
The Suzuki–Miyaura Cross-Coupling as a Versatile Tool for Peptide Diversification and Cyclization


{kind=link}
{kind=link}
{kind=link}
{kind=link}
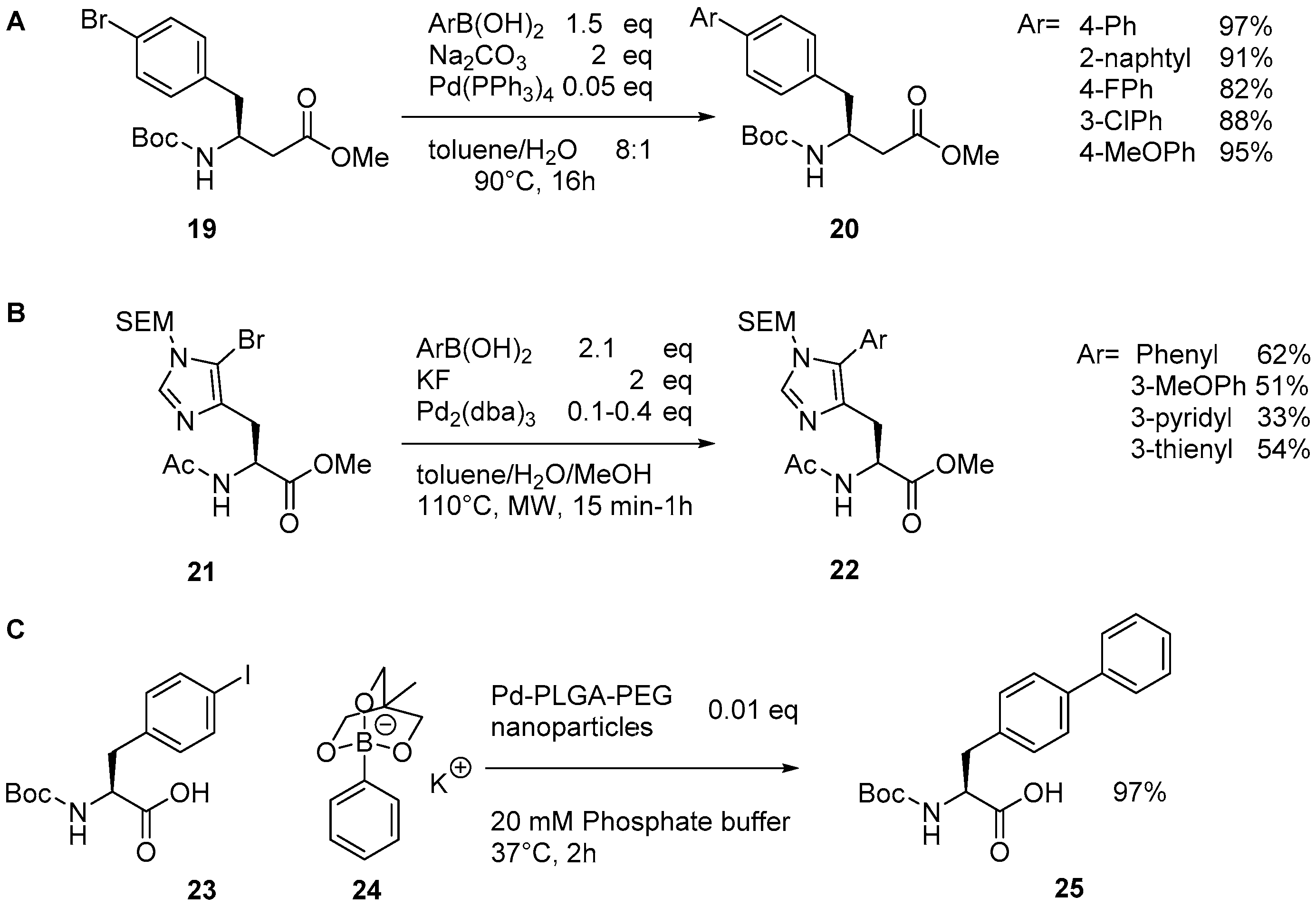{kind=link}
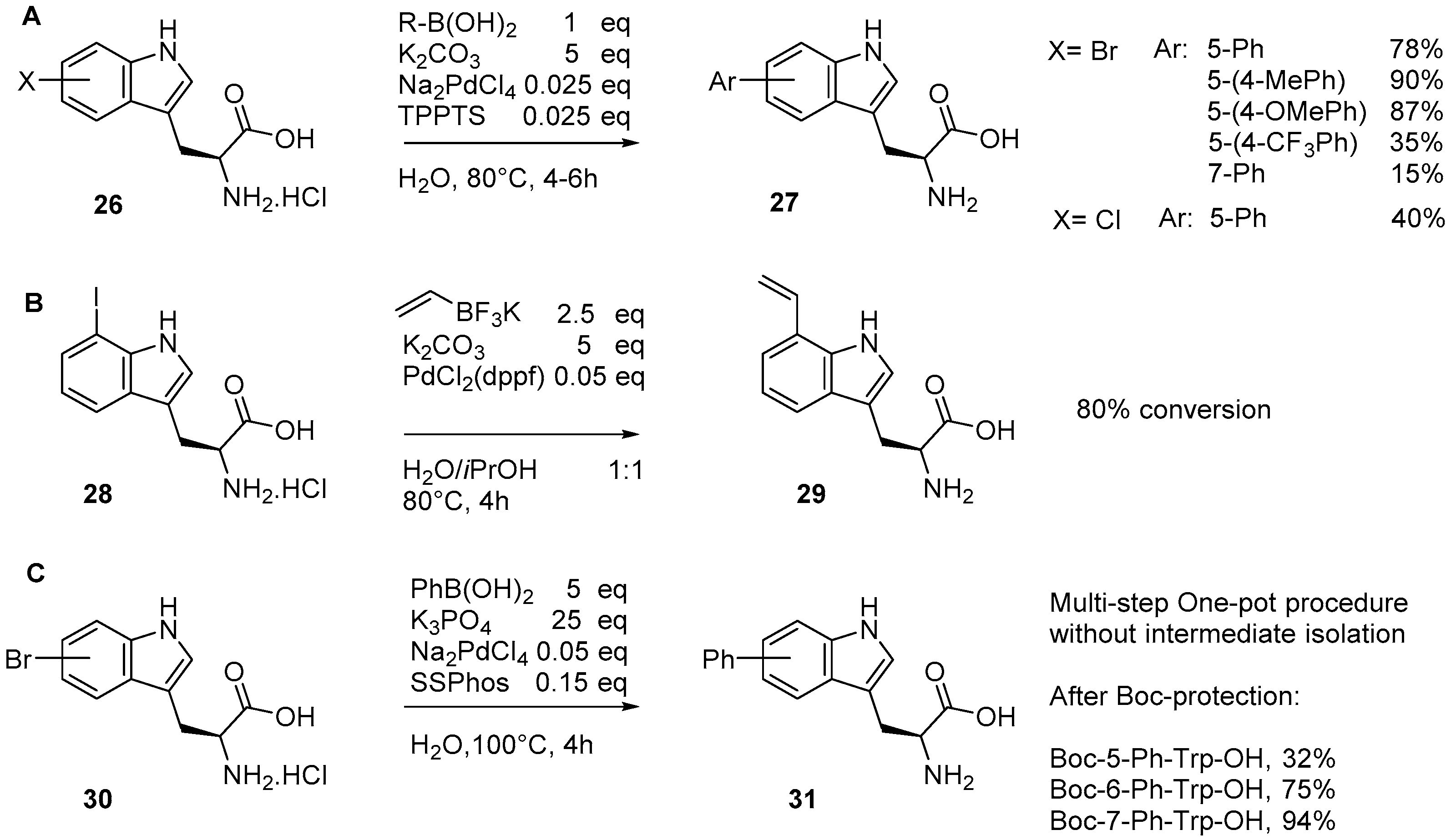{kind=link}
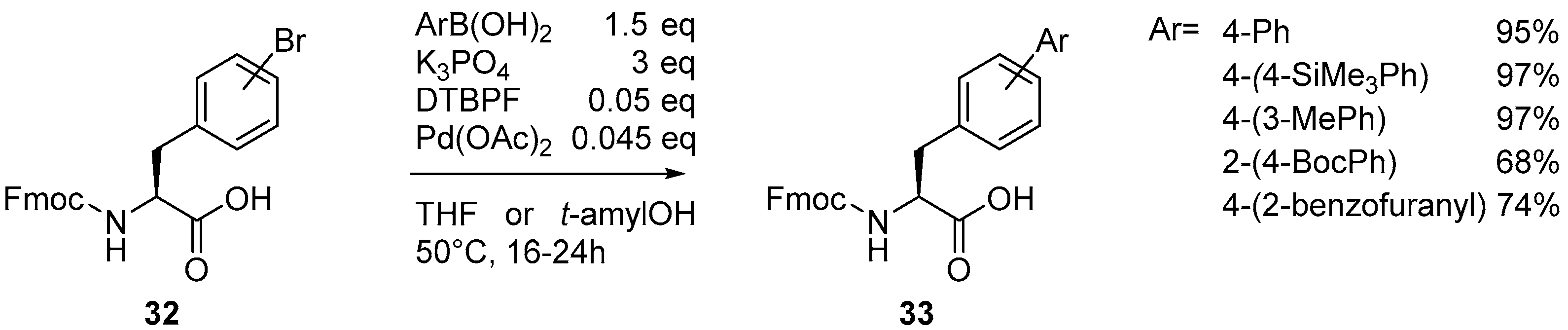{kind=link}
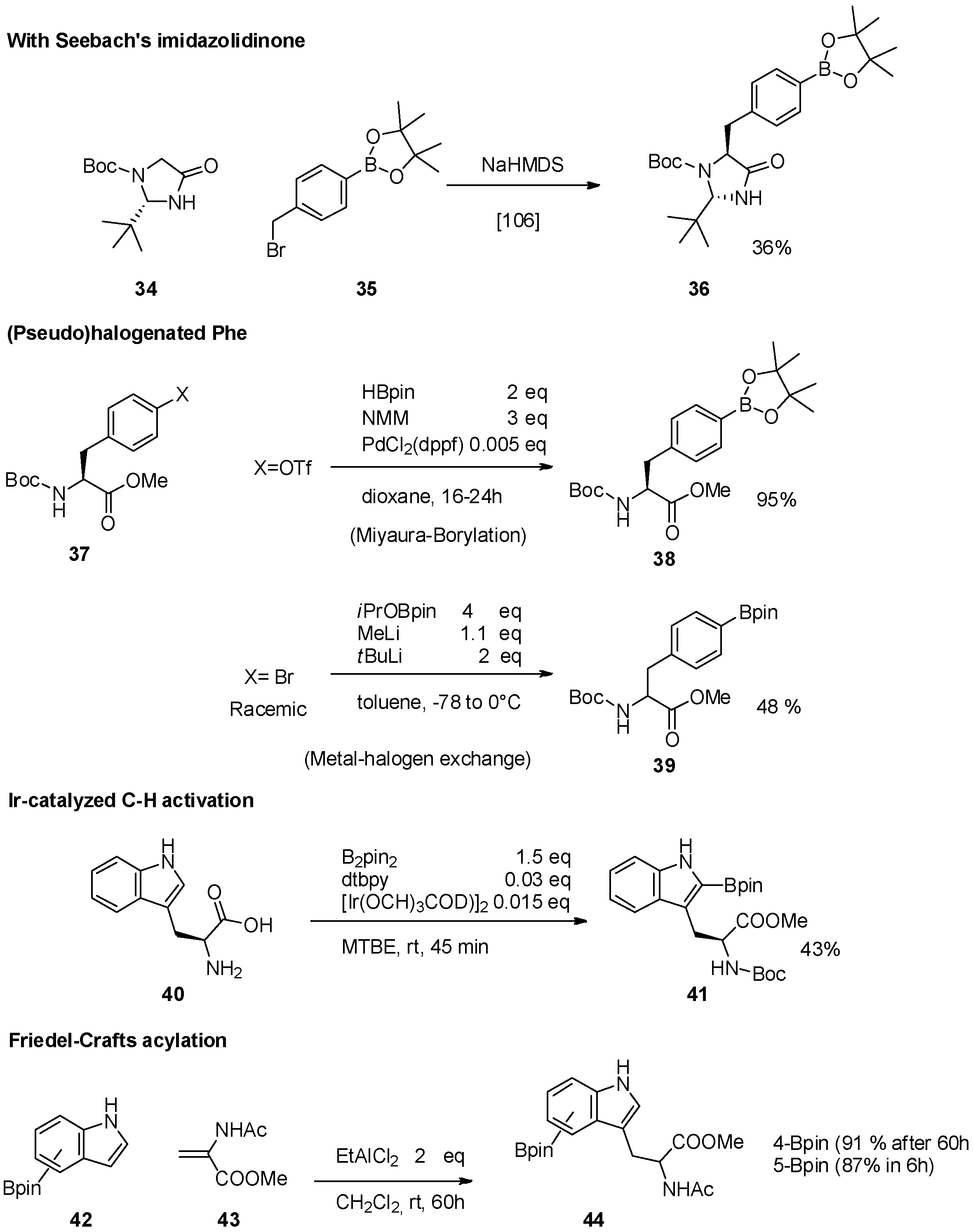{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Derivatization of Amino Acids and Peptides in Solution
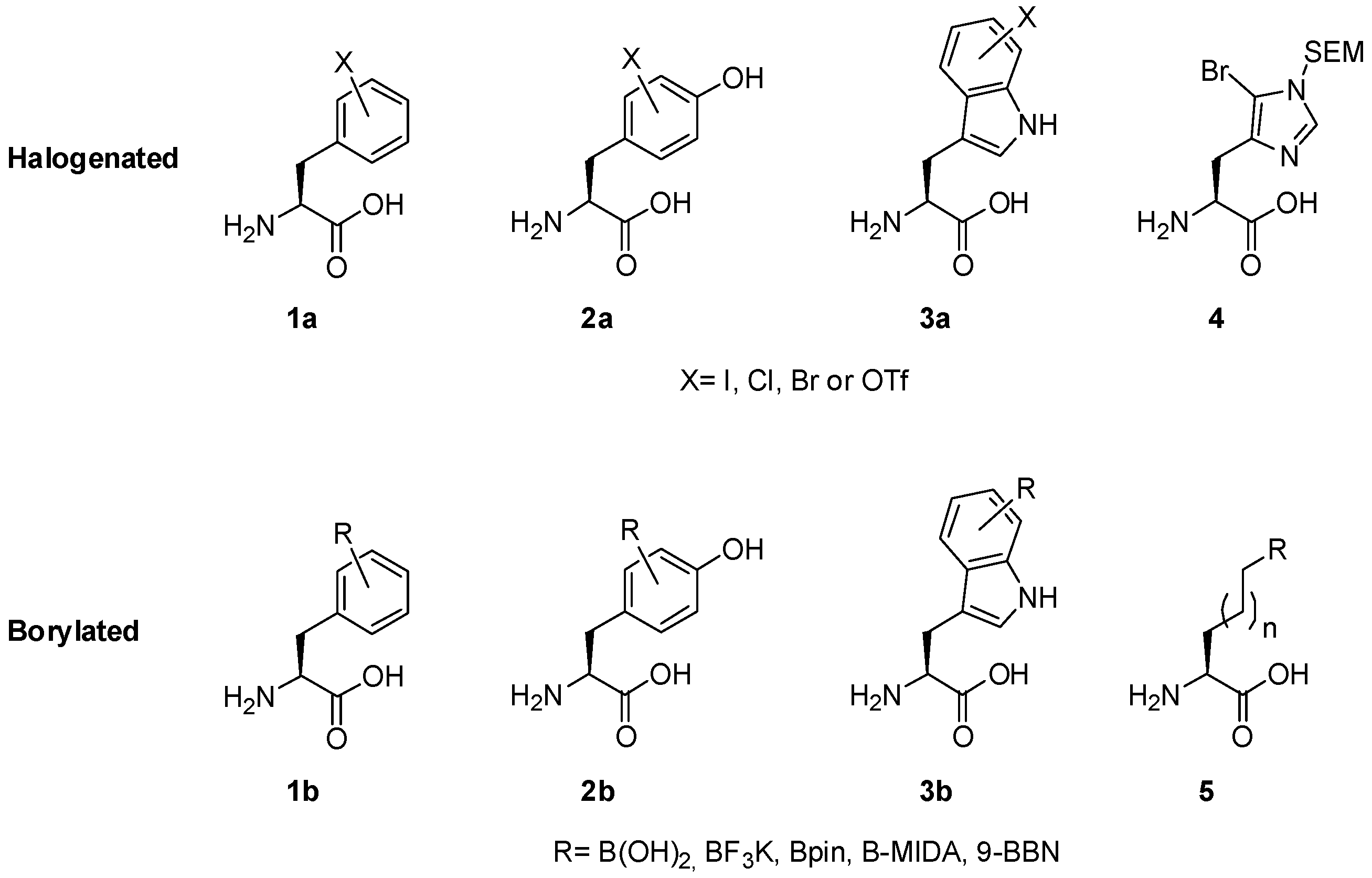
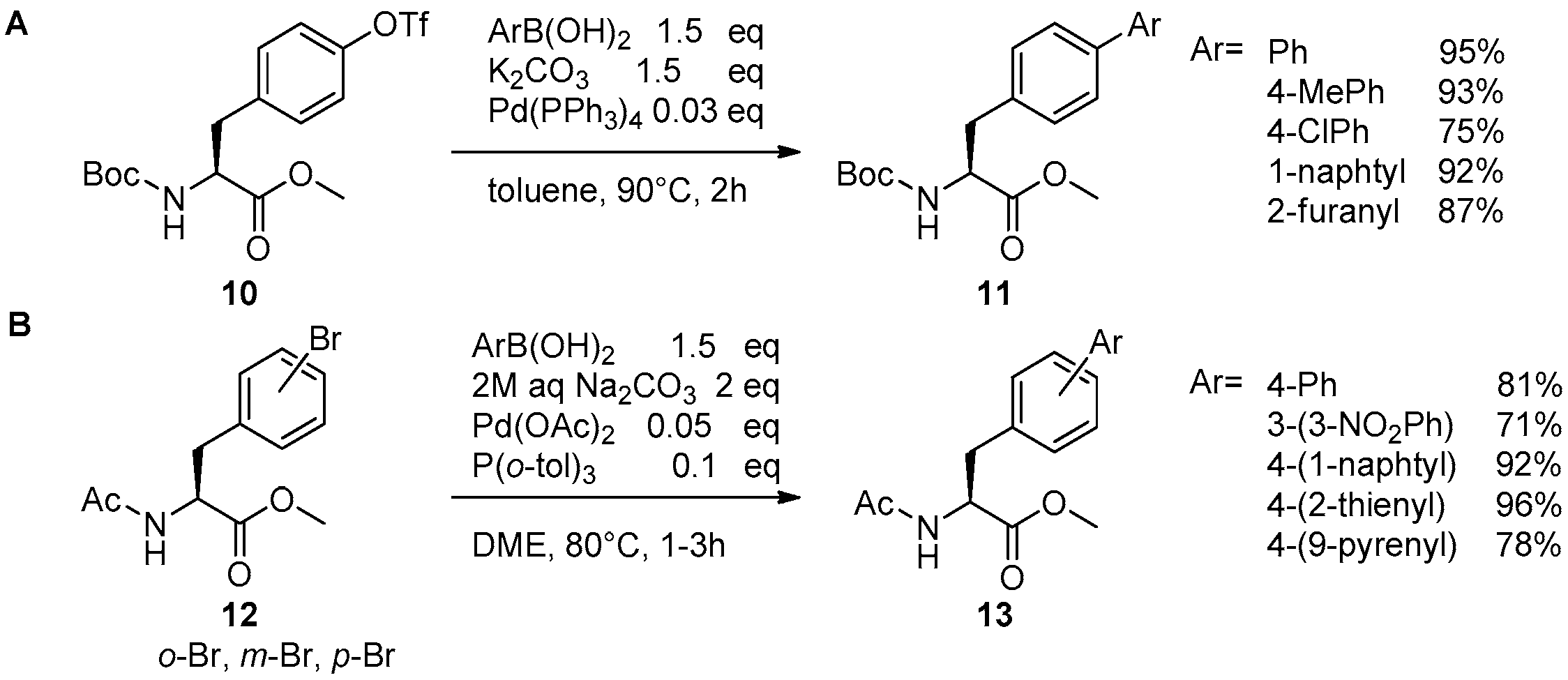
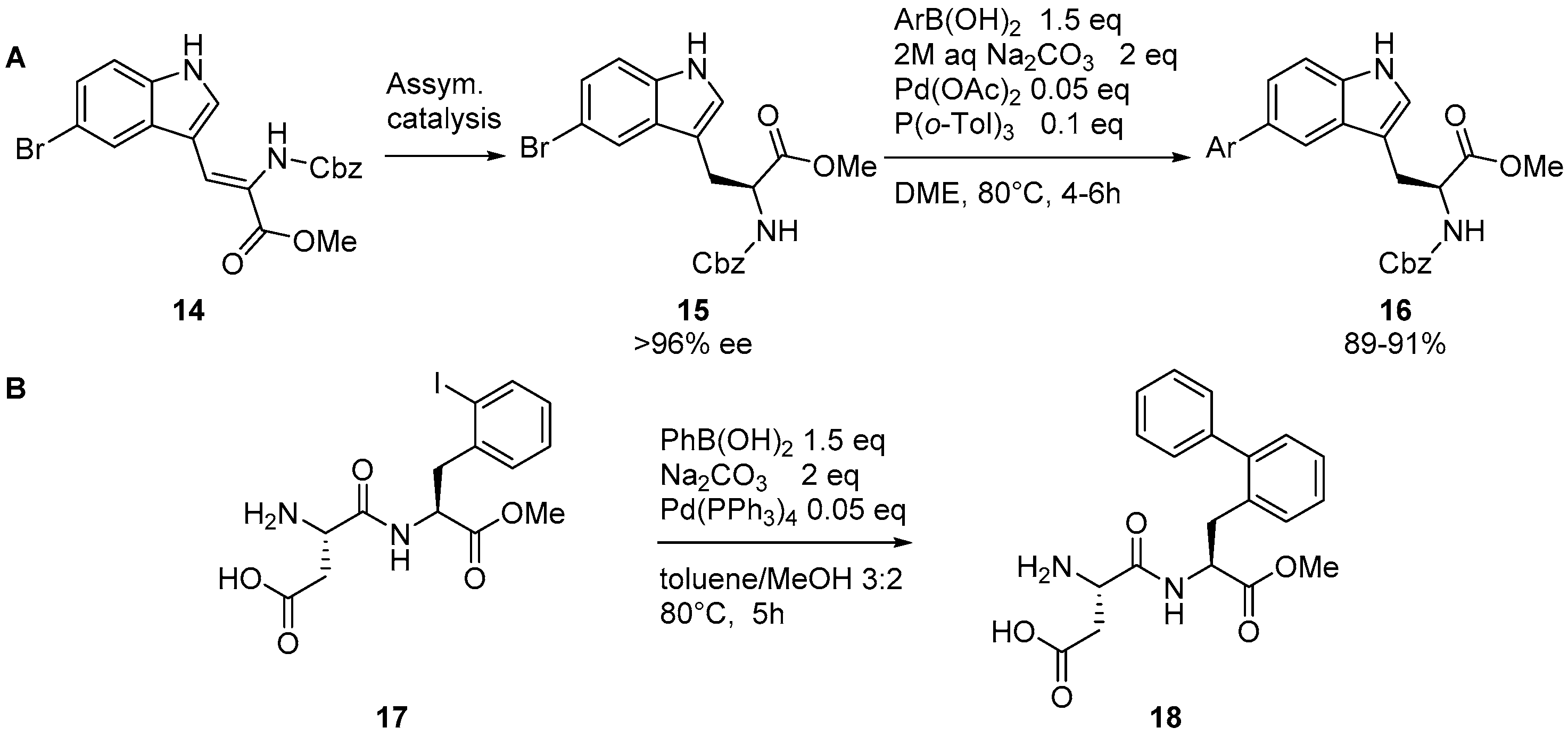
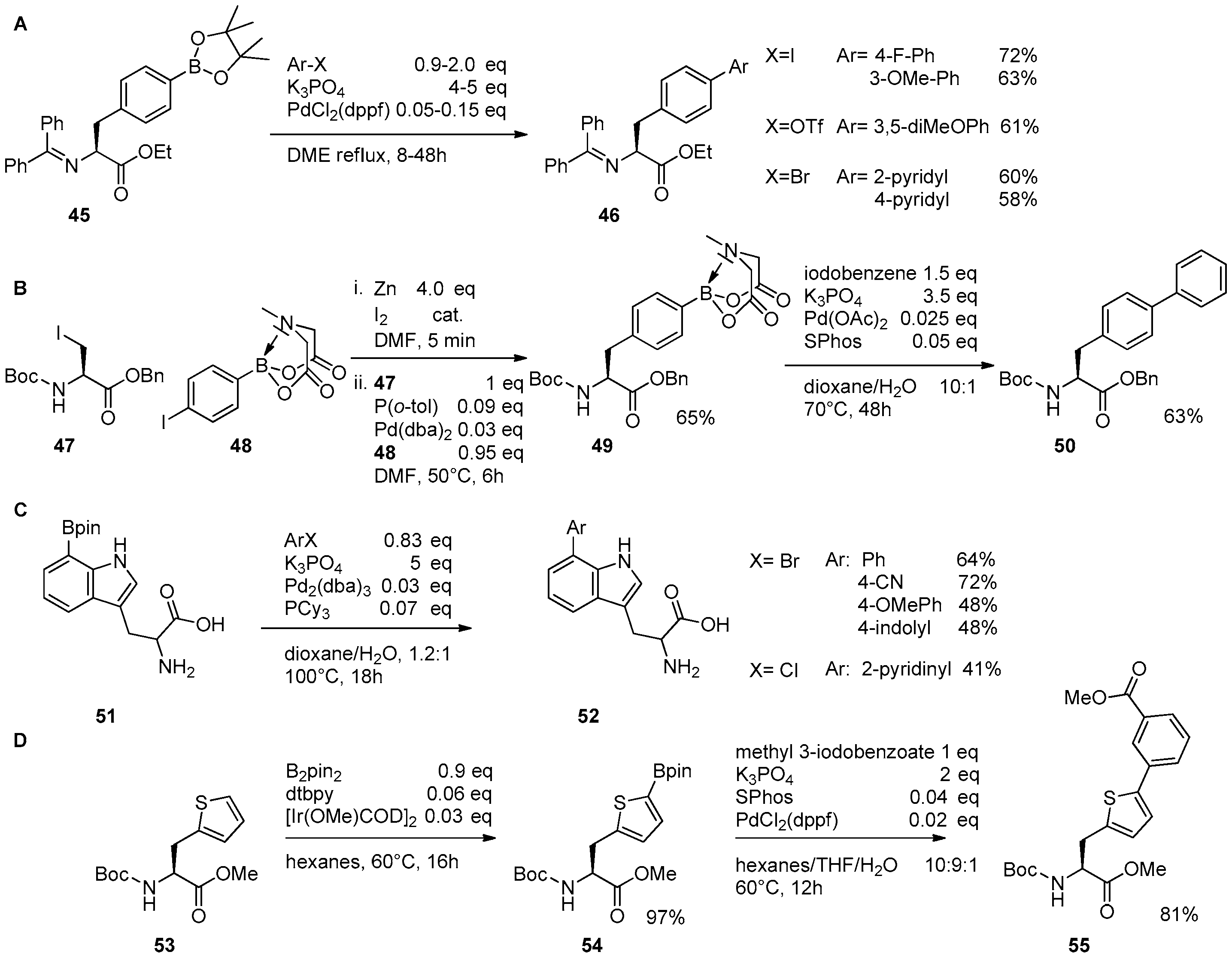
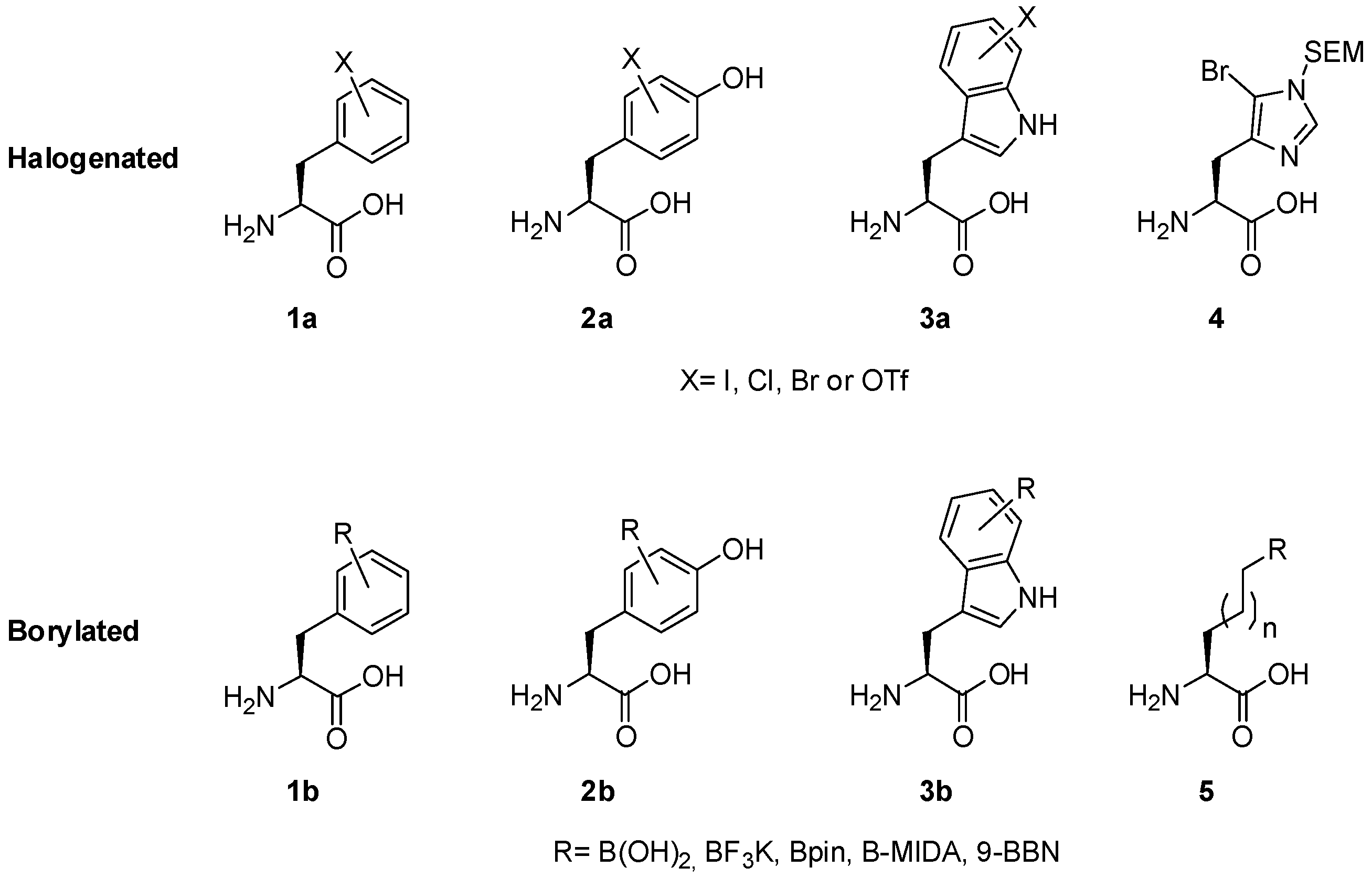
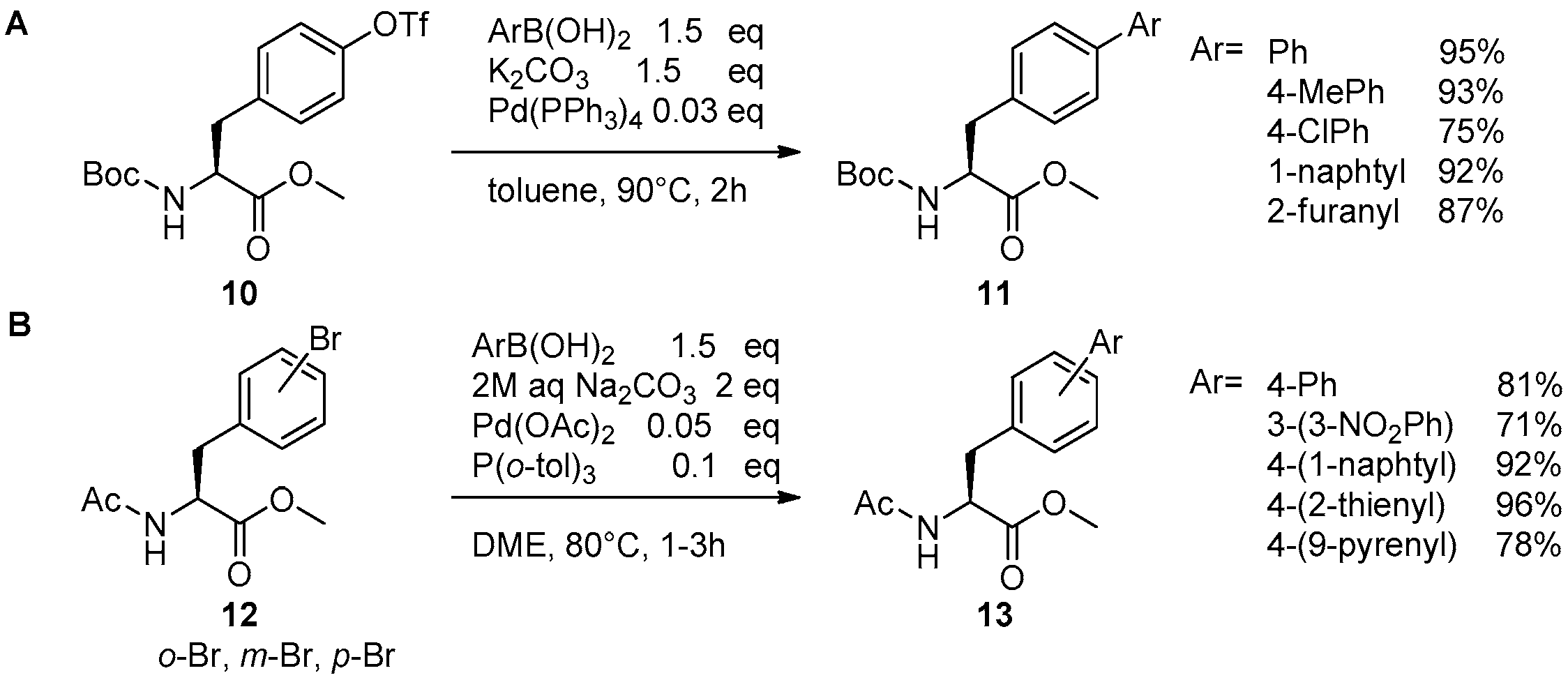
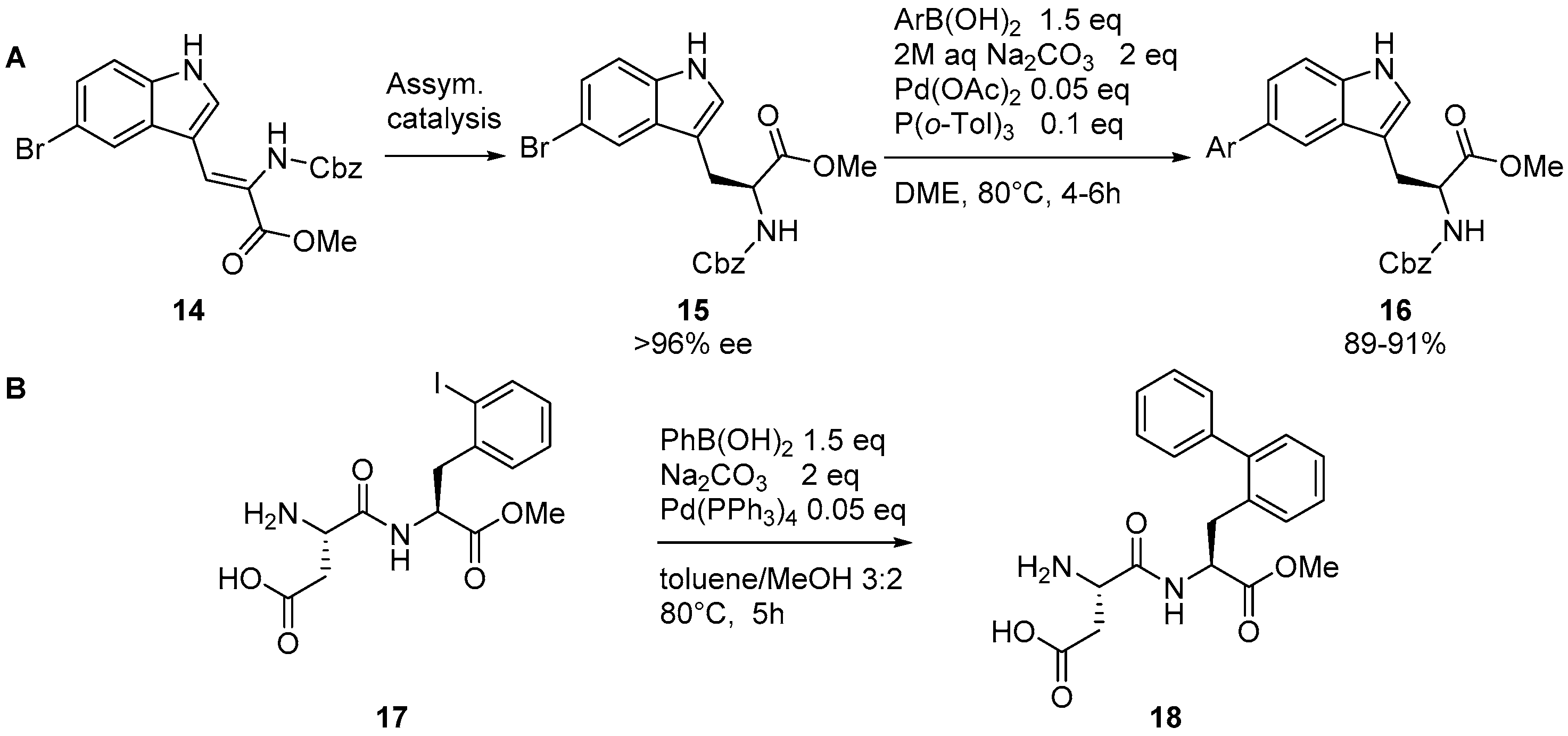
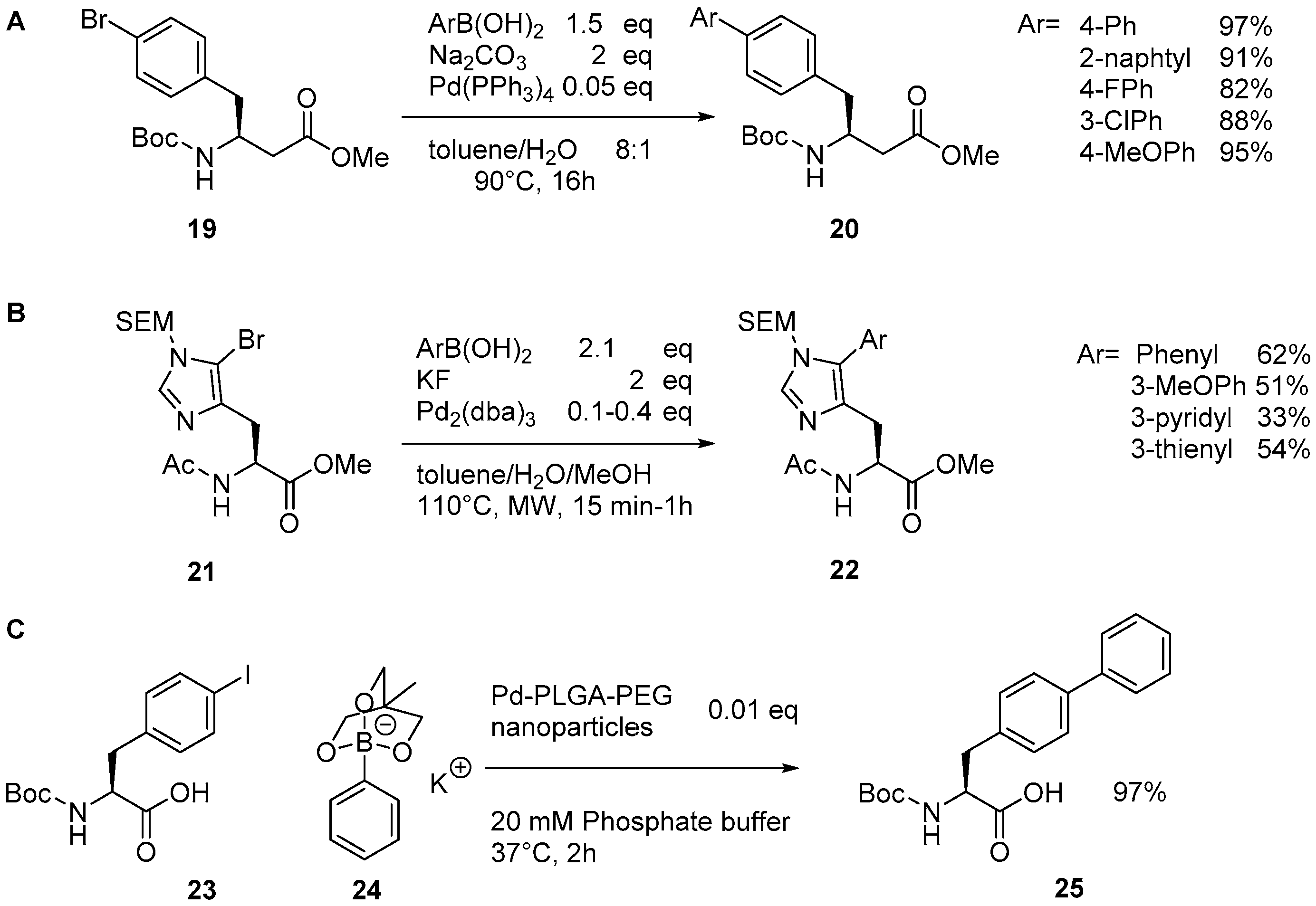
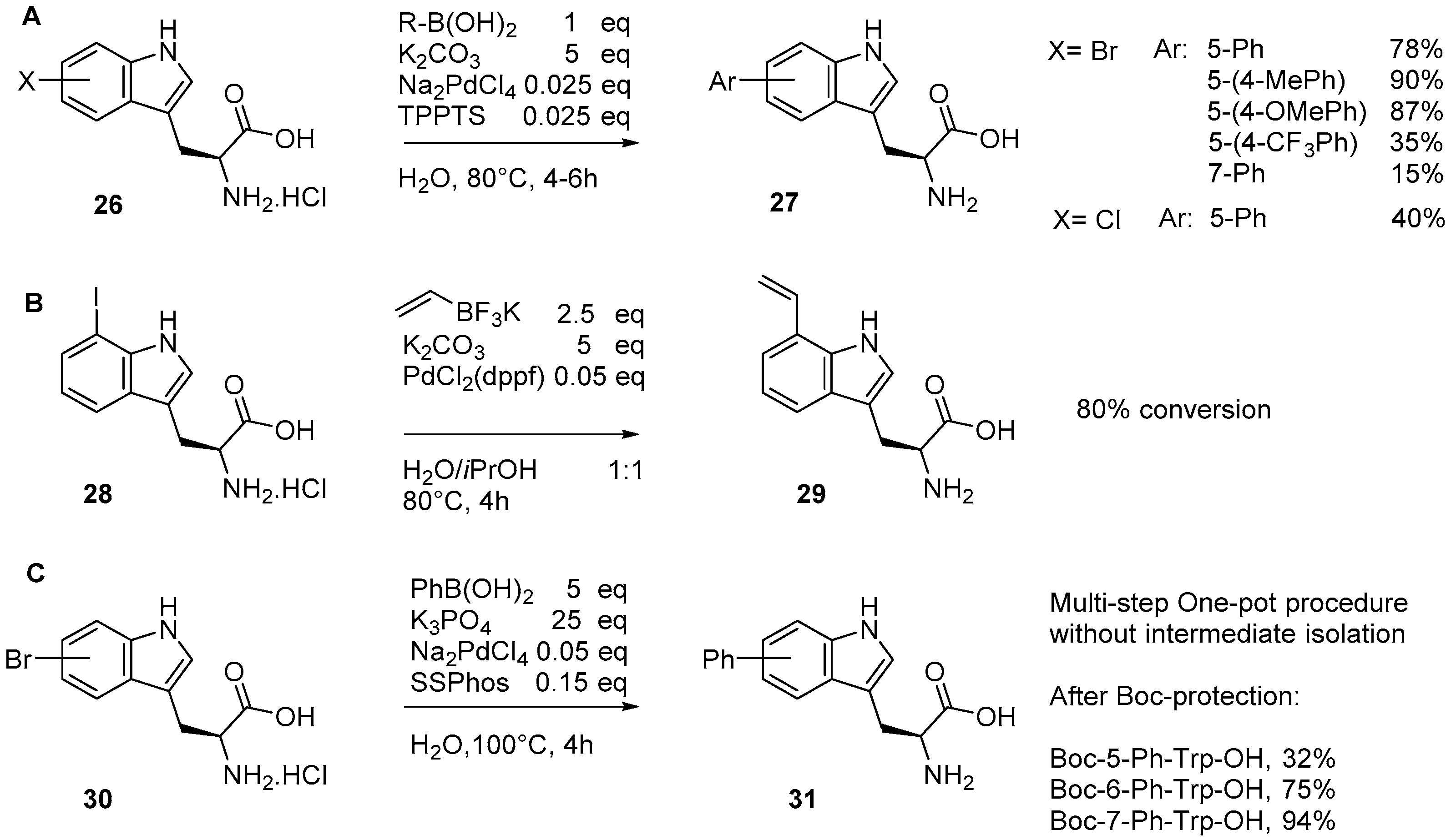
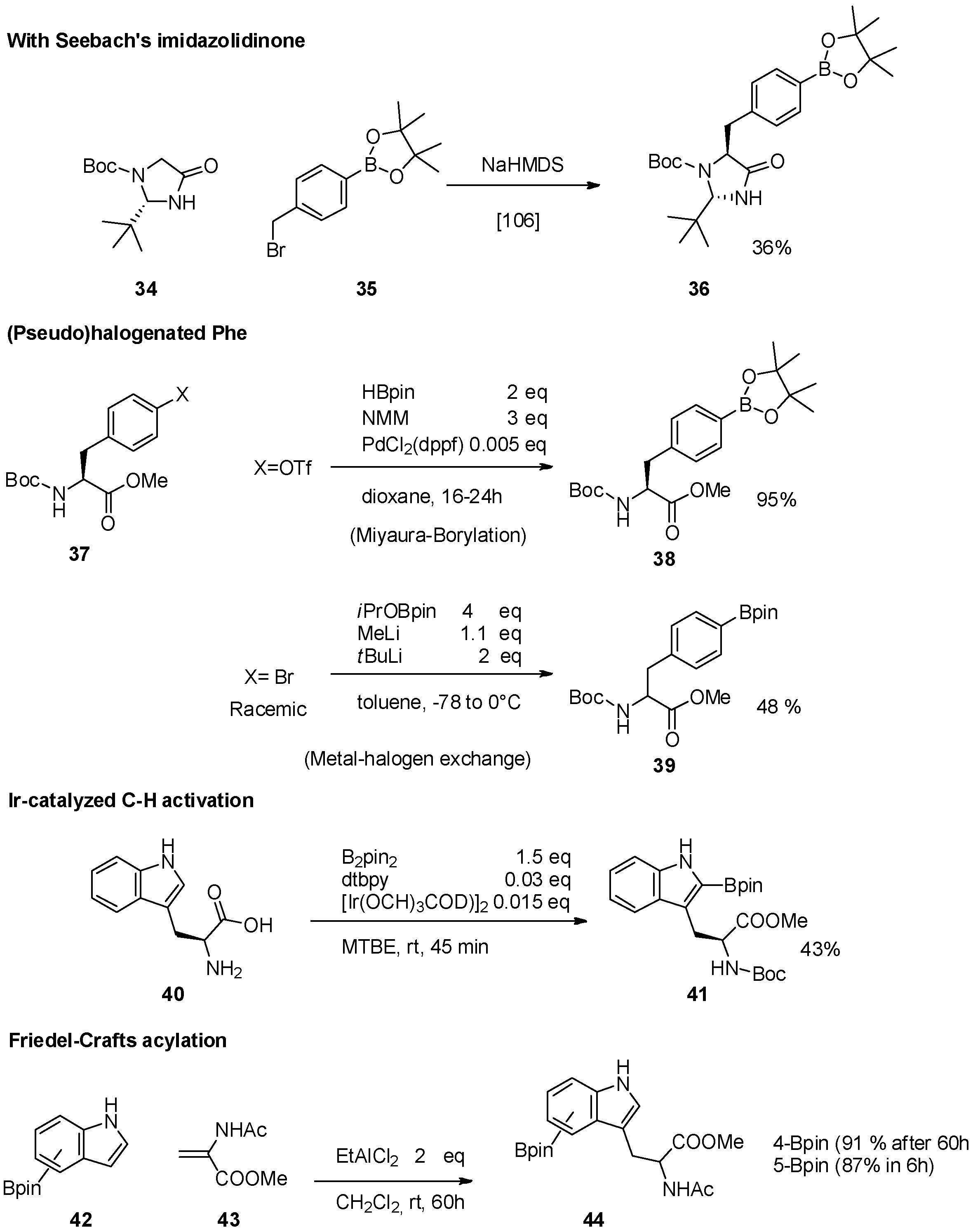
2.1. Access to and Derivatization of (Pseudo)halogenated Aromatic Amino acid Substrates
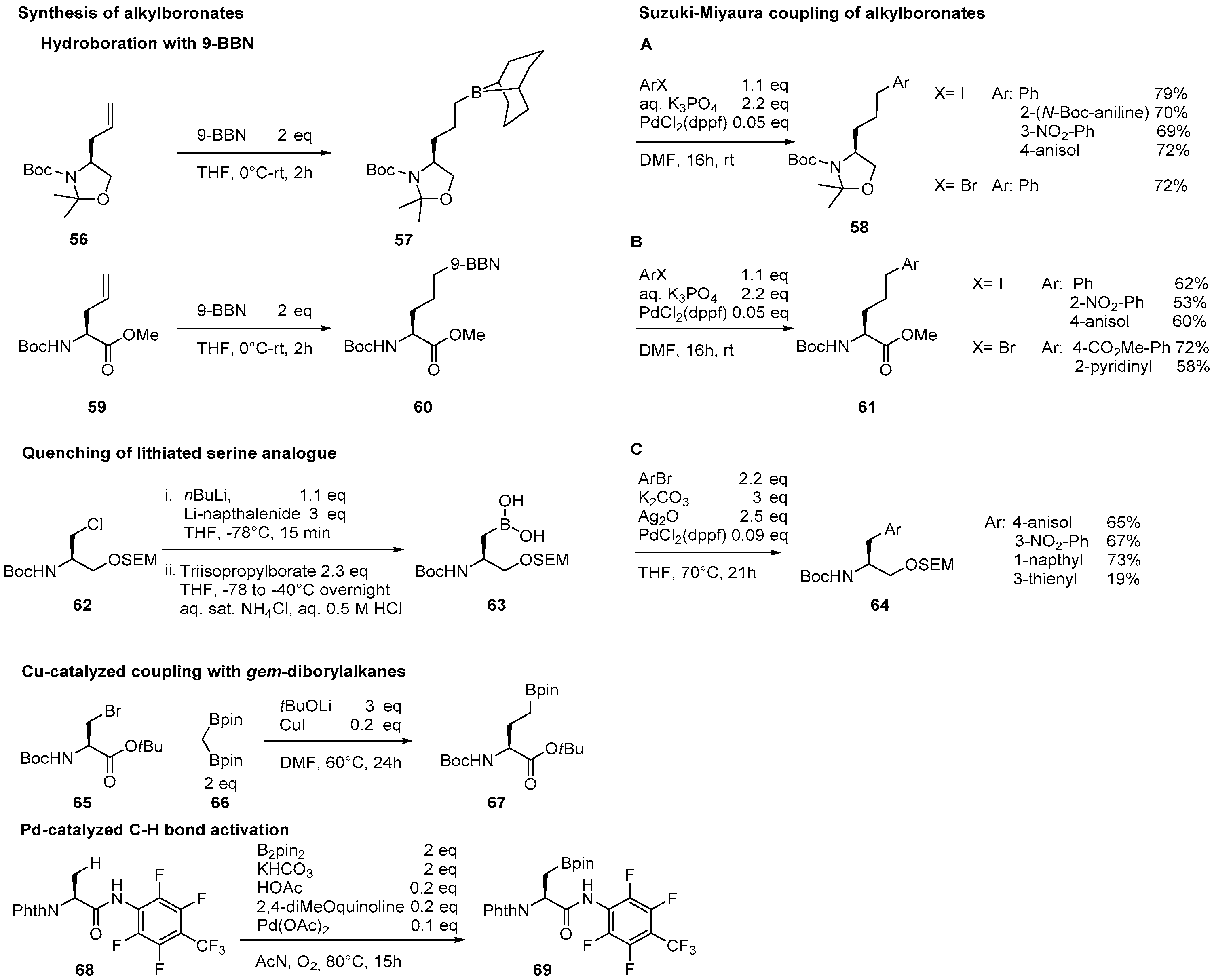
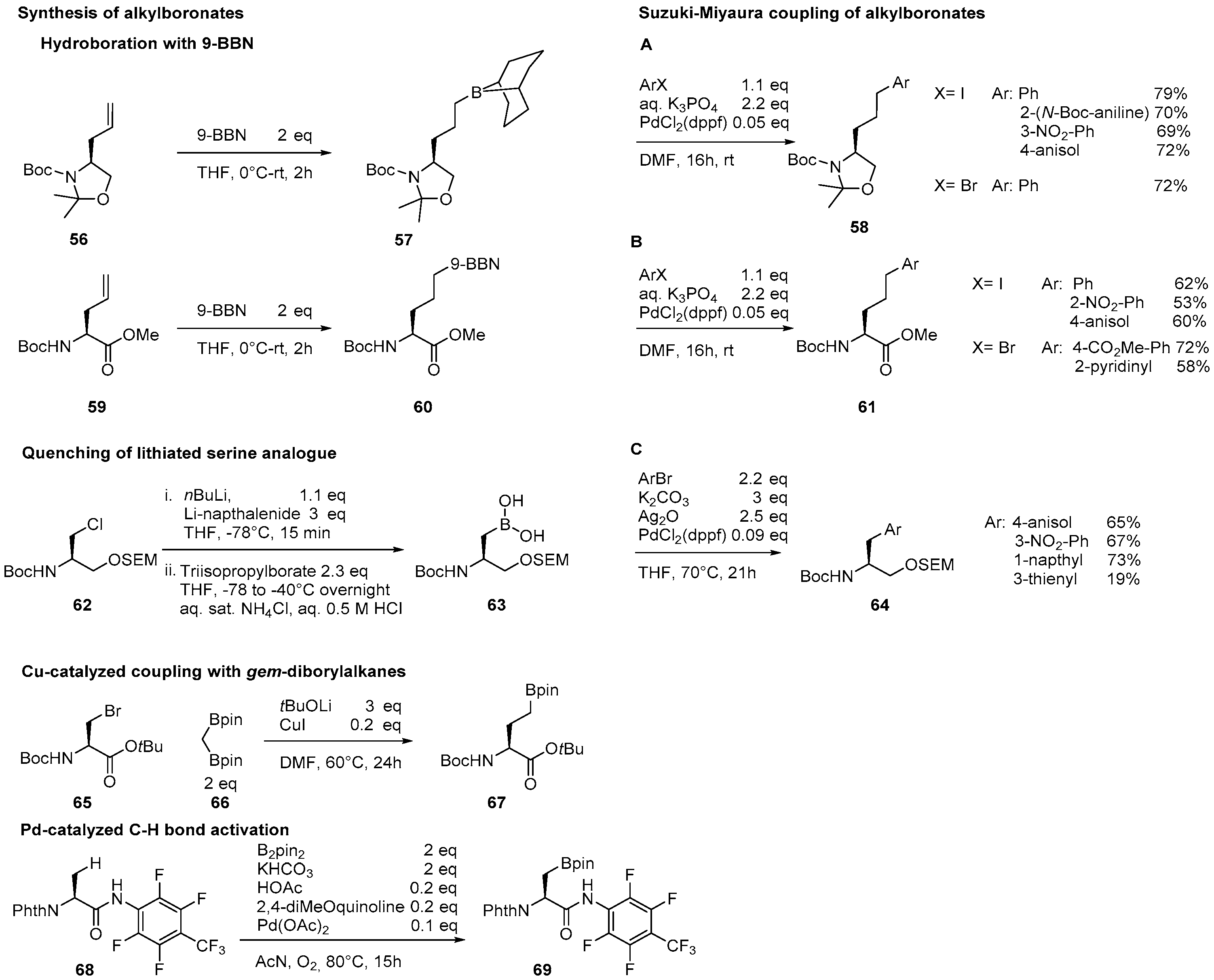
2.2. Synthesis of Borylated Aromatic and Aliphatic Amino Acids and Subsequent Derivatization
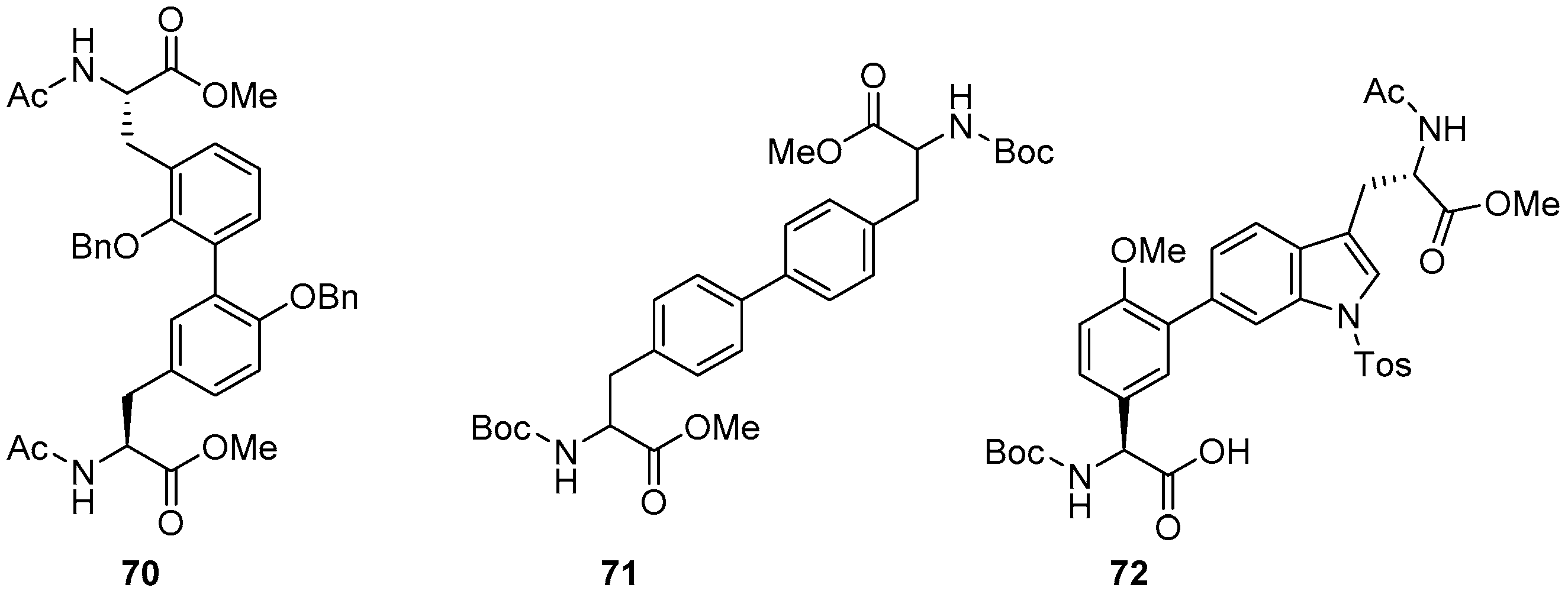
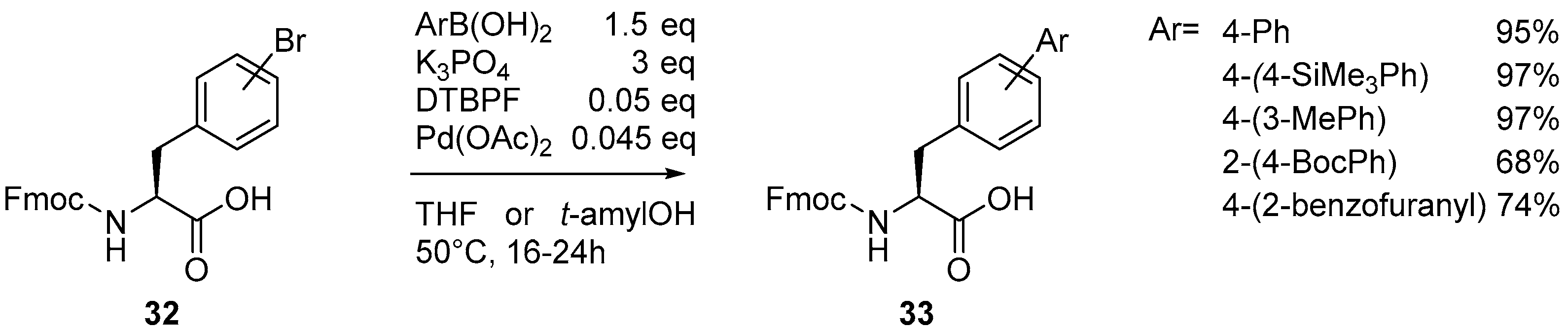
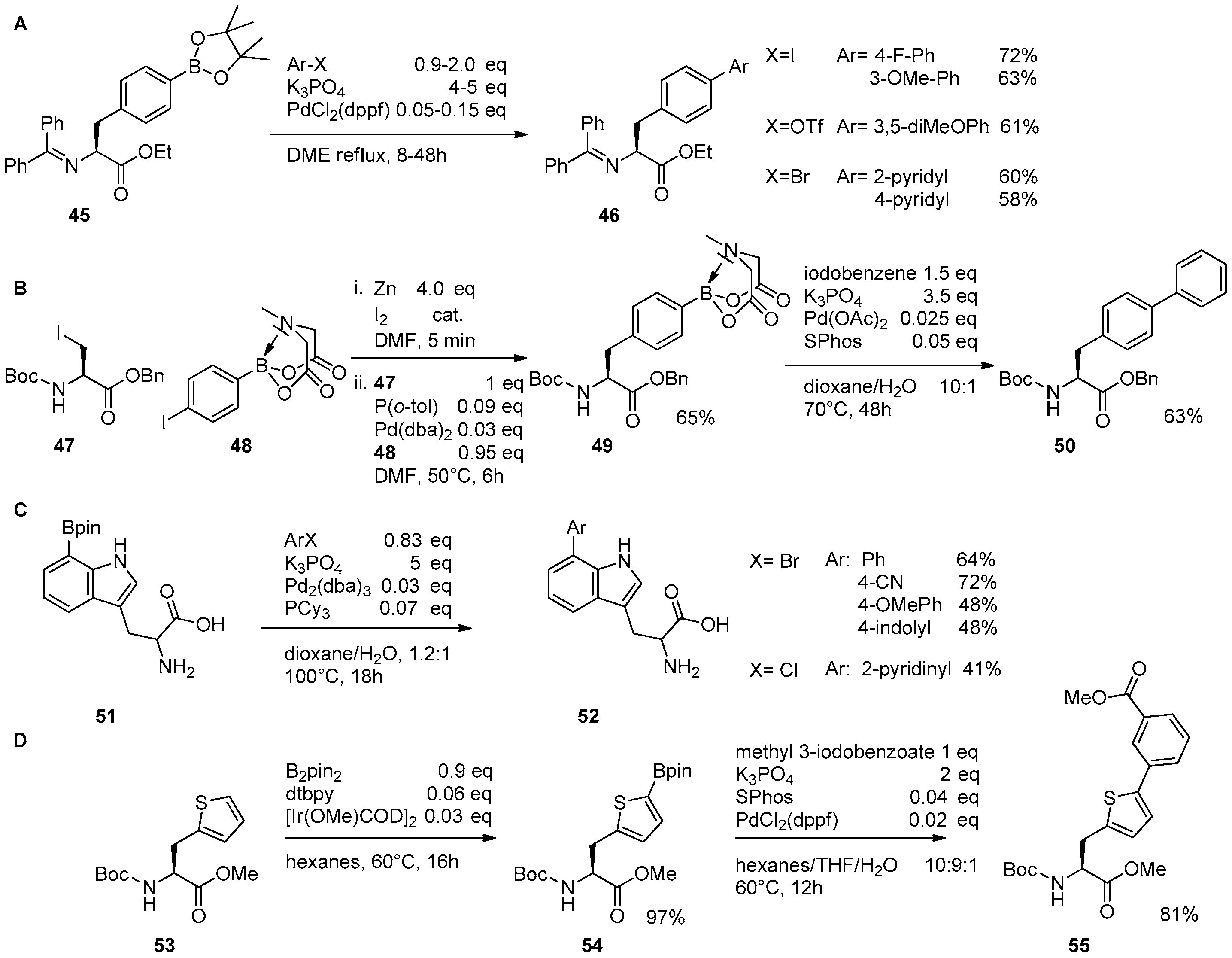
2.3. Coupling of Amino Acids: Synthesis of Biaryl-Bridged Dipeptides
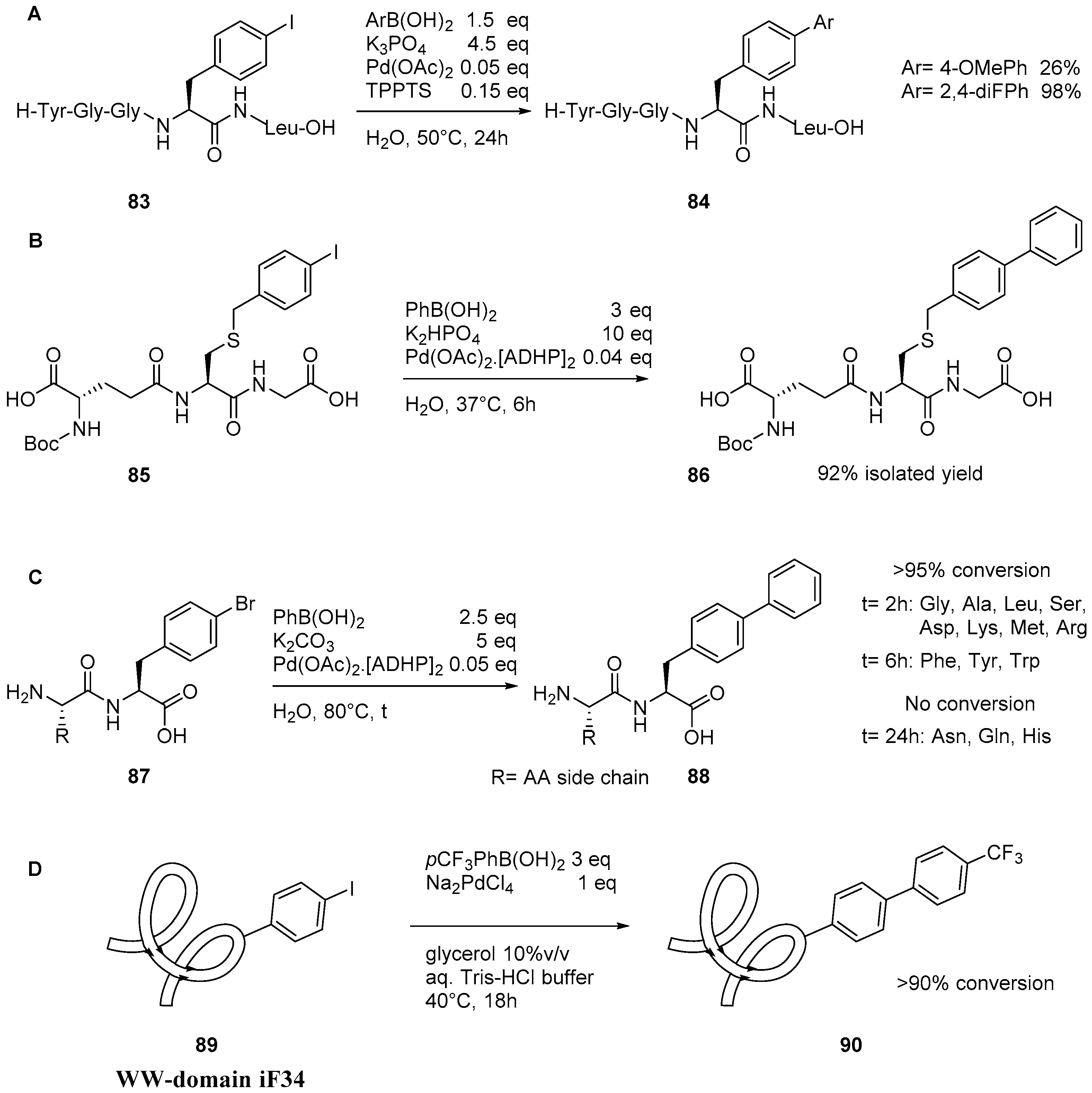
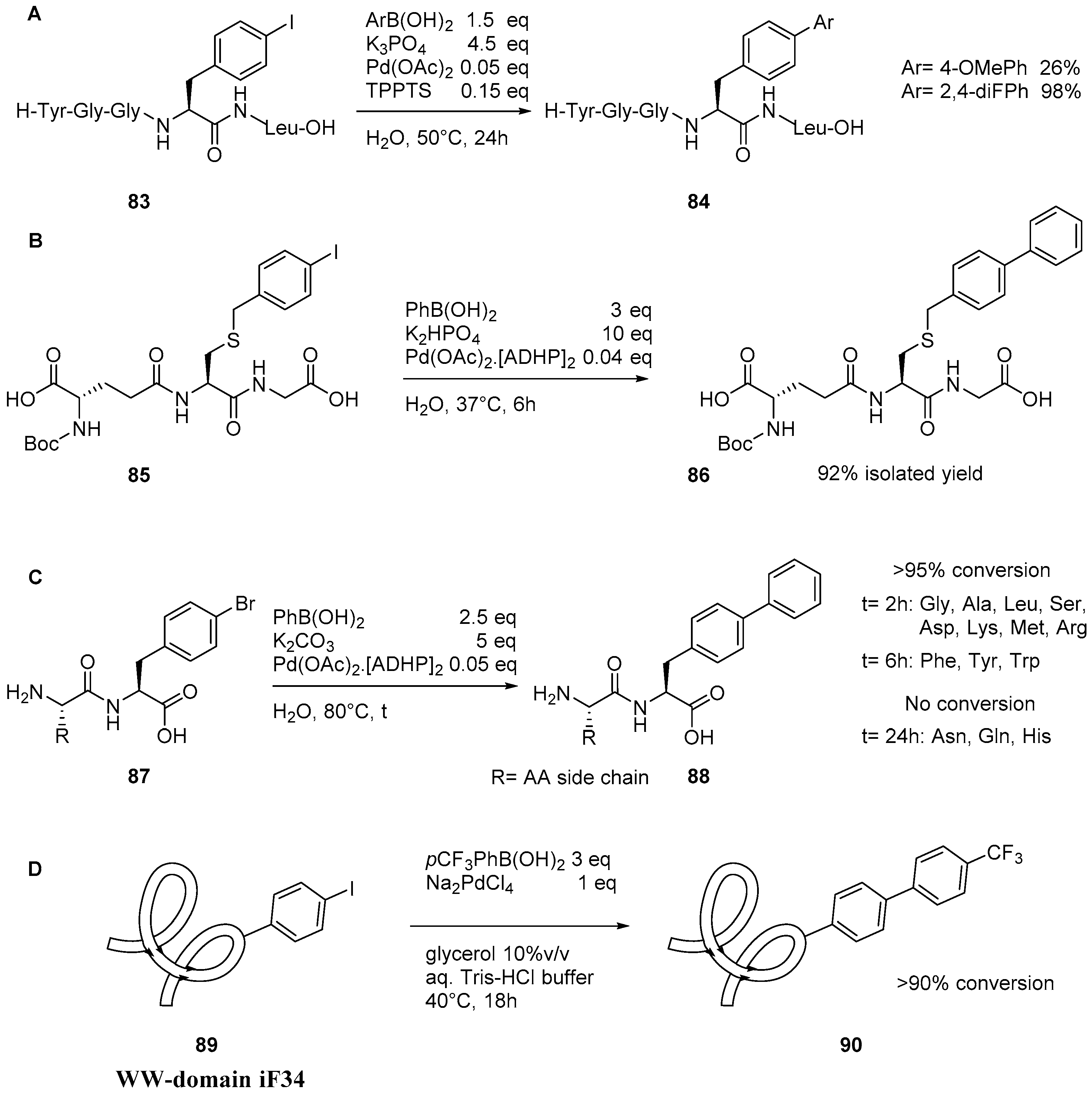
2.4. Suzuki–Miyaura Reaction on Peptidic Substrates
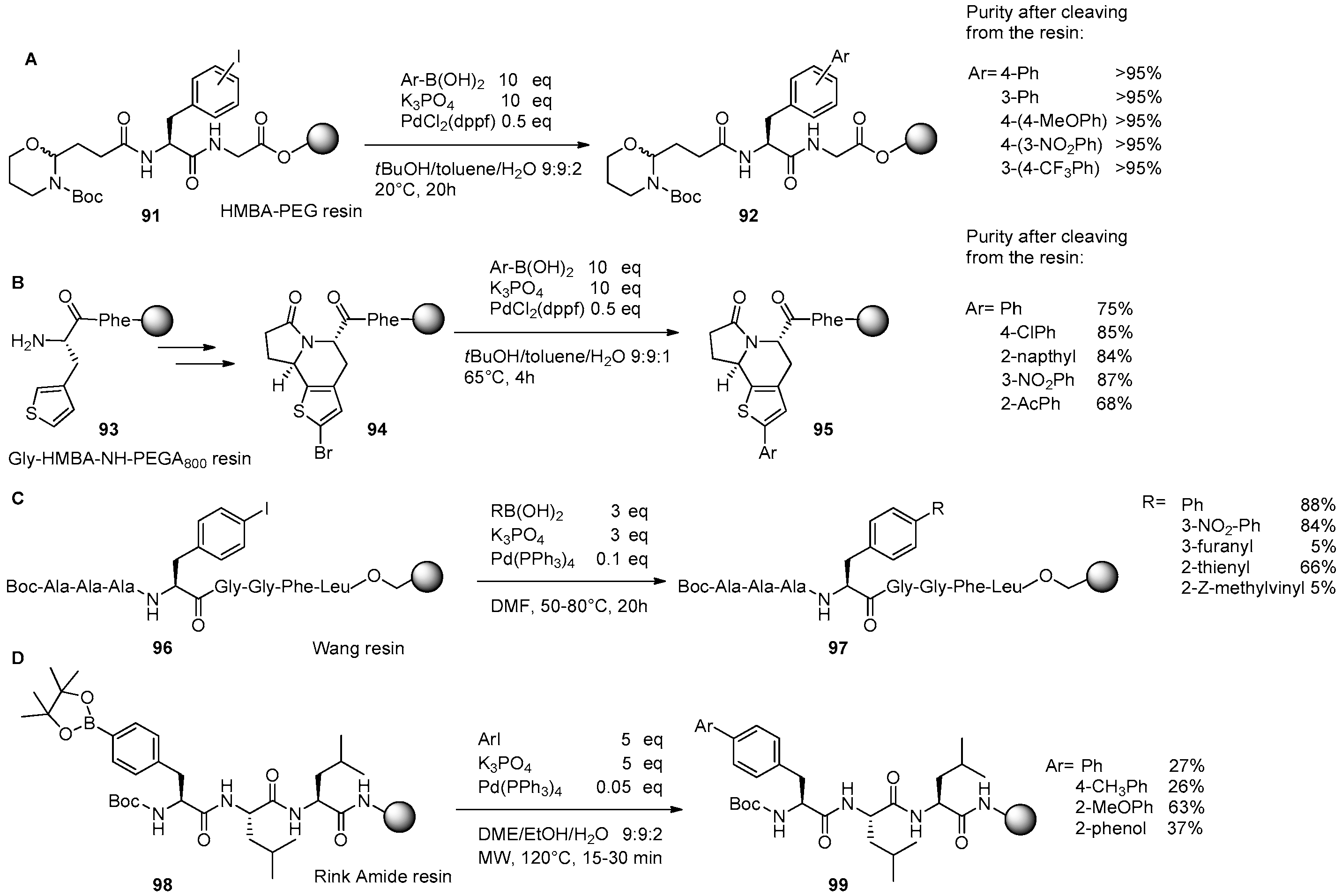
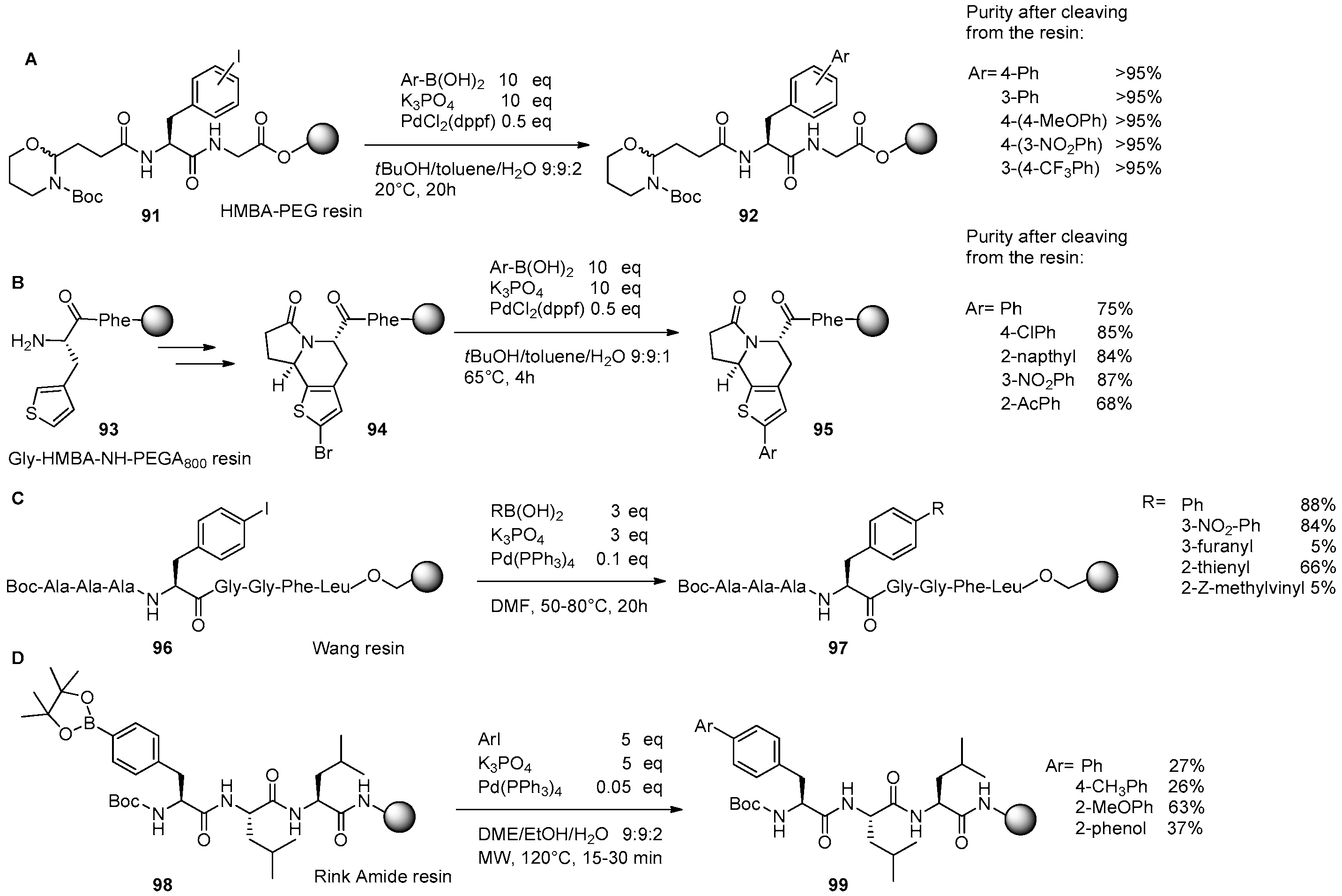
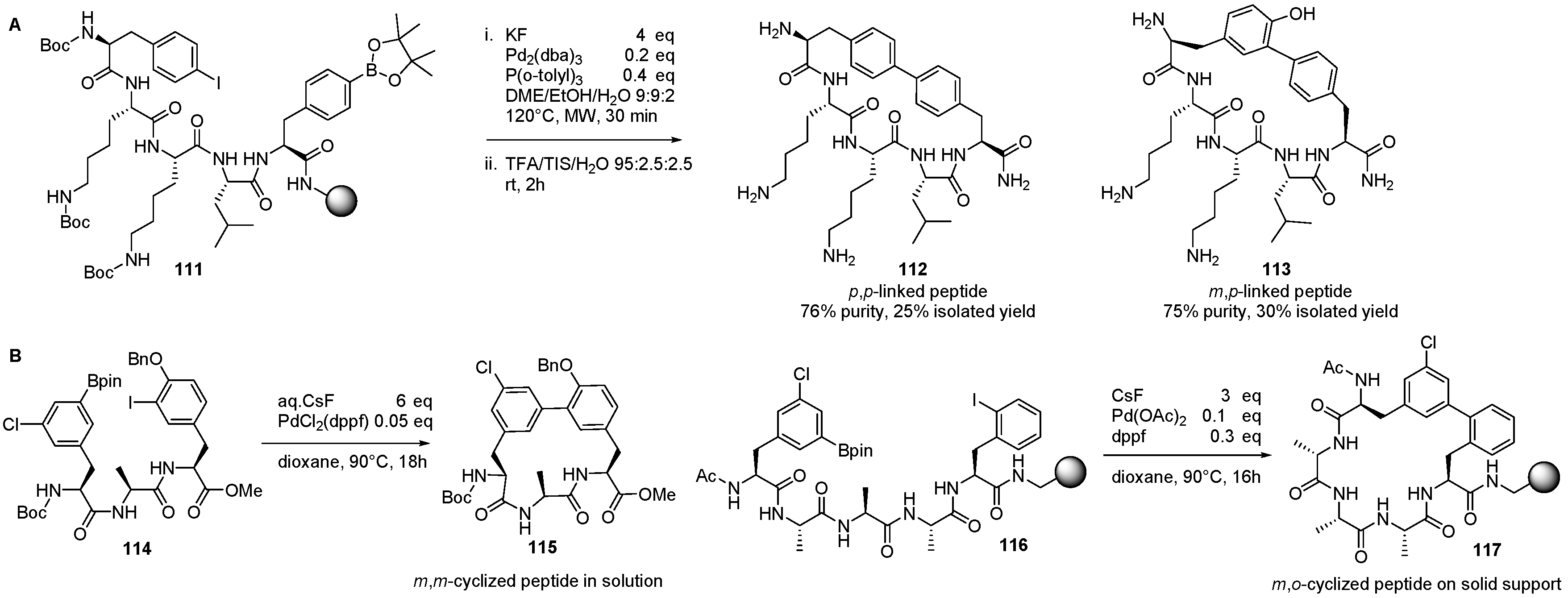
3. Solid-Phase Derivatizations of Peptide Substrates
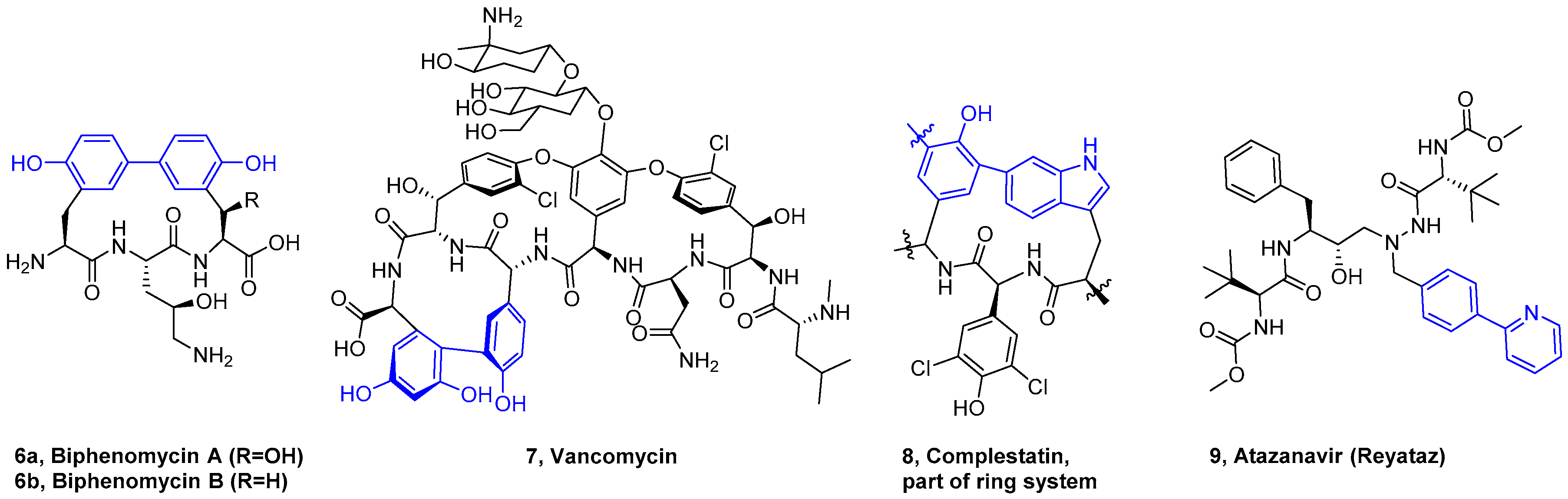
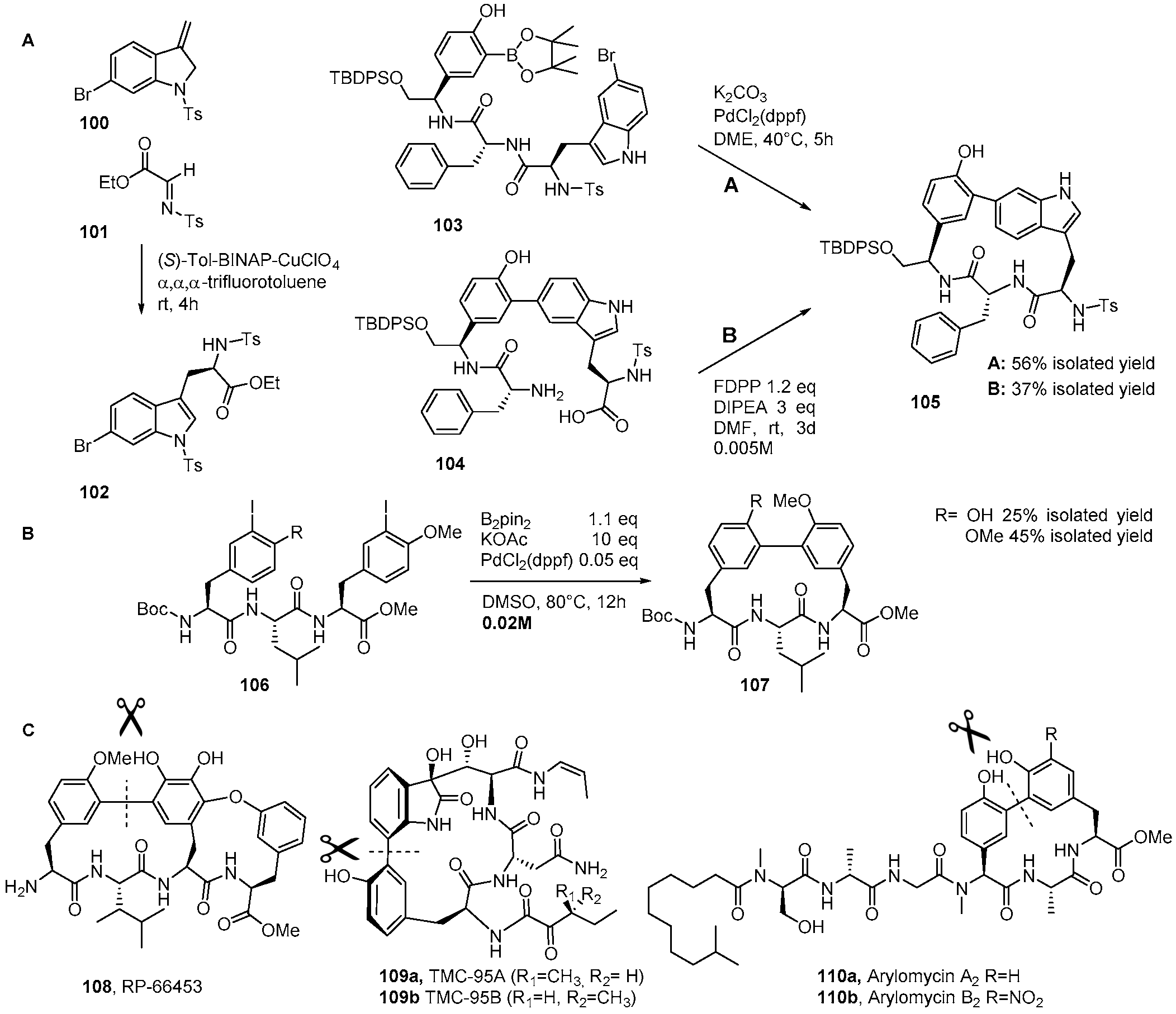
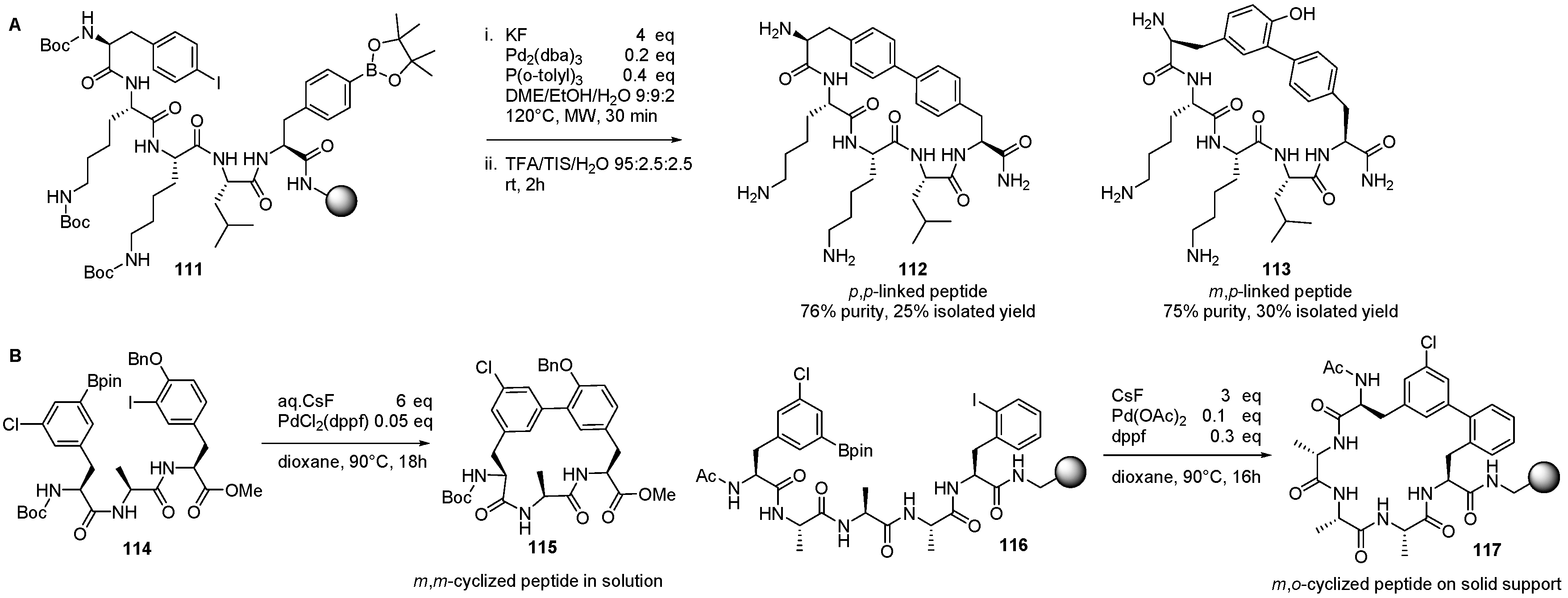
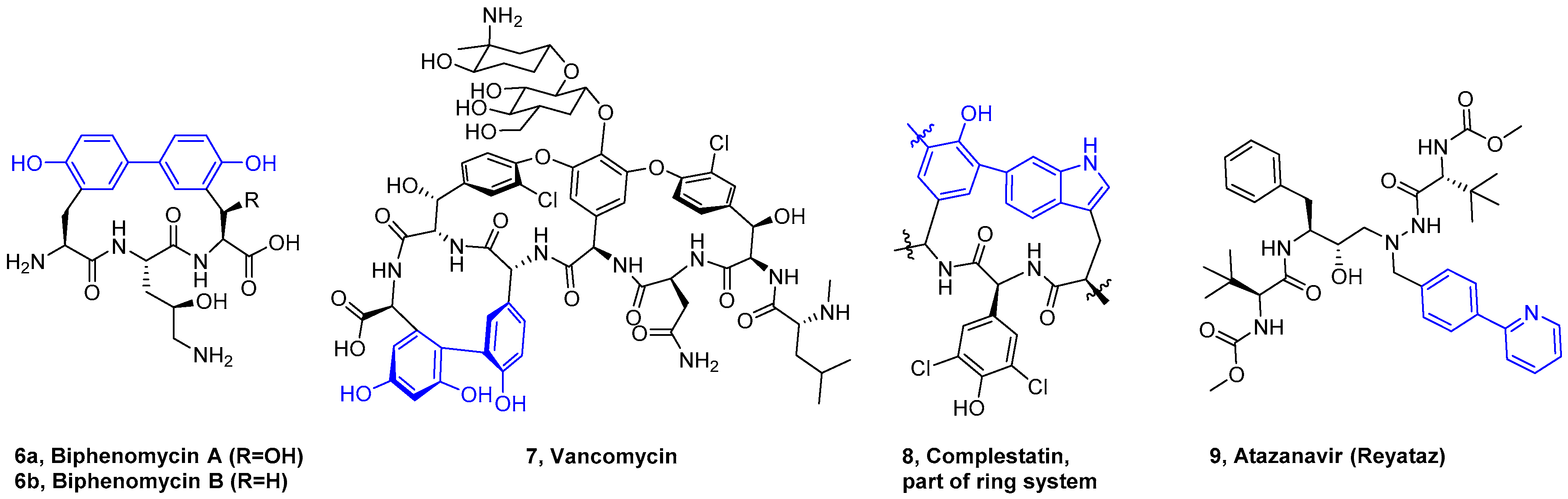
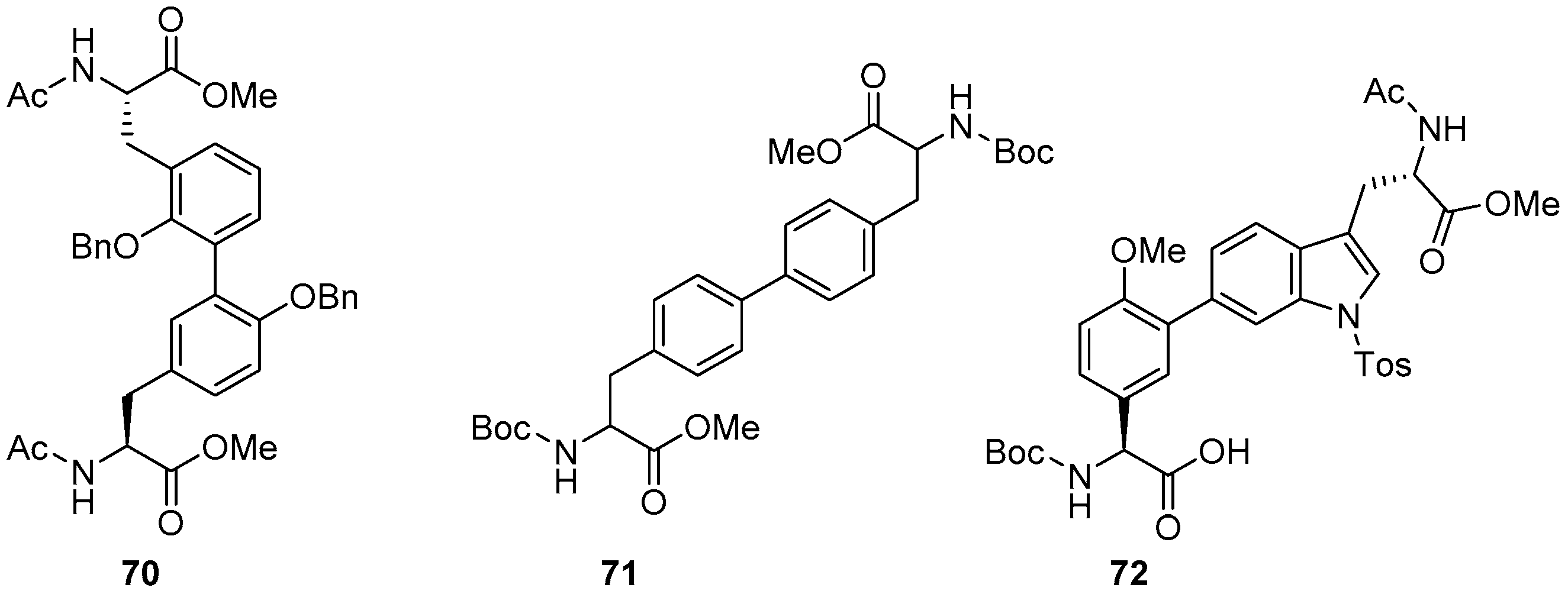
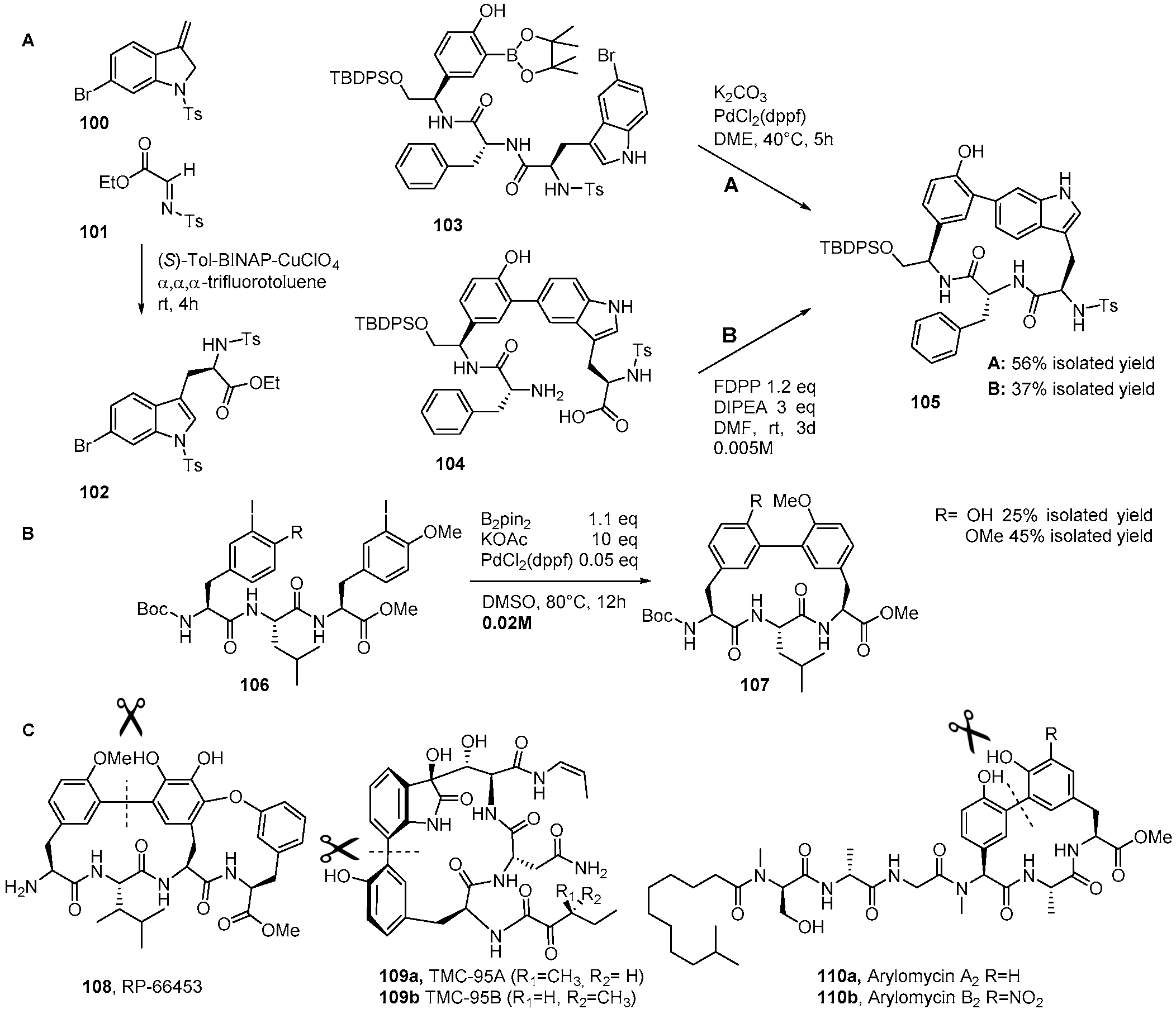
4. Formation of Biaryl-Bridges in Peptidic Natural Products and Peptide Macrocyclics
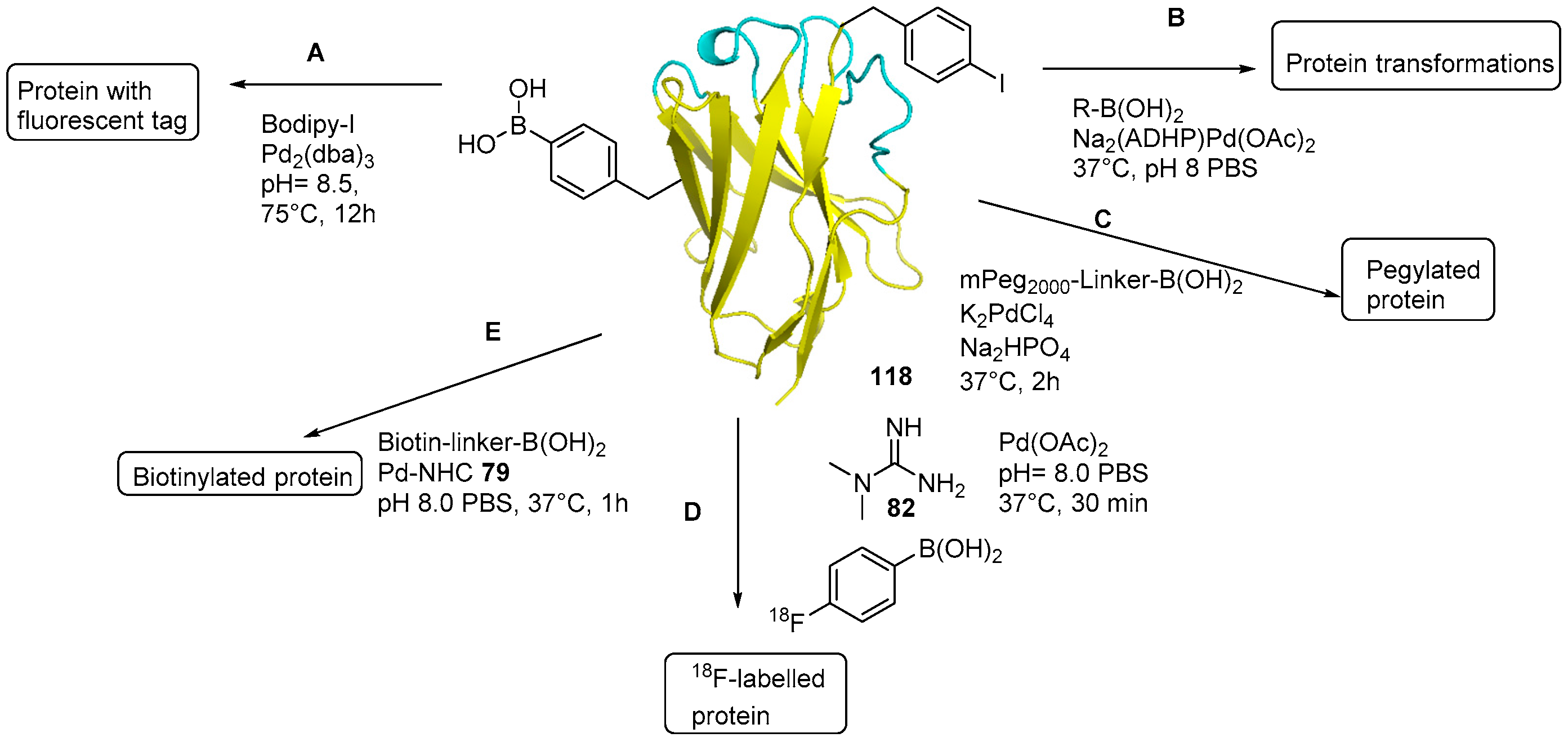
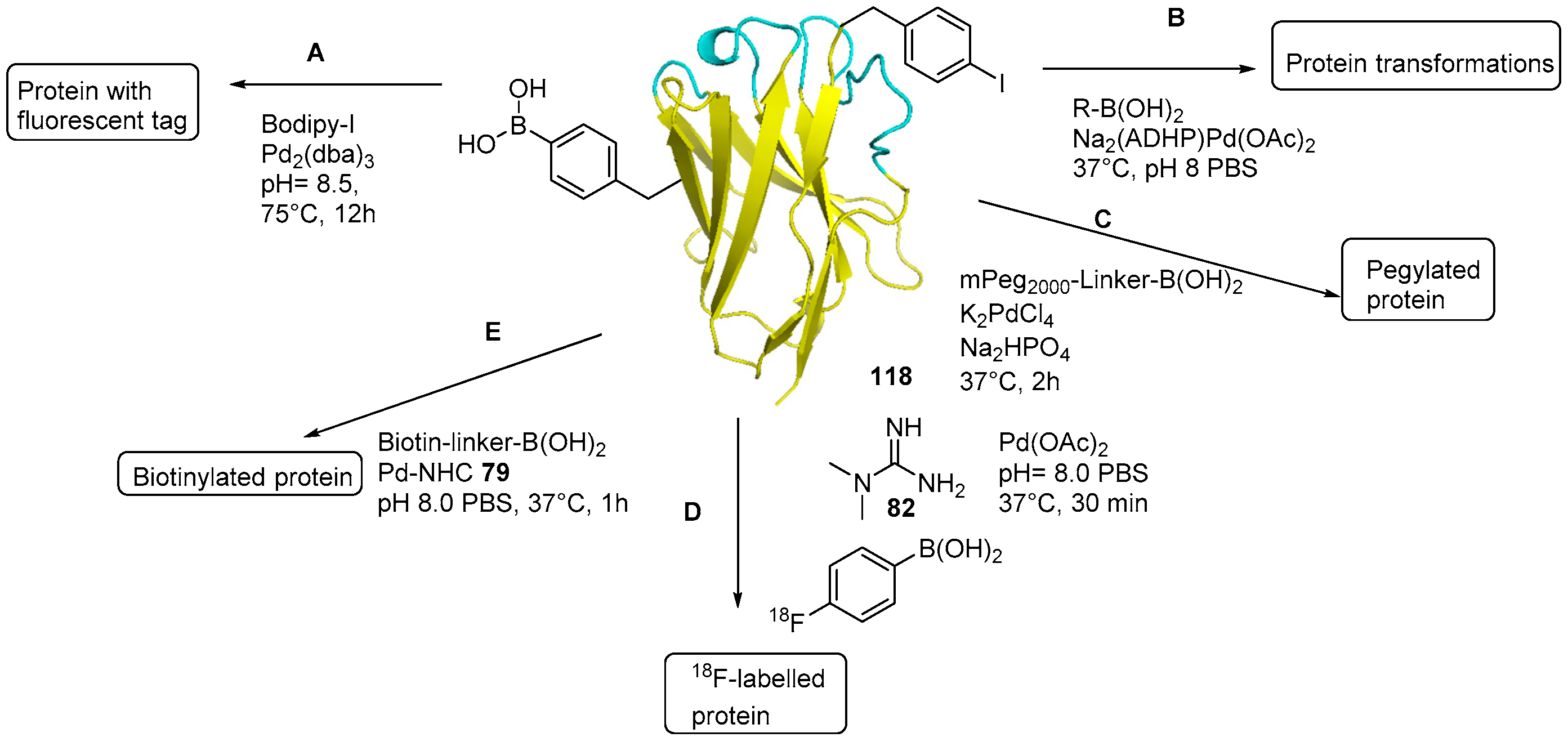
5. Protein Derivatization
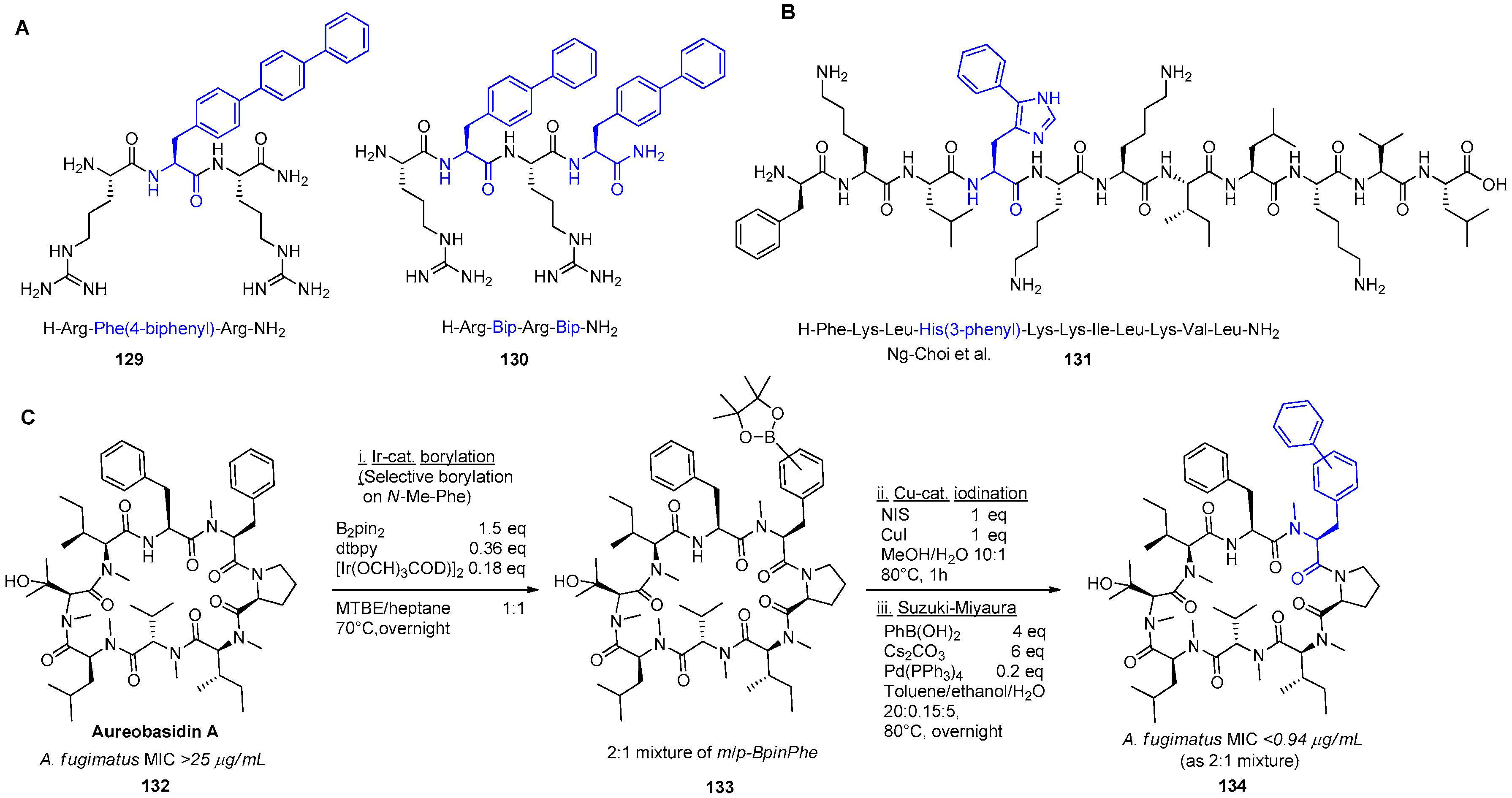
6. The Suzuki–Miyaura Reaction as a Tool towards Improved Biological Activity
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Miyaura, N.; Suzuki, A. Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev. 2015, 95, 2457–2483. [Google Scholar] [CrossRef]
- Suzuki, A. Cross-Coupling Reactions of Organoboranes: An Easy Way to Construct C-C Bonds (Nobel Lecture). Angew. Chem. Int. Ed. 2011, 50, 6722–6737. [Google Scholar] [CrossRef] [PubMed]
- Genet, J.-P.; Savignac, M. Recent developments of palladium(0) catalyzed reactions in aqueous medium. J. Organomet. Chem. 1999, 576, 305–317. [Google Scholar] [CrossRef]
- Lennox, J.J.; Lloyd-Jones, G.C. Preparation of Organotrifluoroborate Salts: Precipitation-Driven Equilibrium under Non-Etching Conditions. Angew. Chem. Int. Ed. 2012, 51, 9385–9388. [Google Scholar] [CrossRef] [PubMed]
- Darses, S.; Genet, J.-P. Potassium Organotrifluoroborates: New Perspectives in Organic Synthesis. Chem. Rev. 2008, 108, 288–325. [Google Scholar] [CrossRef] [PubMed]
- Knapp, D.M.; Gillis, E.P.; Burke, M.D. A General Solution for Unstable Boronic Acids: Slow-Release Cross-Coupling from Air-Stable MIDA Boronates. J. Am. Chem. Soc. 2009, 131, 6961–6963. [Google Scholar] [CrossRef] [PubMed]
- Molander, G.A.; Ellis, N. Organotrifluoroborates: Protected Boronic Acids That Expand the Versatility of the Suzuki Coupling Reaction. Acc. Chem. Res. 2007, 40, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Lennox, J.J.; Lloyd-Jones, G.C. Selection of boron reagents for Suzuki–Miyaura coupling. Chem. Soc. Rev. 2014, 43, 412–443. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.G. Structure, properties, and preparation of boronic acid derivatives. overview of their reactions and applications. In Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine; Hall, D.G., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005; Volume 1, pp. 1–99. [Google Scholar]
- Anderson, K.W.; Buchwald, S.L. General Catalysts for the Suzuki–Miyaura and Sonogashira Coupling Reactions of Aryl Chlorides and for the Coupling of Challenging Substrate Combinations in Water. Angew. Chem. Int. Ed. 2005, 44, 6173–6177. [Google Scholar] [CrossRef] [PubMed]
- Polshettiwar, V.; Decottignies, A.; Len, C.; Fihri, A. Suzuki–Miyaura Cross-Coupling Reactions in Aqueous Media: Green and Sustainable Syntheses of Biaryls. ChemSusChem 2010, 3, 502–522. [Google Scholar] [CrossRef] [PubMed]
- Shaugnessy, K.H. Cross-Coupling reactions in Aqueous Media. In Cross-Coupling Reactions in Aqueous Media, in Palladium-Catalyzed Coupling Reactions: Practical Aspects and Future Developments; Molnár, Á., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; Volume 1, pp. 235–286. [Google Scholar]
- Rossi, R.; Bellina, F.; Lessi, M.; Manzini, C.; Marianetti, G.; Perego, L.A. Recent Applications of Phosphane-based Palladium Catalysts in Suzuki–Miyaura Reactions Involved in Total Syntheses of Natural Products. Curr. Org. Chem. 2015, 19, 1302–1409. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Bulger, P.G.; Sarlah, D. Palladium-Catalyzed Cross-Coupling Reactions in Total Synthesis. Angew. Chem. Int. Ed. 2005, 44, 4442–4489. [Google Scholar] [CrossRef] [PubMed]
- Ronson, T.O.; Taylor, R.J.K.; Fairlamb, I.J.S. Palladium-Catalysed macrocyclisations in the total synthesis of natural products. Tetrahedron 2015, 71, 989–1009. [Google Scholar] [CrossRef]
- Heravi, M.M.; Hashemi, E. Recent applications of the Suzuki reaction in total synthesis. Tetrahedron 2012, 68, 9145–9478. [Google Scholar] [CrossRef]
- Amann, N.; Wagenknecht, H.A. Preparation of pyrenyl-modified nucleosides via Suzuki–Miyaura cross-coupling reactions. Synlett 2002, 5, 687–691. [Google Scholar] [CrossRef]
- Western, E.C.; Daft, J.R.; Johnson, E.M.; Gannett, P.M.; Shaughnessy, K.H. Efficient One-Step Suzuki Arylation of Unprotected Halonucleosides, Using Water-Soluble Palladium Catalysts. J. Org. Chem. 2003, 68, 6767–6774. [Google Scholar] [CrossRef] [PubMed]
- Nencka, R.; Sinnaeve, D.; Karalic, I.; Martins, J.C.; Van Calenbergh, S. Synthesis of C-6-substituted uridine phosphonates through aerobic ligand-free Suzuki–Miyaura cross-coupling. Org. Biomol. Chem. 2010, 8, 5234–5246. [Google Scholar] [CrossRef] [PubMed]
- Omumi, A.; Beach, D.G.; Baker, M.; Gabryelski, W.; Manderville, R.A. Postsynthetic Guanine Arylation of DNA by Suzuki–Miyaura Cross-Coupling. J. Am. Chem. Soc., 2011, 133, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Fresneau, N.; Hiebel, M.-A.; Agrofoglio, L.A.; Berteina-Raboin, S. Efficient Synthesis of Unprotected C-5-Aryl/Heteroaryl-2’-deoxyuridine via a Suzuki–Miyaura Reaction in Aqueous Media. Molecules 2012, 17, 14409–14417. [Google Scholar] [CrossRef] [PubMed]
- De Ornellas, S.; Williams, T.J.; Baumann, C.G.; Fairlamb, I.J.S. Catalytic C-H/C-X bond functionalisation of nucleosides, nucleotides, nucleic acids, amino acids, peptides and proteins. In C–H and C–X Bond Functionalization; Ribas, X., Ed.; Royal Society of Chemistry: Cambridge, UK, 2013; Volume 11, pp. 409–447. [Google Scholar]
- Herve, G.; Sartori, G.; Enderlin, G.; Mackenzie, G.; Len, C. Palladium-catalyzed Suzuki reaction in aqueous solvents applied to unprotected nucleosides and nucleotides. RSC Adv. 2014, 4, 18558–18594. [Google Scholar] [CrossRef]
- Carrow, B.P.; Hartwig, J.F. Distinguishing Between Pathways for Transmetalation in Suzuki–Miyaura Reactions. J. Am. Chem. Soc. 2011, 133, 2116–2119. [Google Scholar] [CrossRef] [PubMed]
- Amatore, C.; Jutand, A.; Le Duc, G. Kinetic Data for the Transmetalation/Reductive Elimination in Palladium-Catalyzed Suzuki–Miyaura Reactions: Unexpected Triple Role of Hydroxide Ions Used as Base. Chem. Eur. J. 2011, 17, 2492–2503. [Google Scholar] [CrossRef] [PubMed]
- Amatore, C.; Le Duc, G.; Jutand, A. Mechanism of Palladium-Catalyzed Suzuki–Miyaura Reactions: Multiple and Antagonistic Roles of Anionic “Bases” and Their Countercations. Chem. Eur. J. 2013, 19, 10082–10093. [Google Scholar] [CrossRef] [PubMed]
- Lennox, A.J.J.; Lloyd-Jones, G.C. Organotrifluoroborate Hydrolysis: Boronic Acid Release Mechanism and an Acid-Base Paradox in Cross-Coupling. J. Am. Chem. Soc. 2012, 134, 7431–7441. [Google Scholar] [CrossRef] [PubMed]
- Lennox, A.J.J.; Lloyd-Jones, G.C. Transmetalation in the Suzuki–Miyaura Coupling: The Fork in the Trail. Angew. Chem. Int. Ed. 2013, 52, 7362–7370. [Google Scholar] [CrossRef] [PubMed]
- Lang, K.; Chin, J.W. Bioorthogonal Reactions for Labeling Proteins. ACS Chem. Biol. 2014, 9, 16–20. [Google Scholar] [CrossRef] [PubMed]
- King, M.; Wagner, A. Developments in the Field of Bioorthogonal Bond Forming Reactions-Past and Present Trends. Bioconj. Chem. 2014, 25, 825–839. [Google Scholar] [CrossRef] [PubMed]
- Spicer, C.D.; Davis, B.G. Selective chemical protein modification. Nat. Commun. 2014, 5, 4740–4753. [Google Scholar] [CrossRef] [PubMed]
- Sletten, M.; Bertozzi, C.R. Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chem. Int. Ed. 2009, 48, 6974–6998. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wu, Y.-W. Selective labelling of proteins. Org. Biomol. Chem. 2016, 14, 5417–5439. [Google Scholar] [CrossRef] [PubMed]
- Dumas, A.; Spicer, C.D.; Gao, Z.; Takehana, T.; Lin, Y.A.; Yasukohchi, T.; Davis, B.G. Self-Liganded Suzuki–Miyaura Coupling for Site-Selective Protein PEGylation. Angew. Chem. Int. Ed. 2013, 52, 3916–3921. [Google Scholar] [CrossRef] [PubMed]
- Keller, S.; Wage, T.; Hohaus, K.; Hölzer, E.E.; van Pée, K.-H. Purification and Partial Characterization of Tryptophan 7-Halogenase (PrnA) from Pseudomonas fluorescens. Angew. Chem. Int. Ed. 2000, 39, 2300–2302. [Google Scholar] [CrossRef]
- Yeh, E.; Garneau, S.; Walsh, C.T. Robust in vitro activity of RebF and RebH, a two-component reductasehalogenase, generating 7-chlorotryptophan during rebeccamycin biosynthesis. Proc. Natl. Acad. Sci. USA 2005, 102, 3960–3965. [Google Scholar] [CrossRef] [PubMed]
- Goss, R.J.M.; Newill, P.L.A. A Convenient Enzymatic Synthesis of L-halotryptophans. Chem. Commun. 2006, 47, 4924–4925. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.R.M.; Grüshow, S.; Goss, R.J.M. Scope and potential of halogenases in biosynthetic applications. Curr. Opin. Chem. Biol. 2013, 17, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, P.F.; Churches, Q.I.; Hutton, C.A. Organoboron Reagents in the Preparation of Functionalized α-Amino Acids. Aust. J. Chem. 2007, 60, 799–810. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Bregadze, V.I. L-4-Boronophenylalanine (all around the one molecule). ARKIVOC 2008, 47–61. [Google Scholar]
- Moreira, I.S.; Fernandes, P.A.; Ramos, M.J. Hot spots—A review of the protein-protein interface determinant amino-acid residues. Proteins: Struct. Funct. Genet. 2007, 68, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Kotha, S.; Goyal, D.; Chavan, A.S. Diversity-Oriented Approaches to Unusual α-Amino Acids and Peptides: Step Economy, Atom Economy, Redox Economy, and Beyond. J. Org. Chem. 2014, 78, 12288–12313. [Google Scholar] [CrossRef] [PubMed]
- Stevenazzi, A.; Marchini, M.; Sandrone, G.; Vergani, B.; Lattanzio, M. Amino acidic scaffolds bearing unnatural side chains: An old idea generates new and versatile tools for the life sciences. Bioorg. Med. Chem. Lett. 2014, 24, 5349–5356. [Google Scholar] [CrossRef] [PubMed]
- Blaskovich, M.A.T. Unusual Amino Acids in Medicinal Chemistry. J. Med. Chem. 2016, 59, 10807–10836. [Google Scholar] [CrossRef] [PubMed]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Adessi, C.; Soto, C. Converting a Peptide into a Drug: Strategies to Improve Stability and Bioavailability. Curr. Med. Chem. 2002, 9, 963–978. [Google Scholar] [CrossRef] [PubMed]
- Van der Poorten, O.; Knuhtsen, A.; Pedersen, D.S.; Ballet, S.; Tourwé, D. Side Chain Cyclized Aromatic Amino Acids: Great Tools as Local Constraints in Peptide and Peptidomimetic Design. J. Med. Chem. 2016, 59, 10865–10890. [Google Scholar] [CrossRef] [PubMed]
- Marsault, E.; Peterson, M.L. Macrocycles Are Great Cycles: Applications, Opportunities, and Challenges of Synthetic Macrocycles in Drug Discovery. J. Med. Chem. 2011, 54, 1961–2004. [Google Scholar] [CrossRef] [PubMed]
- White, C.J.; Yudin, A.K. Contemporary strategies for peptide macrocyclization. Nat. Chem. 2011, 3, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Martí-Centelles, V.; Pandey, M.D.; Burguete, M.I.; Luis, S.V. Macrocyclization Reactions: The Importance of Conformational, Configurational, and Template-Induced Preorganization. Chem. Rev. 2015, 115, 8736–8834. [Google Scholar] [CrossRef] [PubMed]
- Yudin, A.K. Macrocycles: Lessons from the distant past, recent developments, and future directions. Chem. Sci. 2015, 6, 30–49. [Google Scholar] [CrossRef]
- Lau, Y.H.; de Andrade, P.; Wu, Y.; Spring, D.R. Peptide stapling techniques based on different macrocyclisation chemistries. Chem. Soc. Rev. 2015, 44, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Góngora-Benítez, M.; Tulla-Puche, J.; Albericio, F. Multifaceted Roles of Disulfide Bonds. Peptides as Therapeutics. Chem. Rev. 2014, 114, 901–926. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, E.C.; Roy Jackson, W.; Robinson, A.J. Ring-Closing metathesis in peptides. Tetrahedron Lett. 2016, 57, 4325–4333. [Google Scholar] [CrossRef]
- Ahmad Fuaad, A.A.H.; Azmi, F.; Skwarczynski, M.; Toth, I. Peptide Conjugation via CuAAC ‘Click’ Chemistry. Molecules 2013, 18, 13148–13174. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Limberakis, C.; Liras, S.; Price, D.; James, K. Peptidic macrocyclization via palladium-catalyzed chemoselective indole C-2 arylation. Chem. Commun. 2012, 48, 11644–11646. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, B.A.; Smith, G.F.; Sciammetta, N. Synthesis of Cyclic Peptidomimetics via a Pd-Catalyzed Macroamination Reaction. Org. Lett. 2016, 18, 4072–4075. [Google Scholar] [CrossRef] [PubMed]
- Makwana, K.M.; Mahalakshmi, R. Trp-Trp Cross-Linking: A Structure-Reactivity Relationship in the Formation and Design of Hyperstable Peptide β-Hairpin and α-Helix Scaffolds. Org. Lett. 2014, 17, 2498–2501. [Google Scholar] [CrossRef] [PubMed]
- Mendive-Tapia, L.; Preciado, S.; Garciá, J.; Ramón, R.; Kielland, N.; Albericio, F.; Lavilla, R. New peptide architectures through C–H activation stapling between tryptophan-phenylalanine/tyrosine residues. Nat. Comm. 2015, 6, 7160. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Williams, P.; Giralt, E. Atropisomerism, biphenyls and the Suzuki coupling: Peptide antibiotics. Chem. Soc. Rev. 2001, 30, 145–157. [Google Scholar] [CrossRef]
- Feliu, L.; Planas, M. Cyclic Peptides Containing Biaryl and Biaryl Ether Linkages. Int. J. Pept. Res. Ther. 2005, 11, 53–97. [Google Scholar] [CrossRef]
- Shaugnessy, K.H. Hydrophilic Ligands and Their Application in Aqueous-Phase Metal-Catalyzed Reactions. Chem. Rev. 2009, 109, 643–710. [Google Scholar] [CrossRef] [PubMed]
- Chalker, J.M.; Wood, C.S.C.; Davis, B.G. A Convenient Catalyst for Aqueous and Protein Suzuki–Miyaura Cross-Coupling. J. Am. Chem. Soc. 2009, 131, 16346–16347. [Google Scholar] [CrossRef] [PubMed]
- Deb Roy, A.; Goss, R.J.M.; Wagner, G.K.; Winn, M. Development of fluorescent aryltryptophans by Pd mediated cross-coupling of unprotected halotryptophans in water. Chem. Commun. 2008, 39, 4831–4833. [Google Scholar]
- Kodama, K.; Fukuzawa, S.; Nakayama, H.; Sakamoto, K.; Kigawa, T.; Yabuki, T.; Matsuda, N.; Shirouzu, M.; Takio, K.; Yokoyama, S.; et al. Site-Specific Functionalization of Proteins by Organopalladium Reactions. ChemBioChem 2007, 8, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.F.W.; Wishart, N.; Wood, A.; James, K.; Wythes, M.J. Preparation of Enantiomerically Pure Protected 4-Oxo-α-amino Acids and 3-Aryl-α-amino Acids from Serine. J. Org. Chem. 1992, 57, 3397–3404. [Google Scholar] [CrossRef]
- Jackson, R.F.W.; Moore, R.J.; Dexter, C.S. Concise Synthesis of Enantiomerically Pure Phenylalanine, Homophenylalanine, and Bishomophenylalanine Derivatives Using Organozinc Chemistry: NMR Studies of Amino Acid-Derived Organozinc Reagents. J. Org. Chem. 1998, 63, 7875–7884. [Google Scholar] [CrossRef]
- Ross, A.J.; Lang, H.L.; Jackson, R.F.W. Much Improved Conditions for the Negishi Cross- Coupling of Iodoalanine Derived Zinc Reagents with Aryl Halides. J. Org. Chem. 2010, 75, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.J.; Dreiocker, F.; Schäfer, M.; Oomens, J.; Meijer, A.J.H.M.; Pickup, B.T.; Jackson, R.F.W. Evidence for the Role of Tetramethylethylenediamine in Aqueous Negishi Cross-Coupling: Synthesis of Nonproteinogenic Phenylalanine Derivatives on Water. J. Org. Chem. 2011, 76, 1727–1734. [Google Scholar] [CrossRef] [PubMed]
- Bittner, S.; Scherzer, R.; Harlev, E. The five bromotryptophans. Amino Acids 2007, 33, 19–42. [Google Scholar] [CrossRef] [PubMed]
- Konda-Yamada, Y.; Okada, C.; Yoshida, K.; Umeda, Y.; Arima, S.; Sato, N.; Kai, T.; Takayanagi, H.; Harigaya, Y. Convenient synthesis of 7′- and 6′-bromo-D-tryptophan and their derivatives by enzymatic optical resolution using d-aminoacylase. Tetrahedron 2002, 58, 7851–7861. [Google Scholar] [CrossRef]
- Kieffern, M.E.; Repka, L.M.; Reisman, S.E. Enantioselective Synthesis of Tryptophan Derivatives by a Tandem Friedel-Crafts Conjugate Addition/Asymmetric Protonation Reaction. J. Am. Chem. Soc. 2012, 134, 5131–5137. [Google Scholar] [CrossRef] [PubMed]
- Blaser, G.; Sanderson, J.M.; Batsanov, A.S.; Howard, J.A.K. The facile synthesis of a series of tryptophan derivatives. Tetrahedron Lett. 2008, 49, 2795–2798. [Google Scholar] [CrossRef]
- Winn, M.; Roy, A.D.; Grüschow, S.; Parameswaran, R.S.; Goss, R.J.M. A convenient one-step synthesis of L-aminotryptophans and improved synthesis of 5-fluorotryptophan. Bioorg. Med. Chem. Lett. 2008, 18, 4508–4510. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.R.M.; Willemse, T.; Gkotsi, D.S.; Schepens, W.; Maes, B.U.W.; Ballet, S.; Goss, R.J.M. The First One-Pot Synthesis of l-7-Iodotryptophan from 7-Iodoindole and Serine, and an Improved Synthesis of Other l-7-Halotryptophans. Org. Lett. 2014, 16, 2622–2625. [Google Scholar] [CrossRef] [PubMed]
- Seibold, C.; Schnerr, H.; Rumpf, J.; Kunzendorf, A.; Hatscher, C.; Wage, T.; Ernyei, A.J.; Dong, C.; Naismith, J.H.; Van Pée, K.-H. A flavin-dependent tryptophan 6-halogenase and its use in modification of pyrrolnitrin biosynthesis. Biocatal. Biotransformation 2006, 24, 401–408. [Google Scholar] [CrossRef]
- Zehner, S.; Kotzsch, A.; Bister, B.; Süssmuth, R.D.; Méndez, C.; Salas, J.A.; van Pée, K.-H. A Regioselective Tryptophan 5-Halogenase Is Involved in Pyrroindomycin Biosynthesis in Streptomyces rugosporus LL-42D005. Chem. Biol. 2005, 12, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Brock, A.; Herberich, B.; Schultz, P.G. Expanding the Genetic Code of Escherichia coli. Science 2001, 292, 498–500. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Schultz, P.W. At the Interface of Chemical and Biological Synthesis: An Expanded Genetic Code. Cold Spring Harb. Perspect. Biol. 2016, 8, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Brock, A.; Chen, S.; Chen, S.; Schultz, P.G. Genetic incorporation of unnatural amino acids into proteins in mammalian cells. Nat. Meth. 2007, 4, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Mack, A.V.; Brustad, E.M.; Mills, J.H.; Groff, D.; Smider, V.V.; Schultz, P.G. Evolution of Proteins with Genetically Encoded “Chemical Warheads”. J. Am. Chem. Soc. 2009, 131, 9616–9617. [Google Scholar] [CrossRef] [PubMed]
- Kodama, K.; Fukuzawa, S.; Nakayama, H.; Kigawa, T.; Sakamoto, K.; Yabuki, T.; Matsuda, N.; Shirouzu, M.; Takio, K.; Tachibana, K.; et al. Regioselective Carbon–Carbon Bond Formation in Proteins with Palladium Catalysis; New Protein Chemistry by Organometallic Chemistry. ChemBioChem 2006, 7, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Brustad, E.; Bushey, M.L.; Lee, J.W.; Groff, D.; Liu, W.; Schultz, P.G. A Genetically Encoded Boronate-Containing Amino Acid. Angew. Chem. Int. Ed. 2008, 47, 8220–8223. [Google Scholar] [CrossRef] [PubMed]
- Casalnuovo, A.L.; Calabrese, J.C. Palladium-Catalyzed Alkylations in Aqueous Media. J. Am. Chem. Soc. 1999, 112, 4324–4330. [Google Scholar] [CrossRef]
- Tilley, J.W.; Sarabu, R.; Wagner, R.; Mulkerins, K. Preparation of Carboalkoxyalkylphenylalanine Derivatives from Tyrosine. J. Org. Chem. 1990, 55, 906–910. [Google Scholar] [CrossRef]
- Shieh, W.-C.; Carlson, J.A. A Simple Asymmetric Synthesis of 4-Arylphenylalanines via Palladium-Catalyzed Cross-Coupling Reaction of Arylboronic Acids with Tyrosine Triflate. J. Org. Chem. 1992, 57, 379–381. [Google Scholar] [CrossRef]
- Burk, M.J.; Lee, J.R.; Martinez, J.P. A Versatile Tandem Catalysis Procedure for the Preparation of Novel Amino Acids and Peptides J. Am. Chem. Soc. 1994, 116, 10847–10848. [Google Scholar] [CrossRef]
- Kotha, S.; Lahiri, K. A New Approach for Modification of Phenylalanine Peptides by Suzuki–Miyaura Coupling Reaction. Bioorg. Med. Chem. Lett. 2001, 11, 2887–2890. [Google Scholar] [CrossRef]
- Wang, W.; Xiong, C.; Zhang, J.; Hruby, V.J. Practical, assymmetric synthesis of aromatic-substituted bulky and hydrophobic tryptophan and phenylalanine derivatives. Tetrahedron 2002, 58, 3101–3110. [Google Scholar] [CrossRef]
- Espuña, G.; Arsequell, G.; Valencia, G.; Barluenga, J.; Alvarez-Gutiérrez, J.M.; Ballesteros, A.; González, J.M. Regioselective Postsynthetic Modification of Phenylalanine Side Chains of Peptides Leading to Uncommon ortho-Iodinated Analogues. Angew. Chem. Int. Ed. 2004, 43, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Limbach, M.; Löweneck, M.; Schreiber, J.V.; Frackenpohl, J.; Seebach, D. Synthesis of β3-Homophenylalanine-Derived Amino Acids and Peptides by Suzuki Coupling in Solution and on Solid Support. Helv. Chim. Acta 2006, 89, 1427–1441. [Google Scholar] [CrossRef]
- Knör, S.; Laufer, B.; Kessler, H. Efficient Enantioselective Synthesis of Condensed and Aromatic-Ring-Substituted Tyrosine Derivatives. J. Org. Chem. 2006, 71, 5625–5630. [Google Scholar] [CrossRef] [PubMed]
- Cerezo, V.; Afonso, A.; Planas, M.; Feliu, L. Synthesis of 5-arylhistidines via a Suzuki–Miyaura cross-coupling. Tetrahedron 2007, 63, 10445–10453. [Google Scholar] [CrossRef]
- Prieto, M.; Mayor, S.; Rodríguez, K.; Lloyd-Williams, P.; Giralt, E. Racemization in Suzuki Couplings: A Quantitative Study Using 4-Hydroxyphenylglycine and Tyrosine Derivatives as Probe Molecules. J. Org. Chem. 2007, 72, 1047–1050. [Google Scholar] [CrossRef] [PubMed]
- Dumas, A.; Peramo, A.; Desmaële, D.; Couvreur, P. PLGA-PEG-supported Pd Nanoparticles as Efficient Catalysts for Suzuki–Miyaura Coupling Reactions in Water. Chimia 2016, 70, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Deb Roy, A.; Grüshow, S.; Cairns, N.; Goss, R.J.M. Gene Expression Enabling Synthetic Diversification of Natural Products: Chemogenetic Generation of Pacidamycin Analogs. J. Am. Chem. Soc. 2010, 132, 12243–12245. [Google Scholar] [PubMed]
- Willemse, T.; Van Imp, K.; Vlijmen, H.W.T.; Schepens, W.; Goss, R.J.M.; Maes, B.U.W.; Ballet, S. Suzuki–Miyaura Diversification of Amino Acids and Dipeptides in Aqueous Media. ChemCatChem 2015, 7, 2055–2070. [Google Scholar] [CrossRef]
- Frese, M.; Schnepel, C.; Minges, H.; Voß, H.; Feiner, R.; Sewald, N. Modular Combination of Enzymatic Halogenation of Tryptophan with Suzuki–Miyaura Cross-Coupling Reactions. ChemCatChem 2016, 8, 1799–1803. [Google Scholar] [CrossRef]
- Maity, J.; Honcharenko, D.; Strömberg, R. Synthesis of fluorescent D-amino acids with 4-acetamidobiphenyl and 4-N,N-dimethylamino-1,8-naphthalimido containing side chains. Tetrahedron Lett. 2015, 56, 4780–4783. [Google Scholar] [CrossRef]
- Qiao, J.X.; Fraunhoffer, K.J.; Hsiao, Y.; Li, Y.-X.; Wang, C.; Wang, T.C.; Poss, M.A. Synthesis of Fmoc-Protected Arylphenylalanines (Bip Derivatives) via Nonaqueous Suzuki–Miyaura Cross-Coupling Reactions. J. Org. Chem. 2016, 81, 9499–9506. [Google Scholar] [CrossRef] [PubMed]
- Jana, R.; Pathak, T.P.; Sigman, M.S. Advances in Transition Metal (Pd,Ni,Fe)-Catalyzed Cross-Coupling Reactions Using Alkyl-organometallics as Reaction Partners. Chem. Rev. 2011, 111, 1417–1492. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Gao, X.; Wang, B. Boronic Acid Compounds as Potential Pharmaceutical Agents. Med. Res. Rev. 2003, 23, 346–368. [Google Scholar] [CrossRef] [PubMed]
- Satoh, Y.; Gude, C.; Chan, K.; Firooznia, F. Synthesis of 4-Substitured Phenylalanine Derivatives by Cross-Coupling Reaction of p-Boronophenylalanine. Tetrahedron. Lett. 1997, 38, 7645–7648. [Google Scholar] [CrossRef]
- Firooznia, F.; Gude, C.; Chan, K.; Marcopulos, N.; Satoh, Y. Enantioselective Synthesis of 4-Substituted Phenylalanines by Cross-Coupling Reactions. Tetrahedron. Lett. 1999, 40, 213–216. [Google Scholar] [CrossRef]
- Iimura, S.; Wu, W. Palladium-catalyzed borylation of l-tyrosine triflate derivative with pinacolborane: practical route to 4-borono-l-phenylalanine (L-BPA) derivatives. Tetrahedron Lett. 2010, 51, 1353–1355. [Google Scholar] [CrossRef]
- Jung, M.E.; Lazarova, T.I. New Efficient Method for the Total Synthesis of (S,S)-Isodityrosine from Natural Amino Acids. J. Org. Chem. 1999, 64, 2976–2977. [Google Scholar] [CrossRef] [PubMed]
- Malan, C.; Morin, C. A Concise Preparation of 4-Borono-l-phenylalanine (l-BPA) from l-Phenylalanine. J. Org. Chem. 1998, 63, 8019–8020. [Google Scholar] [CrossRef]
- Lépine, R.; Zhu, J. Microwave-Assisted Intramolecular Suzuki–Miyaura Reaction to Macrocycle, a Concise Asymmetric Total Synthesis of Biphenomycin B. Org. Lett. 2005, 7, 2981–2984. [Google Scholar] [CrossRef] [PubMed]
- Wienhold, F.; Claes, D.; Graczyk, K.; Maison, W. Synthesis of Functionalized Benzoboroxoles for the Construction of Boronolectins. Synthesis 2011, 24, 4059–4067. [Google Scholar]
- Kallepalli, V.A.; Shi, F.; Paul, S.; Onyeozili, E.N.; Maleczka, R.E.; Smith, M.R. Boc Groups as Protectors and Directors for Ir-Catalyzed C–H Borylation of Heterocycles. J. Org. Chem. 2009, 74, 9199–9201. [Google Scholar] [CrossRef] [PubMed]
- Meyer, F.-M.; Liras, S.; Guzman-Perez, A.; Perrault, C.; Bian, J.; James, K. Functionalization of Aromatic Amino Acids via Direct C-H Activation: Generation of Versatile Building Blocks for Accessing Novel Peptide Space. Org. Lett. 2010, 12, 3870–3873. [Google Scholar] [CrossRef] [PubMed]
- Audi, H.; Rémond, E.; Eymin, M.-J.; Tessier, A.; Malacea-Kabbara, R.; Jugé, S. Modular Hemisyntheses of Boronato- and Trifluoroborato-Substituted L-NHBoc Amino Acid and Peptide Derivatives. Eur. J. Org. Chem. 2013, 2013, 7960–7972. [Google Scholar] [CrossRef]
- Bartolucci, S.; Bartoccini, F.; Righi, M.; Piersanti, G. Direct, Regioselective, and Chemoselective Preparation of Novel Boronated Tryptophans by Friedel-Crafts Alkylation. Org. Lett. 2011, 14, 600–603. [Google Scholar] [CrossRef] [PubMed]
- Colgin, N.; Flinn, T.; Cobb, S.L. Synthesis and properties of MIDA boronate containing aromatic amino acids: New peptide building blocks. Org. Biomol. Chem. 2011, 9, 1864–1870. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; He, W. Direct Synthesis of Unprotected 4-Aryl Phenylalanines via the Suzuki Reaction under Microwave Irradiation. Org. Lett. 2002, 4, 3803–3805. [Google Scholar] [CrossRef] [PubMed]
- Čapek, P.; Hocek, M. Efficient One-Step Synthesis of Optically Pure (Adenin-8-yl)phenylalanine Nucleosides. Synlett 2005, 19, 3005–3007. [Google Scholar]
- Čapek, P.; Pohl, R.; Hocek, M. Cross-Coupling reactions of unprotected halopurine bases, nucleosides, nucleotides and nucleoside triphosphates with 4-boronophenylalanine in water. Synthesis of (purin-8-yl)- and (purin-6-yl)phenylalanines. Org. Biomol. Chem. 2006, 4, 2278–2284. [Google Scholar] [CrossRef] [PubMed]
- Bartocinni, S.; Bartolucci, M.M.; Piersanti, G. A simple, modular synthesis of C4-substituted tryptophan derivatives. Org. Biomol. Chem. 2016, 14, 10095–10100. [Google Scholar] [CrossRef] [PubMed]
- Loach, R.P.; Fenton, O.S.; Amaike, K.; Siegel, D.S.; Ozkal, E.; Movassaghi, M. C7-Derivatization of C3-Alkylindoles Including Tryptophans and Tryptamines. J. Org. Chem. 2014, 79, 11254–11263. [Google Scholar] [CrossRef] [PubMed]
- Afonso, A.; Feliu, L.; Planas, M. Solid-Phase synthesis of biaryl cyclic peptides by borylation and microwave assisted intramolecular Suzuki–Miyaura reaction. Tetrahedron 2011, 67, 2238–2245. [Google Scholar] [CrossRef]
- Campbell, A.D.; Raynham, T.M.; Taylor, R.J.K. The Synthesis of Novel Amino Acids v/a Hydroboration-Suzuki Cross Coupling. Tetrahedron Lett. 1999, 40, 5263–5266. [Google Scholar] [CrossRef]
- Sabat, M.; Johnson, C.R. Synthesis of Unnatural Amino Acids via Suzuki Cross-Coupling of Enantiopure Vinyloxazolidine Derivatives. Org. Lett. 2000, 2, 1089–1092. [Google Scholar] [CrossRef] [PubMed]
- Collier, P.N.; Campbell, A.D.; Patel, I.; Taylor, R.J.K. The direct synthesis of novel enantiomerically pure α-amino acids in protected form via Suzuki cross-coupling. Tetrahedron Lett. 2000, 41, 7115–7119. [Google Scholar] [CrossRef]
- Rodríguez, A.; Miller, D.D.; Jackson, R.F.W. Combined application of organozinc chemistry and one-pot hydroboration-Suzuki coupling to the synthesis of amino acids. Org. Biomol. Chem. 2003, 1, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.J.; Chandra, J.S.; Reddy, M.V.R. Concise synthesis of ω-borono-α-amino acids. Org. Biomol. Chem. 2007, 5, 889–891. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.E.; Kenworthy, M.N.; Taylor, R.J.K. Synthesis of non-proteinogenic phenylalanine analogues by Suzuki cross-coupling of a serine-derived alkyl boronic acid. Tetrahedron Lett. 2004, 45, 2467–2471. [Google Scholar] [CrossRef]
- Zhang, Z.Q.; Yang, C.T.; Liang, L.J.; Xiao, B.; Lu, X.; Liu, J.-H.; Sun, Y.-Y.; Marder, T.B.; Fu, Y. Copper-Catalyzed/Promoted Cross-coupling of gem-Diborylalkanes with Nonactivated Primary Alkyl Halides: An Alternative Route to Alkylboronic Esters. Org. Lett. 2014, 16, 6342–6345. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Jiang, H.; Takise, R.; Zhu, R.-Y.; Chen, G.; Dai, H.-X.; Murali Dhar, T.G.; Shi, J.; Zhang, H.; Cheng, P.T.W.; et al. Ligand-Promoted Borylation of C(sp3)-H Bonds with Palladium(II) Catalysts. Angew. Chem. Int. Ed. 2016, 55, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Yoburn, J.C.; Van Vranken, D.L. Synthesis of Dityrosine Cross-Linked Peptide Dimers Using the Miyaura-Suzuki Reaction. Org. Lett. 2003, 5, 2817–2820. [Google Scholar] [CrossRef] [PubMed]
- Hutton, C.A.; Skaff, O. A convenient preparation of dityrosine via Miyaura borylation-Suzuki coupling of iodotyrosine derivatives. Tetrahedron Lett. 2003, 44, 4895–4898. [Google Scholar] [CrossRef]
- Kotha, S.; Shah, V.R.; Halder, S.; Vinodkumar, R.; Lahiri, K. Synthesis of bis-armed amino acid derivatives via the alkylation of ethyl isocyanoacetate and the Suzuki–Miyaura cross-coupling reaction. Amino Acids 2007, 32, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Prieto, M.; Mayor, S.; Lloyd-Williams, P.; Giralt, E. Use of the SPhos Ligand to Suppress Racemization in Arylpinacolboronate Ester Suzuki Couplings Involving r-Amino Acids. Synthesis of Biaryl Derivatives of 4-Hydroxyphenylglycine, Tyrosine, and Tryptophan. J. Org. Chem. 2009, 74, 9202–9205. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Prickett, C.D.; Shaughnessy, K.H. Efficient Sonogashira Coupling of Unprotected Halonucleosides in Aqueous Solvents Using Water-Soluble Palladium Catalysts. Eur. J. Org. Chem. 2010, 2010, 3678–3683. [Google Scholar] [CrossRef]
- Roy, S.; Plenio, H. Sulfonated N-Heterocyclic Carbenes for Pd-Catalyzed Sonogashira and Suzuki–Miyaura Coupling in Aqueous Solvents. Adv. Synth. Catal. 2010, 352, 1014–1022. [Google Scholar] [CrossRef]
- Ma, X.; Wang, H.; Chen, W. N-Heterocyclic Carbene-Stabilized Palladium Complexes as Organometallic Catalysts for Bioorthogonal Cross-Coupling Reactions. J. Org. Chem. 2014, 79, 8652–8658. [Google Scholar] [CrossRef] [PubMed]
- Fleckenstein, C.; Roy, S.; Leuthäußer, S.; Plenio, H. Sulfonated N-heterocyclic carbenes for Suzuki coupling in water. Chem. Commun. 2007, 27, 2870–2872. [Google Scholar] [CrossRef] [PubMed]
- Godoy, F.; Segarra, C.; Poyatos, M.; Peris, E. Palladium Catalysts with Sulfonate-Functionalized-NHC Ligands for Suzuki–Miyaura Cross-Coupling Reactions in Water. Organometallics 2011, 30, 684–688. [Google Scholar] [CrossRef]
- Chatterjee, A.; Ward, T.R. Recent Advances in the Palladium Catalyzed Suzuki–Miyaura Cross-Coupling Reaction in Water. Catal. Lett. 2016, 146, 820–840. [Google Scholar] [CrossRef]
- Lipshutz, B.H.; Ghorai, S. “Designer”-Surfactant-Enabled Cross-Couplings in Water at Room Temperature. Aldrichim. Acta 2012, 45, 3–16. [Google Scholar]
- Li, J.-H.; Zhang, X.-D.; Xie, Y.-X. Efficient Pd(OAc)2/pyrimidine catalytic system for Suzuki–Miyaura Cross-Coupling reaction. Synlett 2005, 12, 1897–1900. [Google Scholar] [CrossRef]
- Li, S.; Lin, Y.; Cao, J.; Zhang, S. Guanidine/Pd(OAc)2-Catalyzed Room Temperature Suzuki Cross-Coupling Reaction in Aqueous Media under Aerobic Conditions. J. Org. Chem. 2007, 72, 4067–4072. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Gouverneur, V.; Davis, B.G. Enhanced Aqueous Suzuki–Miyaura Coupling Allows Site-Specific Polypeptide 18F-Labeling. J. Am. Chem. Soc. 2013, 135, 13612–13615. [Google Scholar] [CrossRef] [PubMed]
- Vilaró, M.; Arsequell, G.; Valencia, G.; Ballesteros, A.; Barluenga, J. Arylation of Phe and Tyr Side Chains of Unprotected Peptides by a Suzuki–Miyaura Reaction in Water. Org. Lett. 2008, 10, 3243–3245. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.A.; Chalker, J.M.; Davis, B.G. Olefin Metathesis for Site-Selective Protein Modification. ChemBioChem 2009, 10, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Ai, H.-W.; Shen, W.; Brustad, E.; Schultz, P.G. Genetically Encoded Alkenes in Yeast. Angew. Chem. Int. Ed. 2010, 49, 935–937. [Google Scholar] [CrossRef] [PubMed]
- Ojida, A.; Tsutsumi, H.; Kasagi, N.; Hamachi, I. Suzuki coupling for protein modification. Tetrahedron Lett. 2005, 46, 3301–3305. [Google Scholar] [CrossRef]
- Lorsbach, B.A.; Kurth, M.J. Carbon–Carbon Bond Forming Solid-Phase Reactions. Chem. Rev. 1999, 99, 1549–1581. [Google Scholar] [CrossRef] [PubMed]
- Colombel, V.; Presset, M.; Oehlrich, D.; Rombouts, F.; Molander, G.A. Synthesis and Reactivity of Solid- Supported Organotrifluoroborates in Suzuki Cross-Coupling. Org. Lett. 2012, 14, 1680–1683. [Google Scholar] [CrossRef] [PubMed]
- Yokum, T.S.; Barany, G. Strategy in Solid-Phase Peptide Syntesis. In Solid-Phase Synthesis: A Practical Guide; Kates, S.A., Albericio, F., Eds.; Marcel Dekker, Inc.: New York, NY, USA, 2000; Volume 1, pp. 79–103. [Google Scholar]
- Adams, J.H.; Cook, R.M.; Hudson, D.; Jammalamadaka, V.; Lyttle, M.H.; Songster, M.F. A Reinvestigation of the Preparation, Properties, and Applications of Aminomethyl and 4-Methylbenzhydrylamine Polystyrene Resins. J. Org. Chem. 1998, 63, 3706–3716. [Google Scholar] [CrossRef]
- García-Martín, F.; Quintanar-Audelo, M.; García-Ramos, Y.; Cruz, L.J.; Gravel, C.; Furic, R.; Côté, S.; Tulla-Puche, J.; Albericio, F. ChemMatrix, a Poly(ethylene glycol)-Based Support for the Solid-Phase Synthesis of Complex Peptides. J. Comb. Chem. 2006, 8, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Colombo, A.; Fernàndez, J.-C.; de la Figuera, N.; Fernàndez-Forner, D.; Forns, P.; Albericio, F. Solid-Phase Preparation of a Library Based on a Phenylalanine Scaffold. QSAR Comb. Sci. 2005, 24, 913–922. [Google Scholar] [CrossRef]
- Nielsen, T.E.; Le Quement, S.; Meldal, M. Solid-Phase synthesis of biarylalanines via Suzuki cross-coupling and intramolecular N-acyliminium Pictet-Spengler reactions. Tetrahedron Lett. 2005, 46, 7959–7962. [Google Scholar] [CrossRef]
- Le Quement, S.; Nielsen, T.E.; Meldal, M. Solid-Phase Synthesis of Aryl-Substituted Thienoindolizines: Sequential Pictet-Spengler, Bromination and Suzuki Cross-Coupling Reactions of Thiophenes. J. Comb. Chem. 2008, 10, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Le Quement, S.; Ishoey, M.; Petersen, M.T.; Thastrup, J.; Hagel, G.; Nielsen, T.E. Solid-Phase Synthesis of Smac Peptidomimetics Incorporating Triazoloprolines and Biarylalanines. ACS Comb. Sci. 2011, 13, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Haug, D.E.; Stensen, W.; Svendsen, J.S. Application of the Suzuki–Miyaura cross-coupling to increase antimicrobial potency generates promising novel antibacterials. Bioorg. Med. Chem. Lett. 2007, 17, 2361–2364. [Google Scholar] [CrossRef] [PubMed]
- Doan, N.-D.; Bourgault, S.; Létourneau, M.; Fournier, A. Effectiveness of the Suzuki–Miyaura Cross-Coupling Reaction for Solid-Phase Peptide Modification. J. Comb. Chem. 2008, 10, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Afonso, A.; Rosés, C.; Planas, M.; Feliu, L. Biaryl Peptides from 4-Iodophenylalanine by Solid-Phase Borylation and Suzuki–Miyaura Cross-Coupling. Eur. J. Org. Chem. 2010, 2010, 1461–1468. [Google Scholar] [CrossRef]
- Cerezo, V.; Amblard, M.; Martinez, J.; Verdié, P.; Planas, M.; Feliu, L. Solid-phase synthesis of 5-arylhistidines via a microwave-assisted Suzuki–Miyaura cross-coupling. Tetrahedron 2008, 64, 10538–10545. [Google Scholar] [CrossRef]
- Elder, A.M.; Rich, D.H. Two Syntheses of the 16- and 17-Membered DEF Ring Systems of Chloropeptin and Complestatin. Org. Lett. 1999, 1, 1443–1446. [Google Scholar] [CrossRef] [PubMed]
- Carbonelle, A.-C.; Zhu, J. A Novel Synthesis of Biaryl-Containing Macrocycles by a Domino Miyaura Arylboronate Formation: Intramolecular Suzuki Reaction. Org. Lett. 2000, 2, 3477–3480. [Google Scholar] [CrossRef]
- Boisnard, S.; Carbonelle, A.-C.; Zhu, J. Studies on the Total Synthesis of RP 66453: Synthesis of Fully Functionalized 15-Membered Biaryl-Containing Macrocycle. Org. Lett. 2001, 3, 2061–2064. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, H.; He, Y.-P.; Tan, H.; Arve, L.; Arndt, H.-D. Flexible total synthesis of biphenomycin B. Chem. Commun. 2008, 43, 5562–5564. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Danishefsky, S.J. The Total Synthesis of Proteasome Inhibitors TMC-95A and TMC-95B: Discovery of a New Method to Generate cis-Propenyl Amides. Angew. Chem. Int. Ed. 2002, 41, 512–515. [Google Scholar] [CrossRef]
- Berthelot, A.; Piguel, S.; Le Dour, G.; Vidal, J. Synthesis of Macrocyclic Peptide Analogues of Proteasome Inhibitor TMC-95A. J. Org. Chem. 2003, 68, 9835–9838. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Sakazaki, H.; Furyama, H.; Hirama, M. Total Synthesis of TMC-95A. Angew. Chem. Int. Ed. 2003, 42, 2654–2657. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.; Siciliano, C.; Assfalg-Machleidt, I.; Groll, M.; Milbradt, A.; Moroder, L. Synthesis of a TMC-95A Ketomethylene Analogue by Cyclization via Intramolecular Suzuki Coupling. Org. Lett. 2003, 5, 3435–3437. [Google Scholar] [CrossRef] [PubMed]
- Coste, A.; Bayle, A.; Marrot, J.; Evano, G. A Convergent Synthesis of the Fully Elaborated Macrocyclic Core of TMC-95A. Org. Lett. 2014, 16, 1306–1309. [Google Scholar] [CrossRef] [PubMed]
- Dufour, J.; Neuville, L.; Zhu, J. Intramolecular Suzuki–Miyaura Reaction for the Total Synthesis of Signal Peptidase Inhibitors, Arylomycins A2 and B2. Chem. Eur. J. 2010, 16, 10523–10534. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Bois-Choussy, M.; Zhu, J. Synthesis of Diastereomers of Complestatin and Chloropeptin I: Substrate-Dependent Atropstereoselectivity of the Intramolecular Suzuki–Miyaura Reaction. Angew. Chem. Int. Ed. 2008, 47, 4167–4172. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Bois-Choussy, M.; Jia, Y.; Zhu, J. Total Synthesis of Complestatin (Chloropeptin II). Angew. Chem. Int. Ed. 2010, 49, 2018–2022. [Google Scholar] [CrossRef] [PubMed]
- Moschitto, M.J.; Lewis, C.A. Synthesis of the Rubiyunnanin B Core Aglycon. Eur. J. Org. Chem. 2016, 2016, 4773–4777. [Google Scholar] [CrossRef]
- Afonso, A.; Cussó, O.; Feliu, L.; Planas, M. Solid-Phase Synthesis of Biaryl Cyclic Peptides Containing a 3-Aryltyrosine. Eur. J. Org. Chem. 2012, 2012, 6204–6211. [Google Scholar] [CrossRef]
- Meyer, F.-M.; Collins, J.C.; Borin, B.; Bradow, J.; Liras, S.; Limberakis, C.; Mathiowetz, A.M.; Philippe, L.; Price, D.; Song, K.; et al. Biaryl-Bridged Macrocyclic Peptides: Conformational Constraint via Carbogenic Fusion of Natural Amino Acid Side Chains. J. Org. Chem. 2012, 77, 3099–3114. [Google Scholar] [CrossRef] [PubMed]
- Antos, J.M.; Francis, M.B. Transition metal catalyzed methods for site-selective protein modification. Curr. Opin. Chem. Biol. 2006, 10, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Chankeshwara, S.V.; Indrigo, E.; Bradley, M. Palladium-mediated chemistry in living cells. Curr. Opin. Chem. Biol. 2014, 21, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Sasmal, P.K.; Streu, C.N.; Meggers, E. Metal Complex Catalysis in Living Biological Systems. Chem. Commun. 2013, 49, 1581–1587. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, Y.; Ojida, A.; Hamachi, I. Protein Organic Chemistry and Applications for Labeling and Engineering in Live-Cell Systems. Angew. Chem. Int. Ed. 2013, 52, 4088–4106. [Google Scholar] [CrossRef] [PubMed]
- Garrett, C.E.; Prasad, K. The Art of Meeting Palladium Specifications in Active Pharmaceutical Ingredients Produced by Pd-Catalyzed Reactions. Adv. Synth. Catal. 2004, 346, 889–900. [Google Scholar] [CrossRef]
- Spicer, C.D.; Davis, B.G. Palladium-Mediated site-selective Suzuki–Miyaura protein modification at genetically encoded aryl halides. Chem. Commun. 2011, 47, 1698–1700. [Google Scholar] [CrossRef] [PubMed]
- Magano, J.; Dunetz, J.R. Large-Scale Applications of Transition Metal-Catalyzed Couplings for the Synthesis of Pharmaceuticals. Chem. Rev. 2011, 111, 2177–2250. [Google Scholar] [CrossRef] [PubMed]
- Molnár, Á. Efficient, Selective, and Recyclable Palladium Catalysts in Carbon–Carbon Coupling Reactions. Chem. Rev. 2011, 111, 2251–2320. [Google Scholar] [CrossRef] [PubMed]
- Lamblin, M.; Nasser-Hardy, L.; Hierso, J.-C.; Fouquet, E.; Felpin, F.-X. Recyclable Heterogeneous Palladium Catalysts in Pure Water: Sustainable Developments in Suzuki, Heck, Sonogashira and Tsuji-Trost Reactions. Adv. Synth. Catal. 2010, 352, 33–79. [Google Scholar] [CrossRef]
- Zhang, Q.; Su, H.; Luo, J.; Wei, Y. Recyclable palladium(II) imino-pyridine complex immobilized on mesoporous silica as a highly active and recoverable catalyst for Suzuki–Miyaura coupling reactions in aqueous medium. Tetrahedron 2013, 69, 447–454. [Google Scholar] [CrossRef]
- Shokouhimehr, M.; Lee, J.E.; Han, S.I.; Hyeon, T. Magnetically recyclable hollow nanocomposite catalysts for heterogeneous reduction of nitroarenes and Suzuki reactions. Chem. Commun. 2013, 49, 4779–4781. [Google Scholar] [CrossRef] [PubMed]
- Shokouhimehr, M.; Kim, T.; Jun, S.W.; Shin, K.; Jang, Y.; Kim, B.H.; Kim, J.; Hyeon, T. Magnetically separable carbon nanocomposite catalysts for efficient nitroarene reduction and Suzuki reactions. Appl. Catal. A 2014, 476, 133–139. [Google Scholar] [CrossRef]
- Choi, K.-H.; Shokouhimehr, M.; Sung, Y.-E. Heterogeneous Suzuki Cross-Coupling Reaction Catalyzed by Magnetically Recyclable Nanocatalyst. Bull. Korean Chem. Soc. 2013, 34, 1477–1480. [Google Scholar] [CrossRef]
- Karimi, B.; Akhavan, P.F. Main-Chain NHC-palladium polymer as a recyclable self-supported catalyst in the Suzuki–Miyaura coupling of aryl chlorides in water. Chem. Commun. 2009, 25, 3750–3752. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, M.; Chiarucci, M.; Trombini, C. A recyclable triethylammonium ion-tagged diphenylphosphine palladium complex for the Suzuki–Miyaura reaction in ionic liquids. Green Chem. 2009, 11, 574–579. [Google Scholar] [CrossRef]
- Edwards, G.A.; Trafford, M.A.; Hamilton, A.E.; Buxton, A.M.; Bardeaux, M.C.; Chalker, J.M. Melamine and Melamine-Formaldehyde Polymers as Ligands for Palladium and Application to Suzuki–Miyaura Cross-Coupling Reactions in Sustainable Solvents. J. Org. Chem. 2014, 79, 2094–2104. [Google Scholar] [CrossRef] [PubMed]
- Spicer, C.D.; Triemer, T.; Davis, B.G. Palladium-Mediated Cell-Surface Labeling. J. Am. Chem.Soc. 2012, 134, 800–803. [Google Scholar] [CrossRef] [PubMed]
- Alconcel, S.N.S.; Baas, A.S.; Maynard, H.D. FDA-Approved poly(ethylene glycol)–protein conjugate drugs. Polym. Chem. 2011, 2, 1442–1448. [Google Scholar] [CrossRef]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The future of peptide-based drugs. Chem. Biol. Drug. Des. 2013, 81, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Hagmann, W.K.; Durette, P.L.; Lanza, T.; Kevin, N.J.; de Laszlo, S.E.; Kopka, I.E.; Young, D.; Magriotis, P.A.; Li, B.; Lin, L.S.; et al. The Discovery of Sulfonylated Dipeptides as Potent VLA-4 Antagonists. Bioorg. Med. Chem. Lett. 2001, 11, 2709–2713. [Google Scholar] [CrossRef]
- Yang, G.X.; Chang, L.L.; Truong, Q.; Doherty, G.A.; Magriotis, P.A.; de Laszlo, S.E.; Li, B.; MacCoss, M.; Kidambi, U.; Egger, L.A.; et al. N-Tetrahydrofuroyl-(l)-phenylalanine derivatives as potent VLA-4 antagonists. Bioorg. Med. Chem. Lett. 2002, 12, 1497–1500. [Google Scholar] [CrossRef]
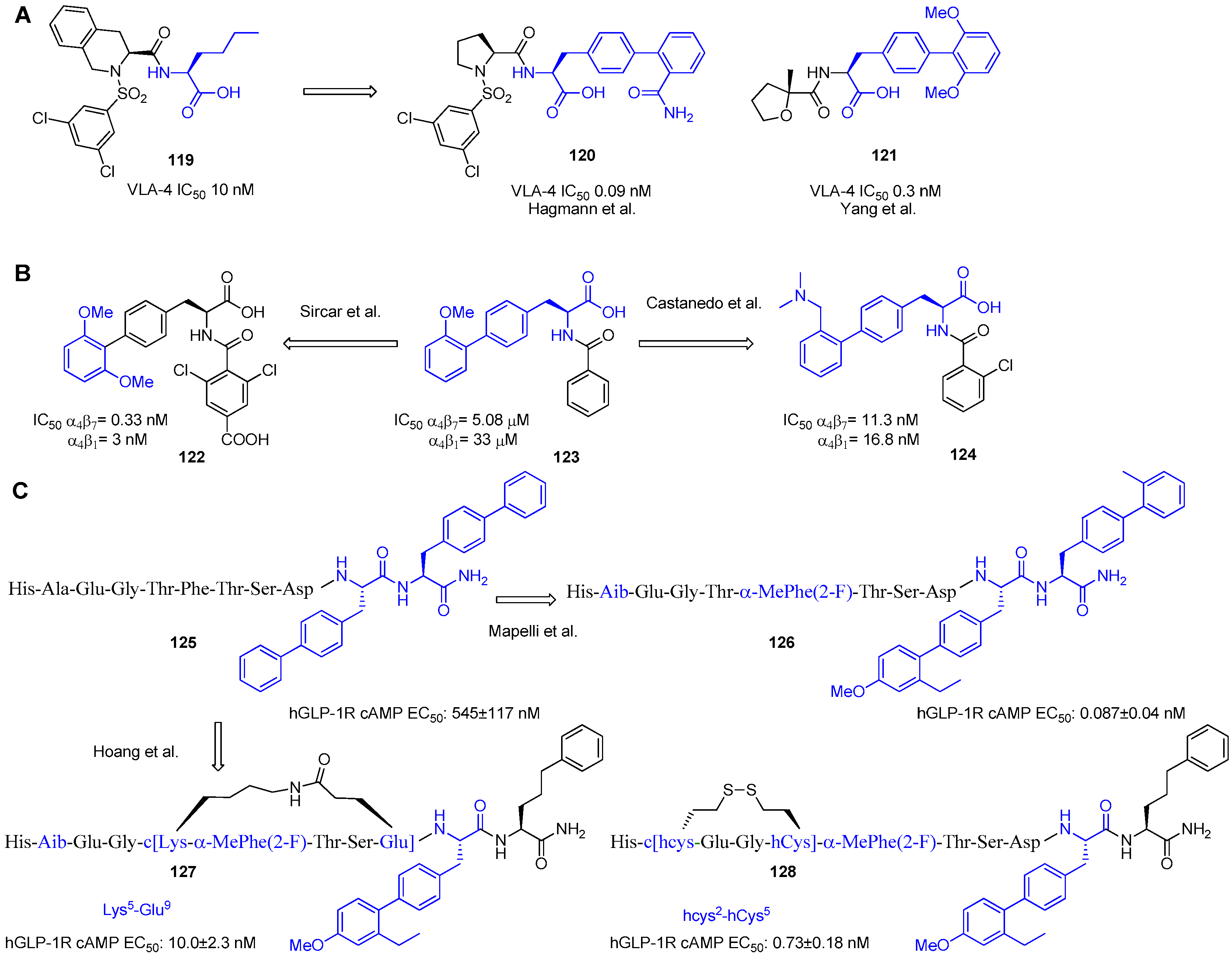
- Sircar, I.; Gudmundsson, K.S.; Martin, R.; Liang, J.; Nomura, S.; Jayakumar, H.; Teegarden, B.R.; Nowlin, D.M.; Cardarelli, P.M.; Mah, J.R.; et al. Synthesis and SAR of N-Benzoyl-l-Biphenylalanine Derivatives: Discovery of TR-14035, A Dual α4β1/α4β7 Integrin Antagonisty. Bioorg. Med. Chem. 2002, 10, 2051–2066. [Google Scholar] [CrossRef]
- Castanedo, G.M.; Sailes, F.C.; Dubree, N.J.P.; Nicholas, J.B.; Caris, L.; Clark, K.; Keating, S.M.; Beresini, M.H.; Chiu, H.; Fong, S.; et al. Solid-Phase Synthesis of Dual α4β1/α4β7 Integrin Antagonists: Two Scaffolds with Overlapping Pharmacophores. Bioorg. Med. Chem. Lett. 2002, 12, 2913–2917. [Google Scholar] [CrossRef]
- Mapelli, C.; Natarajan, S.I.; Meyer, J.-P.; Bastos, M.M.; Bernatowicz, M.S.; Lee, V.G.; Pluscec, J.; Riexinger, D.J.; Sieber-McMaster, E.S.; Constantine, K.L.; et al. Eleven Amino Acid Glucagon-like Peptide-1 Receptor Agonists with Antidiabetic Activity. J. Med. Chem. 2009, 52, 7788–7799. [Google Scholar] [CrossRef] [PubMed]
- Hoang, H.N.; Song, K.; Hill, T.A.; Derksen, D.R.; Edmonds, D.J.; Kok, W.M.; Limberakis, C.; Liras, S.; Loria, P.M.; Mascitti, V.; et al. Short Hydrophobic Peptides with Cyclic Constraints Are Potent Glucagon-like Peptide-1 Receptor (GLP-1R) Agonists. J. Med. Chem. 2015, 58, 4080–4085. [Google Scholar] [CrossRef] [PubMed]
- Haug, B.E.; Skar, M.L.; Svendsen, J.S. Bulky Aromatic Amino Acids Increase the Antibacterial Activity of 15-Residue Bovine Lactoferricin Derivatives. J. Peptide Sci. 2001, 7, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Lau, Q.Y.; Ng, F.M.; Cheong, J.W.D.; Yap, Y.Y.A.; Tan, Y.Y.F.; Jureen, R.; Hill, J.; Chia, C.S.B. Discovery of an ultra-short linear antibacterial tetrapeptide with anti-MRSA activity from a structure-activity relationship study. Eur. J. Med. Chem. 2015, 105, 138–144. [Google Scholar] [CrossRef] [PubMed]
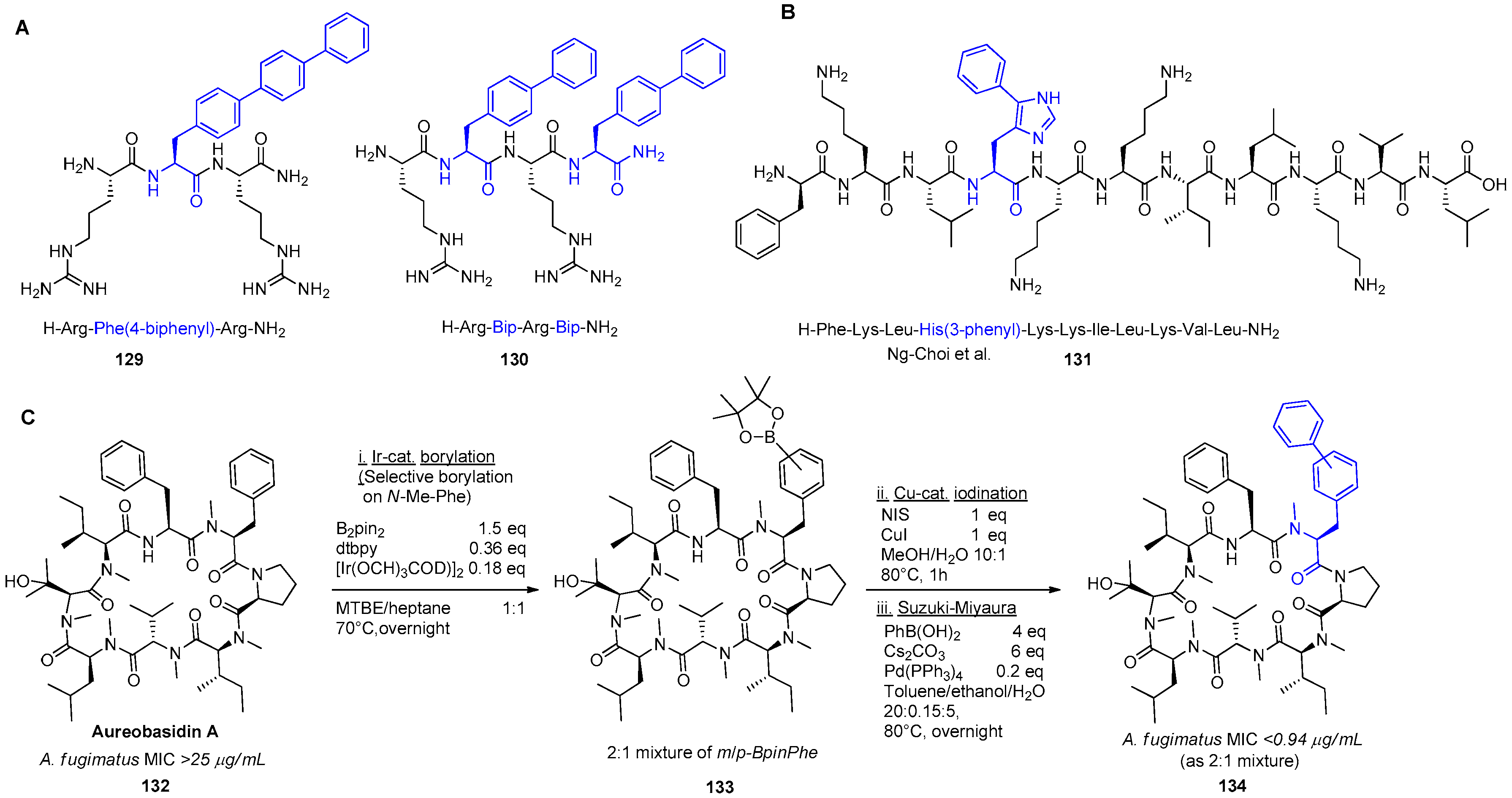
- Ng-Choi, I.; Soler, M.; Cerezo, V.; Badosa, E.; Montesinos, E.; Planas, M.; Feliu, L. Solid-Phase Synthesis of 5-Arylhistidine-Containing Peptides with Antimicrobial Activity Through a Microwave-Assisted Suzuki–Miyaura Cross-Coupling. Eur. J. Org. Chem. 2012, 2012, 4321–4332. [Google Scholar] [CrossRef]
- Wuts, P.G.M.; Simons, L.J.; Metzger, B.P.; Sterling, R.C.; Slightom, J.L.; Elhammer, A.P. Generation of Broad-Spectrum Antifungal Drug Candidates from the Natural Product Compound Aureobasidin A. ACS Med. Chem. Lett. 2015, 6, 645–649. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Willemse, T.; Schepens, W.; Vlijmen, H.W.T.v.; Maes, B.U.W.; Ballet, S. The Suzuki–Miyaura Cross-Coupling as a Versatile Tool for Peptide Diversification and Cyclization. Catalysts 2017, 7, 74. https://doi.org/10.3390/catal7030074
Willemse T, Schepens W, Vlijmen HWTv, Maes BUW, Ballet S. The Suzuki–Miyaura Cross-Coupling as a Versatile Tool for Peptide Diversification and Cyclization. Catalysts. 2017; 7(3):74. https://doi.org/10.3390/catal7030074
Chicago/Turabian StyleWillemse, Tom, Wim Schepens, Herman W. T. van Vlijmen, Bert U. W. Maes, and Steven Ballet. 2017. "The Suzuki–Miyaura Cross-Coupling as a Versatile Tool for Peptide Diversification and Cyclization" Catalysts 7, no. 3: 74. https://doi.org/10.3390/catal7030074
APA StyleWillemse, T., Schepens, W., Vlijmen, H. W. T. v., Maes, B. U. W., & Ballet, S. (2017). The Suzuki–Miyaura Cross-Coupling as a Versatile Tool for Peptide Diversification and Cyclization. Catalysts, 7(3), 74. https://doi.org/10.3390/catal7030074