
Catalytic Ammonia Decomposition over High-Performance Ru/Graphene Nanocomposites for Efficient COx-Free Hydrogen Production

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
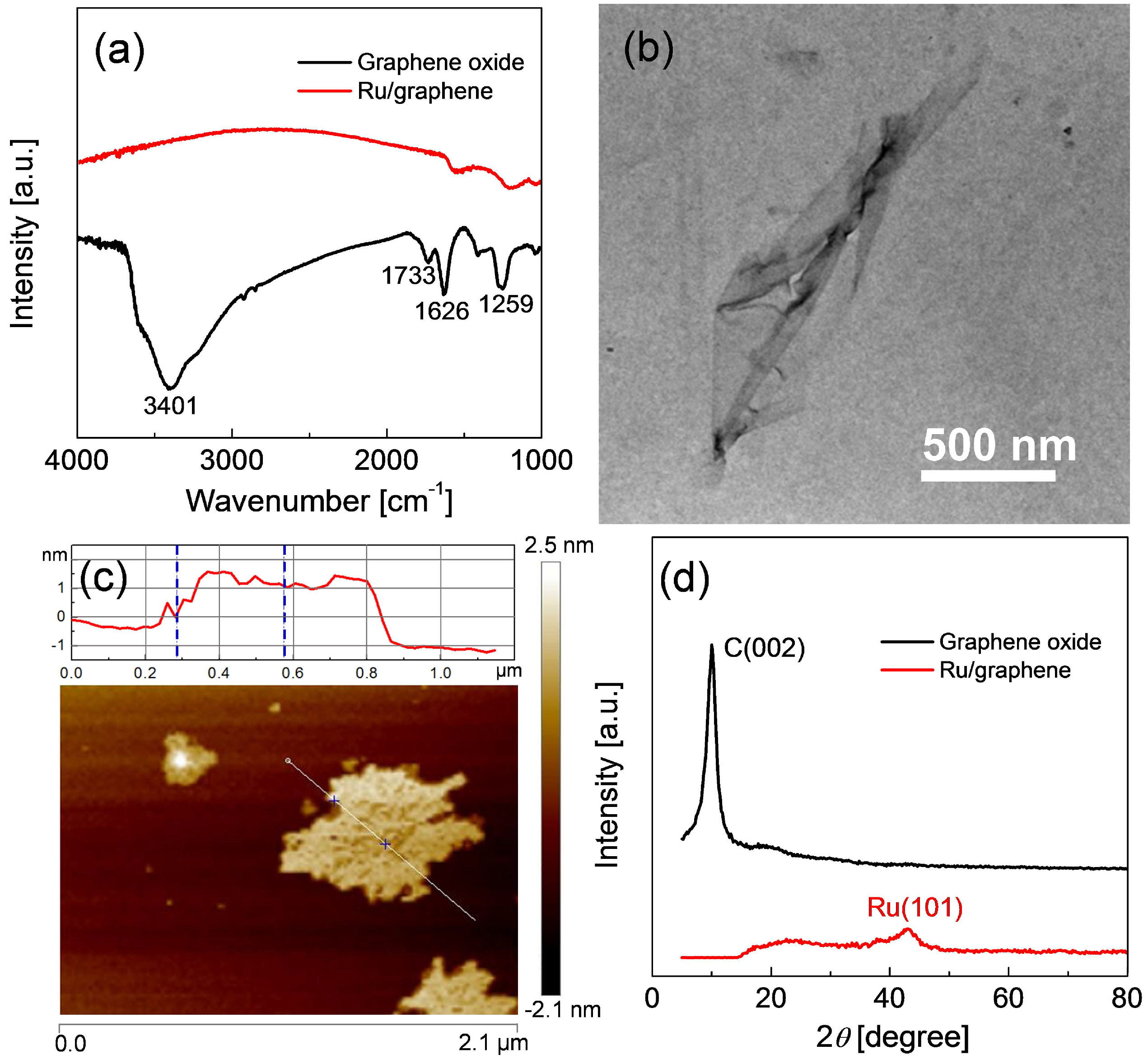
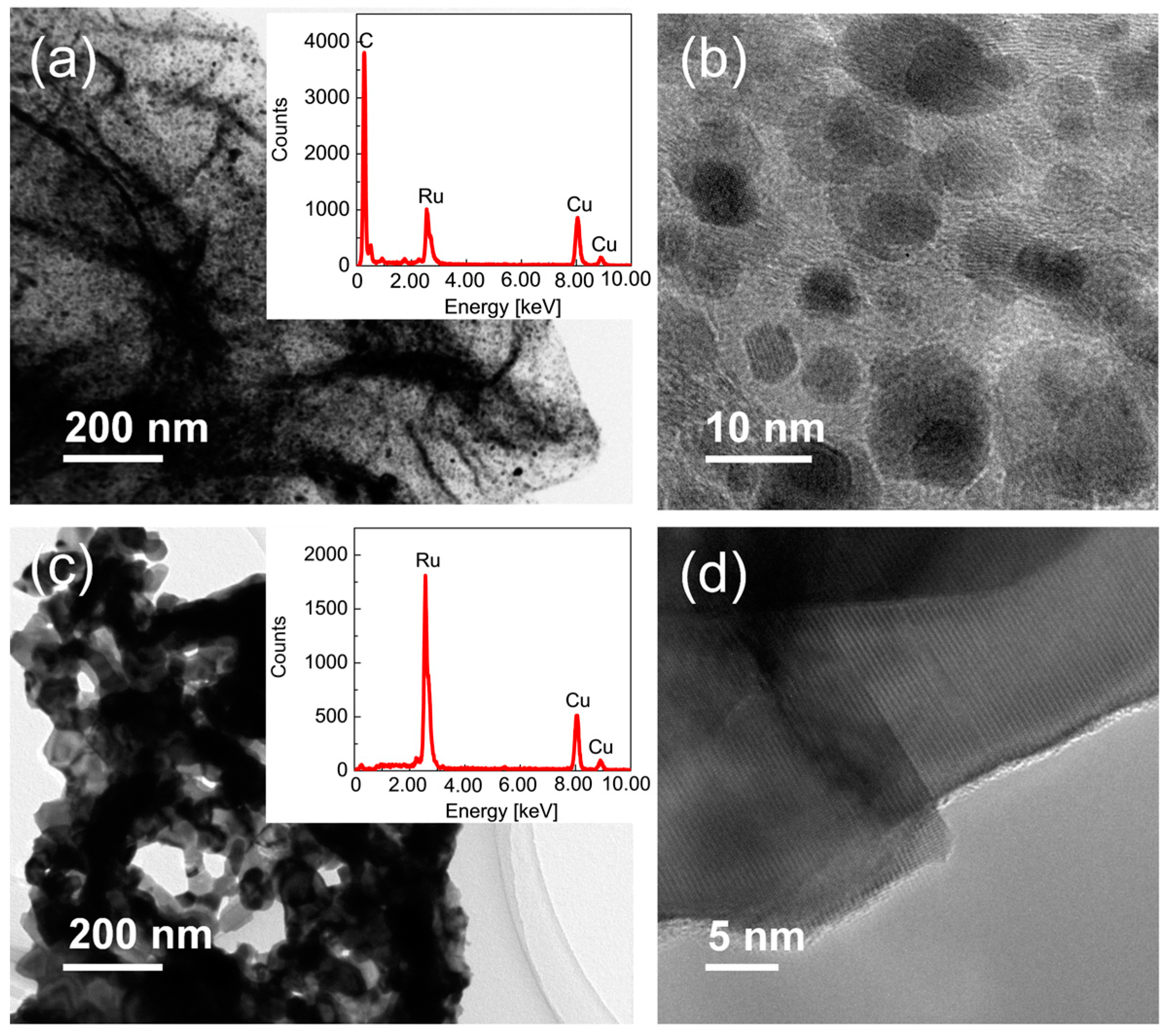
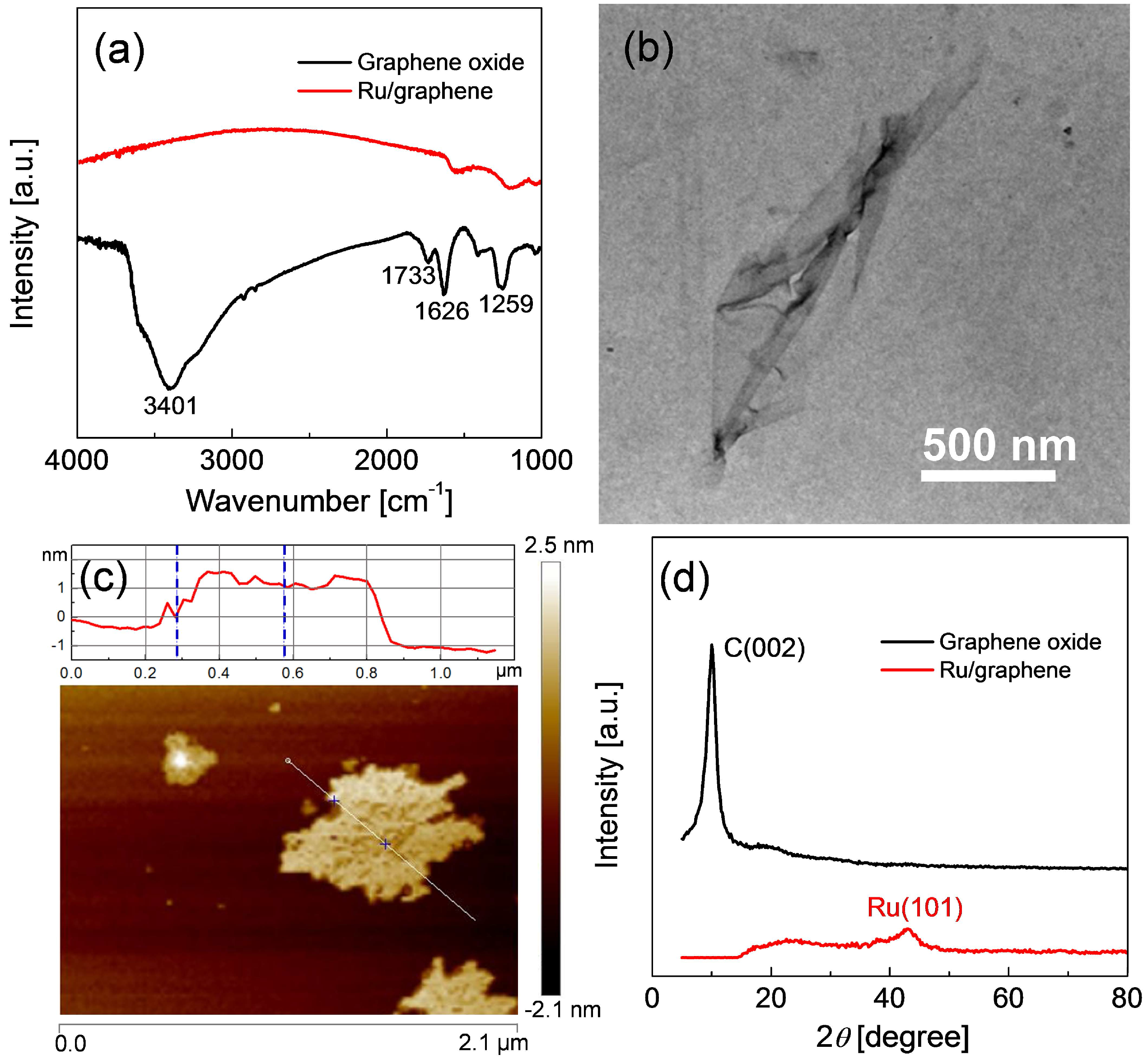
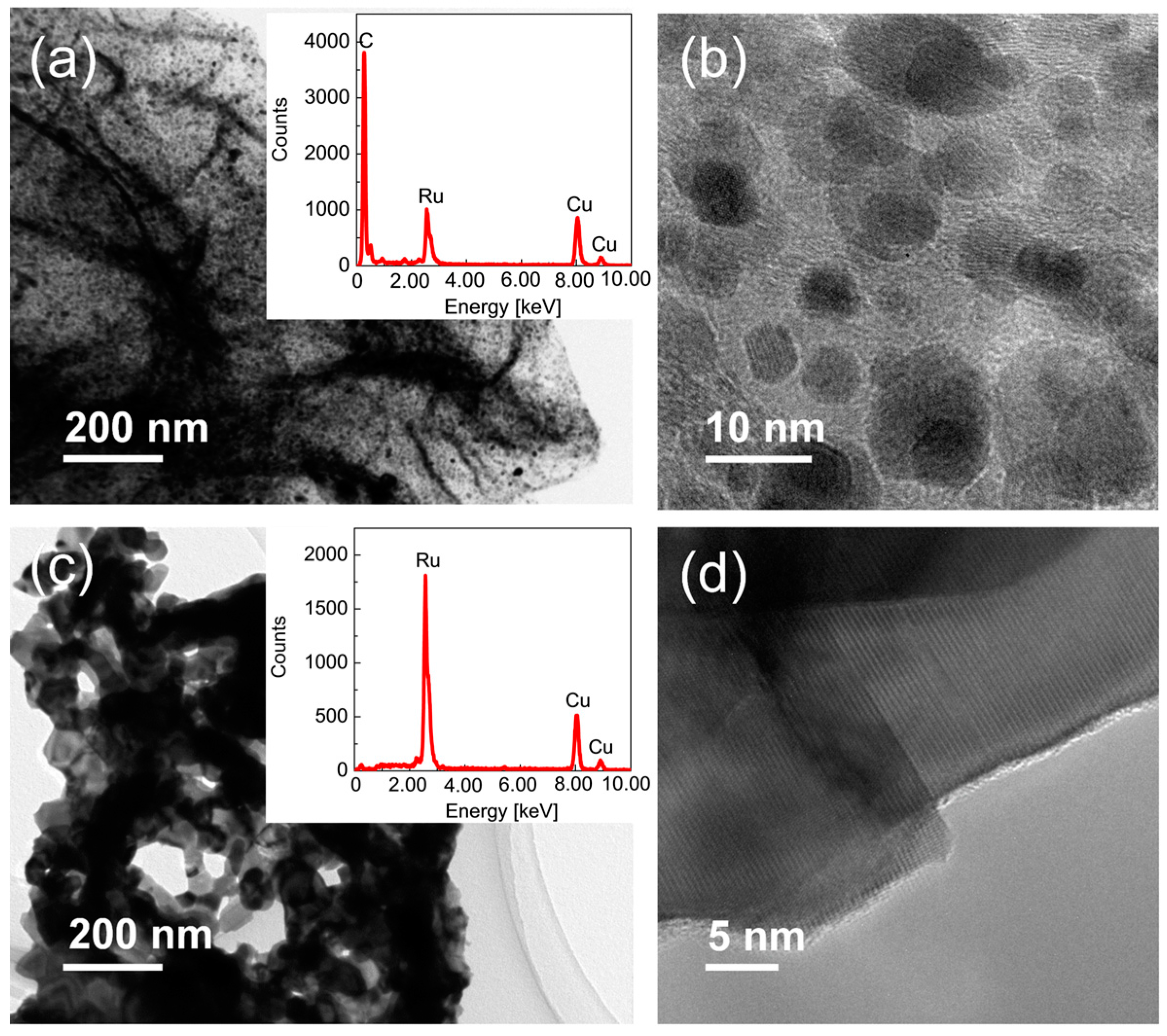
2.1. Preparation of Ru/Graphene Nanocomposites
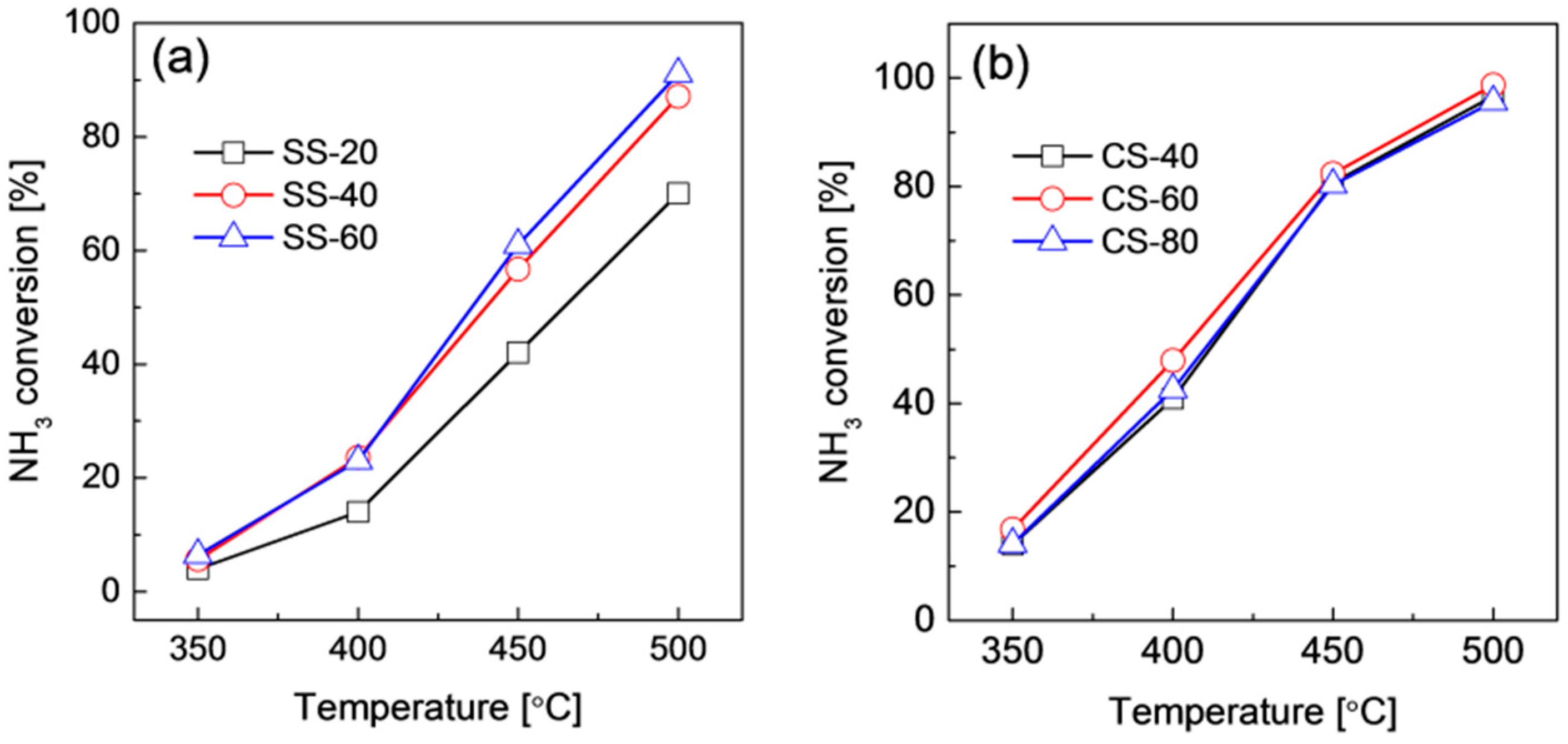
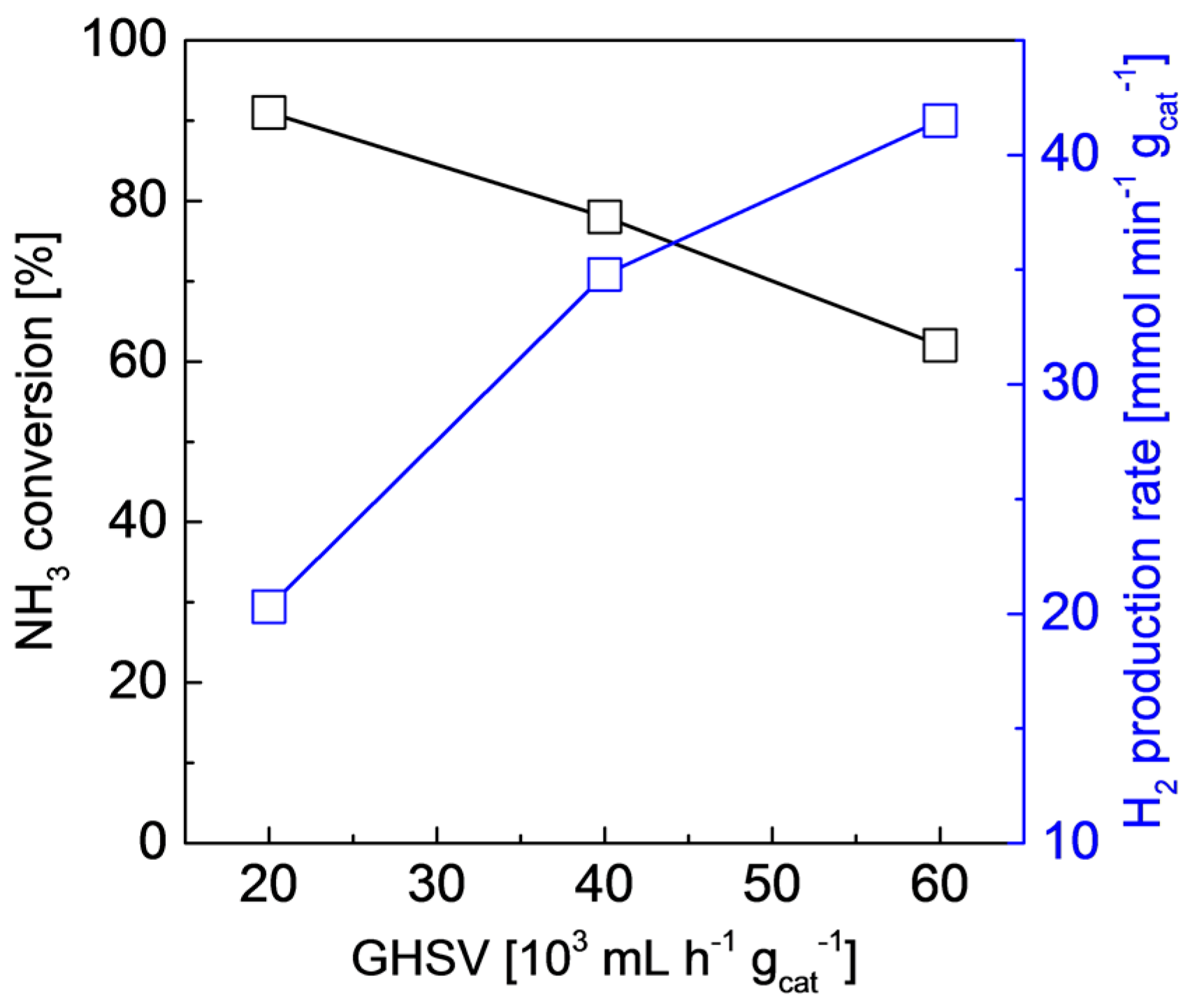
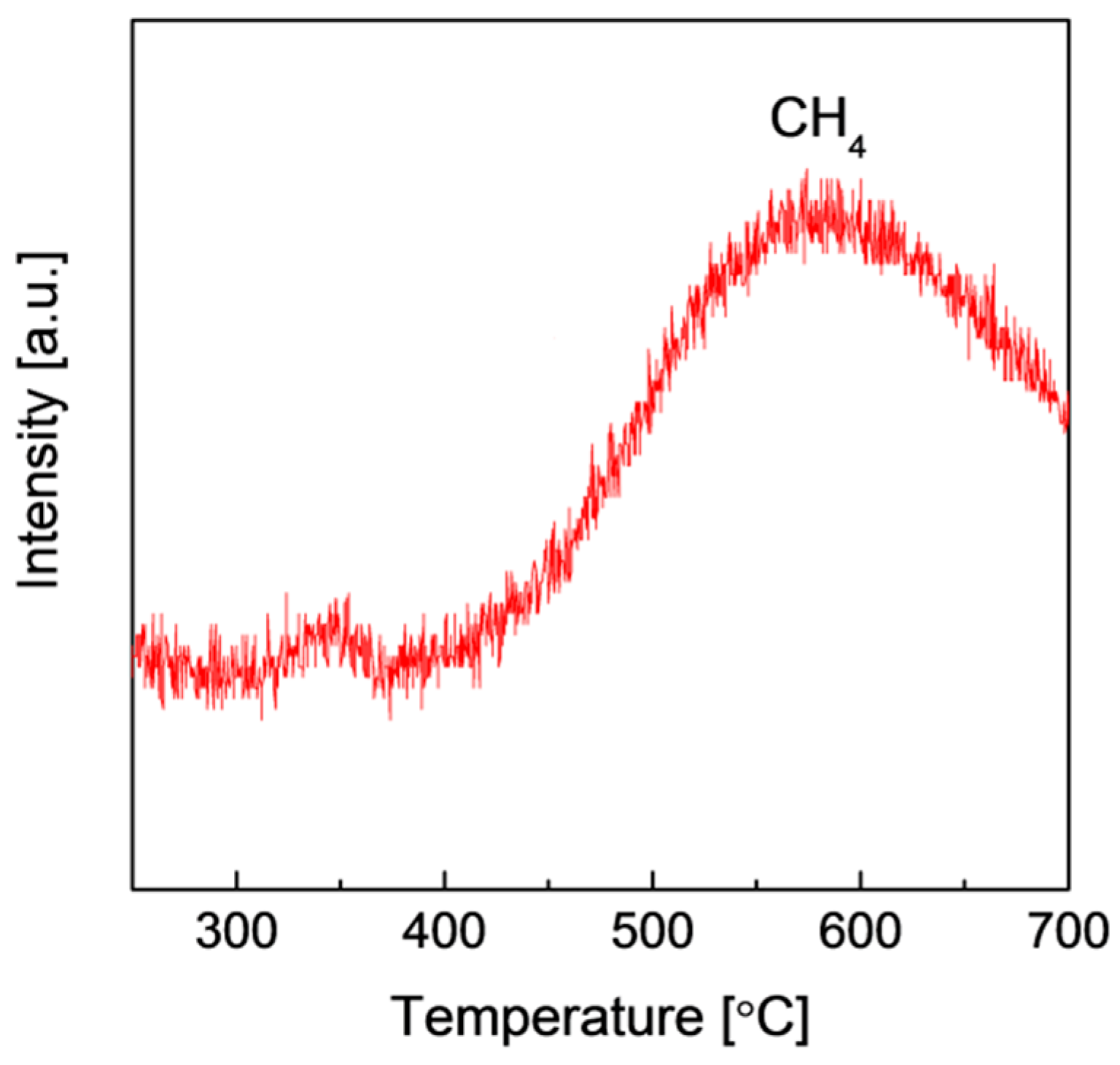
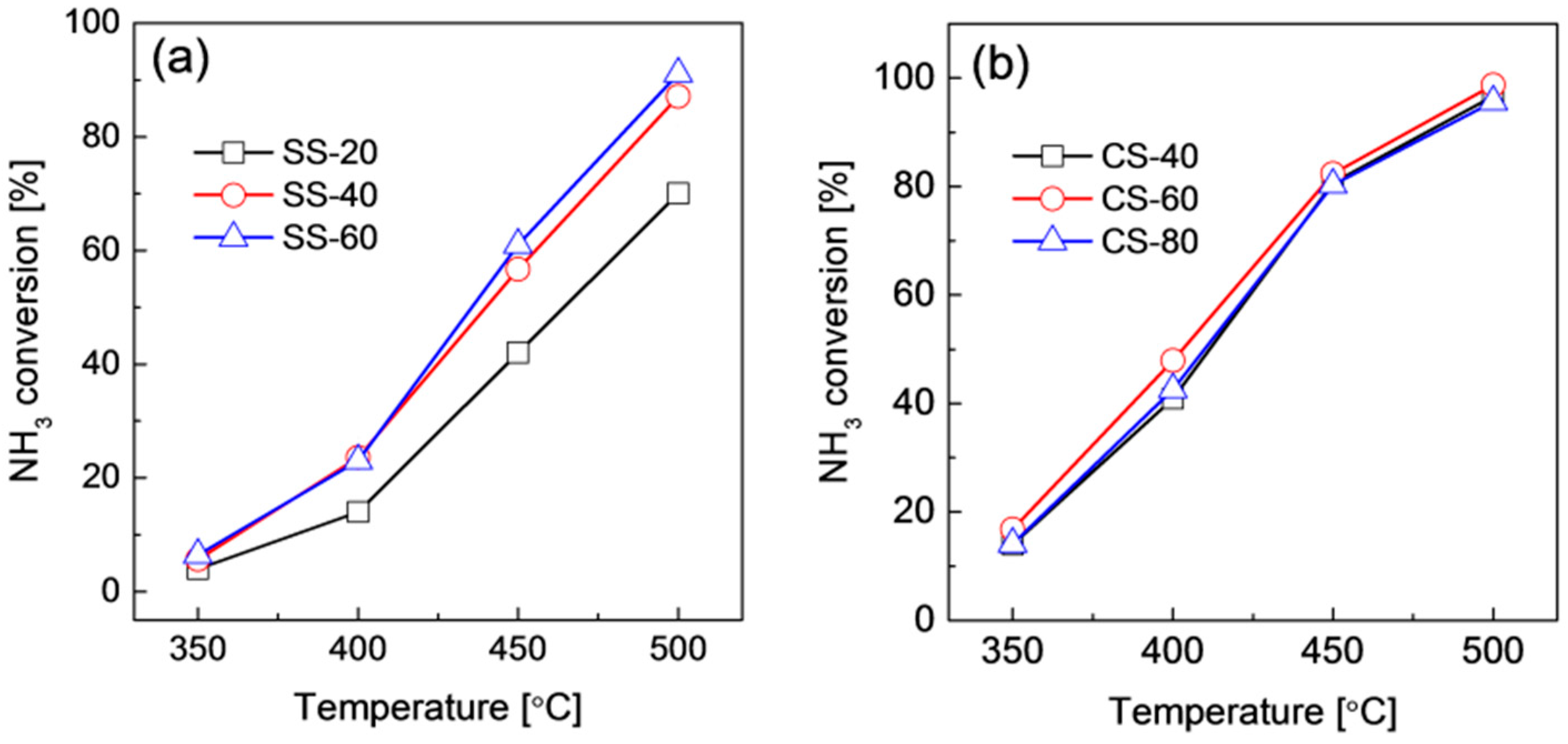
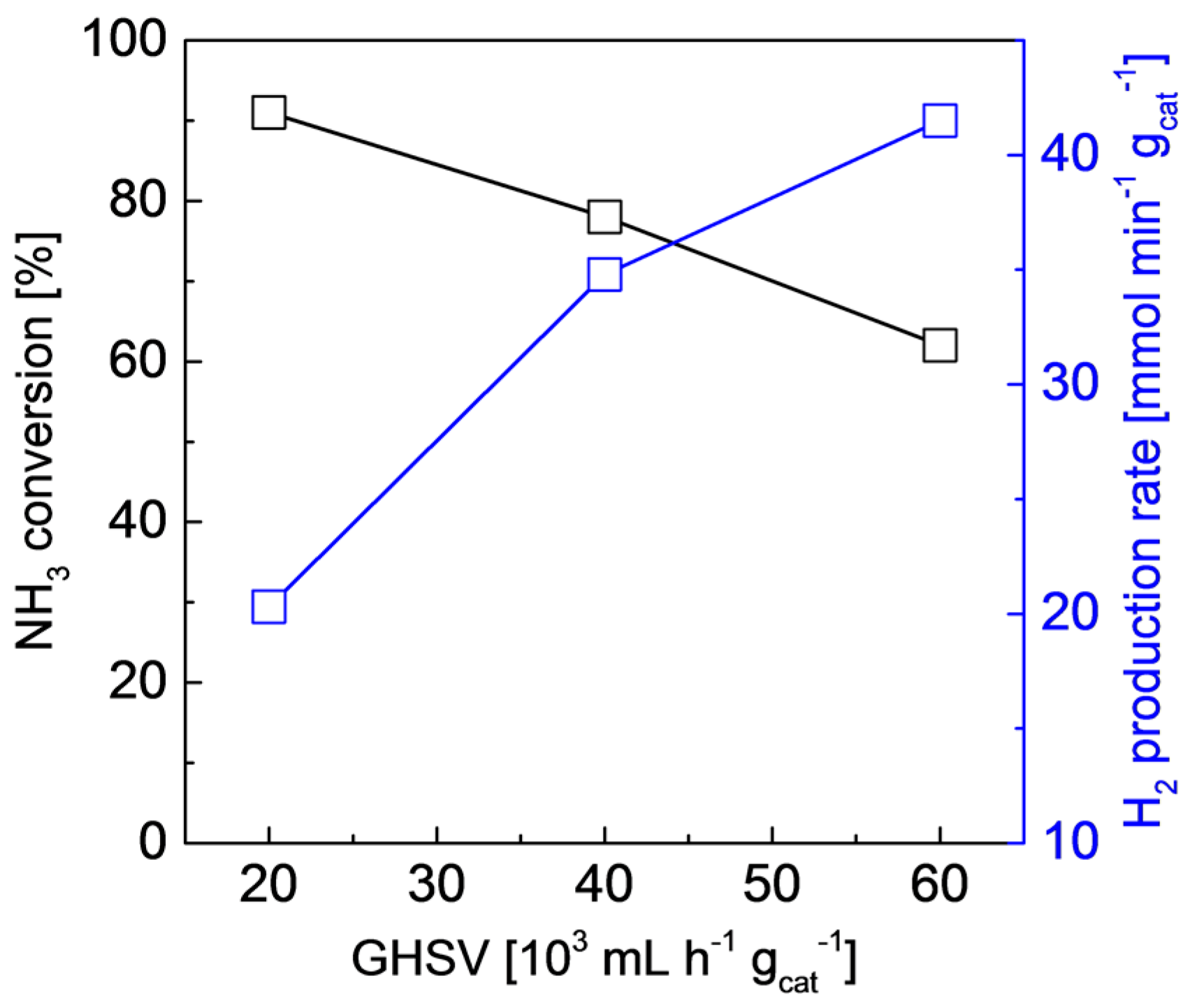
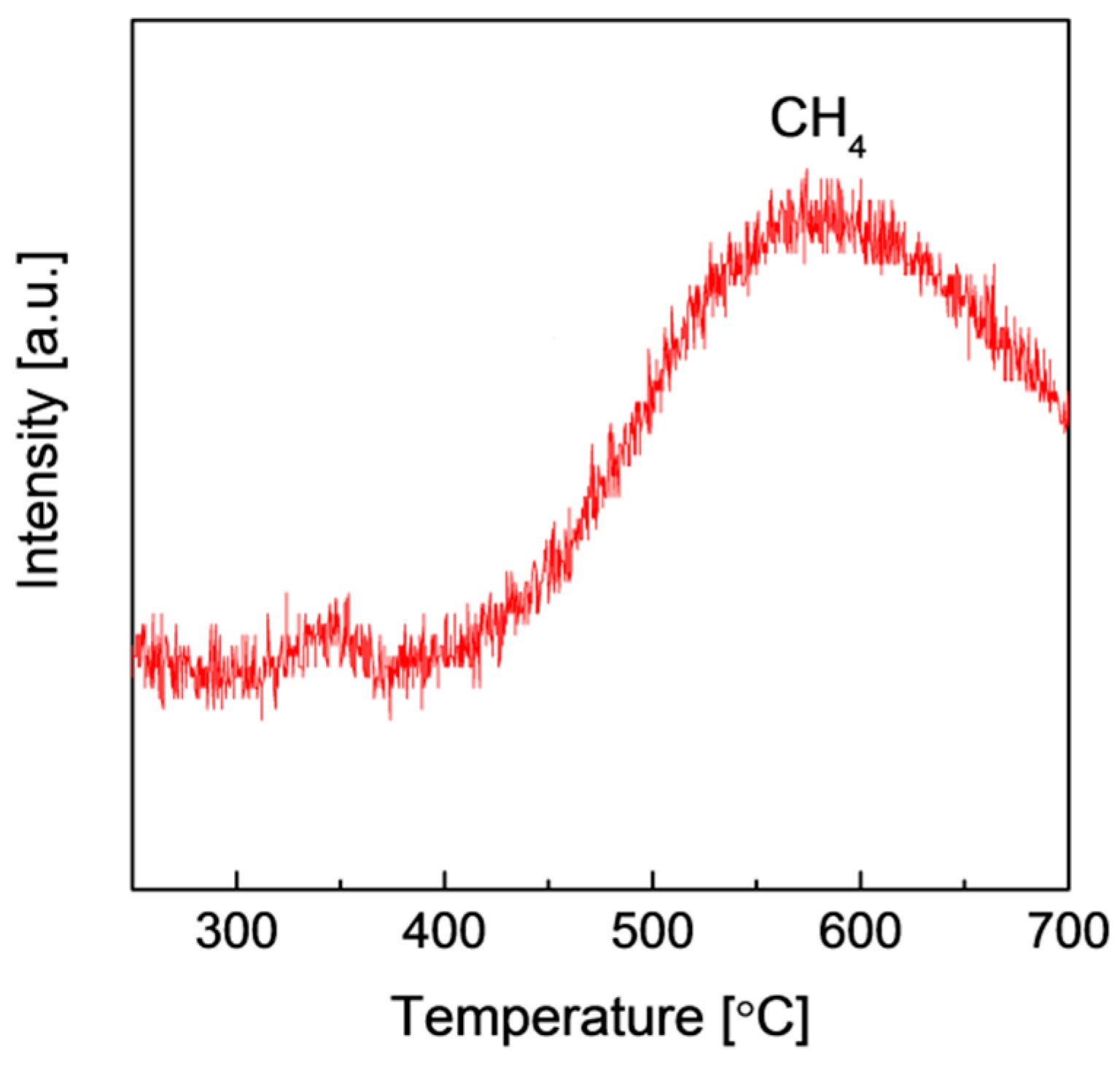
2.2. Structures and Catalytic Activities of Ru/Graphene Nanocomposites for Ammonia Decomposition
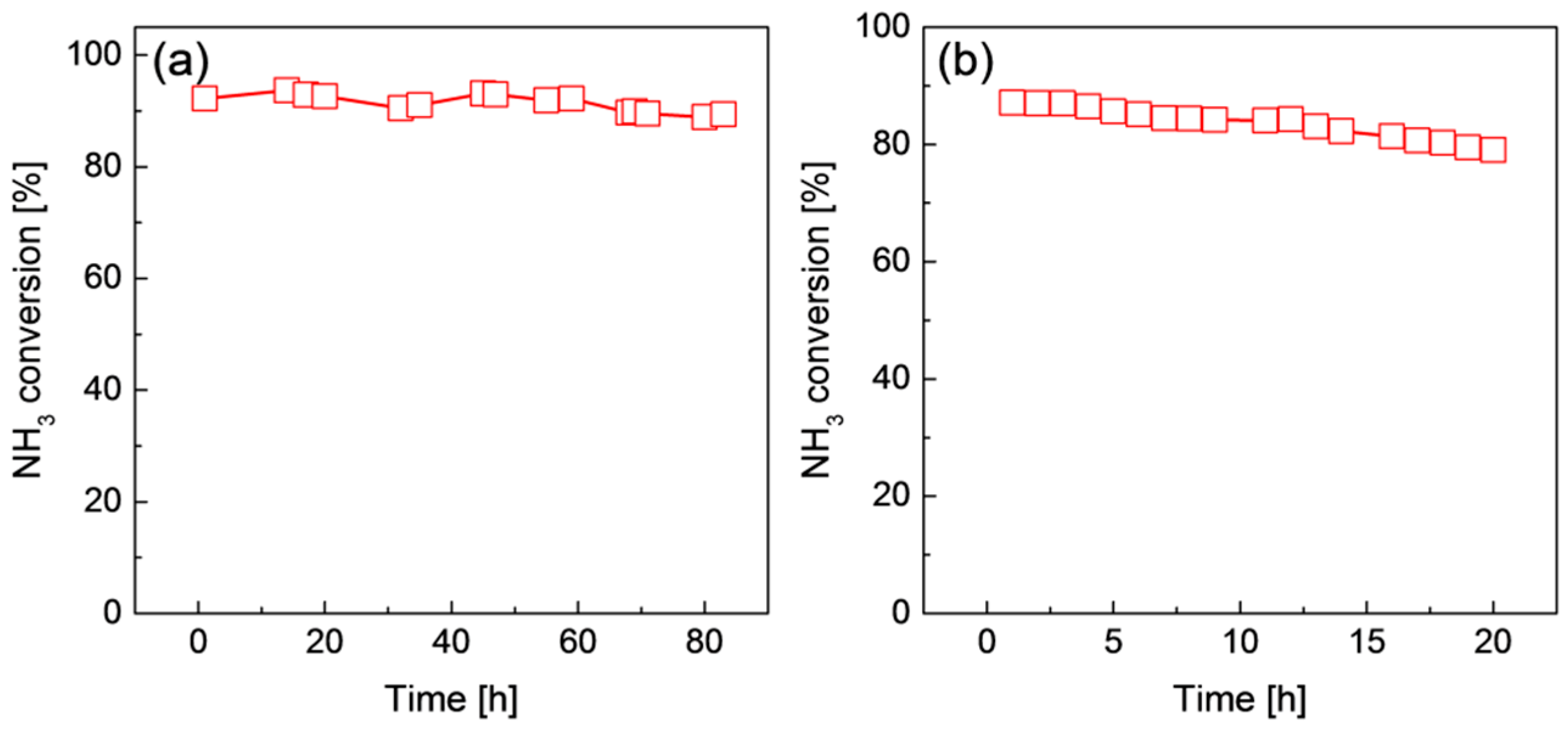
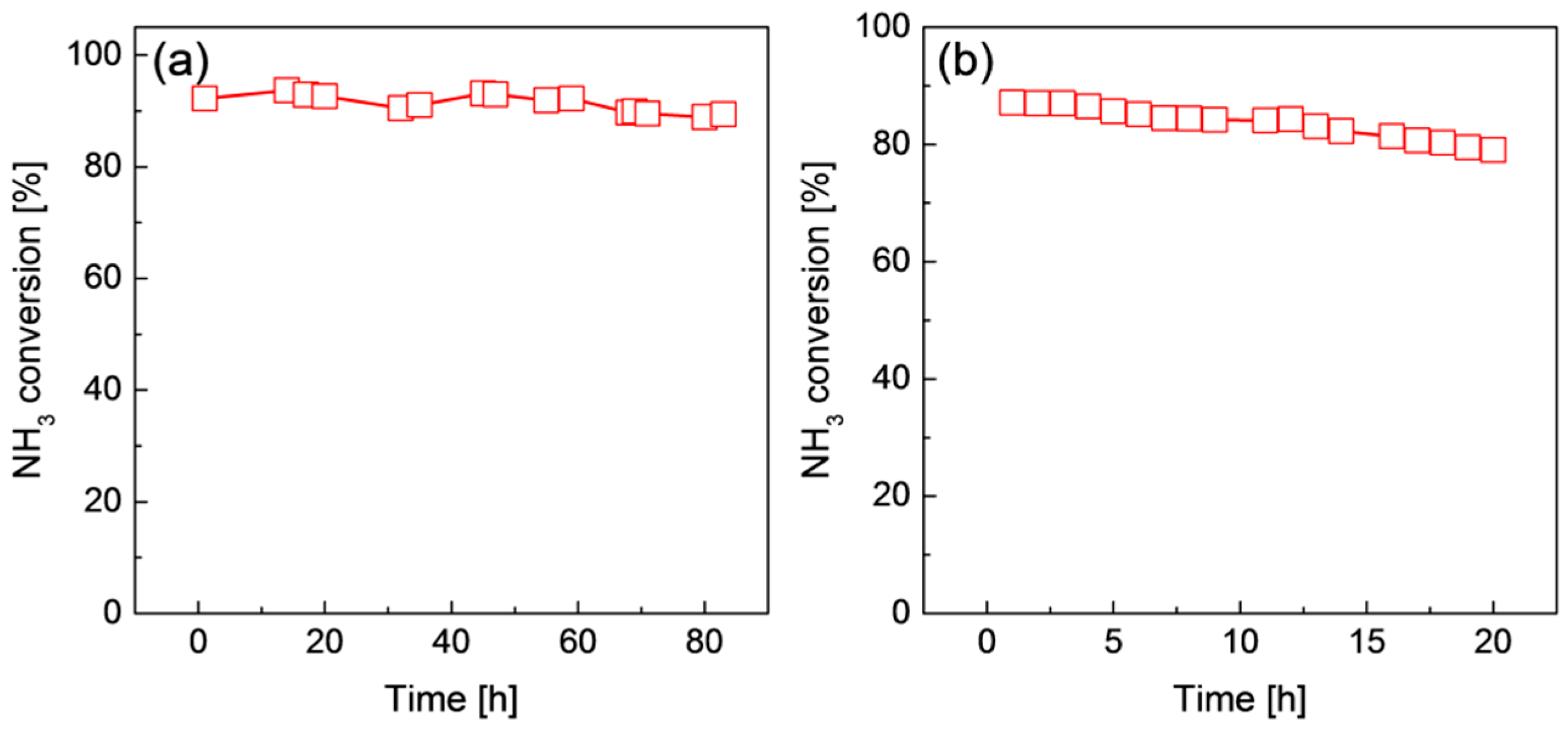
2.3. Stability of Ru/Graphene Nanocomposites for Hydrogen Production from Ammonia
3. Materials and Methods
3.1. Materials
3.2. Preparation and Characterization of Ru/Graphene Nanocomposites
3.3. Catalyst Testing
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Murray, L.J.; Dincă, M.; Long, J.R. Hydrogen storage in metal–organic frameworks. Chem. Soc. Rev. 2009, 38, 1294–1314. [Google Scholar] [CrossRef] [PubMed]
- Eberle, U.; Felderhoff, M.; Schüth, F. Chemical and Physical Solutions for Hydrogen Storage. Angew. Chem. Int. Ed. 2009, 48, 6608–6630. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, C.W.; Baker, R.T.; Staubitz, A.; Manners, I. B–N compounds for chemical hydrogen storage. Chem. Soc. Rev. 2009, 38, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.-L.; Xu, Q. Liquid organic and inorganic chemical hydrides for high-capacity hydrogen storage. Energy Environ. Sci. 2015, 8, 478–512. [Google Scholar] [CrossRef]
- Han, S.S.; Furukawa, H.; Yaghi, O.M.; Goddarlll, W.A. Covalent organic frameworks as exceptional hydrogen storage materials. J. Am. Chem. Soc. 2008, 130, 11580–11581. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.J.; Kim, T.; Im, J.H.; Kim, Y.S.; Lee, K.; Jung, H.; Rae, C. MOF-derived hierarchically porous carbon with exceptional porosity and hydrogen storage capacity. Chem. Mater. 2012, 24, 464–470. [Google Scholar] [CrossRef]
- Zhou, C.; Szpunar, J.A.; Cui, X. Synthesis of Ni/graphene nanocomposite for hydrogen storage. ACS Appl. Mater. Interfaces 2016, 8, 15232–15241. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Szpunar, J.A. Hydrogen storage performance in Pd/graphene nanocomposites. ACS Appl. Mater. Interfaces 2016, 8, 25933–25940. [Google Scholar] [CrossRef] [PubMed]
- Parambhath, V.B.; Nagar, R.; Ramaprabhu, S. Effect of nitrogen doping on hydrogen storage capacity of palladium decorated graphene. Langmuir 2012, 28, 7826–7833. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Langmi, H.W.; Beattie, S.D.; Azenwi, F.F.; McGrady, G.S.; Jensen, C.M. Ti-doped LiAlH4 for hydrogen storage: Synthesis, catalyst loading and cycling performance. J. Am. Chem. Soc. 2011, 133, 15593–15597. [Google Scholar] [CrossRef] [PubMed]
- Filinchuk, Y.E.; Yvon, K. Boron-induced hydrogen localization in the novel metal hydride LaNi3BHx (x = 2.5–3.0). Inorg. Chem. 2005, 44, 4398–4406. [Google Scholar] [CrossRef] [PubMed]
- Alhumaidan, F.; Cresswell, D.; Garforth, A. Hydrogen storage in liquid organic hydrides: Producing hydrogen catalytically for methylcyclohexane. Energy Fuels 2011, 25, 4217–4234. [Google Scholar] [CrossRef]
- Li, G.; Yada, K.; Kanezashi, M.; Yoshioka, T.; Tsuru, T. Methylcyclohexane dehydrogenation in catalytic membrane reactors for efficient hydrogen production. Ind. Eng. Chem. Res. 2013, 52, 13325–13332. [Google Scholar] [CrossRef]
- Teichmann, D.; Arlt, W.; Wasserscheid, P.; Freymann, R. A future energy supply based on liquid organic hydrogen carriers (LOHC). Energy Environ. Sci. 2011, 4, 2767–2773. [Google Scholar] [CrossRef]
- Lan, R.; Irvine, J.T.S.; Tao, S. Ammonia and related chemicals as potential indirect hydrogen materials. Int. J. Hydrog. Energy 2012, 37, 1482–1494. [Google Scholar] [CrossRef]
- Klerke, A.; Christensen, C.H.; Nørskov, J.K.; Vegge, T. Ammonia for hydrogen storage: Challenges and opportunities. J. Mater. Chem. 2008, 18, 2304–2310. [Google Scholar] [CrossRef]
- Vitse, F.; Cooper, M.; Botte, G.G. On the use of ammonia electrolysis for hydrogen production. J. Power Sources 2005, 142, 18–26. [Google Scholar] [CrossRef]
- Christensen, C.H.; Johannessen, T.; Sørensen, R.Z.; Nørskov, J.K. Towards an ammonia-mediated hydrogen economy. Catal. Today 2006, 111, 140–144. [Google Scholar] [CrossRef]
- Li, G.; Kanezashi, M.; Yoshioka, T.; Tsuru, T. Ammonia decomposition in catalytic membrane reactors: Simulation and experimental studies. AIChE J. 2013, 59, 168–179. [Google Scholar] [CrossRef]
- Schüth, F.; Palkovits, R.; Schlögl, R.; Su, D.S. Ammonia as a possible element in an energy infrastructure: Catalysis for ammonia decomposition. Energy Environ. Sci. 2012, 5, 6278–6289. [Google Scholar] [CrossRef]
- Hayashi, F.; Toda, Y.; Kanie, Y.; Kitano, M.; Inoue, Y.; Yokoyama, T.; Hara, M.; Hosono, H. Ammonia decomposition by ruthenium nanoparticles loaded on inorganic electride C12A7:e−. Chem. Sci. 2013, 4, 3124–3130. [Google Scholar] [CrossRef]
- Tüysüz, H.; Schüth, F.; Zhi, L.; Müllen, K. Ammonia decomposition over iron phthalocyanine-based materials. ChemSusChem 2015, 7, 1453–1459. [Google Scholar] [CrossRef]
- Guo, J.; Wang, P.; Wu, G.; Wu, A.; Hu, D.; Xiong, Z.; Wang, J.; Yu, P.; Chang, F.; Chen, Z.; Chen, P. Lithium imide synergy with 3d transition-metal nitrides leading to unprecedented catalytic activities for ammonia decomposition. Angew. Chem. Int. Ed. 2015, 54, 2950–2954. [Google Scholar] [CrossRef] [PubMed]
- Okura, K.; Okanishi, T.; Muroyama, H.; Matsui, T.; Eguchi, K. Additive effect of alkaline earth metals on ammonia decomposition reaction over Ni/Y2O3 catalysts. RSC Adv. 2016, 6, 85142–85148. [Google Scholar] [CrossRef]
- Makepeace, J.W.; Wood, T.J.; Hunter, H.M.A.; Jones, M.O.; David, W.I.F. Ammonia decomposition catalysis using non-stoichiometric lithium imide. Chem. Sci. 2015, 6, 3805–3815. [Google Scholar] [CrossRef]
- Gu, Y.-Q.; Fu, X.-P.; Du, P.-P.; Gu, D.; Jin, Z.; Huang, Y.-Y.; Si, R.; Zheng, L.-Q.; Song, Q.-S.; Jia, C.-J.; et al. In situ X-ray diffraction study of Co-Al nanocomposites as catalysts for ammonia decomposition. J. Phys. Chem. C 2015, 119, 17102–17110. [Google Scholar] [CrossRef]
- Bell, T.E.; Torrente-Murciano, L. H2 production via ammonia decomposition using non-noble metal catalysis: A review. Top Catal. 2016, 59, 1438–1457. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, Y.; Liu, C.; Gong, W.; Guo, H. Plasma driven ammonia decomposition on a Fe-catalyst: Eliminating surface nitrogen poisoning. Chem. Commun. 2013, 49, 3787–3789. [Google Scholar] [CrossRef] [PubMed]
- Hansgen, D.A.; Vlachos, D.G.; Chen, J.G. Using first principles to predict bimetallic catalysts for the ammonia decomposition reaction. Nat. Chem. 2010, 2, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Inokawa, H.; Ichikawa, T.; Miyaoka, H. Catalysis of nickel nanoparticles with high thermal stability for ammonia decomposition. Appl. Catal. A 2015, 491, 184–188. [Google Scholar] [CrossRef]
- Raróg-Pilecka, W.; Miśkiewicz, E.; Szmigiel, D.; Kowalczyk, Z. Structure sensitivity of ammonia synthesis over promoted ruthenium catalysts supported on graphitised carbon. J. Catal. 2005, 231, 11–19. [Google Scholar] [CrossRef]
- García- García, F.R.; Guerrero-Ruiz, A.; Rodríguez-Ramos, I. Roles of B5-type sites in Ru catalysts used for the NH3 decomposition reaction. Top Catal. 2009, 52, 758–764. [Google Scholar] [CrossRef]
- Dahl, S.; Logadottir, A.; Egeberg, R.C.; Larsen, J.H.; Chorkendorff, I.; Törnqvist, E.; Nørskov, J.K. Role of steps in N2 activation on Ru(0001). Phys. Rev. Lett. 1999, 83, 1814–1817. [Google Scholar] [CrossRef]
- Dahl, S.; Sehested, J.; Jacobsen, C.J.H.; Törnqvist, E.; Chorkendorff, I. Surface science based microkinetic analysis of ammonia synthesis over ruthenium catalysts. J. Catal. 2000, 192, 391–399. [Google Scholar] [CrossRef]
- Yin, S.F.; Xu, B.Q.; Zhou, X.P.; Au, C.T. A mini-review on ammonia decomposition catalysts for on-site generation of hydrogen for fuel cell applications. Appl. Catal. A 2004, 277, 1–9. [Google Scholar] [CrossRef]
- García-García, F.R.; Álvarez-Rodríguez, J.; Rodríguez-Ramos, I.; Guerrero-Ruiz, A. The use of carbon nanotubes with and without nitrogen doping as support for ruthenium catalysts in the ammonia decomposition reaction. Carbon 2010, 48, 267–276. [Google Scholar] [CrossRef]
- Yin, S.-F.; Xu, B.-Q.; Ng, C.-F.; Au, C.-T. Nano Ru/CNTs: A highly active and stable catalyst for the generation of COx-free hydrogen in ammonia decomposition. Appl. Catal. B 2004, 48, 237–241. [Google Scholar] [CrossRef]
- Wang, S.J.; Yin, S.F.; Li, L.; Xu, B.Q.; Ng, C.F.; Au, C.T. Investigation on modification of Ru/CNTs catalyst for the generation of COx-free hydrogen from ammonia. Appl. Catal. B 2004, 52, 287–299. [Google Scholar] [CrossRef]
- Li, G.; Nagasawa, H.; Kanezashi, M.; Yoshioka, T.; Tsuru, T. Graphene nanosheets supporting Ru nanoparticles with controlled nanoarchitectures form a high-performance catalyst for COx-free hydrogen production from ammonia. J. Mater. Chem. A 2014, 2, 9185–9192. [Google Scholar] [CrossRef]
- Rossetti, I.; Pernicone, N.; Forni, L. Promoters effect in Ru/C ammonia synthesis catalyst. Appl. Catal. A 2001, 208, 271–278. [Google Scholar] [CrossRef]
- Yang, X.; Wang, X.; Qiu, J. Aerobic oxidation of alcohols over carbon nantube-supported Ru catalysts assembled at the interfaces of emulsion droplets. Appl. Catal. A 2010, 382, 131–137. [Google Scholar] [CrossRef]




© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, G.; Kanezashi, M.; Tsuru, T. Catalytic Ammonia Decomposition over High-Performance Ru/Graphene Nanocomposites for Efficient COx-Free Hydrogen Production. Catalysts 2017, 7, 23. https://doi.org/10.3390/catal7010023
Li G, Kanezashi M, Tsuru T. Catalytic Ammonia Decomposition over High-Performance Ru/Graphene Nanocomposites for Efficient COx-Free Hydrogen Production. Catalysts. 2017; 7(1):23. https://doi.org/10.3390/catal7010023
Chicago/Turabian StyleLi, Gang, Masakoto Kanezashi, and Toshinori Tsuru. 2017. "Catalytic Ammonia Decomposition over High-Performance Ru/Graphene Nanocomposites for Efficient COx-Free Hydrogen Production" Catalysts 7, no. 1: 23. https://doi.org/10.3390/catal7010023
APA StyleLi, G., Kanezashi, M., & Tsuru, T. (2017). Catalytic Ammonia Decomposition over High-Performance Ru/Graphene Nanocomposites for Efficient COx-Free Hydrogen Production. Catalysts, 7(1), 23. https://doi.org/10.3390/catal7010023