Advances in Base-Free Oxidation of Bio-Based Compounds on Supported Gold Catalysts

 ,
, 

Abstract
:1. Introduction
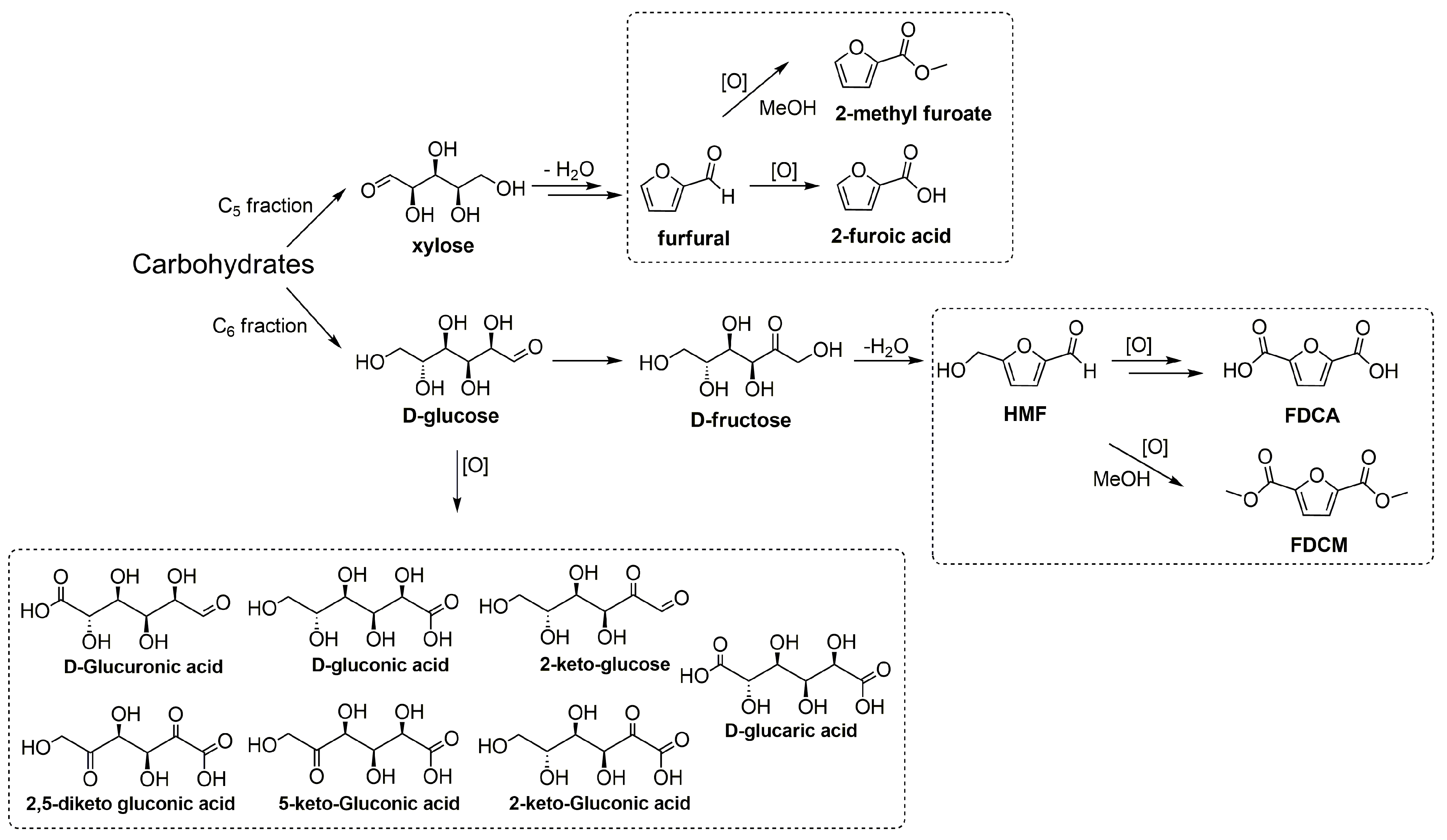
2. Base-Free Carbohydrate Oxidation
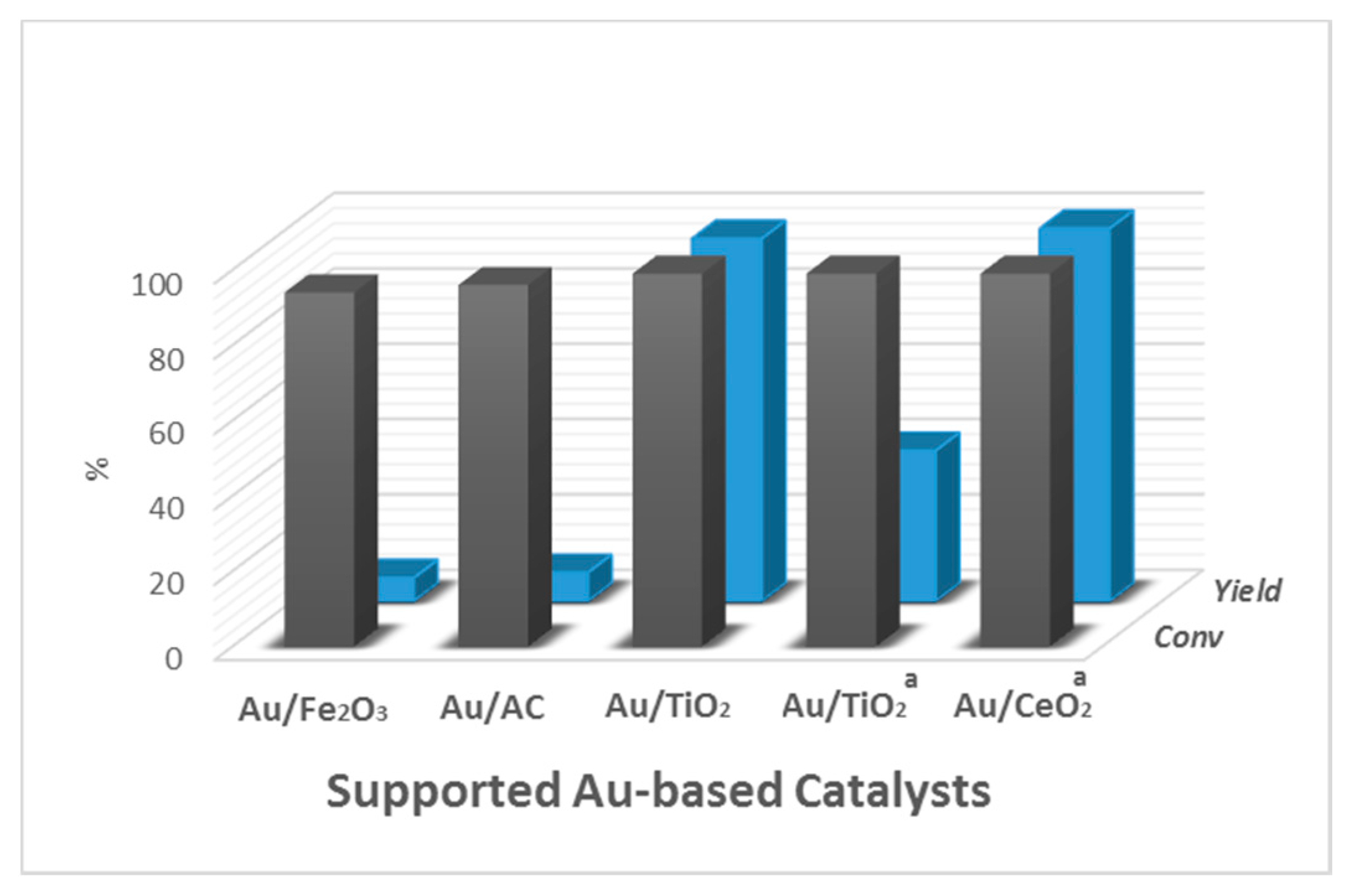
2.1. Supported Au-Based Catalysts
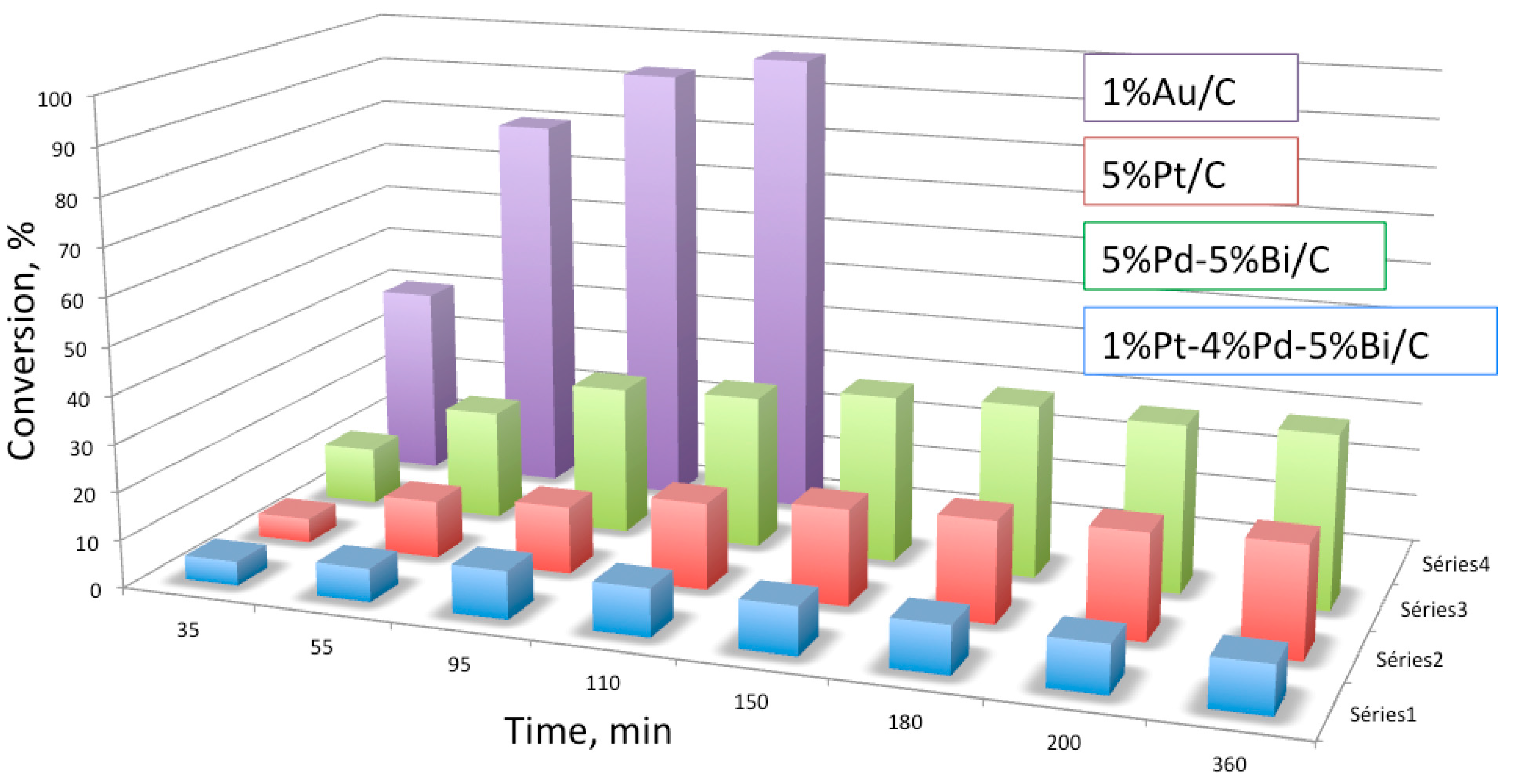
2.2. Multimetallic Au-Based Catalysts
2.3. Mechanism of the Base-Free Oxidation of Carbohydrates
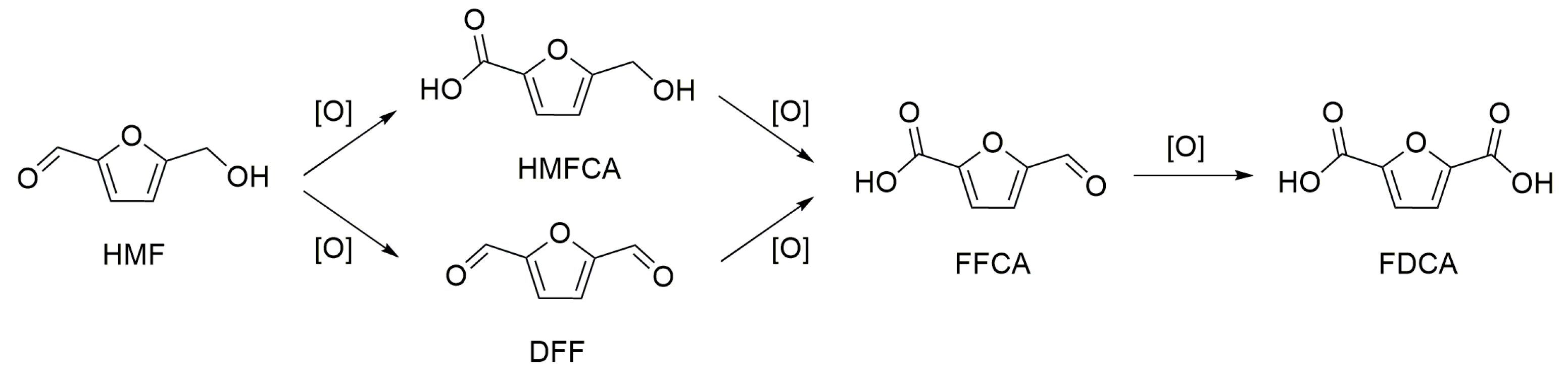
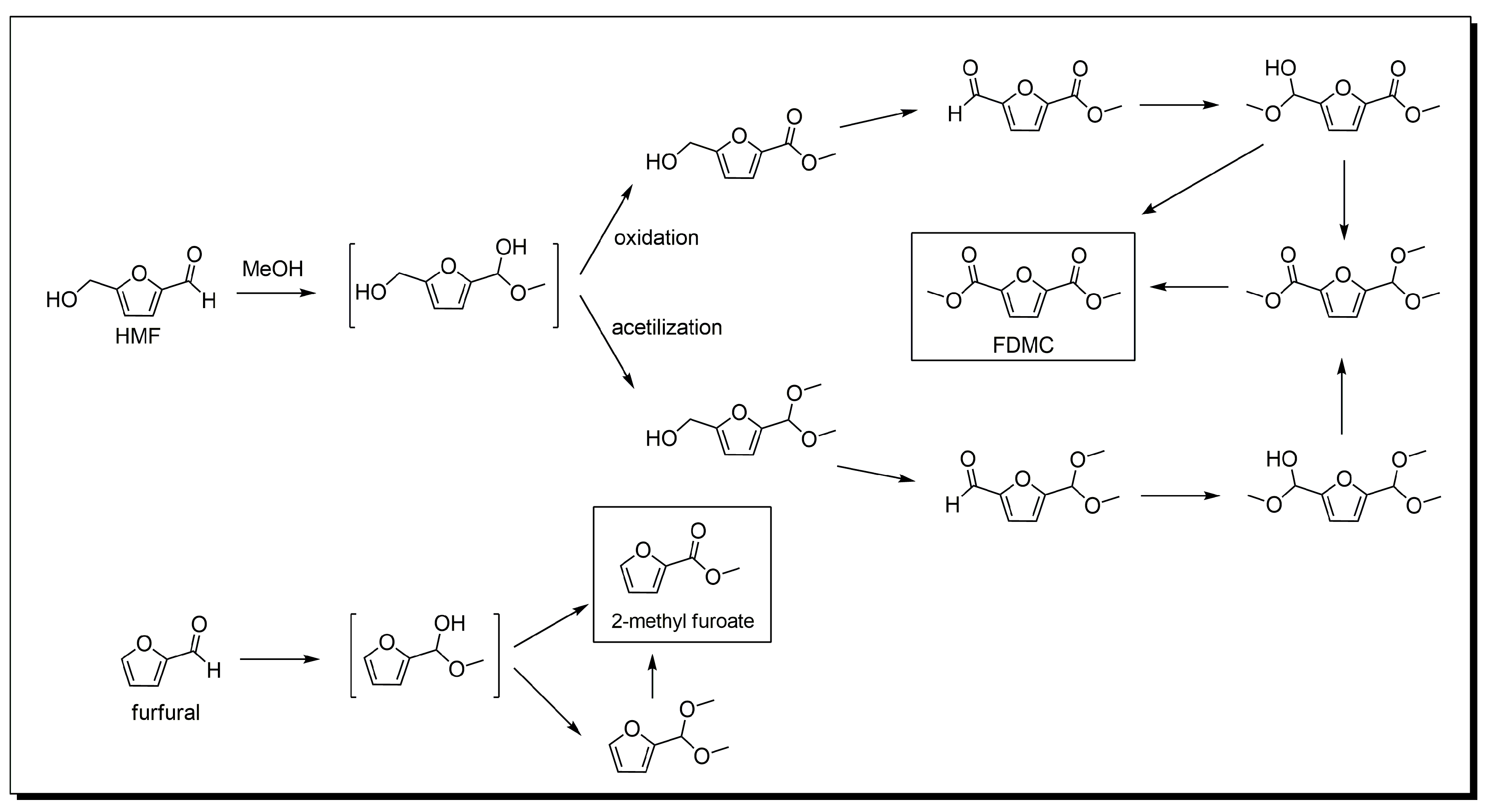
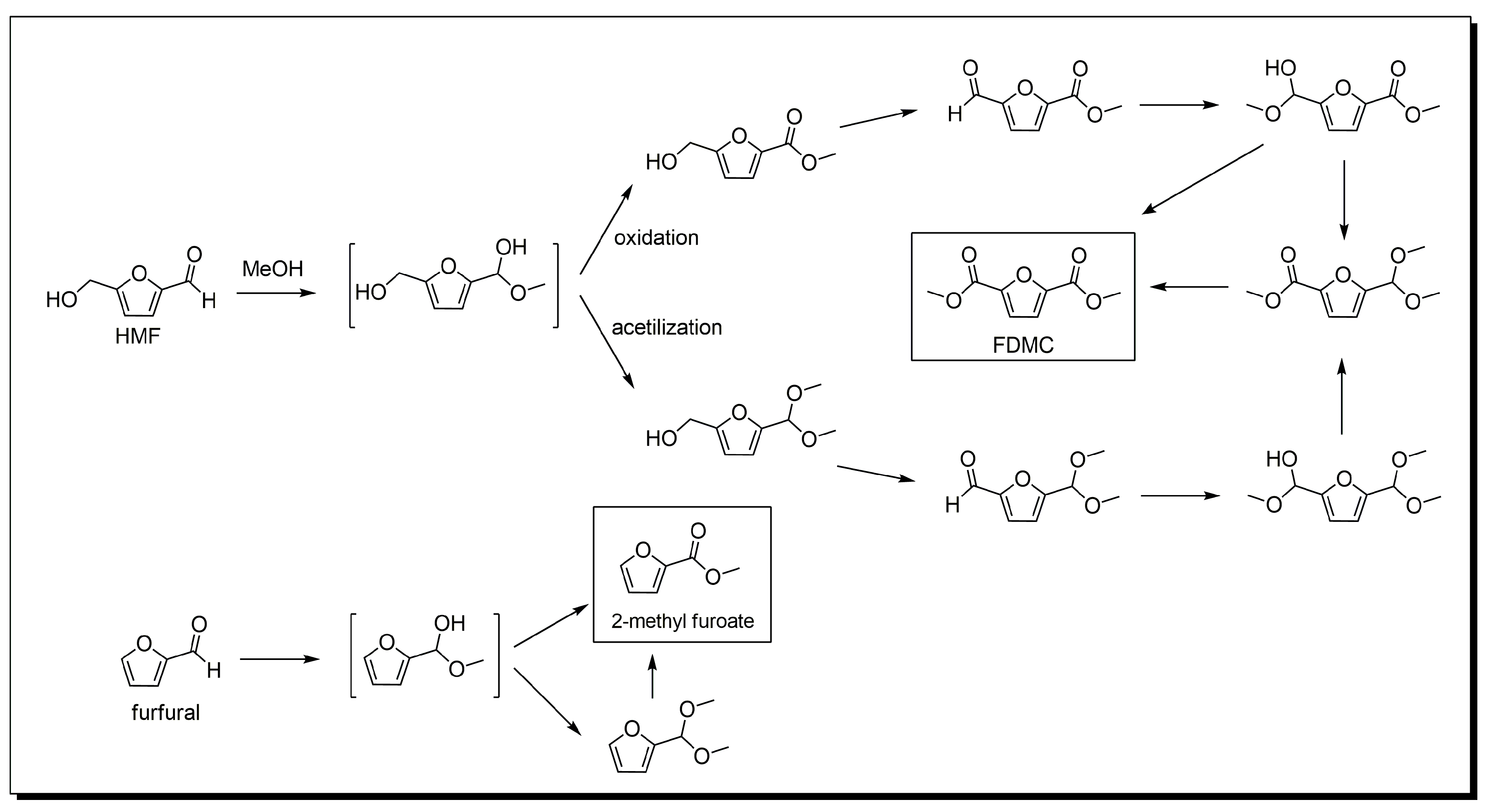
3. Base-Free 5-Hydroxymethylfurfural Oxidation
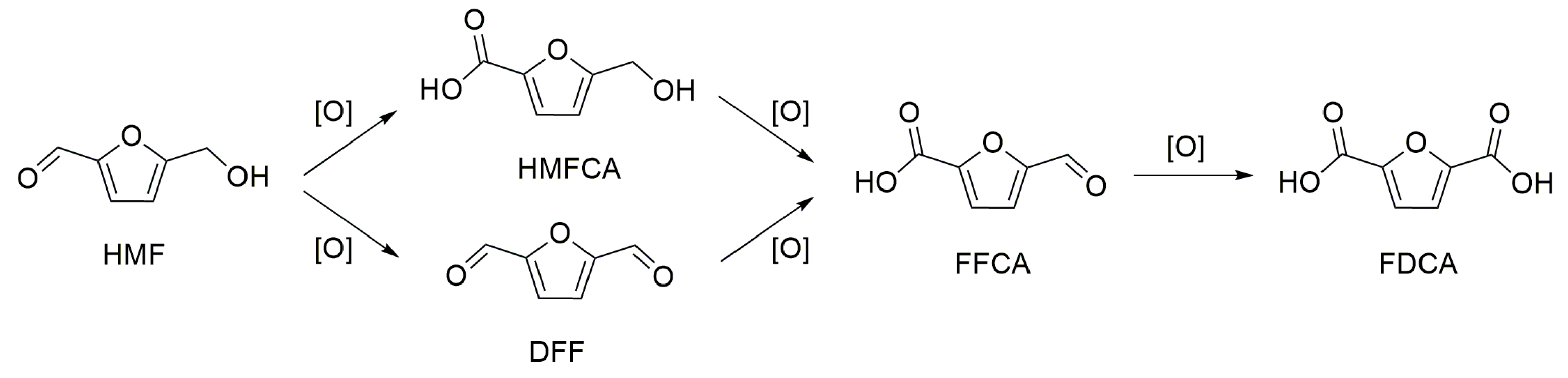
Mechanism of HMF Oxidation
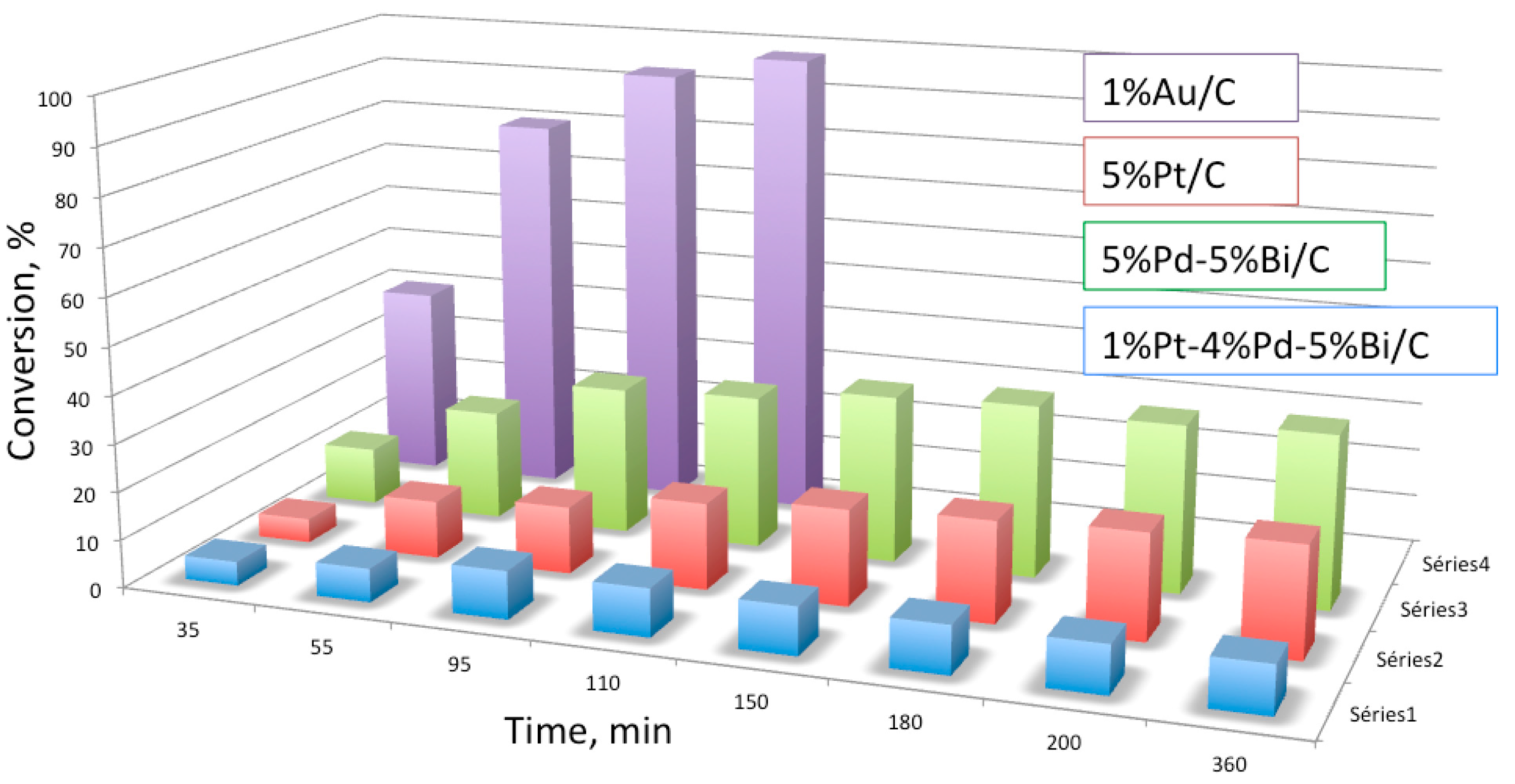
4. Base-Free Furfural Oxidation
Mechanism of Furfural Oxidation
5. Factors Influencing the Activity in the Oxidation on Gold-Based Catalysts
5.1. Effect of the Size of the Gold Particles
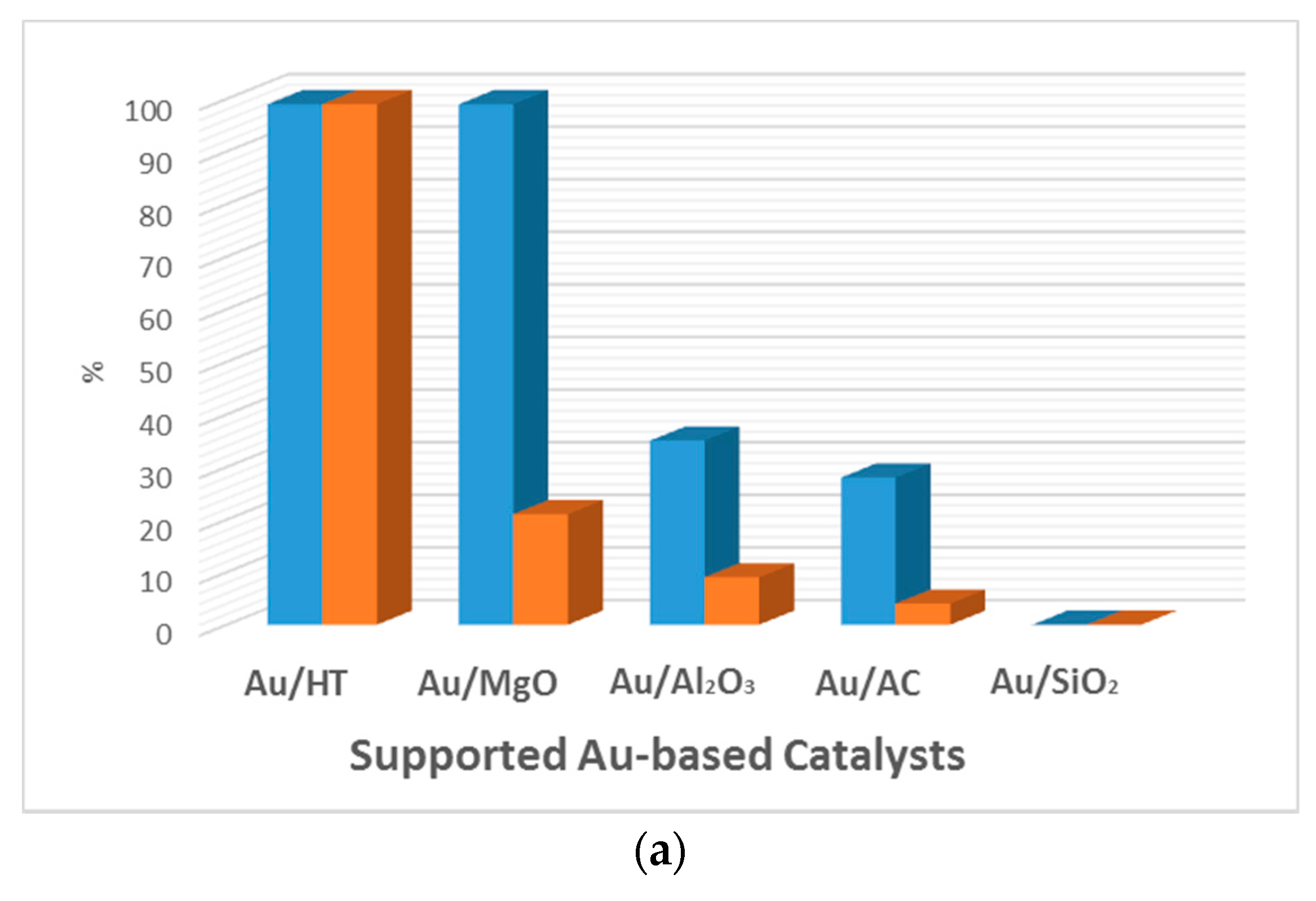
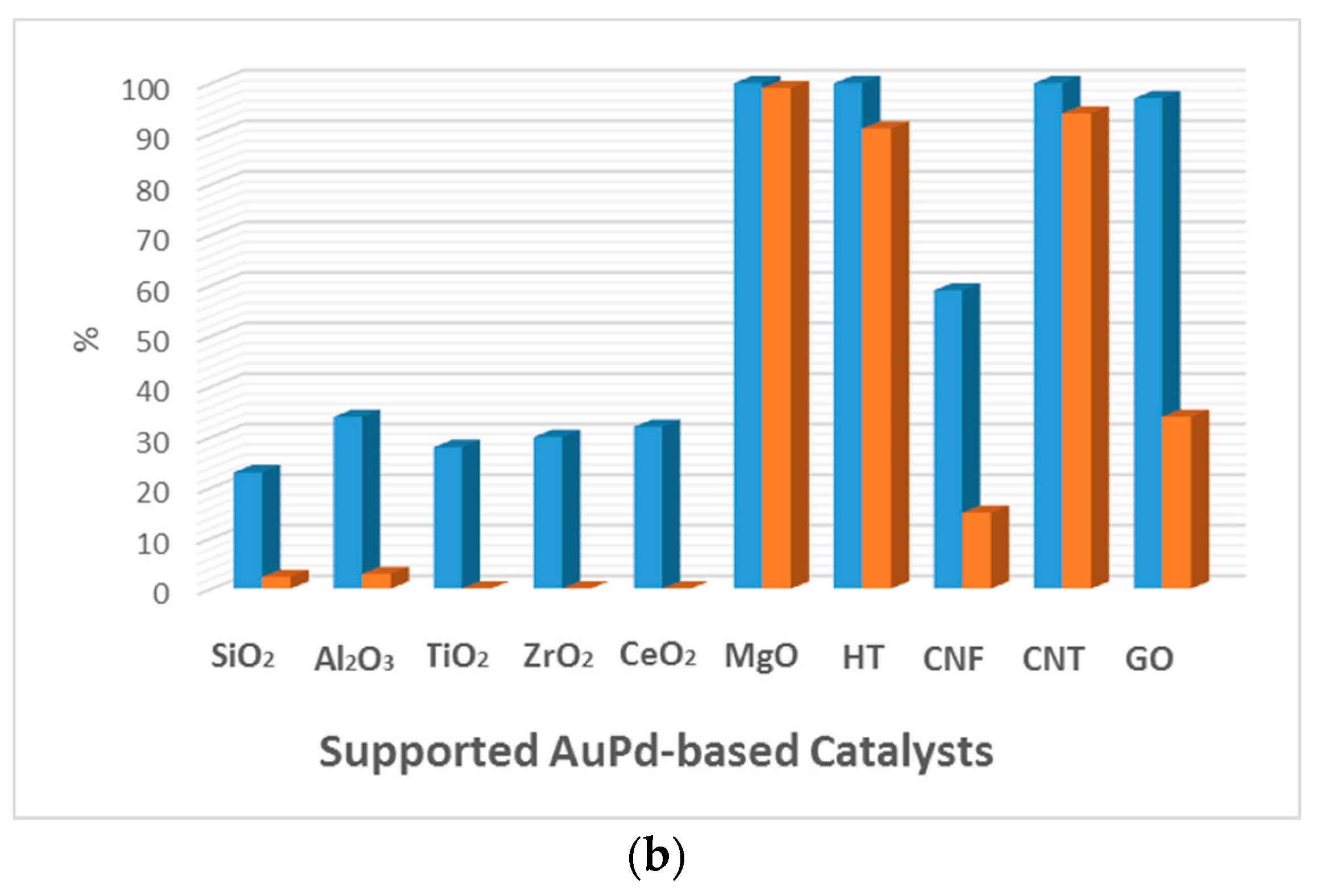
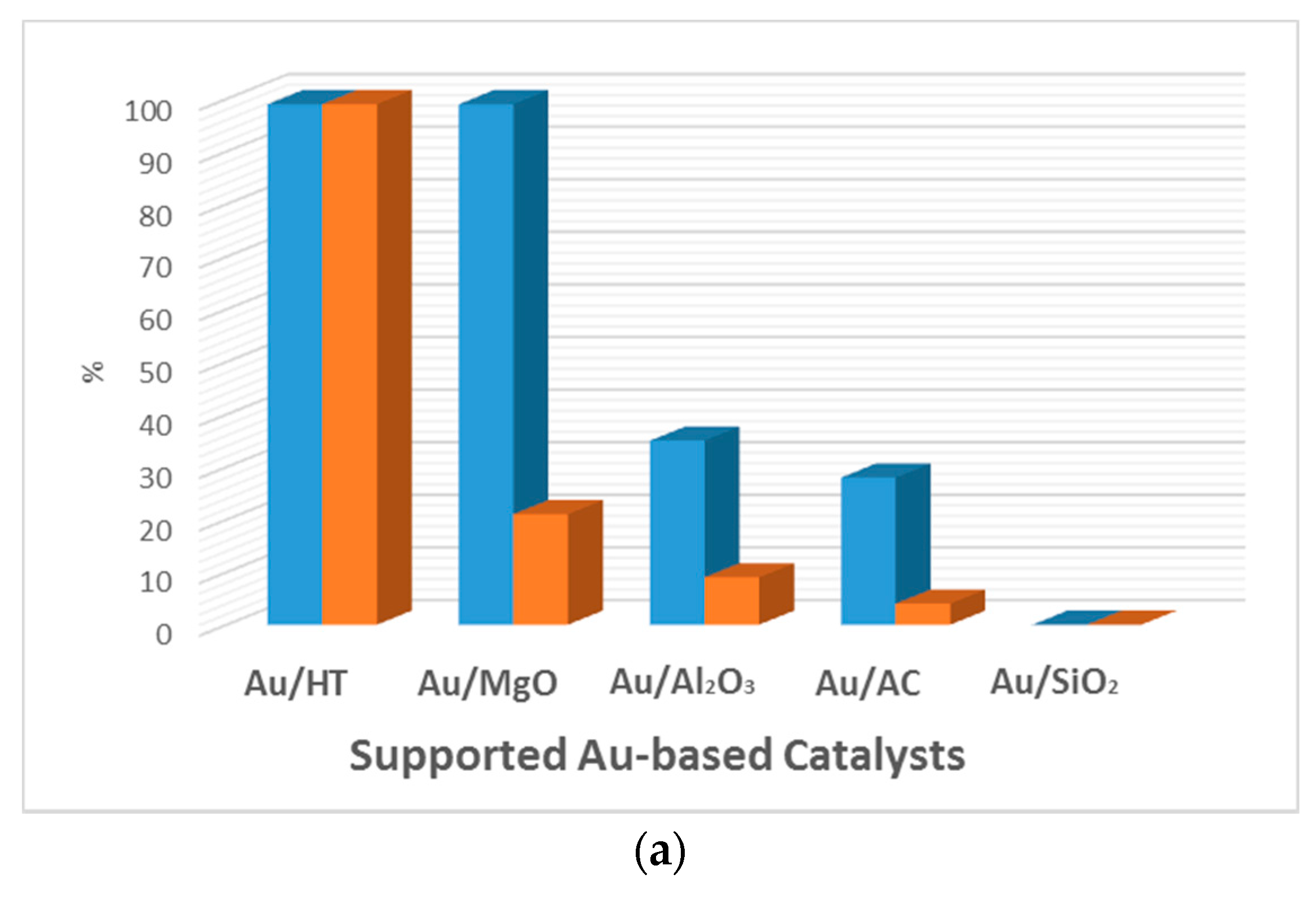
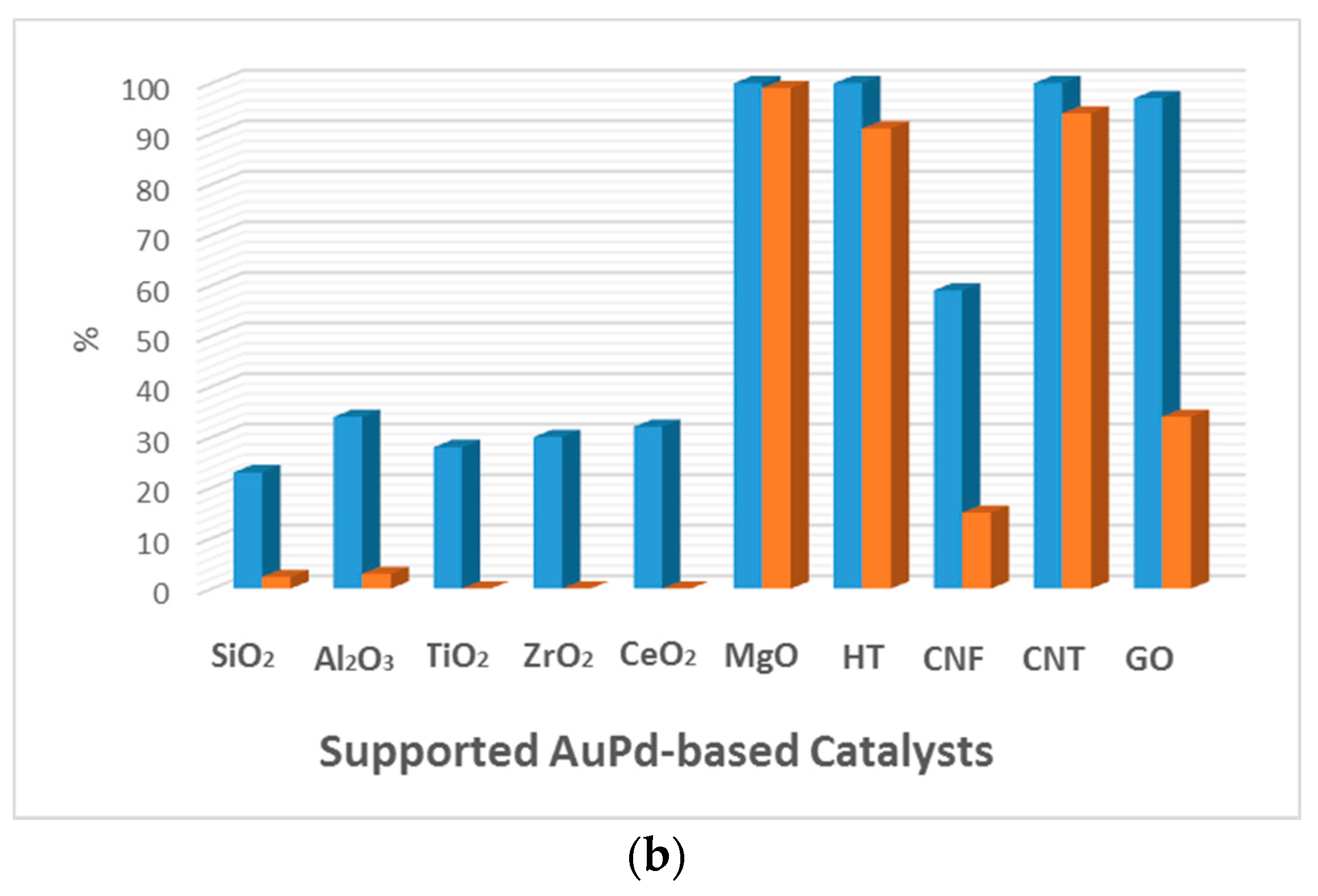
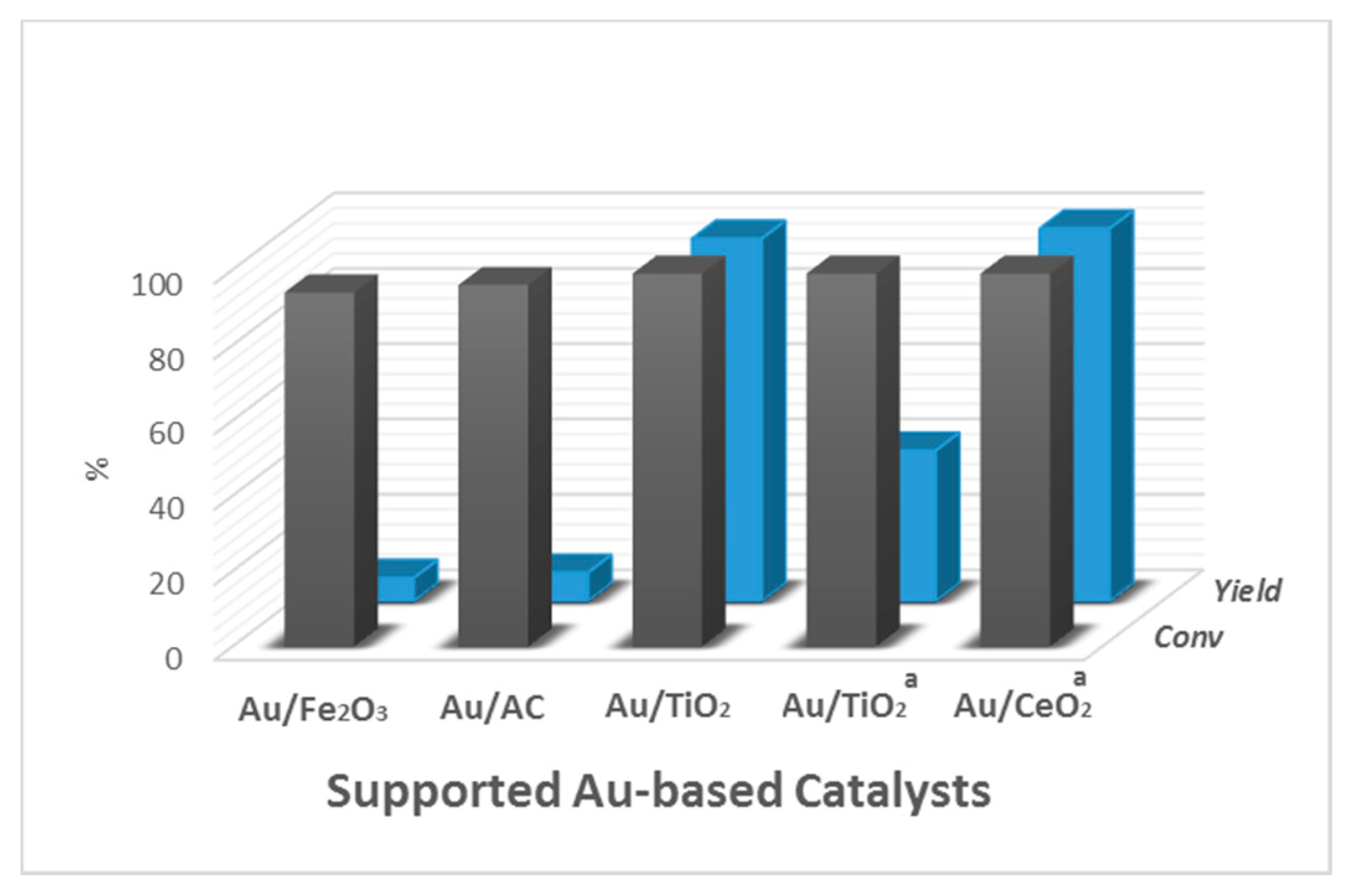
5.2. Effect of the Support
5.3. Stability
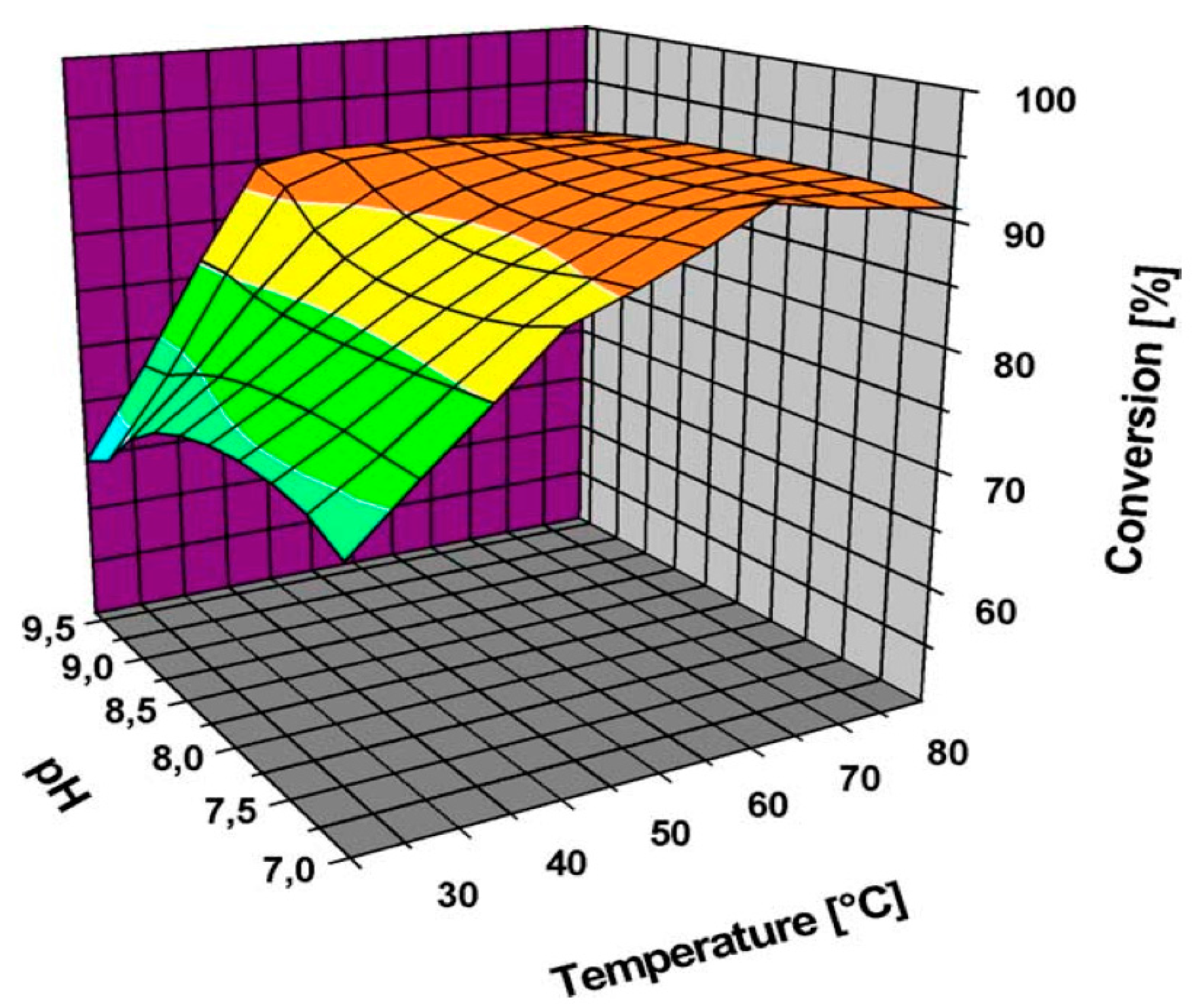
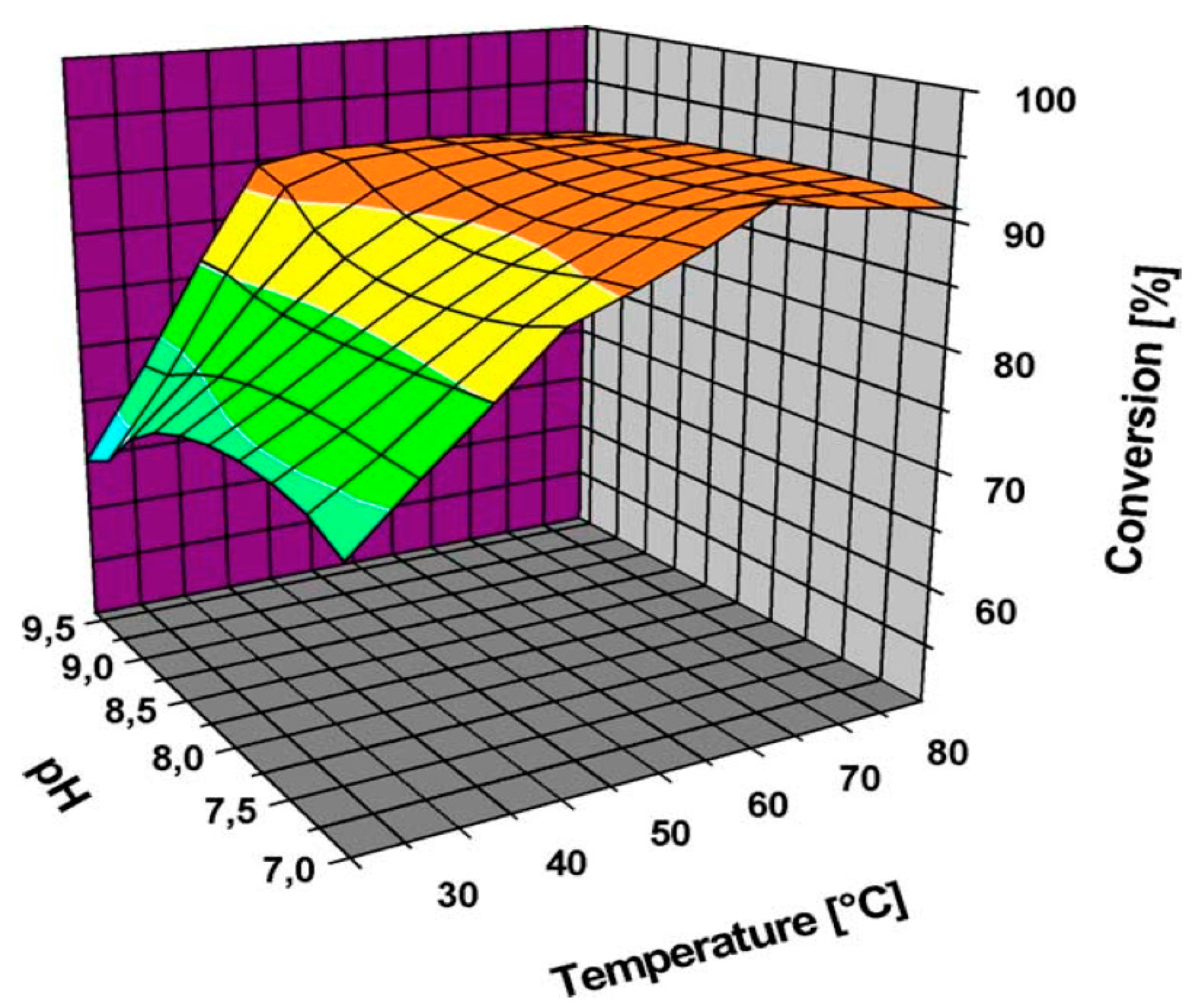
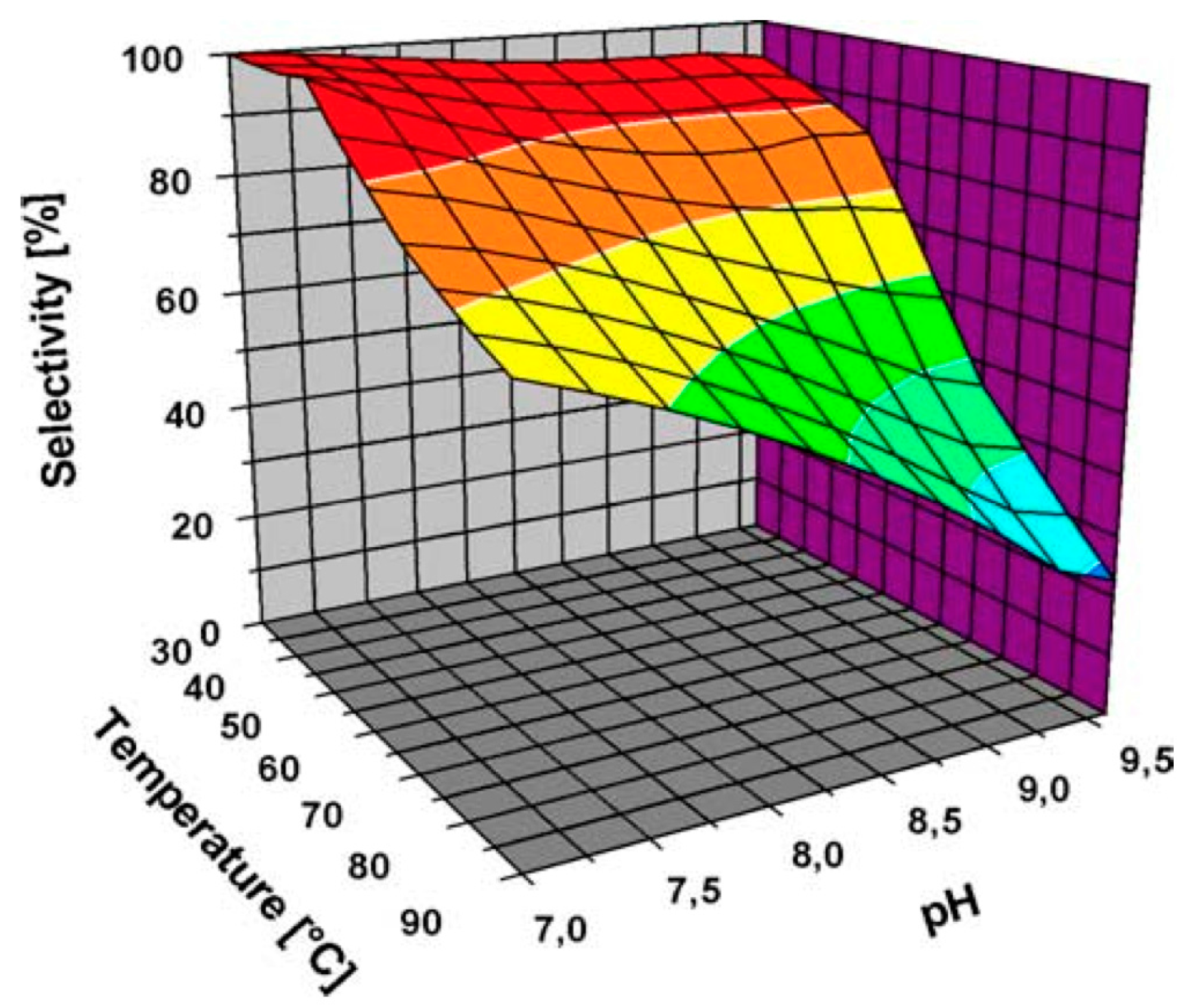
5.4. Effect of the Reaction Temperature
6. Conclusions
Author Contributions
Conflicts of Interest
References
- Chheda, J.N.; Huber, G.W.; Dumesic, J.A. Liquid-Phase Catalytic Processing of Biomass-Derived Oxygenated Hydrocarbons to Fuels and Chemicals. Angew. Chem. Int. Ed. 2007, 46, 7164–7183. [Google Scholar] [CrossRef] [PubMed]
- Corma, A.; Iborra, S.; Velty, A. Chemical Routes for the Transformation of Biomass into Chemicals. Chem. Rev. 2007, 107, 2411–2502. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.E.; Ide, M.S.; Davis, R.J. Selective oxidation of alcohols and aldehydes over supported metal nanoparticles. Green Chem. 2013, 15, 17–45. [Google Scholar] [CrossRef]
- Dumeignil, F.; Capron, M.; Katryniok, B.; Wojcieszak, R.; Löfberg, A.; Girardon, J.-S.; Desset, S.; Araque-Marin, M.; Jalowiecki-Duhamel, L.; Paul, S. Biomass-derived Platform Molecules Upgrading through Catalytic Processes: Yielding Chemicals and Fuels. J. Jpn. Pet. Inst. 2015, 58, 257–273. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, J.; Huang, J.; Zhang, C.; Hong, F.; Zhou, Y.; Li, G.; Haruta, M. Efficient Aerobic Oxidation of Glucose to Gluconic Acid over Activated Carbon-Supported Gold Clusters. ChemSusChem 2017, 10, 1976–1980. [Google Scholar] [CrossRef] [PubMed]
- Miedziak, P.J.; Alshammari, H.; Kondrat, S.A.; Clarke, T.J.; Davies, T.E.; Morad, M.; Morgan, D.J.; Willock, D.J.; Knight, D.W.; Taylor, S.H.; et al. Base-free glucose oxidation using air with supported gold catalysts. Green Chem. 2014, 16, 3132–3141. [Google Scholar] [CrossRef]
- Eloutassi, N.; Louaste, B.; Boudine, L.; Remmal, A. Valorisation de la biomasse lignocellulosique pour la production de bioéthanol de deuxième génération. Rev. Energies Renouv. 2014, 17, 600–609. [Google Scholar]
- Donate, P.M. Green synthesis from biomass. Chem. Biol. Technol. Agric. 2014, 1, 4. [Google Scholar] [CrossRef]
- Dedsuksophon, W.; Faungnawakij, K.; Champreda, V.; Laosiripojana, N. Hydrolysis/dehydration/aldol-condensation/hydrogenation of lignocellulosic biomass and biomass-derived carbohydrates in the presence of Pd/WO3–ZrO2 in a single reactor. Bioresour. Technol. 2011, 102, 2040–2046. [Google Scholar] [CrossRef] [PubMed]
- Tathod, A.; Kane, T.; Sanil, E.S.; Dhepe, P.L. Solid base supported metal catalysts for the oxidation and hydrogenation of sugars. J. Mol. Catal. A Chem. 2014, 388–389, 90–99. [Google Scholar] [CrossRef]
- Guimarães, L.H.S. Carbohydrates from Biomass: Sources and Transformation by Microbial Enzymes. In Carbohydrates-Comprehensive Studies on Glycobiology and Glycotechnology; Intech: London, UK, 2012; Chapter 20; pp. 441–456. [Google Scholar]
- Li, X.; Jia, P.; Wang, T. Furfural: A Promising Platform Compound for Sustainable Production of C4 and C5 Chemicals. ACS Catal. 2016, 6, 7621–7640. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A.; Dumeignil, F. Biorefineries an Introduction; Walter de Gruyter GmbH & Co. KG: Berlin, Germany, 2015. [Google Scholar]
- Zhang, Z.; Deng, K. Recent Advances in the Catalytic Synthesis of 2,5-Furandicarboxylic Acid and Its Derivatives. ACS Catal. 2015, 5, 6529–6544. [Google Scholar] [CrossRef]
- Zhu, Y.; Shen, M.; Xia, Y.; Lu, M. Au/MnO2 nanostructured catalysts and their catalytic performance for the oxidation of 5-(hydroxymethyl)furfural. Catal. Commun. 2015, 64, 37–43. [Google Scholar] [CrossRef]
- Yang, B.; Dai, Z.; Ding, S.-Y.; Wyman, C.E. Enzymatic hydrolysis of cellulosic biomass. Biofuels 2011, 2, 421–449. [Google Scholar] [CrossRef]
- De Wit, D.; Maat, L.; Kieboom, A.P.G. Carbohydrates as industrial raw materials. Ind. Crops Prod. 1993, 2, 1–12. [Google Scholar] [CrossRef]
- Rauter, A.P.; Vogel, P.; Queneau, Y. Carbohydrates in Sustainable Development II; Springer: Brelin, Germany, 2010; p. 204. [Google Scholar]
- Aquino Neto, S.; Milton, R.D.; Crepaldi, L.B.; Hickey, D.P.; de Andrade, A.R.; Minteer, S.D. Co-immobilization of gold nanoparticles with glucose oxidase to improve bioelectrocatalytic glucose oxidation. J. Power Sources 2015, 285, 493–498. [Google Scholar] [CrossRef]
- Sharma, K.K.; Wang, Y.; Roman-Leshkov, Y. Gold supported on nanocrystalline metal oxides as highly effective catalysts for the oxidation of glucose under base-free conditions. In Abstracts of Papers of the American Chemical Society; American Chemical Society: Washington, DC, USA, 2012. [Google Scholar]
- Trombotto, S.; Violet-Courtens, E.; Cottier, L.; Queneau, Y. Oxidation of Two Major Disaccharides: Sucrose and Isomaltulose. Top. Catal. 2004, 27, 31–37. [Google Scholar] [CrossRef]
- Pasta, M.; Ruffo, R.; Falletta, E.; Mari, C.M.; Pina, C.D. Alkaline glucose oxidation on nanostructured gold electrodes. Gold Bull. 2010, 43, 57–64. [Google Scholar] [CrossRef]
- Biella, S.; Prati, L.; Rossi, M. Selective Oxidation of d-Glucose on Gold Catalyst. J. Catal. 2002, 206, 242–247. [Google Scholar] [CrossRef]
- Besson, M.; Gallezot, P.; Pinel, C. Conversion of Biomass into Chemicals over Metal Catalysts. Chem. Rev. 2014, 114, 1827–1870. [Google Scholar] [CrossRef] [PubMed]
- Wojcieszak, R.; Cuccovia, I.M.; Silva, M.A.; Rossi, L.M. Selective oxidation of glucose to glucuronic acid by cesium-promoted gold nanoparticle catalyst. J. Mol. Catal. A Chem. 2016, 422, 35–42. [Google Scholar] [CrossRef]
- Biella, S.; Castiglioni, G.L.; Fumagalli, C.; Prati, L.; Rossi, M. Application of gold catalysts to selective liquid phase oxidation. Catal. Today 2002, 72, 43–49. [Google Scholar] [CrossRef]
- Delidovich, I.V.; Taran, O.P.; Matvienko, L.G.; Simonov, A.N.; Simakova, I.L.; Bobrovskaya, A.N.; Parmon, V.N. Selective Oxidation of Glucose over Carbon-supported Pd and Pt Catalysts. Catal. Lett. 2010, 140, 14–21. [Google Scholar] [CrossRef]
- Solmi, S.; Morreale, C.; Ospitali, F.; Agnoli, S.; Cavani, F. Oxidation of d-Glucose to Glucaric Acid Using Au/C Catalysts. ChemCatChem 2017, 9, 2797–2806. [Google Scholar] [CrossRef]
- Novotný, O.; Cejpek, K.; Velíšek, J. Formation of Carboxylic Acids during Degradation of Monosaccharides. Czech. J. Food Sci. 2008, 26, 117–131. [Google Scholar]
- Moulik, S.P.; Basu, D.; Bhattacharya, P.K. Effects of various conditions on the alkaline degradation of d-fructose and d-glucose. Carbohydr. Res. 1978, 63, 165–172. [Google Scholar] [CrossRef]
- Vuyyuru, K.R.; Strasser, P. Oxidation of biomass derived 5-hydroxymethylfurfural using heterogeneous and electrochemical catalysis. Catal. Today 2012, 195, 144–154. [Google Scholar] [CrossRef]
- Piccolo, A.; Conte, P.; Cozzolino, A. Effects of mineral and monocarboxylic acids on the molecular association of dissolved humic substances. Eur. J. Soil Sci. 1999, 50, 687–694. [Google Scholar] [CrossRef]
- Pasini, T.; Piccinini, M.; Blosi, M.; Bonelli, R.; Albonetti, S.; Dimitratos, N.; Lopez-Sanchez, J.A.; Sankar, M.; He, Q.; Kiely, C.J.; et al. Selective oxidation of 5-hydroxymethyl-2-furfural using supported gold-copper nanoparticles. Green Chem. 2011, 13, 2091–2099. [Google Scholar] [CrossRef]
- Comotti, M.; Della Pina, C.; Matarrese, R.; Rossi, M. The Catalytic Activity of “Naked” Gold Particles. Angew. Chem. Int. Ed. 2004, 43, 5812–5815. [Google Scholar] [CrossRef] [PubMed]
- Baatz, C.; Thielecke, N.; Prüße, U. Influence of the preparation conditions on the properties of gold catalysts for the oxidation of glucose. Appl. Catal. B Environ. 2007, 70, 653–660. [Google Scholar] [CrossRef]
- Önal, Y.; Schimpf, S.; Claus, P. Structure sensitivity and kinetics of d-glucose oxidation to d-gluconic acid over carbon-supported gold catalysts. J. Catal. 2004, 223, 122–133. [Google Scholar] [CrossRef]
- Hashmi, A.S.K. Gold-Catalyzed Organic Reactions. Chem. Rev. 2007, 107, 3180–3211. [Google Scholar] [CrossRef] [PubMed]
- Comotti, M.; Pina, C.D.; Matarrese, R.; Rossi, M.; Siani, A. Oxidation of alcohols and sugars using Au/C catalysts: Part 2. Sugars. Appl. Catal. A Gen. 2005, 291, 204–209. [Google Scholar] [CrossRef]
- Okatsu, H.; Kinoshita, N.; Akita, T.; Ishida, T.; Haruta, M. Deposition of gold nanoparticles on carbons for aerobic glucose oxidation. Appl. Catal. A Gen. 2009, 369, 8–14. [Google Scholar] [CrossRef]
- Ishida, T.; Kinoshita, N.; Okatsu, H.; Akita, T.; Takei, T.; Haruta, M. Influence of the Support and the Size of Gold Clusters on Catalytic Activity for Glucose Oxidation. Angew. Chem. Int. Ed. 2008, 120, 9405–9408. [Google Scholar] [CrossRef]
- Cao, Y.; Liu, X.; Iqbal, S.; Miedziak, P.J.; Edwards, J.K.; Armstrong, R.D.; Morgan, D.J.; Wang, J.; Hutchings, G.J. Base-free oxidation of glucose to gluconic acid using supported gold catalysts. Catal. Sci. Technol. 2016, 6, 107–117. [Google Scholar] [CrossRef]
- Villa, A.; Wang, D.; Veith, G.M.; Vindigni, F.; Prati, L. Sol immobilization technique: A delicate balance between activity, selectivity and stability of gold catalysts. Catal. Sci. Technol. 2013, 3, 3036–3041. [Google Scholar] [CrossRef]
- Qi, P.; Chen, S.; Chen, J.; Zheng, J.; Zheng, X.; Yuan, Y. Catalysis and Reactivation of Ordered Mesoporous Carbon-Supported Gold Nanoparticles for the Base-Free Oxidation of Glucose to Gluconic Acid. ACS Catal. 2015, 5, 2659–2670. [Google Scholar] [CrossRef]
- Wang, Y.; Van de Vyver, S.; Sharma, K.K.; Roman-Leshkov, Y. Insights into the stability of gold nanoparticles supported on metal oxides for the base-free oxidation of glucose to gluconic acid. Green Chem. 2014, 16, 719–726. [Google Scholar] [CrossRef]
- Rautiainen, S.; Lehtinen, P.; Vehkamäki, M.; Niemelä, K.; Kemell, M.; Heikkilä, M.; Repo, T. Microwave-assisted base-free oxidation of glucose on gold nanoparticle catalysts. Catal. Commun. 2016, 74, 115–118. [Google Scholar] [CrossRef]
- Megías-Sayago, C.; Ivanova, S.; López-Cartes, C.; Centeno, M.A.; Odriozola, J.A. Gold catalysts screening in base-free aerobic oxidation of glucose to gluconic acid. Catal. Today 2017, 279, 148–154. [Google Scholar] [CrossRef]
- Comotti, M.; Pina, C.D.; Rossi, M. Mono- and bimetallic catalysts for glucose oxidation. J. Mol. Catal. A Chem. 2006, 251, 89–92. [Google Scholar] [CrossRef]
- Zhang, H.; Toshima, N. Preparation of novel Au/Pt/Ag trimetallic nanoparticles and their high catalytic activity for aerobic glucose oxidation. Appl. Catal. A Gen. 2011, 400, 9–13. [Google Scholar] [CrossRef]
- Cao, Y.; Iqbal, S.; Miedziak, P.J.; Jones, D.R.; Morgan, D.J.; Liu, X.; Wang, J.; Hutchings, G.J. An investigation into bimetallic catalysts for base free oxidation of cellobiose and glucose. J. Chem. Technol. Biotechnol. 2017, 92, 2246–2253. [Google Scholar] [CrossRef]
- Prüße, U.; Heidinger, S.; Baatz, C. Catalytic conversion of renewables: Kinetic and mechanistic aspects of the gold-catalyzed liquid-phase glucose oxidation. Agric. For. Res. 2011, 16, 261–272. [Google Scholar]
- Comotti, M.; DellaPina, C.; Falletta, E.; Rossi, M. Aerobic Oxidation of Glucose with Gold Catalyst: Hydrogen Peroxide as Intermediate and Reagent. Adv. Synth. Catal. 2006, 348, 313–316. [Google Scholar] [CrossRef]
- Beltrame, P.; Comotti, M.; Della Pina, C.; Rossi, M. Aerobic oxidation of glucose: II. Catalysis by colloidal gold. Appl. Catal. A Gen. 2006, 297, 1–7. [Google Scholar] [CrossRef]
- Larew, L.A.; Johnson, D.C. Concentration dependence of the mechanism of glucose oxidation at gold electrodes in alkaline media. J. Electroanal. Chem. Interfacial Electrochem. 1989, 262, 167–182. [Google Scholar] [CrossRef]
- Dirkn, J.M.H.; van der Baan, H.S.; van den Broen, J.M.A.J.J. The preparation of d-glucaric acid by the oxidation of d-gluconic acid catalysed by platinum on carbon. Carbohydr. Res. 1977, 59, 63–72. [Google Scholar] [CrossRef]
- Dirkx, J.M.H.; van der Baan, H.S. The oxidation of gluconic acid with platinum on carbon as catalyst. J. Catal. 1981, 67, 14–20. [Google Scholar] [CrossRef]
- Casanova, O.; Iborra, S.; Corma, A. Biomass into Chemicals: Aerobic Oxidation of 5-Hydroxymethyl-2-furfural into 2,5-Furandicarboxylic Acid with Gold Nanoparticle Catalysts. ChemSusChem 2009, 2, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Albonetti, S.; Lolli, A.; Morandi, V.; Migliori, A.; Lucarelli, C.; Cavani, F. Conversion of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over Au-based catalysts: Optimization of active phase and metal–support interaction. Appl. Catal. B Environ. 2015, 163, 520–530. [Google Scholar] [CrossRef]
- Miao, Z.; Zhang, Y.; Pan, X.; Wu, T.; Zhang, B.; Li, J.; Yi, T.; Zhang, Z.; Yang, X. Superior catalytic performance of Ce1-xBixO2-[small delta] solid solution and Au/Ce1-xBixO2-[small delta] for 5-hydroxymethylfurfural conversion in alkaline aqueous solution. Catal. Sci. Technol. 2015, 5, 1314–1322. [Google Scholar] [CrossRef]
- Cai, J.; Ma, H.; Zhang, J.; Song, Q.; Du, Z.; Huang, Y.; Xu, J. Gold Nanoclusters Confined in a Supercage of Y Zeolite for Aerobic Oxidation of HMF under Mild Conditions. Chem. Eur. J. 2013, 19, 14215–14223. [Google Scholar] [CrossRef] [PubMed]
- Gorbanev, Y.Y.; Klitgaard, S.K.; Woodley, J.M.; Christensen, C.H.; Riisager, A. Gold-Catalyzed Aerobic Oxidation of 5-Hydroxymethylfurfural in Water at Ambient Temperature. ChemSusChem 2009, 2, 672–675. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.E.; Houk, L.R.; Tamargo, E.C.; Datye, A.K.; Davis, R.J. Oxidation of 5-hydroxymethylfurfural over supported Pt, Pd and Au catalysts. Catal. Today 2011, 160, 55–60. [Google Scholar] [CrossRef]
- Ardemani, L.; Cibin, G.; Dent, A.J.; Isaacs, M.A.; Kyriakou, G.; Lee, A.F.; Parlett, C.M.A.; Parry, S.A.; Wilson, K. Solid base catalysed 5-HMF oxidation to 2,5-FDCA over Au/hydrotalcites: Fact or fiction? Chem. Sci. 2015, 6, 4940–4945. [Google Scholar] [CrossRef]
- Albonetti, S.; Pasini, T.; Lolli, A.; Blosi, M.; Piccinini, M.; Dimitratos, N.; Lopez-Sanchez, J.A.; Morgan, D.J.; Carley, A.F.; Hutchings, G.J.; et al. Selective oxidation of 5-hydroxymethyl-2-furfural over TiO2-supported gold–copper catalysts prepared from preformed nanoparticles: Effect of Au/Cu ratio. Catal. Today 2012, 195, 120–126. [Google Scholar] [CrossRef]
- Villa, A.; Schiavoni, M.; Campisi, S.; Veith, G.M.; Prati, L. Pd-modified Au on Carbon as an Effective and Durable Catalyst for the Direct Oxidation of HMF to 2,5-Furandicarboxylic Acid. ChemSusChem 2013, 6, 609–612. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Gao, T.; Fang, W.; Cao, Q. Base-free aerobic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid in water by hydrotalcite-activated carbon composite supported gold catalyst. Mol. Catal. 2017, 439, 171–179. [Google Scholar] [CrossRef]
- Gao, Z.; Xie, R.; Fan, G.; Yang, L.; Li, F. Highly Efficient and Stable Bimetallic AuPd over La-Doped Ca–Mg–Al Layered Double Hydroxide for Base-Free Aerobic Oxidation of 5-Hydroxymethylfurfural in Water. ACS Sustain. Chem. Eng. 2017, 5, 5852–5861. [Google Scholar] [CrossRef]
- Gui, Z.; Cao, W.; Saravanamurugan, S.; Riisager, A.; Chen, L.; Qi, Z. Efficient Aerobic Oxidation of 5-Hydroxymethylfurfural in Aqueous Media with Au–Pd Supported on Zinc Hydroxycarbonate. ChemCatChem 2016, 8, 3636–3643. [Google Scholar] [CrossRef]
- Gupta, N.K.; Nishimura, S.; Takagaki, A.; Ebitani, K. Hydrotalcite-supported gold-nanoparticle-catalyzed highly efficient base-free aqueous oxidation of 5-hydroxymethylfurfural into 2,5-furandicarboxylic acid under atmospheric oxygen pressure. Green Chem. 2011, 13, 824–827. [Google Scholar] [CrossRef]
- Zope, B.N.; Davis, S.E.; Davis, R.J. Influence of Reaction Conditions on Diacid Formation during Au-Catalyzed Oxidation of Glycerol and Hydroxymethylfurfural. Top. Catal. 2012, 55, 24–32. [Google Scholar] [CrossRef]
- Wan, X.; Zhou, C.; Chen, J.; Deng, W.; Zhang, Q.; Yang, Y.; Wang, Y. Base-Free Aerobic Oxidation of 5-Hydroxymethyl-furfural to 2,5-Furandicarboxylic Acid in Water Catalyzed by Functionalized Carbon Nanotube-Supported Au–Pd Alloy Nanoparticles. ACS Catal. 2014, 4, 2175–2185. [Google Scholar] [CrossRef]
- Nielsen, I.S.; Taarning, E.; Egeblad, K.; Madsen, R.; Christensen, C.H. Direct aerobic oxidation of primary alcohols to methyl esters catalyzed by a heterogeneous gold catalyst. Catal. Lett. 2007, 116, 35–40. [Google Scholar] [CrossRef]
- Gallas-Hulin, A.; Kotni, R.K.; Nielsen, M.; Kegnæs, S. Catalytic Oxidation of Allylic Alcohols to Methyl Esters. Top. Catal. 2017. [Google Scholar] [CrossRef]
- Whiting, G.T.; Kondrat, S.A.; Hammond, C.; Dimitratos, N.; He, Q.; Morgan, D.J.; Dummer, N.F.; Bartley, J.K.; Kiely, C.J.; Taylor, S.H.; et al. Methyl Formate Formation from Methanol Oxidation Using Supported Gold–Palladium Nanoparticles. ACS Catal. 2015, 5, 637–644. [Google Scholar] [CrossRef]
- Oliveira, R.L.; Kiyohara, P.K.; Rossi, L.M. Clean preparation of methyl esters in one-step oxidative esterification of primary alcohols catalyzed by supported gold nanoparticles. Green Chem. 2009, 11, 1366–1370. [Google Scholar] [CrossRef]
- Taarning, E.; Nielsen, I.S.; Egeblad, K.; Madsen, R.; Christensen, C.H. Chemicals from Renewables: Aerobic Oxidation of Furfural and Hydroxymethylfurfural over Gold Catalysts. ChemSusChem 2008, 1, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Guzman, J.; Carrettin, S.; Fierro-Gonzalez, J.C.; Hao, Y.; Gates, B.C.; Corma, A. CO Oxidation Catalyzed by Supported Gold: Cooperation between Gold and Nanocrystalline Rare-Earth Supports Forms Reactive Surface Superoxide and Peroxide Species. Angew. Chem. Int. Ed. 2005, 44, 4778–4781. [Google Scholar] [CrossRef] [PubMed]
- Lilga, M.A.; Hallen, R.T.; Gray, M. Production of Oxidized Derivatives of 5-Hydroxymethylfurfural (HMF). Top. Catal. 2010, 53, 1264–1269. [Google Scholar] [CrossRef]
- Casanova, O.; Iborra, S.; Corma, A. Biomass into chemicals: One pot-base free oxidative esterification of 5-hydroxymethyl-2-furfural into 2,5-dimethylfuroate with gold on nanoparticulated ceria. J. Catal. 2009, 265, 109–116. [Google Scholar] [CrossRef]
- Zope, B.N.; Hibbitts, D.D.; Neurock, M.; Davis, R.J. Reactivity of the Gold/Water Interface during Selective Oxidation Catalysis. Science 2010, 330, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Wojcieszak, R.; Santarelli, F.; Paul, S.; Dumeignil, F.; Cavani, F.; Gonçalves, R.V. Recent developments in maleic acid synthesis from bio-based chemicals. Sustain. Chem. Process. 2015, 3, 9. [Google Scholar] [CrossRef]
- Menegazzo, F.; Signoretto, M.; Pinna, F.; Manzoli, M.; Aina, V.; Cerrato, G.; Boccuzzi, F. Oxidative esterification of renewable furfural on gold-based catalysts: Which is the best support? J. Catal. 2014, 309, 241–247. [Google Scholar] [CrossRef]
- Ampelli, C.; Barbera, K.; Centi, G.; Genovese, C.; Papanikolaou, G.; Perathoner, S.; Schouten, K.J.; van der Waal, J.K. On the nature of the active sites in the selective oxidative esterification of furfural on Au/ZrO2 catalysts. Catal. Today 2016, 278, 56–65. [Google Scholar] [CrossRef]
- Signoretto, M.; Menegazzo, F.; Contessotto, L.; Pinna, F.; Manzoli, M.; Boccuzzi, F. Au/ZrO2: An efficient and reusable catalyst for the oxidative esterification of renewable furfural. Appl. Catal. B Environ. 2013, 129, 287–293. [Google Scholar] [CrossRef]
- Menegazzo, F.; Fantinel, T.; Signoretto, M.; Pinna, F.; Manzoli, M. On the process for furfural and HMF oxidative esterification over Au/ZrO2. J. Catal. 2014, 319, 61–70. [Google Scholar] [CrossRef]
- Pinna, F.; Olivo, A.; Trevisan, V.; Menegazzo, F.; Signoretto, M.; Manzoli, M.; Boccuzzi, F. The effects of gold nanosize for the exploitation of furfural by selective oxidation. Catal. Today 2013, 203, 196–201. [Google Scholar] [CrossRef]
- Manzoli, M.; Menegazzo, F.; Signoretto, M.; Cruciani, G.; Pinna, F. Effects of synthetic parameters on the catalytic performance of Au/CeO2 for furfural oxidative esterification. J. Catal. 2015, 330, 465–473. [Google Scholar] [CrossRef]
- Daniel, M.-C.; Astruc, D. Gold Nanoparticles: Assembly, Supramolecular Chemistry, Quantum-Size-Related Properties, and Applications toward Biology, Catalysis, and Nanotechnology. Chem. Rev. 2004, 104, 293–346. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Jiang, X.; Dong, S.; Wang, E. One-Step Synthesis and Size Control of Dendrimer-Protected Gold Nanoparticles: A Heat-Treatment-Based Strategy. Macromol. Rapid Commun. 2003, 24, 1024–1028. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, T.; Zhang, L.; Wang, Q.; Zhang, R.; Ding, B. Morphological transition of gold nanostructures induced by continuous ultraviolet irradiation. Nanotechnology 2006, 17, 5639–5643. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.; Mahima, S.; Kakade, B.A.; Pasricha, R.; Mandale, A.B.; Vijayamohanan, K. Solvent-Assisted One-Pot Synthesis and Self-Assembly of 4-Aminothiophenol-Capped Gold Nanoparticles. J. Phys. Chem. B 2004, 108, 13280–13286. [Google Scholar] [CrossRef]
- Roy, P.; Dhara, K.; Manassero, M.; Banerjee, P. A new organic compound for the synthesis of gold nanoparticles. Polyhedron 2008, 27, 3085–3090. [Google Scholar] [CrossRef]
- Kim, S.M.; Kim, G.S.; Lee, S.Y. Effects of PVP and KCl concentrations on the synthesis of gold nanoparticles using a solution plasma processing. Mater. Lett. 2008, 62, 4354–4356. [Google Scholar] [CrossRef]
- Luo, W.; Zhu, C.; Su, S.; Li, D.; He, Y.; Huang, Q.; Fan, C. Self-Catalyzed, Self-Limiting Growth of Glucose Oxidase-Mimicking Gold Nanoparticles. ACS Nano 2010, 4, 7451–7458. [Google Scholar] [CrossRef] [PubMed]
- Megías-Sayago, C.; Santos, J.L.; Ammari, F.; Chenouf, M.; Ivanova, S.; Centeno, M.A.; Odriozola, J.A. Influence of gold particle size in Au/C catalysts for base-free oxidation of glucose. Catal. Today. [CrossRef]
- Ishida, T.; Haruta, M. Gold Catalysts: Towards Sustainable Chemistry. Angew. Chem. Int. Ed. 2007, 46, 7154–7156. [Google Scholar] [CrossRef] [PubMed]
- Morawa Eblagon, K.; Pereira, M.F.R.; Figueiredo, J.L. One-pot oxidation of cellobiose to gluconic acid. Unprecedented high selectivity on bifunctional gold catalysts over mesoporous carbon by integrated texture and surface chemistry optimization. Appl. Catal. B Environ. 2016, 184, 381–396. [Google Scholar] [CrossRef]
- An, D.; Ye, A.; Deng, W.; Zhang, Q.; Wang, Y. Selective Conversion of Cellobiose and Cellulose into Gluconic Acid in Water in the Presence of Oxygen, Catalyzed by Polyoxometalate-Supported Gold Nanoparticles. Chem. Eur. J. 2012, 18, 2938–2947. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Particle Size (nm) | Preparation Method | Glucose/Au | T a (°C) | Time (h) | Oxidant | Conversion (%) | Gluconic Acid Selectivity (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1% Au/C | 4.4 | SIM e | 1000 | 100 | 6 | 0.3 MPa O2 | 100 | 100 | [23] |
| 2 | 0.5% Au/nCeO2 f | 1.8 | DP e | 140 | 65 | 2 | 0.23 MPa O2 | 74 | 95 | [44] |
| 3 | 1.4% Au/nCeO2 | 2.5 | DP | 140 | 65 | 2 | 0.23 MPa O2 | 75 | 99 | |
| 4 | 1.6% Au/nZrO2 | 1.7 | DP | 140 | 65 | 2 | 0.23 MPa O2 | 89 | 99 | |
| 5 | 1.3% Au/nTiO2 | 3.0 | DP | 140 | 65 | 2 | 0.23 MPa O2 | 91 | 99 | |
| 6 | 0.6% Au/μCeO2 f | 2.0 | DP | 140 | 65 | 2 | 0.23 MPa O2 | 76 | 96 | |
| 7 | 1.1% Au/μCeO2 | 2.9 | DP | 140 | 65 | 2 | 0.23 MPa O2 | 77 | 98 | |
| 8 | 0.8% Au/μZrO2 | 2.1 | DP | 140 | 65 | 2 | 0.23 MPa O2 | 65 | 98 | |
| 9 | 1.2% Au/μTiO2 | 2.0 | DP | 140 | 65 | 2 | 0.23 MPa O2 | 89 | 98 | |
| 10 | 0.5% Au/MgO | SIM | 1600 | 60 | 24 | air b | 57 | 100 | [6] | |
| 11 | 0.5% Au–Pd/MgO | 6.5 | SIM | 1600 | 60 | 24 | air b | 62 | 100 | |
| 12 | 1% Au/CMK-3 c | 3.0 | See Ref. | 1000 | 110 | 2 | 0.3 MPa O2 | 92 | 88 | [43] |
| 13 | 1% Au/SBA-15 | 3.6 | See Ref. | 100 | 110 | 2 | 0.3 MPa O2 | 67 | 92 | |
| 14 | 1% Au/CNTs | 2.9 | See Ref. | 100 | 110 | 2 | 0.3 MPa O2 | 62 | 83 | |
| 15 | 1% Au/graphene | 3.2 | See Ref. | 100 | 110 | 2 | 0.3 MPa O2 | 56 | 74 | |
| 16 | 1% Au/graphite | 3.7 | See Ref. | 100 | 110 | 2 | 0.3 MPa O2 | 55 | 84 | |
| 17 | 1% Au/AC g | 3.5 | See Ref. | 100 | 110 | 2 | 0.3 MPa O2 | 21 | 91 | |
| 18 | 1% Au/ZrO2 | 2.5 | See Ref. | 100 | 110 | 2 | 0.3 MPa O2 | 13 | 92 | |
| 19 d | 2.3% Au/MgAl2O4 | 3.8 | DP | 870 | 120 | 1/6 | 2.2 eq H2O2 | 54 | 93 | [45] |
| 20 d | 1.8% Au/Al2O3 | 2.4 | DP | 1110 | 120 | 1/6 | 2.2 eq H2O2 | 83 | 87 | |
| 21 | 2% Au/Al h | 5.1 ± 1.5 | DAE e | 100 | 120 | 18 | 0.1MPa O2 | 76 | 95 | [46] |
| 22 | 2% Au/Ce/Al h | 3.4 ± 0.8 | DAE | 100 | 120 | 18 | 0.1 MPa O2 | 78 | 96 | |
| 23 | 2% Au/Ce h | 3.9 ± 1.5 | DAE | 100 | 120 | 18 | 0.1 MPa O2 | 73 | 90 | |
| 24 | 2% Au/Ce25/Zr h | 3.4 ± 0.8 | DAE | 100 | 120 | 18 | 0.1 MPa O2 | 75 | 86 | |
| 25 | 2% Au/Ce50/Zr h | 3.4 ± 0.8 | DAE | 100 | 120 | 18 | 0.1 MPa O2 | 77 | 87 | |
| 26 | 1% Au/TiO2 | 2.1 | SIM | 438 | 160 | 1 | 0.3 MPa O2 | 88 | 95 | [41] |
| Catalyst | PostSynthesis Treatment | Glucose Conversion (%) | Yield (%) |
|---|---|---|---|
| 0.5% Au–0.5% Pd/TiO2 | no | 65 | 60 |
| 0.5% Au–0.5% Pd/TiO2 | air | 70 | 63 |
| 0.5% Au–0.5% Pd/TiO2 | reflux a | 48 | 40 |
| 0.5% Au–0.5% Pt/TiO2 | no | 80 | 70 |
| 0.5% Au–0.5% Pt/TiO2 | air | 68 | 62 |
| 0.5% Au–0.5% Pt/TiO2 | reflux | 71 | 65 |
| Entry | Catalyst | Particle Size (nm) | T Calc. (°C) | T a (°C) | Time (h) | Oxidant (MPa) | Conversion (%) | MF Select. (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1.2% Au/TiO2 | 4.2 | 300 | 120 | 1.5 | 0.6 | 20 | 92 | [81] |
| 2 | 2.2% Au/CeO2 | 2.0 | 650 | 120 | 1.5 | 0.6 | 66 | 92 | |
| 3 | 1.0% Au/ZrO2 | 2.3 | 650 | 120 | 1.5 | 0.6 | 82 | 66 | |
| 4 | 0.4% Au/ZrO2 | 2–3 | 200 | 120 | 6 | 0.6 | 63 | 10 | [82] |
| 5 | 0.4% Au/ZrO2 | 2–3 | 400 | 120 | 6 | 0.6 | 99 | 95 | |
| 6 | 1.0% Au/TiO2 § | 2–3 | 250 | 120 | 6 | 0.6 | 73 | 31 | |
| 7 | 1.5% Au/ZrO2 | 1.8 | 150 | 120 | 3 | 0.6 | 99 | 94 | [83] |
| 8 | 1.5% Au/ZrO2 | 1.9 | 300 | 120 | 3 | 0.6 | 98 | 94 | |
| 9 | 1.5% Au/ZrO2 | 2.7 | 500 | 120 | 3 | 0.6 | 100 | 98 | |
| 10 | 1.5% Au/ZrO2 | 4.9 | 600 | 120 | 3 | 0.6 | 57 | 76 | |
| 11 | 1.2% Au/ZrO2 | - | 400 | 130 | 1.5 | 0.3 | 90 | 99 | [84] |
| 12 | 1.5% Au/TiO2 WGC | - | - | 130 | 1.5 | 0.3 | 63 | 62 | |
| 13 | 1.5% Au/ZrO2 | 1.7 | 180 | 120 | 1.5 | 0.6 | 100 | 100 | [85] |
| 14 | 1.5% Au/ZrO2 | 5.0 | 600 | 120 | 3.0 | 0.6 | 40 | 70 | |
| 15 | 1.5% Au/TiO2 WGC | 3.8 | 400 | 120 | 3.0 | 0.6 | 90 | 65 | |
| 16 | 2.1% Au/CeO2 (90 °C) | 3.1 | 300 | 120 | 1.5 | 0.6 | 29 | 100 | [86] |
| 17 | 1.3% Au/CeO2 (110 °C) | 7.7 | 300 | 120 | 1.5 | 0.6 | 29 | 100 | |
| 18 | 1.4% Au/CeO2 (300 °C) | 7.7 | 300 | 120 | 1.5 | 0.6 | 54 | 100 | |
| 19 | 2.1% Au/CeO2 (90 °C) | 3.2 | 500 | 120 | 1.5 | 0.6 | 6 | 100 | |
| 20 | 1.3% Au/CeO2 (110 °C) | 7.9 | 500 | 120 | 1.5 | 0.6 | 28 | 100 | |
| 21 | 1.5% Au/CeO2 (300 °C) | 8.4 | 500 | 120 | 1.5 | 0.6 | 74 | 100 |
| Support | Al2O3 a | Al2O3 b | ZrO2 a | ZrO2 b | TiO2 b | CeO2 b | CeO2 a | NPC a | AC a |
|---|---|---|---|---|---|---|---|---|---|
| Au particles size/nm | 2.6 | 4.4 | 3.3 | 3.6 | 2.9 | 4 | 4.1 | 2.4 | 5.4 |
| TOF/molglucosemolsurfaceAu−1s−1 | 41 | 7 | 45 | 28.5 | 24 | 19 | 22 | 3 | 10 |
| Catalyst | First Run | Second Run | Third Run | Fourth Run | Ref. |
|---|---|---|---|---|---|
| AuPd/MgO | 62.0 | 24.4 | 18.7 | - | [6] |
| Au/CMK-3 | 92.0 | - | - | 70.0 | [4] |
| Au/nCeO2 | 88.0 | 69.0 | 62.0 | 56.0 | [44] |
| Au/CeO2 | 88.0 | 53.0 | 40.1 | 31.1 | [44] |
| Temperature (°C) | Time (min) | Conversion (%) | Acid Yield (%) |
|---|---|---|---|
| 70 | 240 | 12 | 12 |
| 110 | 240 | 65 | 53 |
| 130 | 240 | 89 | 48 |
| 150 | 240 | 100 | 23 |
| Temperature (°C) | Time (min) | Conversion (%) | Acid Yield (%) |
|---|---|---|---|
| 130 | 60 | 32 | 7 |
| 140 | 60 | 52 | 21 |
| 150 | 60 | 52 | 28 |
| 160 | 60 | 40 | 15 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wojcieszak, R.; Ferraz, C.P.; Sha, J.; Houda, S.; Rossi, L.M.; Paul, S. Advances in Base-Free Oxidation of Bio-Based Compounds on Supported Gold Catalysts. Catalysts 2017, 7, 352. https://doi.org/10.3390/catal7110352
Wojcieszak R, Ferraz CP, Sha J, Houda S, Rossi LM, Paul S. Advances in Base-Free Oxidation of Bio-Based Compounds on Supported Gold Catalysts. Catalysts. 2017; 7(11):352. https://doi.org/10.3390/catal7110352
Chicago/Turabian StyleWojcieszak, Robert, Camila P. Ferraz, Jin Sha, Sarah Houda, Liane M. Rossi, and Sébastien Paul. 2017. "Advances in Base-Free Oxidation of Bio-Based Compounds on Supported Gold Catalysts" Catalysts 7, no. 11: 352. https://doi.org/10.3390/catal7110352
APA StyleWojcieszak, R., Ferraz, C. P., Sha, J., Houda, S., Rossi, L. M., & Paul, S. (2017). Advances in Base-Free Oxidation of Bio-Based Compounds on Supported Gold Catalysts. Catalysts, 7(11), 352. https://doi.org/10.3390/catal7110352