis and efficient electrocatalysts for oxygen evolution reaction. Chem. Commun. 2016, 52, 3793–3796. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Sui, Y.; Xiao, G.; Yang, X.; Wei, Y.; Zou, G.; Zou, B. Synthesis of Cu-Ir nanocages with enhanced electrocatalytic activity for the oxygen evolution reaction. J. Mater. Chem. A 2015, 3, 19669–19673. [Google Scholar] [CrossRef]
- Yoon, D.; Bang, S.; Park, J.; Kim, J.; Baik, H.; Yang, H.; Lee, K. One pot synthesis of octahedral {111} CuIr gradient alloy nanocrystals with a Cu-rich core and an Ir-rich surface and their usage as efficient water splitting catalyst. CrystEngComm 2015, 17, 6843–6847. [Google Scholar] [CrossRef]
- Nong, H.N.; Gan, L.; Willinger, E.; Teschner, D.; Strasser, P. IrOx core-shell nanocatalysts for cost- and energy-efficient electrochemical water splitting. Chem. Sci. 2014, 5, 2955–2963. [Google Scholar] [CrossRef]
- Nong, H.N.; Oh, H.-S.; Reier, T.; Willinger, E.; Willinger, M.-G.; Petkov, V.; Teschner, D.; Strasser, P. Oxide-supported IrNiOx core-shell particles as efficient, cost-effective, and stable catalysts for electrochemical water splitting. Angew. Chem. Int. Ed. 2015, 54, 2975–2979. [Google Scholar] [CrossRef] [PubMed]
- Reier, T.; Pawolek, Z.; Cherevko, S.; Bruns, M.; Jones, T.; Teschner, D.; Selve, S.; Bergmann, A.; Nong, H.N.; Schlögl, R.; et al. Molecular insight in structure and activity of highly efficient, low-Ir Ir-Ni oxide catalysts for electrochemical water splitting (OER). J. Am. Chem. Soc. 2015, 137, 13031–13040. [Google Scholar] [CrossRef] [PubMed]
- Ponce De León, C.; Walsh, F.C.; Bessette, R.R.; Patrissi, C.J.; Medeiros, M.G.; Rose, A.; Browning, D.; Lakeman, J.B.; Reeve, R.W. Recent developments in borohydride fuel cells. ECS Trans. 2008, 15, 25–49. [Google Scholar]
- Demirci, U.B. Direct borohydride fuel cell: Main issues met by the membrane-electrodes-assembly and potential solutions. J. Power Sources 2007, 172, 676–687. [Google Scholar] [CrossRef]
- Gyenge, E. Electrooxidation of borohydride on platinum and gold electrodes: Implications for direct borohydride fuel cells. Electrochim. Acta 2004, 49, 965–978. [Google Scholar] [CrossRef]
- Chatenet, M.; Micoud, F.; Roche, I.; Chainet, E. Kinetics of sodium borohydride direct oxidation and oxygen reduction in sodium hydroxide electrolyte. Part I. BH4− electro-oxidation on Au and Ag catalysts. Electrochim. Acta 2006, 51, 5459–5467. [Google Scholar] [CrossRef]
- Heng, H.; Scott, K. Determination of kinetic parameters for borohydride oxidation on a rotating Au disk electrode. Electrochim. Acta 2006, 51, 3429–3433. [Google Scholar]
- Hosseini, M.G.; Abdolmaleki, M.; Nasirpouri, F. Investigation of the porous nanostructured Cu/Ni/AuNi electrode for sodium borohydride electrooxidation. Electrochim. Acta 2013, 114, 215–222. [Google Scholar] [CrossRef]
- Song, C.; Zhang, D.; Wang, B.; Cai, Z.; Yan, P.; Sun, Y.; Ye, K.; Cao, D.; Cheng, K.; Wang, G. Uniformly grown PtCo-modified Co3O4 nanosheets as a highly efficient catalyst for sodium borohydride electrooxidation. Nano Res. 2016, 9, 3322–3333. [Google Scholar] [CrossRef]
- Valiollahi, R.; Ojani, R. Pt hollow nanospheres/graphene electrocatalytic ability toward sodium borohydride oxidation: A study of morphology effect on electrocatalytic activity. J. Appl. Electrochem. 2017, 47, 205–212. [Google Scholar] [CrossRef]
- Brown, B.; Wolter, S.D.; Stoner, B.R.; Glass, J.T. Alloying effects of cosputtered gold-platinum thin films on the oxygen reduction reaction in acidic electrolyte. J. Electrochem. Soc. 2008, 155, B852–B859. [Google Scholar] [CrossRef]
- Ruban, A.V.; Skriver, H.L.; Nørskov, J.K. Surface segregation energies in transition-metal alloys. Phys. Rev. B 1999, 59, 15990–16000. [Google Scholar] [CrossRef]
- Vanrenterghem, B.; Papaderakis, A.; Sotiropoulos, S.; Tsiplakides, D.; Balomenou, S.; Bals, S.; Breugelmans, T. The reduction of benzylbromide at Ag-Ni deposits prepared by galvanic replacement. Electrochim. Acta 2016, 196, 756–768. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | E0 vs. SHE/V | Reaction | E0 vs. SHE/V |
|---|---|---|---|
| RuCl3+ 3e− ↔ Ru + 3Cl− PdCl42− + 2e− ↔ Pd + 4Cl− PtCl62− + 4e− ↔ Pt + 6Cl− PtCl42− + 2e− ↔ Pt + 4Cl− Ag+ + e− ↔ Ag IrCl62− + 4e− ↔ Ir + 6Cl− AuCl4− + 3e−↔ Au + 4Cl− | +0.386 +0.620 +0.744 +0.730 +0.799 +0.860 +1.002 | Cu+ + e− ↔ Cu Cu2+ + 2e− ↔ Cu CuCl + e− ↔ Cu + Cl− Fe3+ + 3e− ↔ Fe RuOH + e− + H+ ↔ Ru + H2O Pb2++ 2e− ↔ Pb Sn2++ 2e− ↔ Sn Ni2++ 2e− ↔ Ni Co2++ 2e− ↔ Co Ni(OH)3 + 3e− ↔ Ni + 3OH− Fe2+ + 2e− ↔ Fe Zn2++ 2e− ↔ Zn TiO2 + 4e− + 4H+ ↔ Ti + 2H2O Cr(OH)3 + 3e−↔ Cr + 3OH− Al(OH)3 + 3e−↔ Al + 3OH− | +0.520 +0.340 +0.121 −0.040 −0.050 −0.126 −0.136 −0.257 −0.277 −0.320 −0.440 −0.763 −1.095 −1.126 −2.300 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papaderakis, A.; Mintsouli, I.; Georgieva, J.; Sotiropoulos, S. Electrocatalysts Prepared by Galvanic Replacement. Catalysts 2017, 7, 80. https://doi.org/10.3390/catal7030080
Papaderakis A, Mintsouli I, Georgieva J, Sotiropoulos S. Electrocatalysts Prepared by Galvanic Replacement. Catalysts. 2017; 7(3):80. https://doi.org/10.3390/catal7030080
Chicago/Turabian StylePapaderakis, Athanasios, Ioanna Mintsouli, Jenia Georgieva, and Sotiris Sotiropoulos. 2017. "Electrocatalysts Prepared by Galvanic Replacement" Catalysts 7, no. 3: 80. https://doi.org/10.3390/catal7030080
APA StylePapaderakis, A., Mintsouli, I., Georgieva, J., & Sotiropoulos, S. (2017). Electrocatalysts Prepared by Galvanic Replacement. Catalysts, 7(3), 80. https://doi.org/10.3390/catal7030080