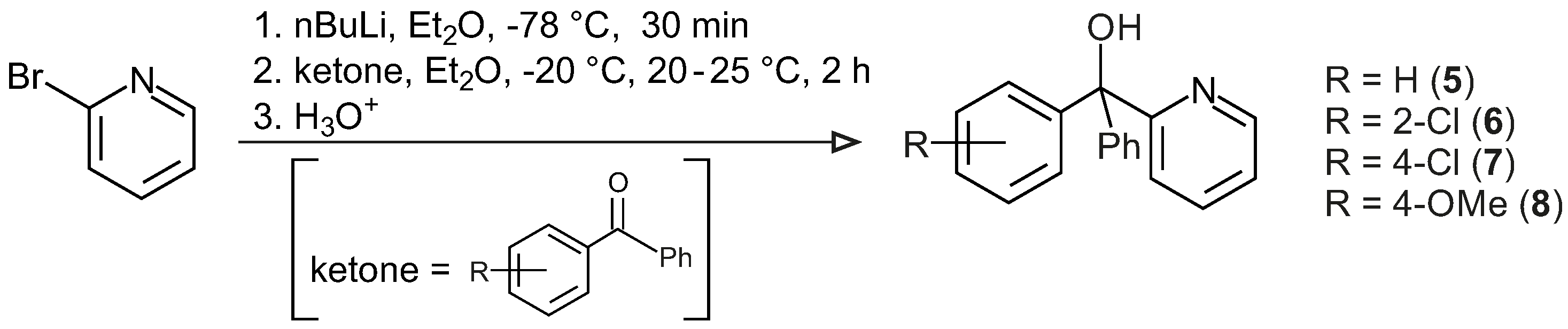
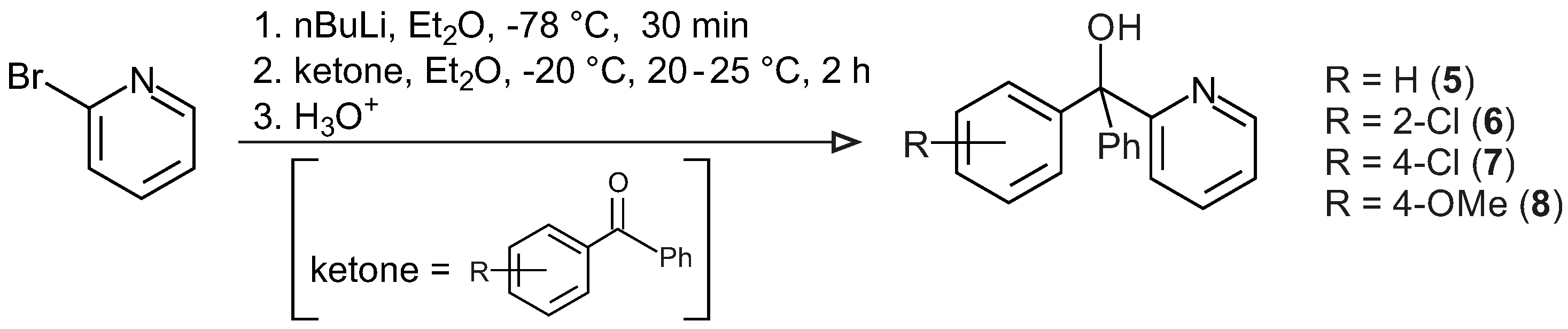
3.3.1. General Procedure for the Synthesis of Ligands 5–8
Diethyl ether (50 mL) was added to a three-neck round-bottom flask (200 mL) in argon and cooled to −78 °C. To this, nBuLi (50 mmol, 2.5 M in hexane) was added slowly, which then was followed by the drop-wise addition of 2-bromopyridine (47.5 mmol, in 12.5 mL diethyl ether) at −78 °C, and the mixture was stirred for 15 min, resulting in a dark-red solution. The reaction temperature was raised to −20 °C, and mono ortho- and/or para-chloro and/or para-methoxy substituted benzophenone/benzophenone (52.5 mmol, in 20 mL diethyl ether) was added slowly to the reaction mixture and the mixture stirred for 2 h. The temperature was raised to room temperature, and the reaction mixture was hydrolysed carefully. The organic phase was separated from the aqueous phase and then extracted with 2 M HCl (5 × 10 mL). The aqueous phase was neutralized with a 2 M NaOH solution and extracted with diethyl ether. The organic phases were combined, dried with MgSO4, and the solvent was slowly evaporated to obtain a white crystalline solid product.
1,1-diphenyl-1-(2′-pyridinyl)-methanol, 5: Yield 5.96 g, 48%, white crystalline solid, m.p. (melting point): 105 °C, 1H NMR (CDCl3, 600 MHz, CDCl3); δH = 8.59 (d, J = 4.9 Hz, 1H, H-6 of C6H4N), 7.62 (td, J1 = 7.7 Hz, J2 = 1.8 Hz, 1H, H-4 of C6H4N), 7.45–7.19 (m, 11H, H-5 of C5H4N & 10H of 2Ph), 7.12 (d, J = 7.9 Hz, 1H, H-3 of C6H4N), 6.30 (s, 1H, OH); 13C NMR (CDCl3, 150 MHz): δC = 163.2 (qC, C-2 C6H4N), 147.7 (CH, C-6 C6H4N), 146.1 (2qC, C-1 Ph), 136.4 (CH, C-4 C6H4N), 128.1 (4CH, C-3 & C-5 Ph), 127.3 (4CH, C-2 & C-6 Ph), 122.9 (2CH, C-4 Ph & C-3 C6H4N), 122.3 (CH, C-5 C6H4N), 80.8 (C–OH); M+ (APCI, m/z): calculated 261.1153 (C18H15NO), found 262.1191 (C18H16NO), 261.1117 (C18H15NO), 260.1045 (C18H14NO), 244.1121 (C18H14N). IR (neat): v(OH) = 3342 cm−1, v(C–H, aromatic) = 3075 cm−1, 3018 cm−1, 756 cm−1, v(C=C & N=C, aromatic) = 1591 cm−1, 1571 cm−1, v(C–O, aliphatic) = 1168 cm−1, 1126 cm−1.
1-(2′-chlorophenyl),1-phenyl-1-(2′-pyridinyl)-methanol, 6: Yield 9.81 g, 89%, white crystalline solid, m.p.: 122 °C, 1H NMR (CDCl3, 600 MHz, CDCl3); δH = 8.54 (d, 1H, J = 4.64 Hz, H-6 of C6H4N), 7.60 (dt, 1H, J = 7.73, 1.69 Hz, H-4 of C6H4N), 7.12 (d, 1H, J = 7.87 Hz, H-3 of C6H4N), 7.04 (t, 1H, J = 7.67 Hz, H-4 of substituted Ph), 6.76 (t, 1H, J = 7.87 Hz, H-3 of substituted Ph), 7.15–7.36 (m, 8H, H-5 of C6H4N and Ph); 13C NMR (CDCl3, 150 MHz): δC = 162.5 (q C-2 C6H4N), 147.8 (CH, C-6 C6H4N), 136.5 (CH, C-4 C6H4N), 122.6 (CH, C-3 C6H4N), 122.3 (CH, C-5 C6H4N), 144.5 (qC, C-1 substituted Ph), 134.7 (qC, C-2 substituted Ph), 131.7 (CH, C-2 substituted Ph), 130.9 (CH, C-3 substituted Ph), 127.2 (CH, C-3 substituted Ph), 127.5 (CH, C-4 substituted Ph), 142.7 (qC, Ph), 128.0 (2CH, C-2 Ph), 129.1 (2CH, C-3 Ph), 125.9 (CH, C-4 Ph), 81.0 (qC, COH); M+ (APCI, m/z): calculated 295.0763 (C18H14ClNO), found 295.0757 (C18H14ClNO), 296.0826 (C18H15ClNO), 278.0739 (C18H13ClN); IR (neat): v(OH) = 3347 cm−1, v(C–H, aromatic) = 3059 cm−1, 3005 cm−1, 753 cm−1, v(C=C & N=C, aromatic) = 1590 cm−1, 1568 cm−1, v(C–O, aliphatic) = 1169 cm−1, 1138cm−1.
1-(4′-chlorophenyl),1-phenyl-1-(2′-pyridinyl)-methanol, 7: Yield 11.23 g, 80%, white crystalline solid, m.p.: 110 °C, 1H NMR (CDCl3, 600 MHz, CDCl3); δH = 8.58 (d, 1H, J = 4.56 Hz, H-6 of C6H4N), 7.64 (dt, 1H, J = 8.02, 1.64 Hz, H-4 of C6H4N), 7.07 (d, 1H, J = 8.97 Hz, H-3 of C6H4N), 7.21–7.31 (m, 10H, H-5 of C6H4N & 2Ph), 6.27 (s, 1H, OH); 13C NMR (CDCl3, 150 MHz): δC = 162.6 (qC, C-2 C6H4N), 147.8 (CH, C-6 C6H4N), 136.5 (CH, C-4 C6H4N), 122.7 (CH, C-3 C6H4N), 122.5 (CH, C-5 C6H4N), 145.6 (qC, C-1 substituted Ph), 131.3 (qC, C-4 substituted Ph), 129.5 (2CH, C-2 substituted Ph), 128.0 (6CH, C-3 substituted Ph, C-2 & C-3 Ph), 144.7 (qC, C-1 of Ph), 127.5 (CH, C-4 Ph), 80.4 (qC, COH); M+ (APCI, m/z): calculated 295.0763 (C18H14ClNO), found 295.0745 (C18H14ClNO), 296.0800 (C18H15ClNO), 294.0674 (C18H13ClNO), 278.0727 (C18H13ClN); IR (neat): v(OH) = 3373 cm−1, v(C–H, aromatic) = 3072 cm−1, 3021 cm−1, 816 cm−1, v(C=C & N=C, aromatic) = 1589 cm−1, 1570 cm−1, v(C–O, aliphatic) = 1169 cm−1, 1093 cm−1.
1-(4′-methoxyphenyl),1-phenyl-1-(2′-pyridinyl)-methanol, 8: Yield 9.5 g, 70%, white crystalline solid, m.p.: 121.6 °C, 1H NMR (CDCl3, 600 MHz, CDCl3); δH = 8.57 (d, 1H, J = 4.85 Hz, 6-H of C6H4N), 7.62 (dt, 1H, J = 7.81, 1.58 Hz, H-4 of C6H4N), 7.08 (d, 1H, J = 7.19 Hz, H-3 of C6H4N), 7.17 (d, 2H, J = 8.87 Hz, H-2 & H-6 of substituted Ph), 6.81 (d, 2H, J = 8.65 Hz, H-3 & H-5 of substituted Ph), 7.19–7.28 (m, 6H, H-5 of C6H4N & Ph), 6.21 (s, 1H, OH), 3.77 (s, 3H, OCH3); 13C NMR (CDCl3, 150 MHz): δC = 158.7 (qC, C-2 C6H4N), 147.6 (CH, C-6 C6H4N), 138.3 (CH, C-4 C6H4N), 122.7 (CH, C-3 C6H4N), 122.2 (CH, C-5 C6H4N), 163.4 (qC, C-4 substituted Ph), 136.3 (qC, C-1 substituted Ph), 129.3 (2CH, C-2 substituted Ph), 113.2 (2CH, C-3 substituted Ph), 146.2 (qC, C-1 Ph), 128.0 (2CH, C-3 Ph), 127.8 (2CH, C-2 Ph), 127.2 (CH, C-4 Ph), 88.1 (qC, COH), 55.2 (OCH3); M+ (APCI, m/z): calculated 291.1259 (C19H17NO2), found 291.1219 (C19H17NO2), 274.1223 (C19H16NO), 184.0735 (C13H12O); IR (neat): v(OH) = 3403 cm−1, v(C–H, aromatic) = 3059 cm−1, 3014 cm−1, 842 cm−1, v(C–H, aliphatic) = 2962 cm−1, 2930 cm−1, 2905 cm−1, 2833 cm−1, v(C=C & N=C, aromatic) 1614 cm−1, 1587 cm−1, 1569 cm−1, 1510 cm−1, v(C–O, aliphatic) = 1167 cm−1, 1027 cm−1, v(C–O, phenol) = 1248 cm−1.
3.3.3. General Procedure for the Synthesis of Complexes 4 and 13–15
The lithium salt of pyridin-2-yl-methanol (0.6 mmol, in 5 mL THF) was added to a Schlenk tube containing Grubbs second generation catalyst (0.6 mmol, in 5–10 mL THF) under inert atmosphere. The reaction mixture was stirred for 2–53 h at room temperature to 40 °C, depending on the reaction rate. Progress of the reaction was followed by TLC (thin layer chromatography) (6:1 hexane:ethyl acetate) until all of the Grubbs catalyst was consumed. The THF was removed from the resulting dark-green Grubbs 2-type precatalysts under reduced pressure in an inert condition. Toluene (7 mL) was added to the tarry black residue in the Schlenk tube, and all of the contents of the Schlenk tube were collected by gastight syringe and filtered into another Schlenk tube using a syringe filter, in an inert condition. Toluene was removed under reduced pressure; THF (1 mL) and pentane (5–10 mL) were added to the tarry black residue in the Schlenk tube, and the mixture was left for 20–30 min to form a precipitate. The pentane was removed by gastight syringe, and the product was washed with pentane (5 mL) using an ultrasonic bath for 5 min once and then four times without the ultrasonic bath. The washed pentane was removed by a gastight syringe, and the final product was dried under reduced pressure in an inert condition.
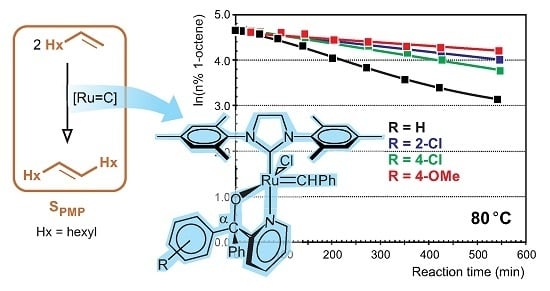
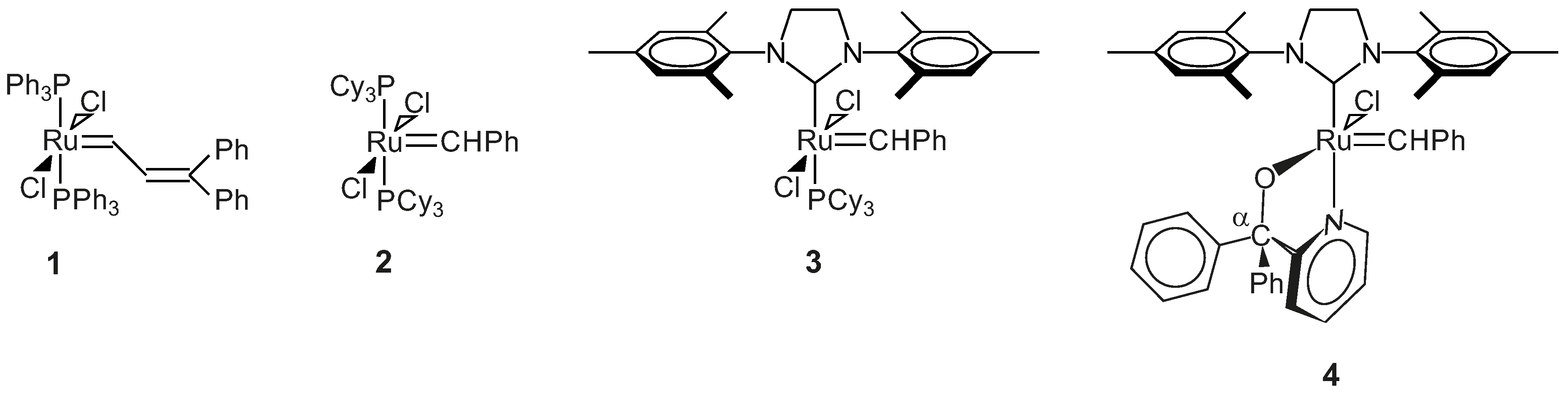
Benzylidene-chloro(1,3-bis-(2,4,6-trimethyl phenyl)-2-imidazolidinylidene)-[1-(2′-pyridinyl)-1,1-diphenyl-methanolato]ruthenium, 4: Yield 0.458 g, 98%, green powder, dec.p. (decomposition point): 114 °C, 1H NMR (CDCl3, 600 MHz, CDCl3): δH = 17.10 (s, 1H, carbene), 9.61 (d, 1H, J = 5.48 Hz, H-6 of C5H4N), 7.62 (t, 1H, J = 7.58 Hz, H-4 of C5H4N), 7.31–7.19 (m, 11H, H-3 of C5H4N & all H’s of 2Ph), 7.17–7.06 (m, 6H, all H’s of carbene Ph & H-5 of C5H4N), 6.64 (s, 4H, H-3 of mesityl), 3.99 (m, 4H, H of N(CH2)2N), 2.61/2.27/2.19 (3s, 18H, 6CH3 of mesityl); 31P NMR (CDCl3, 242 MHz): no signal; M+ (MALDI, m/z): calculated 793.2372 (C46H46ClN3ORu), found 793.2362 (C46H46ClN3ORu). IR (neat): v(C–H, aromatic) = 3055 cm−1, 3019 cm−1, 760 cm−1, v(CH3) = 2921 cm−1, 2849 cm−1, 1379 cm−1, v(C=N, & C=C, aromatic) = 1592 cm−1, 1568 cm−1, v(C–N) = 1262 cm−1, v(C–O, aliphatic) = 1165 cm−1, 1024 cm−1; 13C NMR (CDCl3, 150 MHz): δC = 291.70, 214.44, 171.21, 151.55, 149.88, 149.75, 143.58, 139.21, 137.32, 136.62, 133.94, 129.63, 128.84, 128.39, 127.89, 126.97, 126.84, 126.58, 126.29, 126.11, 122.39, 120.94, 93.94, 51.33, 20.95, 19.07, 18.89.
Benzylidene-chloro(1,3-bis-(2,4,6-trimethylphenyl)-2-imidazolidinylidene)-[1-(2′-pyridinyl)-1-(2′-chlorophenyl),1-phenyl-methanolato]ruthenium, 13: Yield 0.33 g, 66%, light green powder, dec.p.: 210 °C, 1H NMR (CDCl3, 600 MHz, CDCl3): δH = 17.34/17.26 (2s, 1H, carbene H), 9.76 (d, 1H, J = 5.47 Hz, H-6 of C5H4N), 7.65 (t, 1H, J = 7.37 Hz, H-4 of C5H4N), 7.36–7.40 (m, 4H, H of carbene Ph), 7.32 (t, 2H, J = 7.89 Hz, H-4 of carbene Ph & 5-H of C5H4N), 7.27 (d, 1H, J = 7.89 Hz, 3-H of C5H4N), 7.19–7.25 (m, 6H, H of unsubstituted Ph & 6-H of substituted Ph), 7.09 (m, 3H, 3-, 4- & 5-H of substituted Ph), 6.79–6.88 (2d, 4H, J = 7.88 Hz, H-3 of mesityl), 3.96 (m, 4H, N(CH2)2N), 2.65/2.52/2.21/2.14 (4s, 18H, 6CH3); 31P NMR (CDCl3, 242 MHz): no signal; M+ (MALDI, m/z): calculated 827.1983 (C46H45Cl2N3ORu), found 827.1972 (C46H45Cl2N3ORu); IR (neat): v(=C–H, aromatic) = 3057 cm−1, 3020 cm−1, 756 cm−1, v(CH3) = 2918 cm−1, 2853 cm−1, v(C=N & C=C, aromatic) = 1627 cm−1, 1591 cm−1, 1568 cm−1, v(C–N, aliphatic) = 1263 cm−1, v(C–O, aliphatic) = 1162 cm−1, 1026 cm−1; 13C NMR (CDCl3, 150 MHz): δC = 292.70, 214.17, 171.86, 150.01, 149.84, 148.01, 138.87, 137.53, 136.71, 134.30, 131.05, 130.69, 129.45, 129.42, 128.96, 128.58, 127.31, 127.27, 126.95, 126.90, 126.43, 126.27, 125.16, 123.31, 121.16, 94.31, 51.51, 20.94, 18.93, 18.80.
Benzylidene-chloro(1,3-bis-(2,4,6-trimethylphenyl)-2-imidazolidinylidene)-[1-(2′-pyridinyl)-1-(4′-chlorophenyl),1-phenyl-methanolato]ruthenium, 14: Yield 0.46 g, 94%, light green powder, dec.p.: 88 °C, 1H NMR (CDCl3, 600 MHz, CDCl3): δH = 17.11/17.09 (2s, 1H, carbene H), 9.55–9.70 (2d, 1H, J = 5.32 Hz, 6-H of C5H4N), 7.63 (t, 1H, J = 7.56 Hz, 4-H of C5H4N), 7.20–7.30 (m, 11H, 3-H of C5H4N, 2Ph), 7.15 (t, 2H, J = 7.28 Hz, meta-H of para-ClPh), 7.09 (t, 1H, J = 7.56 Hz, 5-H of C5H4N), 7.02 (d, 2H, J = 8.12 Hz, ortho-H of para-ClPh), 6.97/6.75 (2d, 4H, J = 8.12 Hz, mesityl H), 3.98 (m, 4H, N(CH2)2N), 2.60/2.28–2.19 (3s, 18H, 6CH3); 31P NMR (CDCl3, 242 MHz): no signal; M+ (ESI, m/z): calculated 827.1983 (C46H45Cl2N3ORu), found 827.1978 (C46H45Cl2N3ORu); IR (neat): IR: v(=C–H, aromatic) = 3055 cm−1, 3019 cm−1, 817 cm−1, v(CH3) = 2920 cm−1, 2848 cm−1, v(C=N & C=C, aromatic) = 1589–1444 cm−1, v(C–N, aliphatic) = 1260 cm−1, v(C–O, aliphatic) = 1164 cm−1, 1090 cm−1; 13C NMR (CDCl3, 150 MHz): δC = 292.35, 214.20, 170.84, 151.41, 149.19, 143.26, 139.15, 137.52, 137.37, 129.80, 129.67, 129.25, 128.93, 128.31, 126.97, 126.92, 126.78, 126.60, 126.52, 126.32, 122.27, 121.14, 92.72, 51.31, 20.95, 19.09, 18.85.
Benzylidene-chloro(1,3-bis-(2,4,6-trimethylphenyl)-2-imidazolidinylidene)-[1-(2′-pyridinyl)-1-(4′-methoxyphenyl),1-phenyl-methanolato]ruthenium, 15: Yield 0.46 g, 95.8%, dark green powder, dec.p.: 73.5 °C, 1H NMR (CDCl3, 600 MHz, CDCl3): δH = 17.10/17.08 (2s, 1H, carbene-H), 9.63–9.60 (2d, 1H, J = 5.74 Hz, H-6 of C5H4N), 7.61 (t, 1H, J = 7.74 Hz, H-4 of C5H4N), 7.11–7.28 (m, 12H, 3-, 5-H of C5H4N, & all H of 2Ph), 7.08 (d, 2H, J = 8.16 Hz, H-2 & H-6 of substituted Ph), 6.94 (m, 2H, H-3 & H-5 of substituted Ph), 6.73–6.81 (2d, 4H, J = 8.55 Hz, mesityl-H), 4.01 (m, 4H, N(CH2)2N), 3.77 (s, 3H, OCH3), 2.62/2.28/2.20 (3s, 18H, 6CH3 of mesityl); 31P NMR (CDCl3, 242 MHz): no signal; M+ (ESI, m/z): calculated 823.2478 (C47H48ClN3O2Ru), found 823.2407 (C47H48ClN3O2Ru); IR (neat): v(=C-H, aromatic) = 3055 cm−1, 3017 cm−1, 828 cm−1, v(CH3) = 2927 cm−1, 2851 cm−1, v(C=N & C=C, aromatic) = 1606 cm−1, 1590 cm−1, 1507 cm−1, v(C–N, aliphatic) = 1245 cm−1, v(C–O, aliphatic) = 1164 cm−1, 1029 cm−1, v(C–O, phenol) = 1297 cm−1; 13C NMR (CDCl3, 150 MHz): δC = 291.16, 214.47, 171.48, 151.50, 149.87, 139.23, 137.20, 136.63, 135.74, 133.92, 129.65, 128.85, 128.44, 126.94, 126.83, 126.59, 126.37, 126.28, 126.24, 120.88, 112.30, 92.77, 55.23, 51.32, 20.95, 19.09, 18.90.


 ,
, 

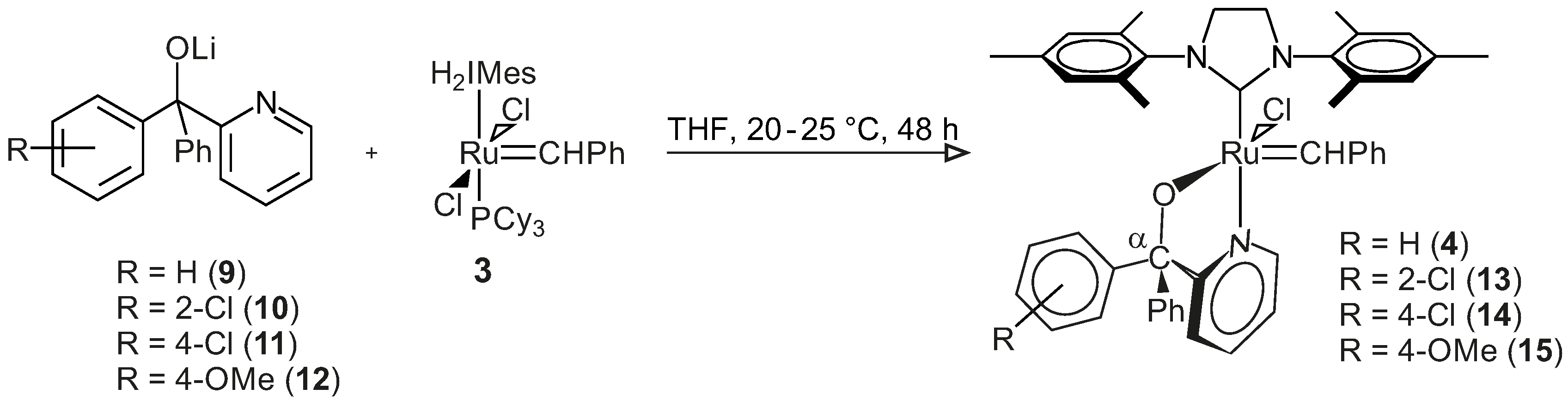


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
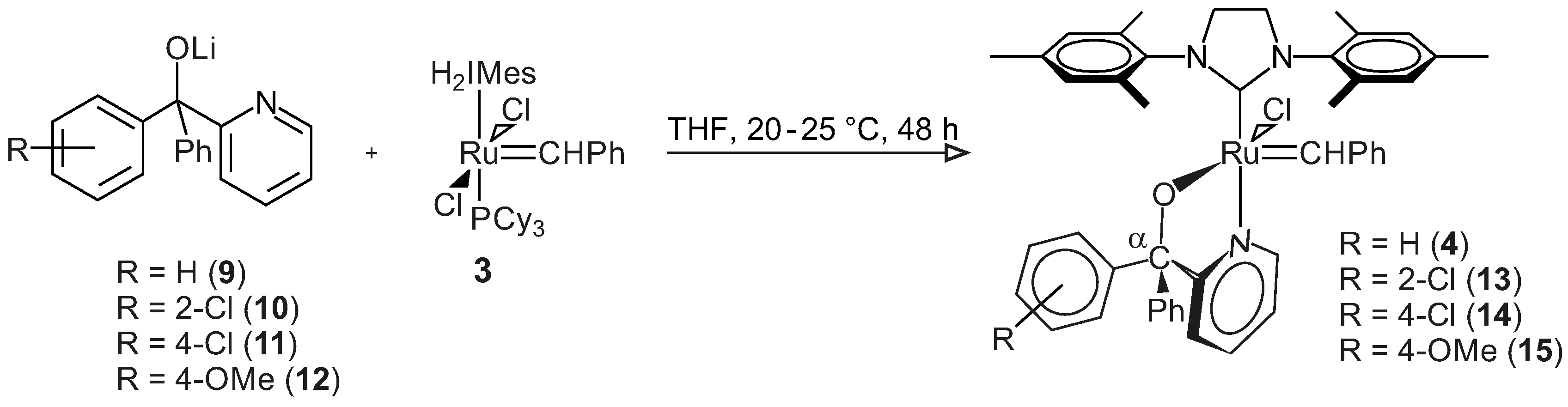{kind=link}