
Facile Synthesis of Vanadium Oxide/Reduced Graphene Oxide Composite Catalysts for Enhanced Hydroxylation of Benzene to Phenol



Abstract
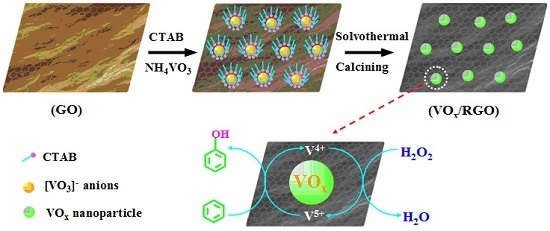
:
1. Introduction
2. Results and Discussion
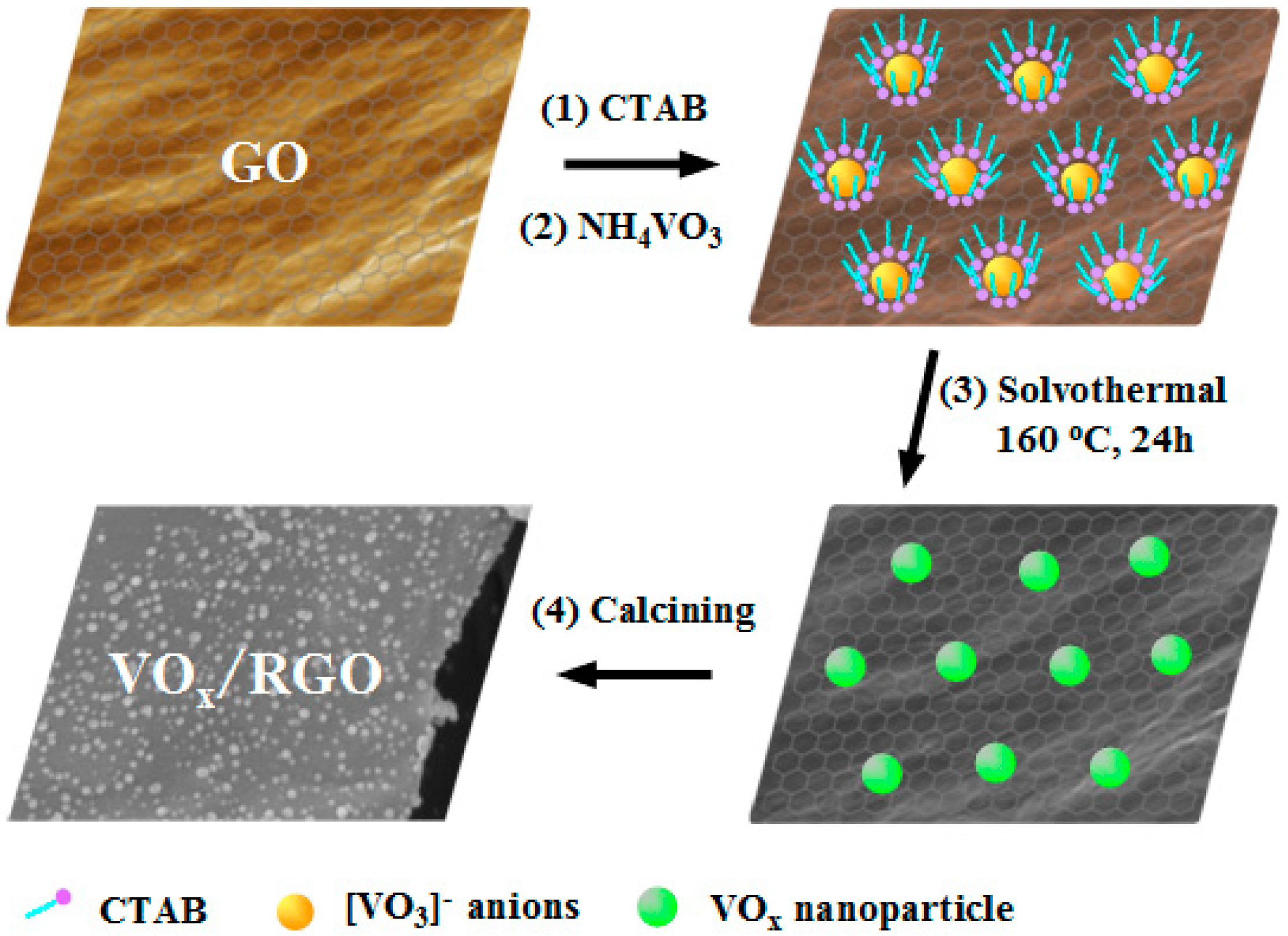
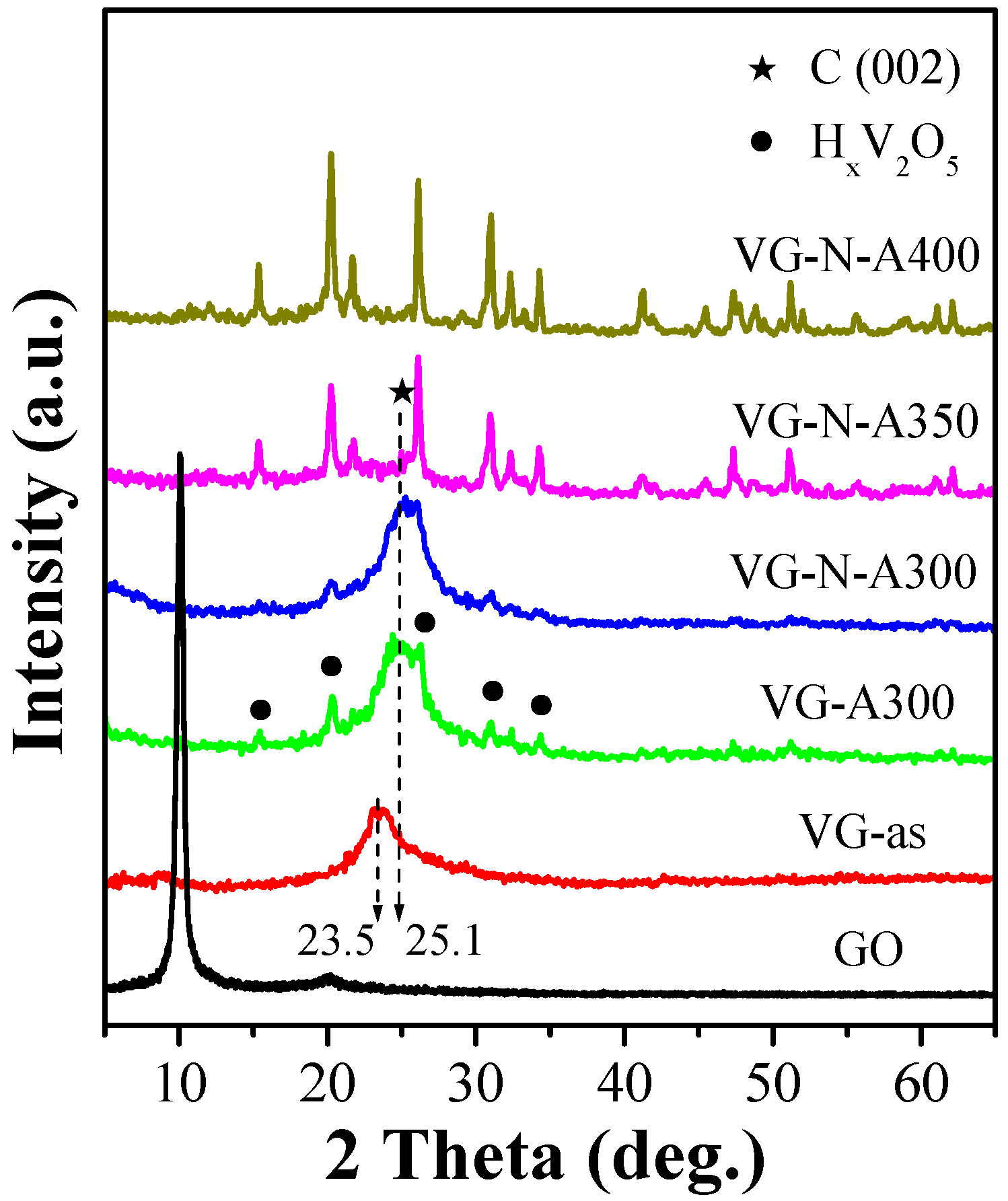
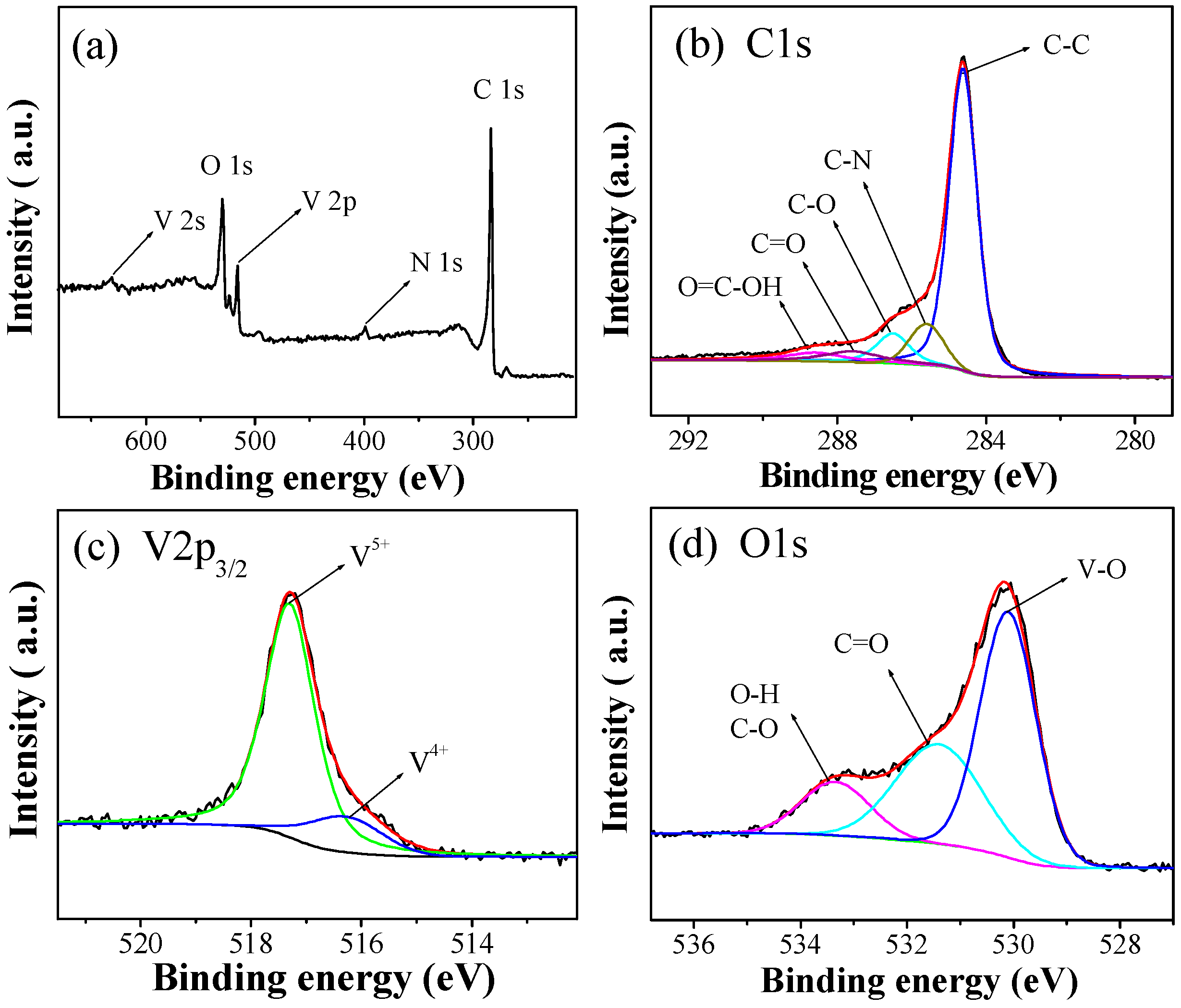
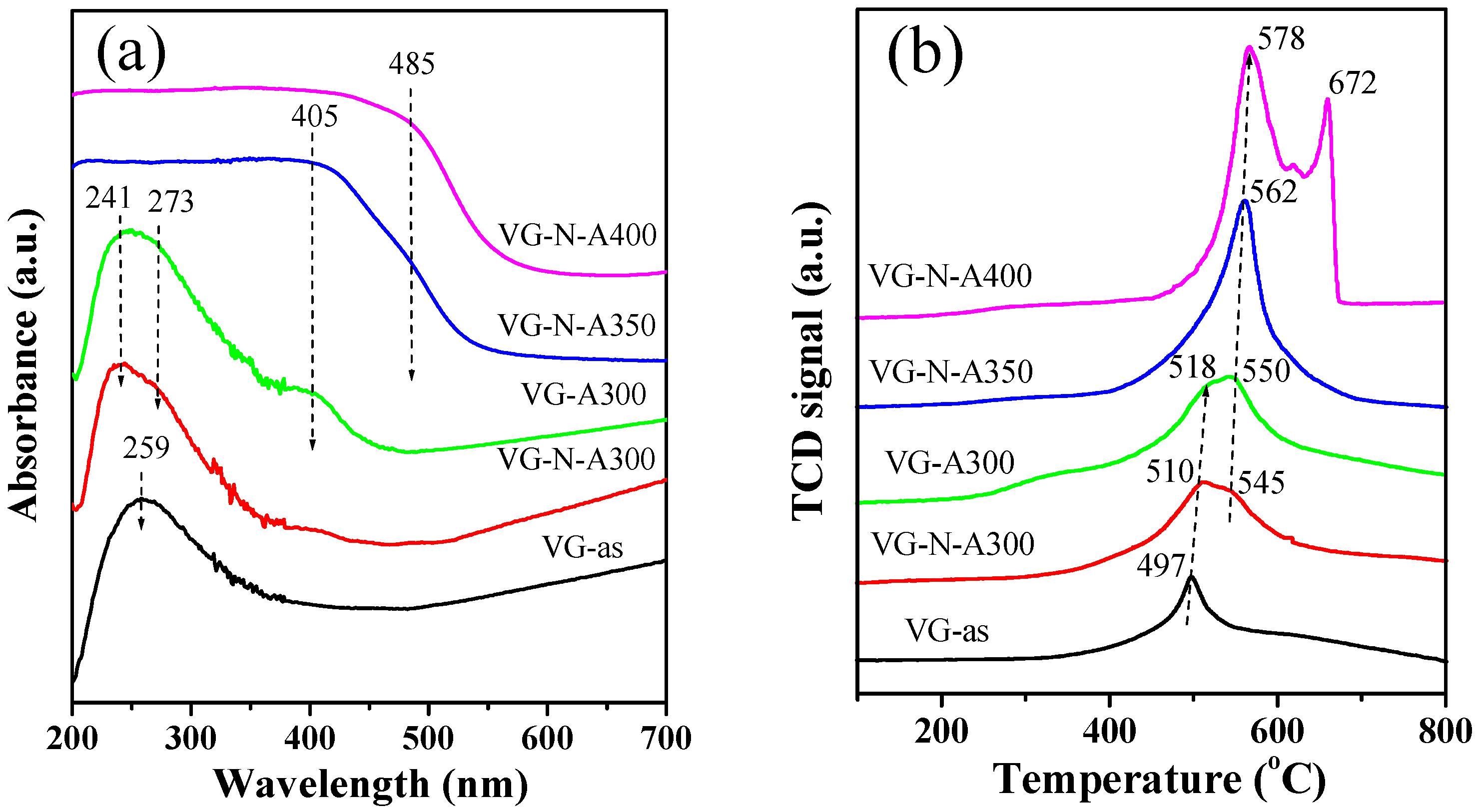
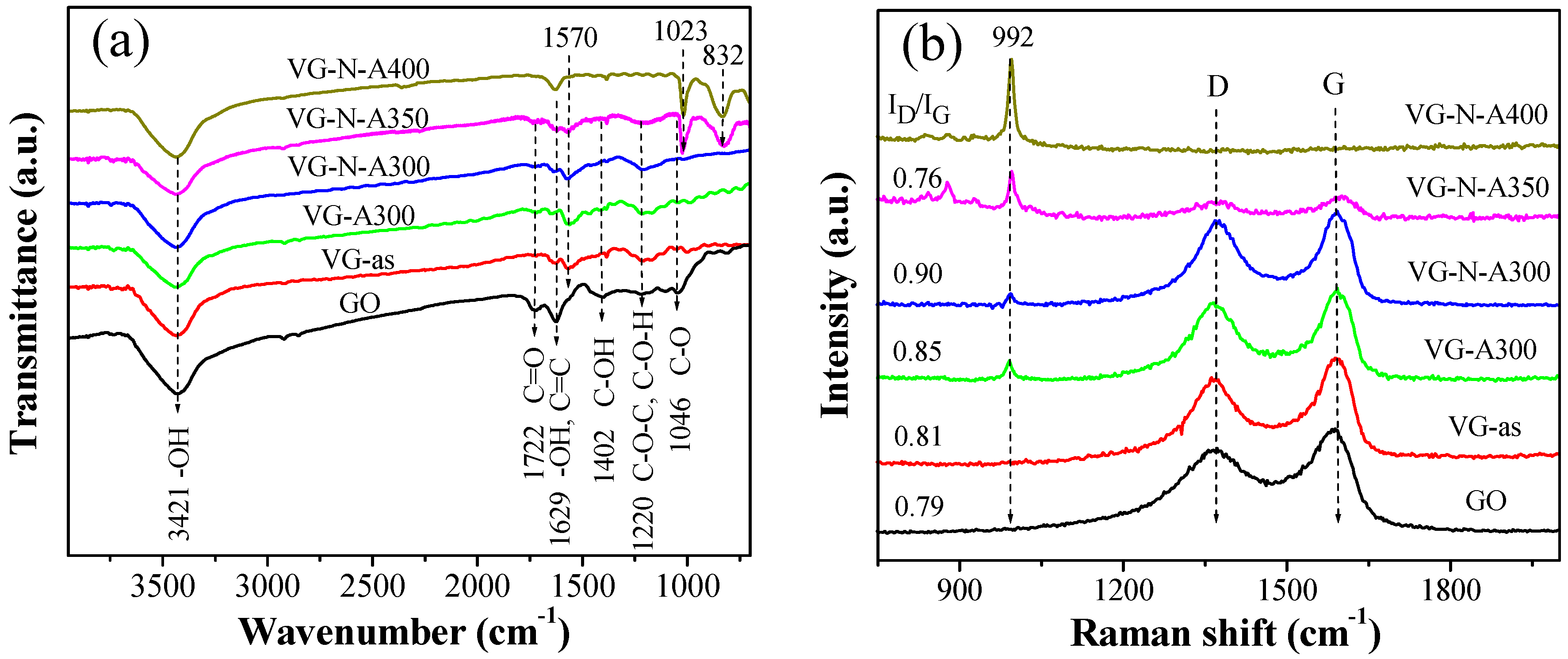
2.1. Synthesis and Characterization of VOx/RGO Composites
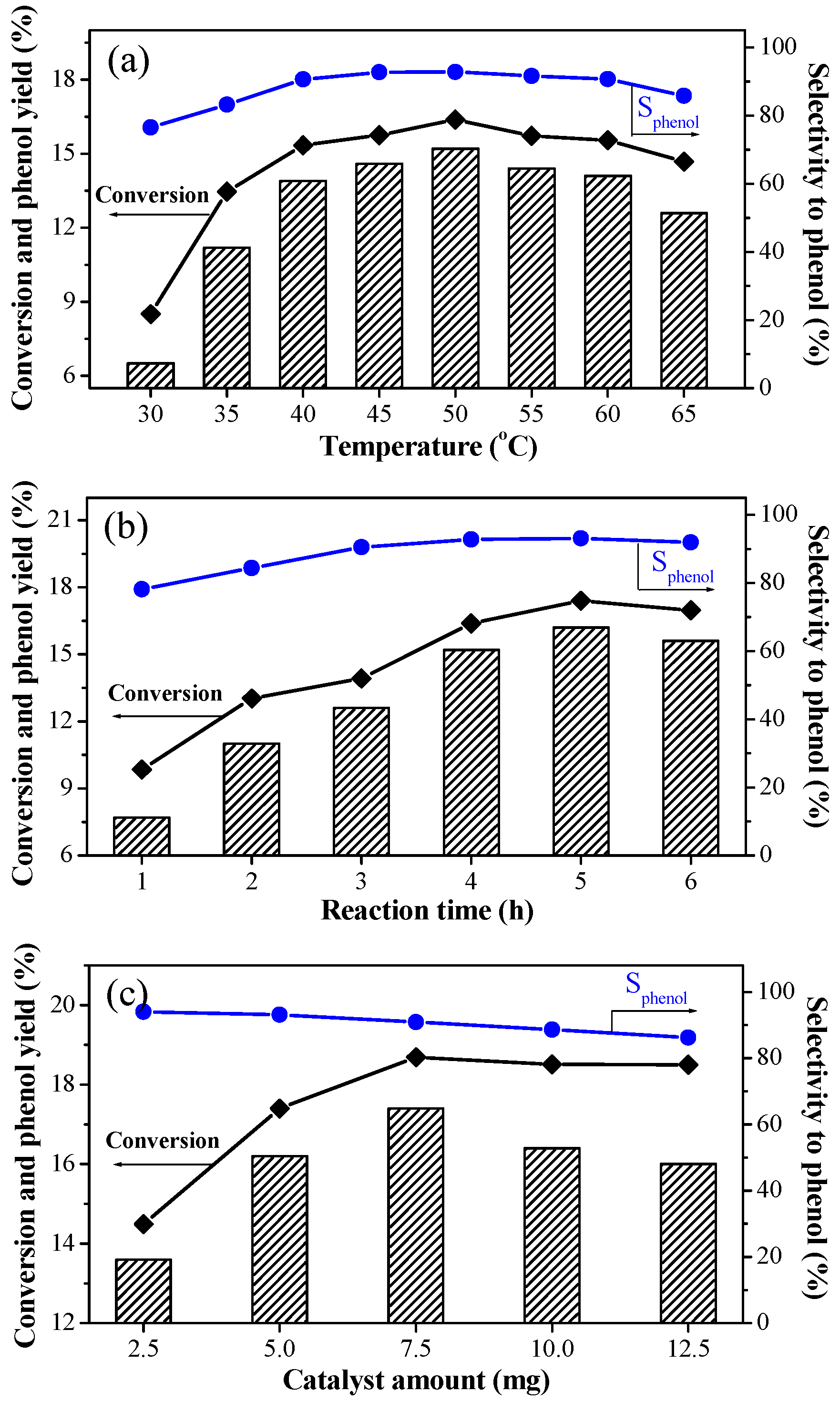
2.2. Hydroxylation of Benzene
3. Experimental Section
3.1. Materials
3.2. Synthesis of Graphite Oxide
3.3. Synthesis of VOx/RGO
3.4. Characterization
3.5. Catalytic Evaluation
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schmidt, R.J. Industrial catalytic processes-phenol production. Appl. Catal. A 2005, 280, 89–103. [Google Scholar] [CrossRef]
- Balducci, L.; Bianchi, D.; Bortolo, R.; D’Aloisio, R.; Ricci, M.; Tassinari, R.; Ungarelli, R. Direct oxidation of benzene to phenol with hydrogen peroxide over a modified titanium silicalite. Angew. Chem. Int. Ed. 2003, 42, 4937–4940. [Google Scholar] [CrossRef] [PubMed]
- Yuranov, I.; Bulushev, D.A.; Renken, A.; Kiwi-Minsker, L. Benzene to phenol hydroxylation with N2O over Fe-β and Fe-ZSM-5: Comparison of activity per Fe-Site. Appl. Catal. A 2007, 319, 128–136. [Google Scholar] [CrossRef]
- Li, Y.; Feng, Z.; van Santen, R.A.; Hensen, E.J.M.; Li, C. Surface functionalization of SBA-15-ordered mesoporous silicas: Oxidation of benzene to phenol by nitrous oxide. J. Catal. 2008, 255, 190–196. [Google Scholar] [CrossRef]
- Borah, P.; Ma, X.; Nguyen, K.T.; Zhao, Y.L. A vanadyl complex grafted to periodic mesoporous organosilica: A green catalyst for selective hydroxylation of benzene to phenol. Angew. Chem. Int. Ed. 2012, 51, 7756–7761. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.L.; Zhan, X.L.; Niu, X.Y.; Li, J.; Yuan, F.L.; Zhu, Y.J.; Fu, H.G. Facile Synthesis of Co-SBA-16 Mesoporous Molecular Sieves with EISA Method and Their Applications for Hydroxylation of Benzene. Microporous Mesoporous Mater. 2014, 185, 97–106. [Google Scholar] [CrossRef]
- Bal, R.; Tada, M.; Sasaki, T.; Iwasawa, Y. Direct phenol synthesis by selective oxidation of benzene with molecular oxygen on an interstitial-N/Re cluster/zeolite catalyst. Angew. Chem. Int. Ed. 2006, 45, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.H.; Lv, X.H.; Wang, X.W.; Yan, S.; Gao, X.H.; Xu, J.; Ma, H.; Jiao, Y.J.; Li, F.Y.; Chen, J.Z. Direct hydroxylation of benzene to phenol with molecular oxygen over vanadium oxide nanospheres and study of its mechanism. RSC Adv. 2015, 5, 94164–94170. [Google Scholar] [CrossRef]
- Niwa, S.; Eswaramoorthy, M.; Nair, J.; Raj, A.; Itoh, N.; Shoji, H.; Namba, T.; Mizukami, F. A one-step conversion of benzene to phenol with a palladium membrane. Science 2002, 295, 105–107. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Naito, H.; Hibino, T. Electrochemical oxidation of benzene to phenol. Angew. Chem. Int. Ed. 2012, 51, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.K.; Li, G.Y.; Zhu, L.F.; Yan, Y.; Wu, G.; Hu, C.W. Low temperature hydroxylation of benzene to phenol by hydrogen peroxide over Fe/activated carbon catalyst. J. Mol. Catal. A 2007, 272, 169–173. [Google Scholar] [CrossRef]
- Ohkubo, K.; Kobayashi, T.; Fukuzumi, S. Direct oxygenation of benzene to phenol using quinolinium ions as homogeneous photocatalysts. Angew. Chem. Int. Ed. 2011, 50, 8652–8655. [Google Scholar] [CrossRef] [PubMed]
- Shoji, O.; Kunimatsu, T.; Kawakami, N.; Watanabe, Y. Highly selective hydroxylation of benzene to phenol by wild-type cytochrome P450BM3 assisted by decoy molecules. Angew. Chem. Int. Ed. 2013, 52, 6606–6610. [Google Scholar] [CrossRef] [PubMed]
- Elkasabi, Y.; Mullen, C.A.; Boateng, A.A. Aqueous extractive upgrading of bio-oils created by tail-gas reactive pyrolysis to produce pure hydrocarbons and phenols. ACS Sustain. Chem. Eng. 2015, 3, 2809–2816. [Google Scholar] [CrossRef]
- Mukarakate, C.; McBrayer, J.D.; Evans, T.J.; Budhi, S.; Robichaud, D.J.; Iisa, K.; ten Dam, J.; Watson, M.J.; Baldwin, R.M.; Nimlos, M.R. Catalytic fast pyrolysis of biomass: The reactions of water and aromatic intermediates produces phenols. Green Chem. 2015, 17, 4217–4227. [Google Scholar] [CrossRef]
- Ding, G.D.; Wang, W.T.; Jiang, T.; Han, B.X.; Fan, H.L.; Yang, G.Y. Highly selective synthesis of phenol from benzene over a vanadium-doped graphitic carbon nitride catalyst. ChemCatChem 2013, 5, 192–200. [Google Scholar] [CrossRef]
- Kharat, A.N.; Moosavikia, S.; Jahromi, B.T.; Badiei, A. Liquid phase hydroxylation of benzene to phenol over vanadium substituted keggin anion supported on amine functionalized SBA-15. J. Mol. Catal. A 2011, 348, 14–19. [Google Scholar] [CrossRef]
- Xu, J.; Jiang, Q.; Shang, J.K.; Wang, Y.; Li, Y.X. A Schiff-base-type vanadyl complex grafted on mesoporous carbon nitride: A new efficient catalyst for hydroxylation of benzene to phenol. RSC Adv. 2015, 5, 92526–92533. [Google Scholar] [CrossRef]
- Guo, C.; Du, W.D.; Chen, G.; Shi, L.; Sun, Q. Influence of temperature on hydroxylation of benzene to phenol using molecular oxygen catalyzed by V/SiO2. Catal. Commun. 2013, 37, 19–22. [Google Scholar] [CrossRef]
- Gao, X.H.; Xu, J. A new application of clay-supported vanadium oxide catalyst to selective hydroxylation of benzene to phenol. Appl. Clay Sci. 2006, 33, 1–6. [Google Scholar] [CrossRef]
- Guo, B.; Zhu, L.F.; Hu, X.K.; Zhang, Q.; Tong, D.M.; Li, G.Y.; Hu, C.W. Nature of vanadium species on vanadium silicalite-1 zeolite and their stability in hydroxylation reaction of benzene to phenol. Catal. Sci. Technol. 2011, 1, 1060–1067. [Google Scholar] [CrossRef]
- Shang, S.S.; Chen, B.; Wang, L.Y.; Dai, W.; Zhang, Y.; Gao, S. High-performance recyclable V–N–C catalysts for the direct hydroxylation of benzene to phenol using molecular oxygen. RSC Adv. 2015, 5, 31965–31971. [Google Scholar] [CrossRef]
- Zhu, Y.J.; Dong, Y.L.; Zhao, L.N.; Yuan, F.L. Preparation and characterization of mesoporous VOx/SBA-16 and their application for the direct catalytic hydroxylation of benzene to phenol. J. Mol. Catal. A 2010, 315, 205–212. [Google Scholar] [CrossRef]
- Hu, L.Y.; Yue, B.; Chen, X.Y.; He, H.Y. Direct hydroxylation of benzene to phenol on Cu-V bimetal modified HMS catalysts. Catal. Commun. 2014, 43, 179–183. [Google Scholar] [CrossRef]
- Xu, J.Q.; Liu, H.H.; Yang, R.G.; Li, G.Y.; Hu, C.W. Hydroxylation of benzene by activated carbon catalyst. Chin. J. Catal. 2012, 33, 1622–1630. [Google Scholar] [CrossRef]
- Song, S.Q.; Yang, H.X.; Rao, R.C.; Liu, H.D.; Zhang, A.M. Defects of multi-walled carbon nanotubes as active sites for benzene hydroxylation to phenol in the presence of H2O2. Catal. Commun. 2010, 11, 783–787. [Google Scholar] [CrossRef]
- Yang, J.H.; Sun, G.; Gao, Y.J.; Zhao, H.B.; Tang, P.; Tan, J.; Lu, A.H.; Ma, D. Direct catalytic oxidation of benzene to phenol over metal-free graphene-based catalyst. Energy Environ. Sci. 2013, 6, 793–798. [Google Scholar] [CrossRef]
- Jin, M.M.; Yang, R.G.; Zhao, M.F.; Li, G.Y.; Hu, C.W. Application of Fe/activated carbon catalysts in the hydroxylation of phenol to dihydroxybenzenes. Ind. Eng. Chem. Res. 2014, 53, 2932–2939. [Google Scholar] [CrossRef]
- Zhu, J.J.; Kailasam, K.; Fischer, A.; Thomas, A. Supported cobalt oxide nanoparticles as catalyst for aerobic oxidation of alcohols in liquid phase. ACS Catal. 2011, 1, 342–347. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, H.H.; Li, G.Y.; Hu, C.W. Continuous flow reactor for hydroxylation of benzene to phenol by hydrogen peroxide. Chin. J. Chem. Phys. 2012, 25, 585–591. [Google Scholar] [CrossRef]
- Zhang, H.B.; Pan, X.L.; Han, X.W.; Liu, X.M.; Wang, X.F.; Shen, W.L.; Bao, X.H. Enhancing chemical reactions in a confined hydrophobic environment: An NMR study of benzene hydroxylation in carbon nanotubes. Chem. Sci. 2013, 4, 1075–1078. [Google Scholar] [CrossRef]
- Wang, X.Y.; Zhang, X.F.; He, X.L.; Ma, A.; Le, L.J.; Lin, S. Facile electrodeposition of flower-like PMo12-Pt/rGO composite with enhanced electrocatalytic activity towards methanol oxidation. Catalysts 2015, 5, 1275–1288. [Google Scholar] [CrossRef]
- Wang, W.T.; Ding, G.D.; Jiang, T.; Zhang, P.; Wu, T.B.; Han, B.X. Facile one-pot synthesis of VxOy@C catalysts using sucrose for the direct hydroxylation of benzene to phenol. Green Chem. 2013, 15, 1150–1154. [Google Scholar] [CrossRef]
- Song, S.Q.; Jiang, S.J.; Rao, R.C.; Yang, H.X.; Zhang, A.M. Bicomponent VO2-Defects/MWCNT catalyst for hydroxylation of benzene to phenol: Promoter effect of defects on catalytic performance. Appl. Catal. A 2011, 401, 215–219. [Google Scholar] [CrossRef]
- Wang, C.; Hu, L.Y.; Hu, Y.C.; Ren, Y.H.; Chen, X.Y.; Yue, B.; He, H.Y. Direct hydroxylation of benzene to phenol over metal oxide supported graphene oxide catalysts. Catal. Commun. 2015, 68, 1–5. [Google Scholar] [CrossRef]
- Li, D.; Kaner, R.B. Graphene-based materials. Science 2008, 320, 1170–1171. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, R.K.; Soinb, N.; Roy, S.S. Role of graphene/metal oxide composites as photocatalysts, adsorbents and disinfectants in water treatment: A review. RSC Adv. 2014, 4, 3823–3851. [Google Scholar] [CrossRef]
- Song, W.N.; He, C.Y.; Zhang, W.; Gao, Y.C.; Yang, Y.X.; Wu, Y.Q.; Chen, Z.M.; Li, X.C.; Dong, Y.L. Synthesis and nonlinear optical properties of reduced graphene oxide hybrid material covalently functionalized with zinc phthalocyanine. Carbon 2014, 77, 1020–1030. [Google Scholar] [CrossRef]
- Chen, S.Q.; Wang, Y. Microwave-assisted synthesis of a Co3O4-graphene sheet-on-sheet nanocomposite as a superior anode material for Li-ion batteries. J. Mater. Chem. 2010, 20, 9735–9739. [Google Scholar] [CrossRef]
- Yan, H.J.; Tian, C.G.; Sun, L.; Wang, B.; Wang, L.; Yin, J.; Wu, A.P.; Fu, H.G. Small-sized and highly dispersed WN from [SiO4(W3O9)4]4− clusters loading on GO-derived graphene as promising carriers for methanol electro-oxidation. Energy Environ. Sci. 2014, 7, 1939–1949. [Google Scholar] [CrossRef]
- Wang, P.; Wang, J.; Wang, X.; Yu, H.G.; Yu, J.G.; Lei, M.; Wang, Y.G. One-step synthesis of easy-recycling TiO2-rGO nanocomposite photocatalysts with enhanced photocatalytic activity. Appl. Catal. B 2013, 132–133, 452–459. [Google Scholar] [CrossRef]
- Hu, J.; Dong, Y.L.; Chen, X.J.; Zhang, H.J.; Zheng, J.M.; Wang, Q.; Chen, X.G. A highly efficient catalyst: In situ growth of Au nanoparticles on graphene oxide-Fe3O4 nanocomposite support. Chem. Eng. J. 2014, 236, 1–8. [Google Scholar] [CrossRef]
- Grigorieva, A.V.; Badalyan, S.M.; Goodilin, E.A.; Rumyantseva, M.N.; Gaskov, A.M.; Birkner, A.; Tretyakov, Y.D. Synthesis, structure, and sensor properties of vanadium pentoxide nanorods. Eur. J. Inorg. 2010, 2010, 5247–5253. [Google Scholar] [CrossRef]
- Stankovich, S.; Dikin, D.A.; Piner, R.D.; Kohlhaas, K.A.; Kleinhammes, A.; Jia, Y.; Wu, Y.; Nguyen, S.T.; Ruoff, R.S. Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide. Carbon 2007, 45, 1558–1565. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, Y.; Liu, G.; Xu, L.Q.; Chen, J.N.; Zhu, C.X.; Neoh, K.G.; Kang, E.T. Push-pull archetype of reduced graphene oxide functionalized with polyfluorene for nonvolatile rewritable memory. J. Polym. Sci. A 2012, 50, 378–387. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, S.B.; Lv, L.P.; Lieberwirth, I.; Zhang, L.C.; Ding, C.X.; Chen, C.H. A composite film of reduced graphene oxide modified vanadium oxide nanoribbons as a free standing cathode material for rechargeable lithium batteries. J. Power Source 2013, 241, 168–172. [Google Scholar] [CrossRef]
- Zhao, L.N.; Dong, Y.L.; Zhan, X.L.; Cheng, Y.; Zhu, Y.J.; Yuan, F.L.; Fu, H.G. One-pot hydrothermal synthesis of mesoporous V-SBA-16 with a function of the pH of the initial gel and its improved catalytic performance for benzene hydroxylation. Catal. Lett. 2012, 142, 619–626. [Google Scholar] [CrossRef]
- Hummers, W.S.; Offeman, R.E. Preparation of graphitic oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | V b (wt %) | Benzene Conversion (%) | Selectivity to Phenol (%) | Selectivity to BQ (%) | Selectivity to HQ (%) | Yield of Phenol (%) | TON c |
|---|---|---|---|---|---|---|---|
| RGO | - | 0.5 | 75.8 | 24.2 | - | 0.4 | - |
| V2O5 | - | 12.9 | 71.5 | 3.6 | 24.9 | 9.2 | - |
| VG-as | 9.6 | 12.0 | 93.0 | - | 7.0 | 11.2 | 39.9 |
| VG-A300 | 12.0 | 13.7 | 89.6 | 1.7 | 8.7 | 12.3 | 35.2 |
| VG-N-A300 | 10.8 | 15.7 | 92.7 | - | 7.3 | 14.6 | 46.3 |
| VG-N-A350 | 32.2 | 13.7 | 86.8 | 2.3 | 10.9 | 11.9 | 12.6 |
| VG-N-A400 | 40.8 | 13.6 | 78.5 | 3.4 | 18.1 | 10.7 | 9.0 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, Y.; Niu, X.; Song, W.; Wang, D.; Chen, L.; Yuan, F.; Zhu, Y. Facile Synthesis of Vanadium Oxide/Reduced Graphene Oxide Composite Catalysts for Enhanced Hydroxylation of Benzene to Phenol. Catalysts 2016, 6, 74. https://doi.org/10.3390/catal6050074
Dong Y, Niu X, Song W, Wang D, Chen L, Yuan F, Zhu Y. Facile Synthesis of Vanadium Oxide/Reduced Graphene Oxide Composite Catalysts for Enhanced Hydroxylation of Benzene to Phenol. Catalysts. 2016; 6(5):74. https://doi.org/10.3390/catal6050074
Chicago/Turabian StyleDong, Yongli, Xiaoyu Niu, Weina Song, Dong Wang, Liqiang Chen, Fulong Yuan, and Yujun Zhu. 2016. "Facile Synthesis of Vanadium Oxide/Reduced Graphene Oxide Composite Catalysts for Enhanced Hydroxylation of Benzene to Phenol" Catalysts 6, no. 5: 74. https://doi.org/10.3390/catal6050074
APA StyleDong, Y., Niu, X., Song, W., Wang, D., Chen, L., Yuan, F., & Zhu, Y. (2016). Facile Synthesis of Vanadium Oxide/Reduced Graphene Oxide Composite Catalysts for Enhanced Hydroxylation of Benzene to Phenol. Catalysts, 6(5), 74. https://doi.org/10.3390/catal6050074