Abstract
The O–H bond breaking in H2O molecules on metal surfaces covered with pre-adsorbed oxygen atoms is an important topic in heterogeneous catalysis. The adsorption configurations of H2O and relevant dissociation species on clean and O-pre-adsorbed Fe(100) surfaces were investigated by density functional theory (DFT). The preferential sites for H2O, HO, O, and H were investigated on both surfaces. Both the first H abstraction from adsorbed H2O and the subsequent OH dissociation are exothermic on the O-pre-adsorbed Fe(100) surface. However, the pre-adsorbed O significantly reduces the kinetics energy barriers for both reactions. Our results confirmed that the presence of pre-adsorbed oxygen species could significantly promote H2O dissociation.
1. Introduction
Water (H2O) is one of the most widespread resources involved in many chemical processes [1]. It has also been proposed as the significant feedstock to produce H2 in steam and oxidative reforming technologies [2,3,4,5,6,7,8], which involve adsorption and dissociation of H2O molecules. A fundamental study of water-metal surface interactions will assist in understanding the reaction mechanisms of this heterogeneous catalytic reaction. Noble metals such as Ru [9,10], Rh [10,11], Pd [10], Ir [10,11,12,13], and Pt [10,14] are active for steam and oxidative reforming but are not preferred in conventional industrial reformers due to their high costs. This motivates studies on non-precious metal catalysts, such as Ni- [15,16,17], Cu- [18,19], Co- [20,21], and Fe- [22,23,24,25,26,27] based materials. Due to extremely low cost and high catalytic activity, Fe-based catalysts have caught great attentions.
Adsorbed hydroxyl, atomic oxygen and hydrogen are the simplest dissociation products of water on metal surfaces. Also, hydroxyl can further dissociate, giving rise to adsorbed atomic hydrogen and oxygen. Some experimental studies and theoretical calculations on adsorption/dissociation of water on the Fe(100) surface have been reported [28,29,30,31,32,33,34,35,36,37]. Eder and Terakura [33] demonstrated that water preferred to adsorb at the bridge site via an upright molecular adsorption configuration. However, it was unstable leading to a spontaneous dissociation into H and OH species. The same conclusion was achieved by Jung and Kang [34] using density functional theory (DFT) calculations. The proposed upright H2O configuration at the bridge site by these authors, however, is not in line with the flat-lying configuration on the top site on other metals, such as Ru, Rh, Pd, Pt, Al, Cu, Ag, and Au [38,39,40,41].
The promotion effect of pre-adsorbed O atoms on the surfaces of the catalysts has been discovered and investigated by both experimental and theoretical approaches. The pre-adsorbed O atoms have various degrees of effect towards H2O dissociation on different metals. A comprehensive review on this topic has been provided by Thiel et al. [42] and Henderson [43]. The positive effect of pre-adsorbed O atoms on water dissociation on Pd single crystals and thin films has been reported [44,45,46,47,48]. On the other hand, the water dissociation on the clean Pd surface has not been observed, in consistent with the DFT calculations [49]. Shavorskiy et al. [50] also found the formation of OH when water reacted with co-adsorbed O on the missing-row reconstructed Pt{110}(1 × 2) surface. A similar promotion effect on the Fe(111) surface has been reported [51]. Hung et al. [52] found that adsorbed oxygen facilitated water dissociation via the hydrogen transfer process on the pre-oxidized Fe(100) surface. Liu et al. [37] studied the effect of the O atom which came from the dissociation of the first H2O molecule on the consequent dissolution of the second H2O molecule. The O atom assisted H2O dissociation (O + H2O = 2OH) was kinetically favorable, and further OH dissociation was roughly thermo-neutral. In that case, the co-adsorption of H coming from the dissociation of the first H2O molecule on the Fe(100) might have an effect on the reaction pathway. In our manuscript, we studied the effect of two O atoms which came from the dissociation of O2 molecule on the adsorption and dissociation of water molecule.
Herein, we report a systematic DFT study of the adsorption of H2O and its dissociation fragments (OH, H and O) on clean and O-pre-adsorbed Fe(100) surfaces. The effect of two O atoms which came from the dissociation of O2 molecule on the adsorption and dissociation of water molecule has not been reported previously. The dissociation pathways on both surfaces were also discussed based on the calculation results to understand the effect from the pre-adsorbed O atoms.
2. Results and Discussion
2.1. Adsorption of H2O, OH, O and H on the Clean Fe(100) Surface
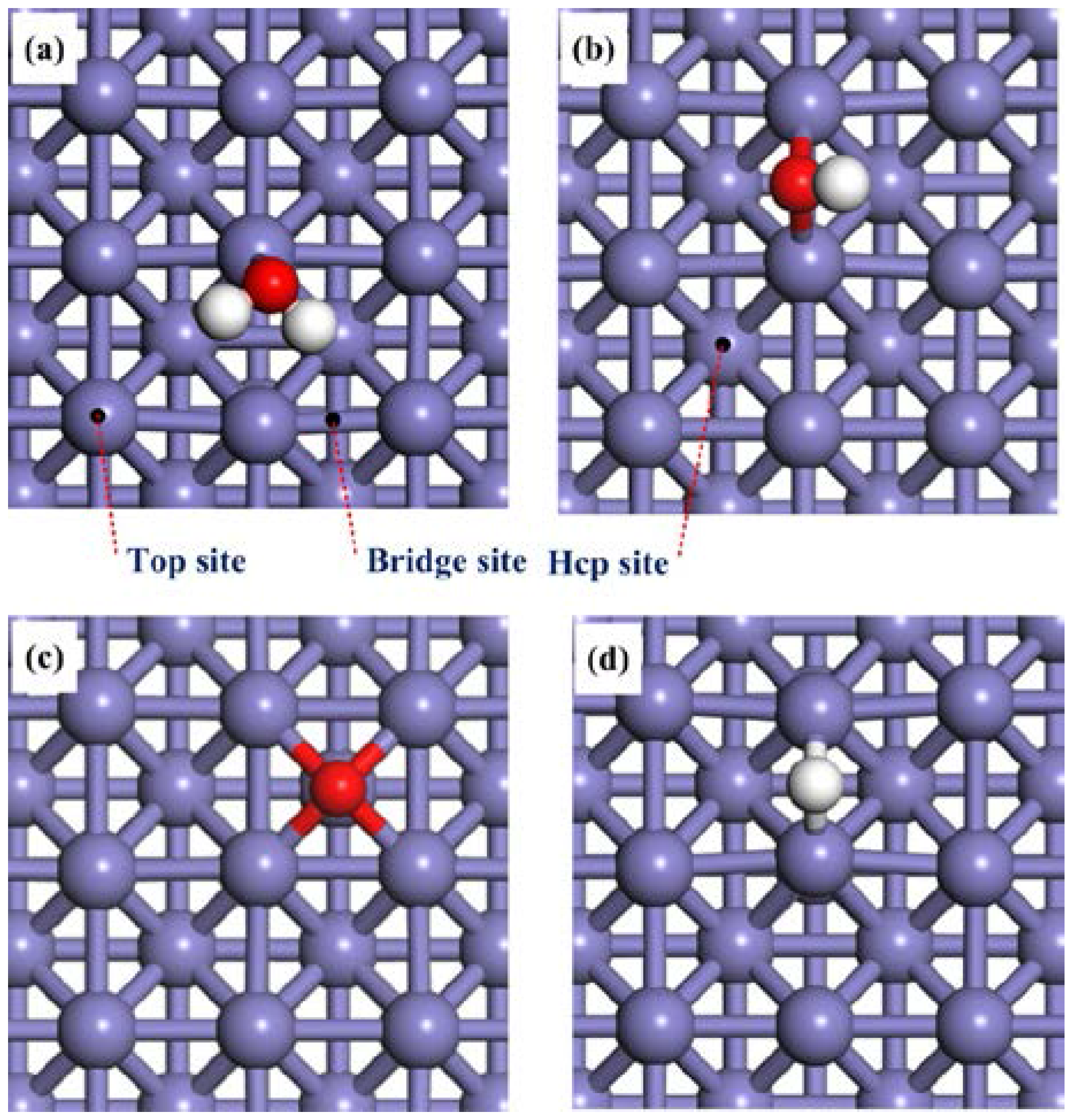
The adsorption properties of H2O and the dissociation products H, O, and OH species on a clean Fe(100) surface will be discussed first. The most stable adsorption configurations of these species are presented in Figure 1. The corresponding adsorption energies and structural details are summarized in Table 1 and Table 2. On the Fe(100) surface, H2O was found to be preferentially adsorbed on a top site via the oxygen atom with the H2O plane being almost parallel to the Fe surface (Figure 1a). The interaction between the Fe(100) surface and H2O molecule is weak, evidenced by a small adsorption energy (−0.65 eV). The O–H bond length of 0.98 Å and the ∠HOH bond angle of 105.4°, as listed in Table 2, are almost identical to those of a free H2O molecule (0.98 Å and 104.4°, respectively). Similar adsorption behaviors of H2O on Ni(111) [53,54,55], Ni(100) [55], Ni(110) [55], Cu(111) [56], Cu(100) [57] and Cu(110) [58,59] were observed.
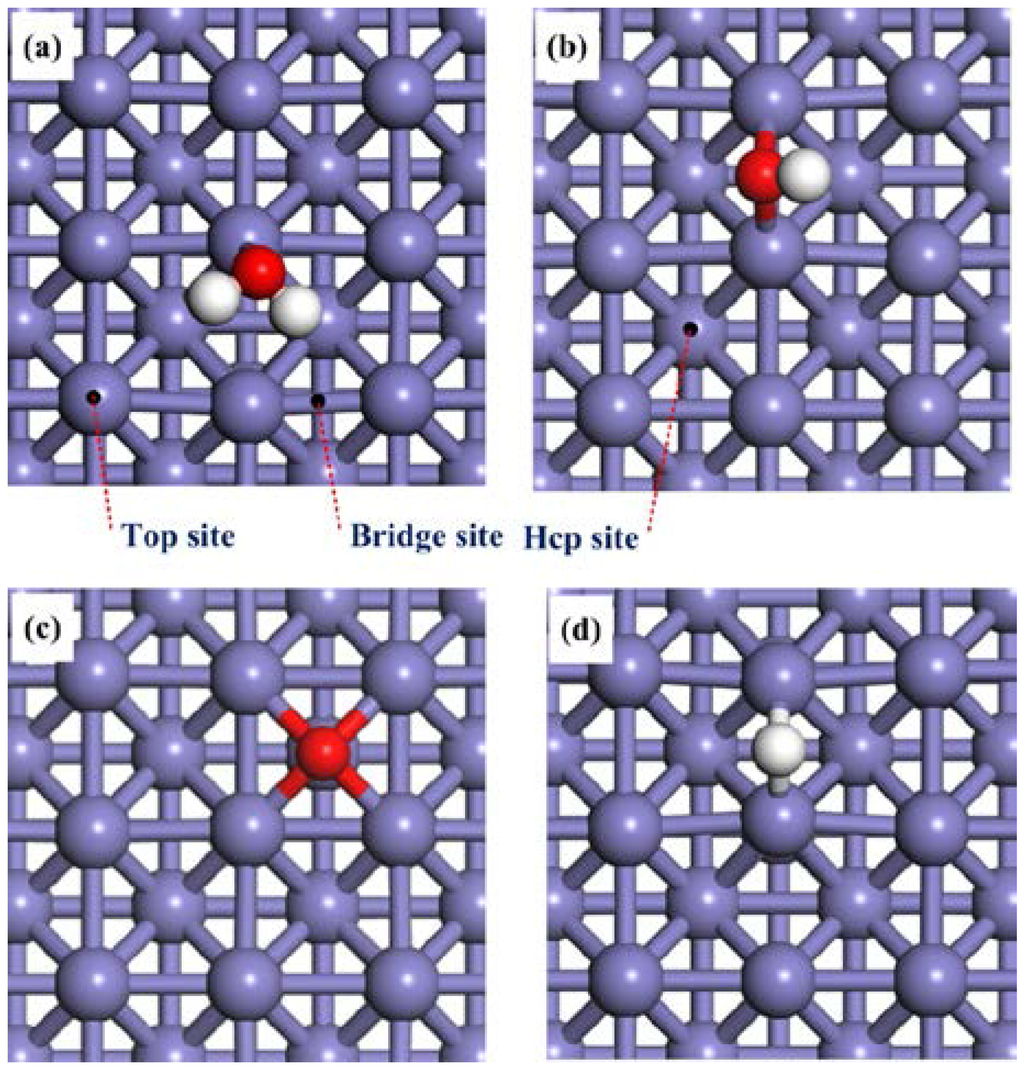
Figure 1.
Top views of the adsorption configurations of (a) H2O; (b) OH; (c) O; and (d) Hon clean Fe(100) surfaces. The slate blue, red and white balls stand for Fe, O and H atoms, respectively.

Table 1.
Adsorption energies (Eads, in eV) of H2O, OH, O and H on clean and O-pre-adsorbed Fe(100) surfaces.
Table 2.
Geometrical parameters for H2O, OH, O, and H adsorbed on clean and O-pre-adsorbed Fe(100) surfaces.
The adsorption energies of the OH species at the bridge and hcp sites are −3.93 eV and −3.84 eV, respectively. The adsorption at the hollow hcp site is locally unstable. These results suggest that the most stable adsorption site for OH on Fe(100) is at the bridge, consistent with the previous study [28,33]. The O–Fe bond length is1.99 Å, which is shorter than that of H2O adsorbed on clean Fe (100) surface (2.17 Å). Based on these results, one may conclude that the OH can be strongly adsorbed on the Fe(100) surface.
For the O species, Govender et al. [35] have found that the hcp and bridge sites were equally stable. On the other hand, Błoński et al. [60] and Lu et al. [30] suggested that O atom only occupied the hollow hcp sites. We found that the adsorption energy of O atom at the hollow hcp is higher than that at the bridge site (−3.67 eV at the former vs. −3.00 eV at the latter). On contrast, the top site is not stable for O adsorption. Thus, our results echo that reported by Błoński et al. [60] and Lu et al. [30].
Finally, for the H atom, the adsorption energies at the bridge and hollow hcp sites are −3.99 eV and −3.86 eV, respectively. The top site is not stable for H adsorption. This result suggested that the bridge is the most stable adsorption site for H atoms, which is in good agreement with previous DFT calculation results [61].
Based on the calculated adsorption energies in Table 1, it can be concluded that the interaction with the Fe(100) surface increases in the order of H2O < O < OH <H. In the following sections, we will discuss the dissociation of H2O on the Fe(100) surface based on these results.
2.2. Adsorption of H2O and OH on the O-Pre-adsorbed Fe(100) Surface
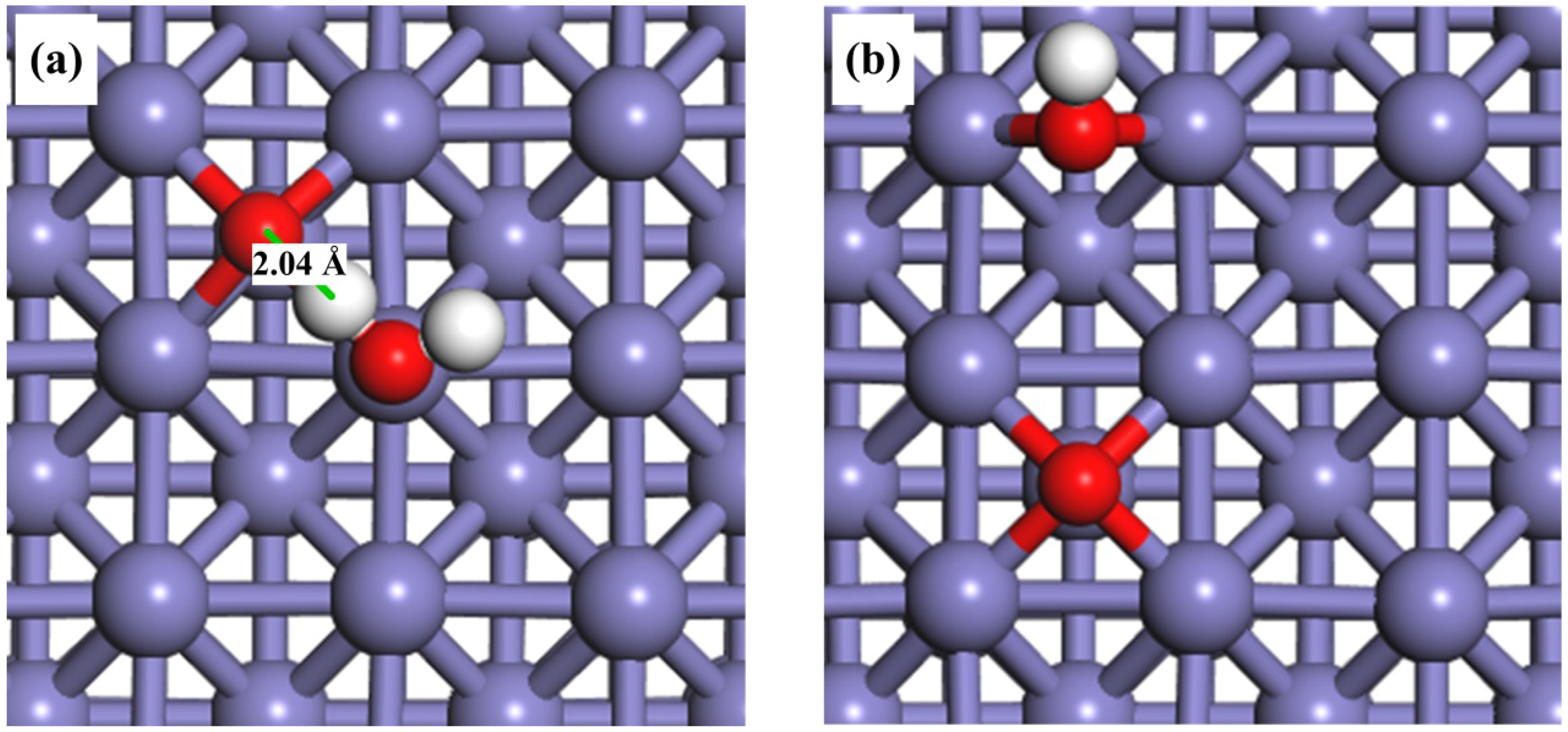
As mentioned in the Introduction, the pre-adsorbed oxygen atoms could play an important role in the activation of O–H bond. Therefore, it is of interest to investigate the adsorption behavior of H2O on O-pre-adsorbed Fe(100) surface. We found that the H2O molecule could also be adsorbed on the top site on the O-pre-adsorbed Fe surface, with a higher interaction energy (−1.13 eV) than that on a clean surface (−0.65 eV). The adsorption configuration was listed in Table 2. The pre-adsorbed electronegative O atom may increase the acidity of neighboring Fe atoms [62], and the adsorption energy of H2O via through-space electronic interaction. Another interesting finding is that the pre-adsorbed O atom and the H atom in the H2O may form a hydrogen bond with a bond distance of 2.04 Å in the co-adsorption configuration, as shown in Figure 2a.
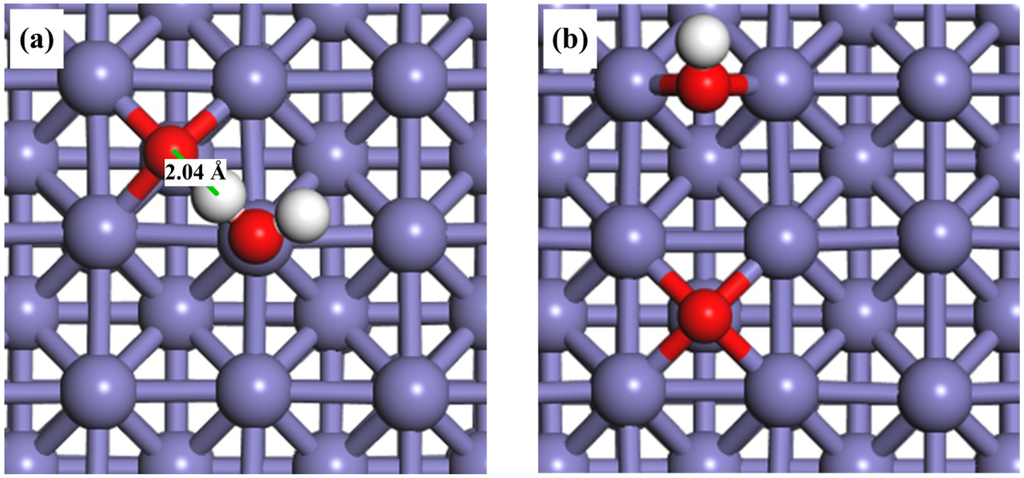
Figure 2.
Top views of the adsorption configurations of (a) H2O + O; and (b) OH+ O on O-pre-adsorbed Fe(100) surfaces.
As for the adsorption of OH on O-pre-adsorbed Fe(100) surface, both OH and O are placed on their favorable bridge sites. The most stable structure of OH co-adsorbed with O atom is shown in Figure 2b. The distance between the O atom of hydroxyl group and the nearest Fe atom remains 1.98 Å. However, the adsorption energy changes from −3.93 eV to −4.02 eV, suggesting a stronger interaction with the O-pre-adsorbed Fe(100) surface than with a clean surface. The bond distance of O–H is 0.98 Å, identical to that on the clean surface. Similar effects from the pre-adsorbed-O atom were also found on an Au(100) surface [63].
2.3. Reaction Mechanisms
2.3.1. H2O Dissociation on the Clean Fe(100) Surface
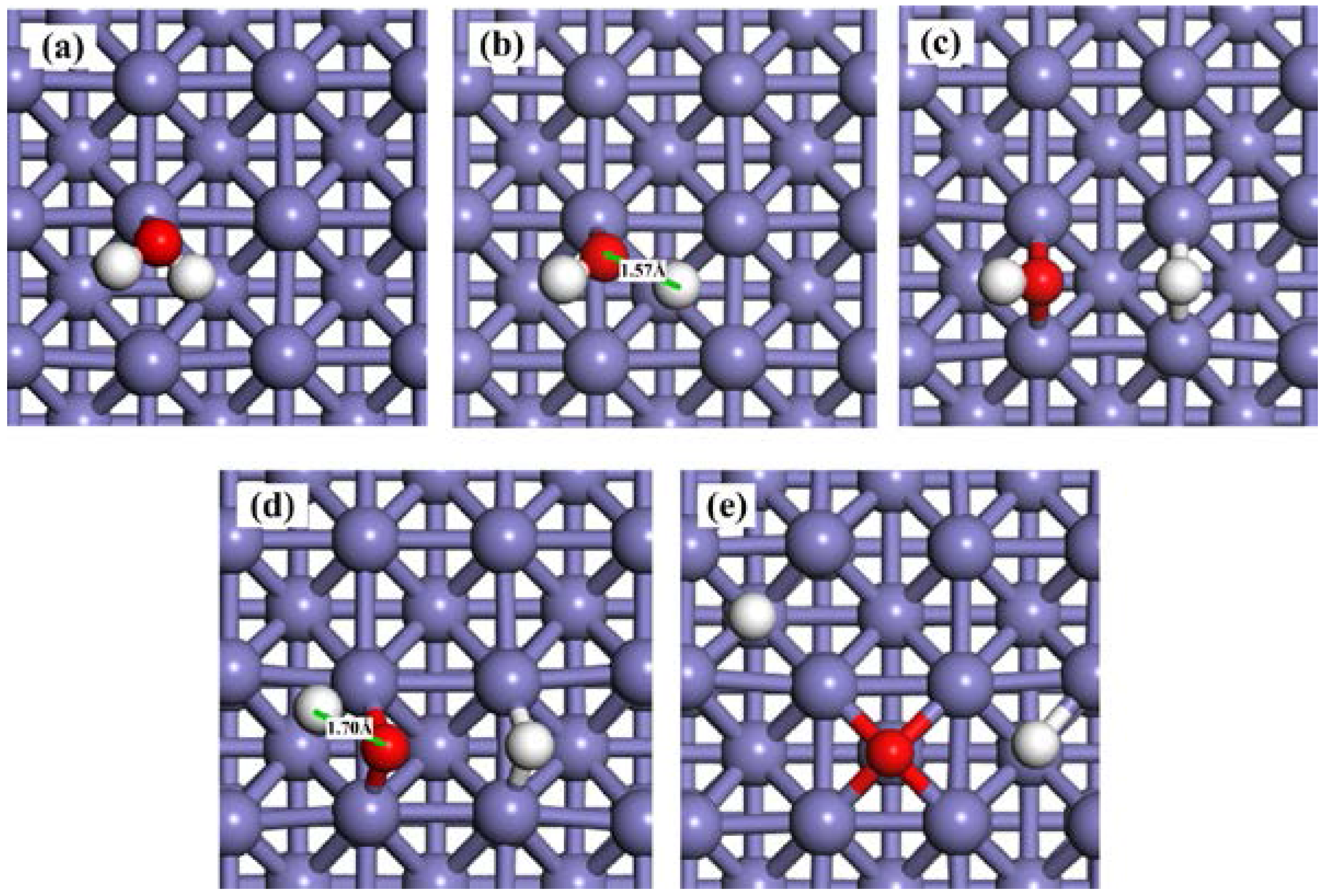
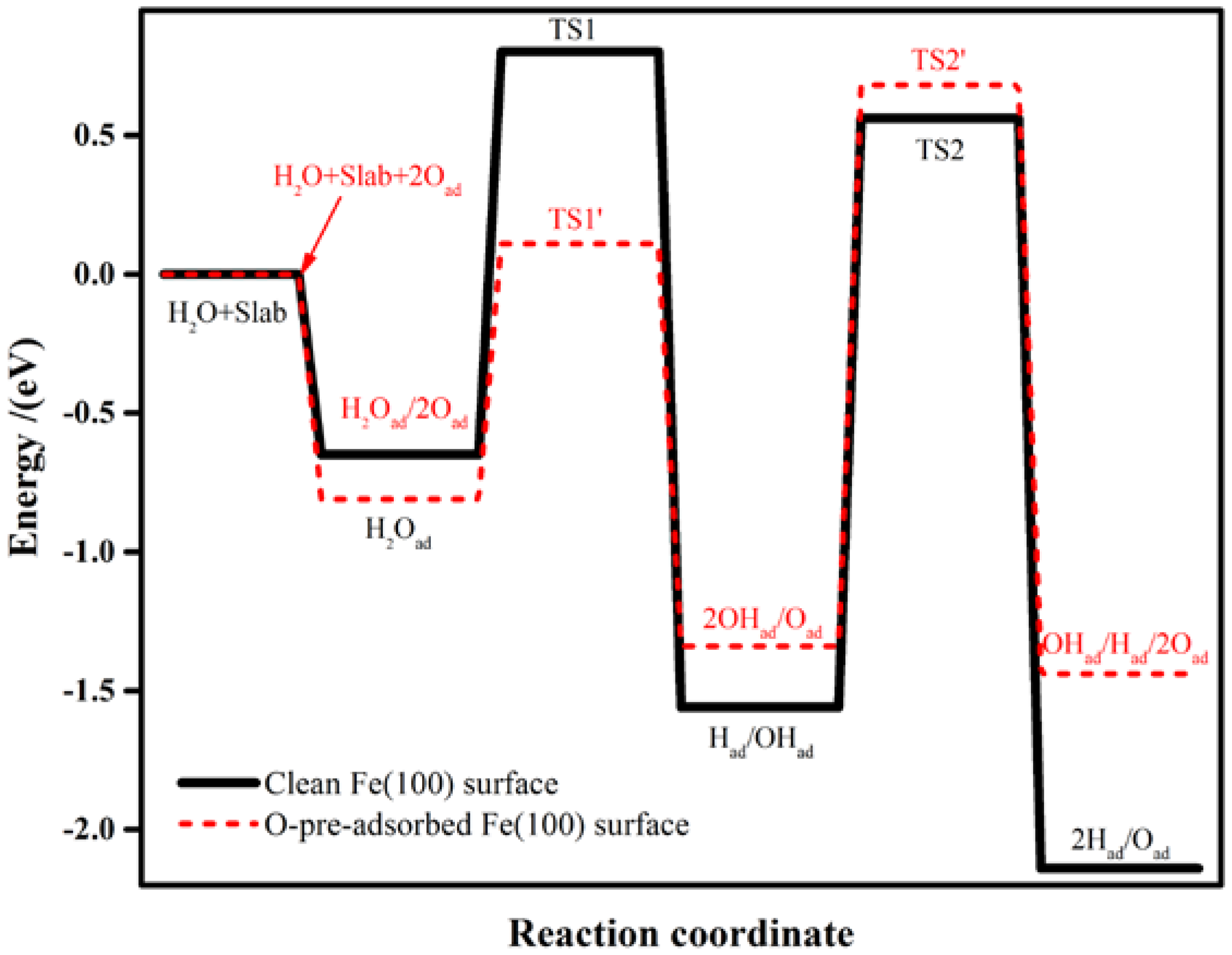
Water may partially dissociate to produce OHad and Had (Equation (1)), and follow the dissociation of OHad to form Had and Oad (Equation (2)). The calculated reaction energies and activation barriers for these two reaction steps are shown in Table 3. The associated transition states (TSs) are displayed in Figure 3. In the first step (Equation (1)), molecular water is adsorbed on the clean Fe(100) surface at the top site as discussed in Section 2.1 (Figure 3a). At TS1, one of the H atoms was stripped from the H2O molecule (Figure 3b). The bond distance between this H atom and O atom in the OHad is 1.57 Å. The energy barrier and reaction energy for this step are 1.45 eV and −0.91 eV (Figure 4), respectively. At the equilibrium state, both the H and OH radicals are adsorbed at the bridge sites (Figure 3c).
H2Oad → Had + OHad
Had + OHad → Oad + 2Had
Table 3.
Energy of reaction (ΔE) and energy of barrier (Ea) for the dissociation of H2O on clean and O-pre-adsorbed Fe(100) surfaces.
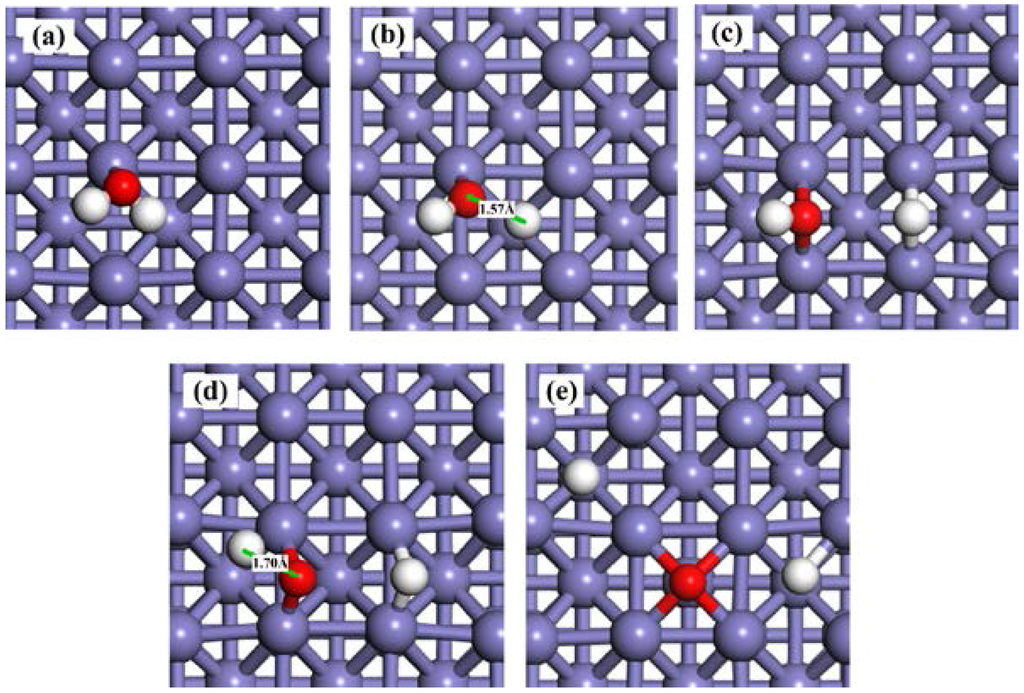
Figure 3.
Snapshots of H2O dissociation process on a clean Fe(100) surface. (a) H2Oad; (b) TS1; (c) Had + OHad; (d) OHad; (e) TS2; and (f) Oad + Had.
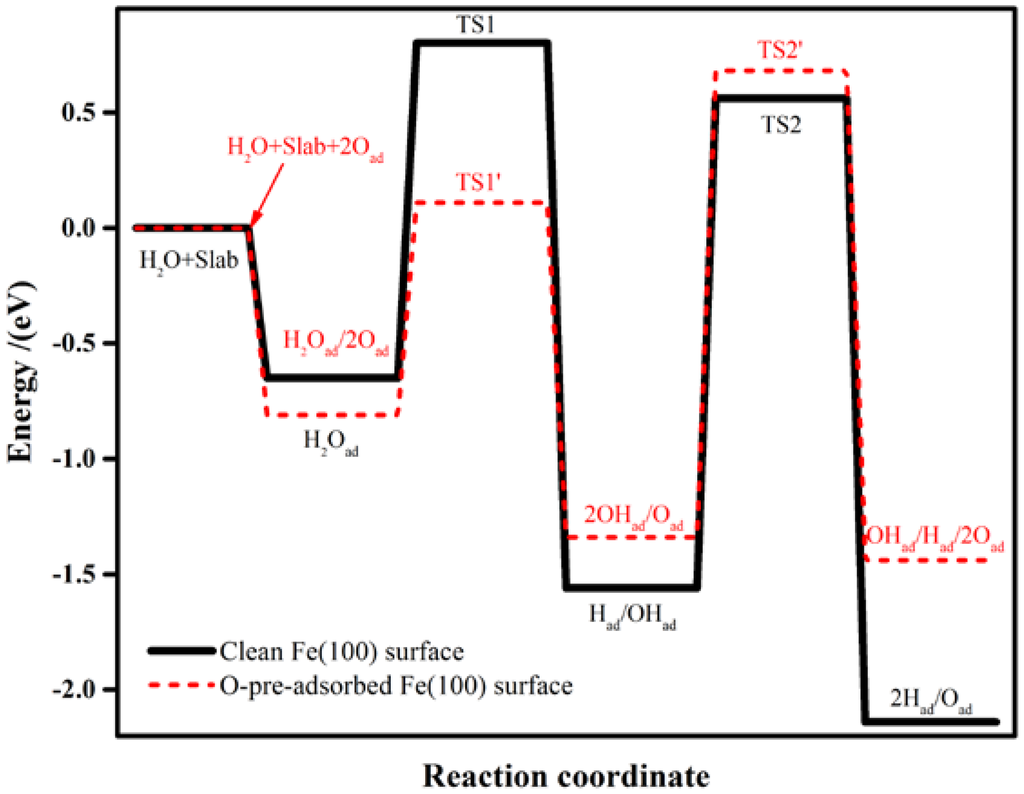
Figure 4.
Schematic energy diagram for the dissociation of H2O on clean (black lines) and O-pre-adsorbed (red lines) Fe(100) surfaces.
In the second step of dissociation (Equation (2)), the O atom remains at the bridge site while the H atom diffuses away and forms a new Fe–H bond (TS2 in Figure 3d). The distance between this H atom and the O atom is 1.70 Å, which is much longer than that of the initial value (0.98 Å). The Fe–H bond is not stable and H continues to move towards the nearby bridge site (Figure 3e). The energy barrier of the second step is 2.12 eV, which is almost 1.5 times of that of the first step. The reaction energy is −0.58 eV, indicating that the second step is an exothermic reaction. According to the activation energies for TS1 and TS2, both steps require significant energies to overcome the energy barriers.
2.3.2. H2O Dissociation on the O-Preadsorbed Fe(100) Surface
The dissociation mechanisms of H2O on the O-pre-adsorbed metal surfaces are significantly different from those on a clean surface. The process of H2O dissociation on an iron surface, promoted by adsorbed oxygen, can be expressed as Equations (3) and (4).
H2Oad + 2Oad → 2OHad + Oad
2OHad + Oad → OHad + 2Oad + Had
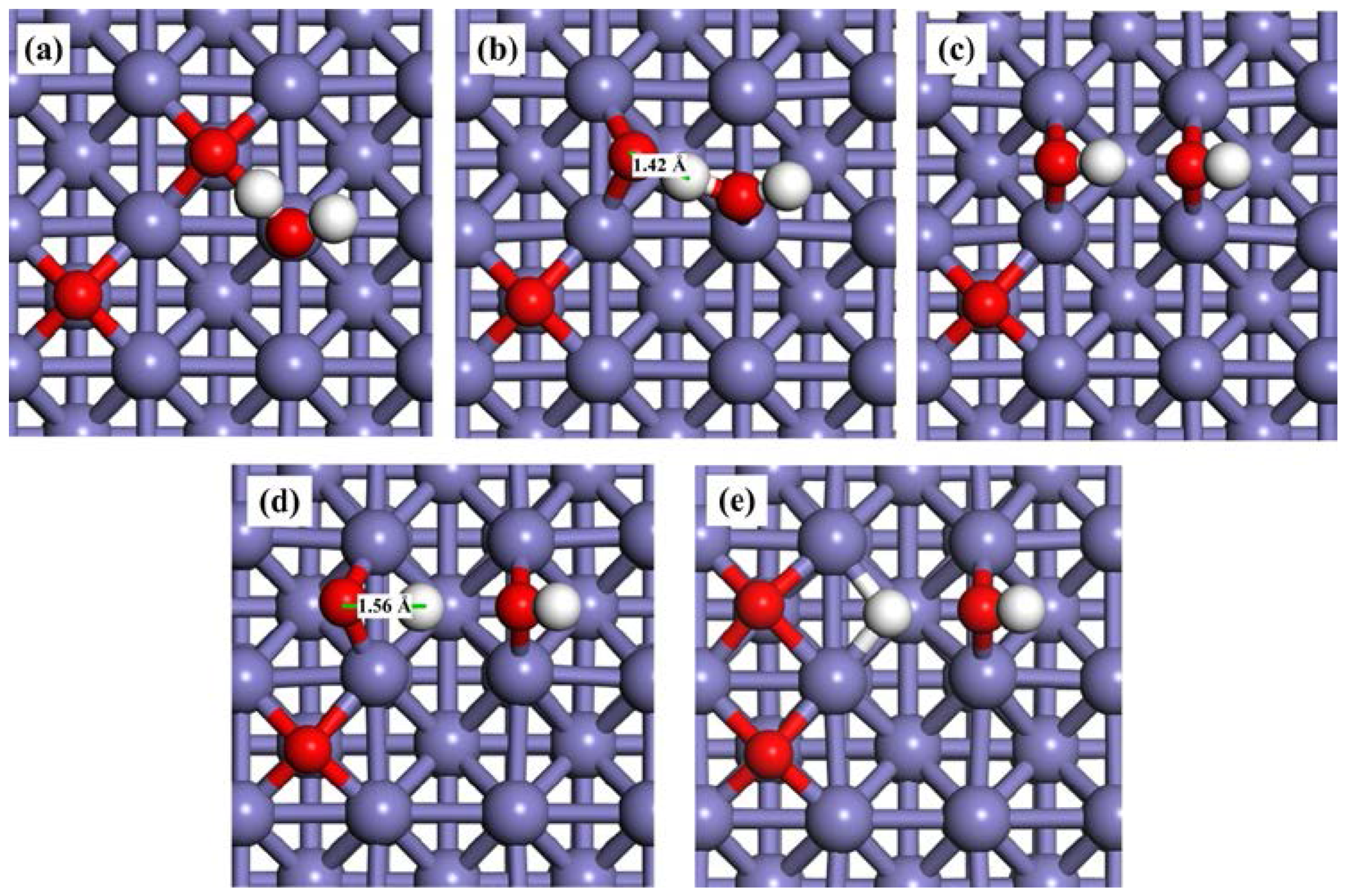
The pre-adsorbed O atom may act as a reactant. The reaction energies and activation barriers for Equation (3) and the consequent Equation (4) are also calculated and shown in Table 3. The corresponding reaction steps are illustrated in Figure 4. On the O-pre-adsorbed Fe(100) surface, molecular water is adsorbed at the top site, similar to that on the clean surface (2.20 Å on the O-covered surface vs. 2.17 Å on the clean surface). The pre-adsorbed O atom forms a hydrogen bond with one of the H atom in the adsorbed H2O with a bond distance of 2.04 Å (Figure 2a). Due to the interaction with the pre-adsorbed O atom, the adsorption of H2O molecule is slightly away from the top site (Figure 5a). Next, the pre-adsorbed O pulls away the hydrogen atom to form a new OHad species adsorbed at the short bridge site, leaving the remaining OH at another bridge site (Figure 5b,c). The energy barrier and heat energy for this step were 0.92 eV and −0.53 eV, respectively. Therefore, the energy barrier of H2O splitting in the presence of O atom on the Fe(100) surface is much lower than that on the clean surface. The strong promotion effect of pre-adsorbed-O atoms on the Fe(100) surface may help design more active catalyst by considering partially oxidizing the metal surfaces. The decrease of the bond length of O–H from 1.57 Å in TS1 to 1.42 Å in TS1’ leads to the reduction of the water dissociation barrier from 1.45 eV on a clean surface to 0.92 eV on an O-pre-adsorbed one. We may conclude that the pre-adsorbed O can significantly promote H2O dissociation.
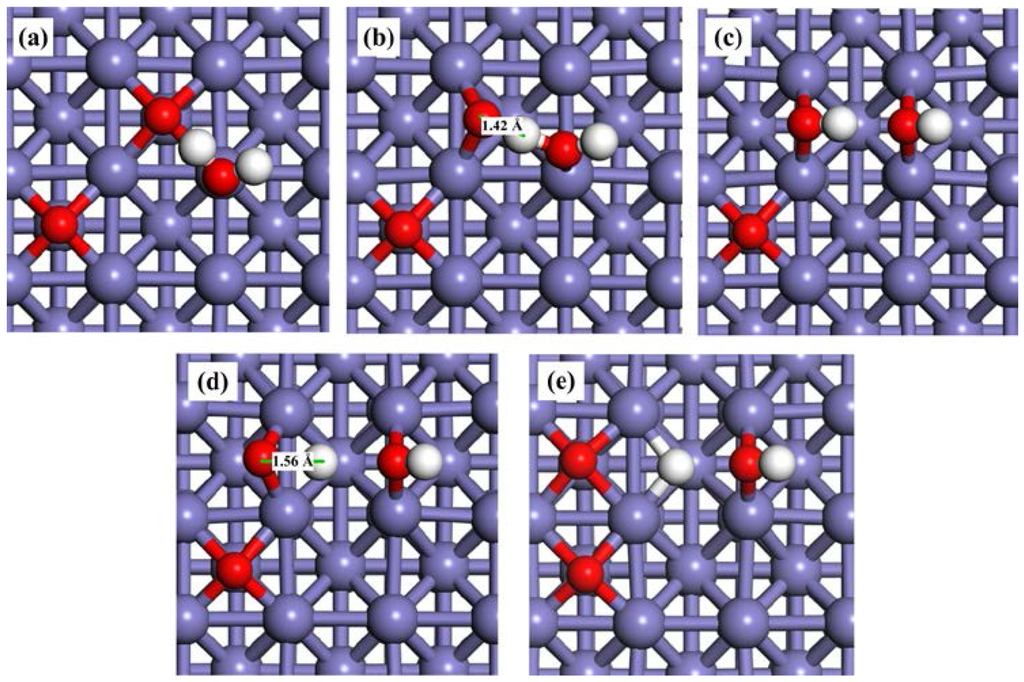
Figure 5.
Snapshots of H2O dissociation process on a O-pre-adsorbed Fe(100) surface. (a) Oad + H2Oad; (b) TS1’; (c) 2OHad; (d) Oad + OHad; (e) TS2’; and (f) 2Oad + Had.
The energy barrier for Equation (4) reported in our work (0.92 eV) is significantly higher than that reported by Liu et al. (0.18 eV) [37]. One of the reasons is the different computation methods used. In the current study, GGA-PBE functional in CASTEP code was used, rather than GGA-PBE-D2 functional in VASP code that includes the long-range dispersion correction for van der Waals (vdW) interactions [37].
OH is also difficult to dissociate on clean Fe surfaces due to the high energy barrier (2.12 eV). In the presence of a pre-adsorbed O atom, the energy barrier is reduced to 2.02 eV. According to Figure 5d, the bond length of O–H at TS2’ (1.56 Å) is shorter than that on clean Fe surfaces (1.70 Å), suggesting that the O atom promotes the dissociation of hydroxyl. These results are also elucidated on Au(100) [61] and Pd (111) [64] surfaces. The high energy barriers of OH dissociation on the clean and O-pre-adsorbed Fe(100) surfaces make the occurrence of this reaction kinetically difficult. Consequently, the decomposition of the O-H bond in the OHad (Equations (2) and (4)) are the rate-determining steps of the whole dissociation reaction of H2O.
3. Computational Methods
Spin-polarized periodic DFT calculations were performed using the Cambridge Sequential Total Energy Package (CASTEP) program in the Materials Studio 6.0 package (Accelrys Software Inc., San Diego, CA, USA). All calculations were performed using ultrasoft pseudopotentials, with kinetic energy cutoff of 400 eV. Electronic exchange and correlation effects were described within the generalized gradient approximation (GGA) [65] using Perdew-Burke-Ernzerhof (PBE) [66] functionals.
The optimized lattice parameter of bulk Fe was calculated using a bcc unit cell sampled with a 15 × 15 × 15 Monkhorst-Pack k-point grid. The calculated value was 2.816 Å, which was comparable to the most accepted experimental value of 2.87 Å [67], with a difference of less than 2.0%.
A (3 × 3) surface unit cell with a slab of four layers’ thickness was selected as the model. This slab was repeated periodically with a 15 Å of vacuum region between the slabs. Only one H2O molecule per super cell was adsorbed on one side of the slab to reduce lateral interactions between adsorbates. The geometry optimization including all degrees of freedom of the adsorbates and the two topmost metal layers were considered. The total energy calculation and the surface structural relaxation were performed by sampling the Brillouin zone with a 6 × 6 × 1 Monkhorst-Pack grid. All of the considered geometries were fully relaxed so that the forces became smaller than 0.01 eV·Å−1. A 0.1 eV Fermi smearing was used and the convergence criteria for the geometry optimizations were 10−5 eV for the total energy. The computed lattice constant (2.643 Å) and magnetic moment (2.22 μB) are close to the experimental values (2.866 Å [68] and 2.22 μB [69]).
As usually defined [70], the adsorption energy (Ead) was calculated by Equation (5):
where ET, EFe and ES are the total energy of the system, the energy associated to the isolated surface, and the energy of the isolated species (H, O, OH and H2O in the present study), respectively. A negative value of Ead indicates an exothermic chemisorption process. The linear synchronous transit (LST) calculation [71] combined with a quadratic synchronous transit (QST) calculation and conjugate gradient refinements [72] were used to obtain the transition state (TS). A LST optimization was performed in the calculations of LST/QST, while QST maximization was used to obtain the TS approximation. The same conjugate gradient minimization was repeated until a stationary point was obtained.
Ead = ET − EFe − ES
We carried the Hubbard correction for H2O adsorption on the Fe(110) surface using the DFT+U method. DFT + U implementation in the code was based on the formalism summarized in Ref. [73]. Only on-site Coulomb repulsion was used and all higher-order multipolar terms were neglected. The effect of the Hubbard correction was found to be negligible for adsorption energies of H2O, O, OH and H on the Fe(100) surface (Table 4).
Table 4.
Comparison of adsorption energies of H2O, OH, O and H on Fe(110) with and without Hubbard correction.
4. Conclusions
The adsorption configurations of H2O and relevant dissociation species on clean and O-pre-adsorbed Fe(100) surfaces were investigated by DFT. It was found that H2O is preferably adsorbed on the top site, O is absorbed on the bridge and hollow hcp site, while OH and H are adsorbed on the bridge site. The calculated adsorption energies revealed that the interactions between the adsorbates and the Fe surface increase in the order of H2O < OH < H < O. In addition, we found that both the first H abstraction from adsorbed H2O and the subsequent OH dissociation are exothermic on both clean Fe(100) and O-pre-adsorbed Fe(100) surfaces. However, the pre-adsorbed O significantly reduces the kinetics energy barriers for both reactions. Our results confirmed that the presence of pre-adsorbed oxygen species significantly promotes H2O dissociation and will help in the design of better catalysts for water dissociation.
Acknowledgments
This work is financially supported by National Natural Science Foundation of China with Grant No. 21206074 and 61304137, Natural Science Foundation of Jiangsu province with Grant No. BK2012406, China Postdoctoral Science Foundation Funded Project with Grant No. 2014M561649, Research Fund for the Doctoral Program of Higher Education of China with Grant No. 20123219120033, the Fundamental Research Funds for the Central Universities with Grant No. 30920140112004, and Zijin Intelligent Program of Nanjing University of Science and Technology with Grant No. 2013-ZJ-0103. MS acknowledges a startup fund from the Hong Kong University of Science and Technology.
Author Contributions
W.J. Wang did the simulation work, wrote the manuscript; G.P. Wang revised the first version of the manuscript; M.H. Shao is a supervisor, made editing and improvement corrections of manuscript, and worked on all topics listed above.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Henniker, J.C. The depth of the surface zone of a liquid. Rev. Mod. Phys. 1949, 21, 322–341. [Google Scholar] [CrossRef]
- Bockris, J.O.M.; Veziroglu, T.N. Estimates of the price of hydrogen as a medium for wind and solar sources. Int. J. Hydrogen Energy 2007, 32, 1605–1610. [Google Scholar] [CrossRef]
- Suh, M.P.; Park, H.J.; Prasad, T.K.; Lim, D.W. Hydrogen storage in metal-organic frameworks. Chem. Rev. 2012, 112, 782–835. [Google Scholar] [CrossRef] [PubMed]
- Eberle, U.; Felderhoff, M.; Schüth, F. Chemical and physical solutions for hydrogen storage. Angew. Chem. Int. Ed. 2009, 48, 6608–6630. [Google Scholar] [CrossRef] [PubMed]
- Dicks, A.L. Hydrogen generation from natural gas for the fuel cell systems of tomorrow. J. Power Sources 1996, 61, 113–124. [Google Scholar] [CrossRef]
- Dybkjaer, I. Tubular reforming and autothermal reforming of natural gas—An overview of available processes. Fuel Process. Technol. 1995, 42, 85–107. [Google Scholar] [CrossRef]
- Takezawa, N.; Iwasa, N. Steam reforming and dehydrogenation of methanol: Difference in the catalytic functions of copper and group VIII metals. Catal. Today 1997, 36, 45–56. [Google Scholar] [CrossRef]
- Haryanto, A.; Fernando, S.; Murali, N.; Adhikari, S. Current status of hydrogen production techniques by steam reforming of ethanol: A review. Energy Fuels 2005, 19, 2098–2106. [Google Scholar] [CrossRef]
- Wei, J.M.; Iglesia, E. Reaction pathways and site requirements for the activation and chemical conversion of methane on Ru-based catalysts. J. Phys. Chem. B 2004, 108, 7253–7262. [Google Scholar] [CrossRef]
- Rostrupnielsen, J.R.; Hansen, J.H.B. CO2-reforming of methane over transition metal. J. Catal. 1993, 144, 38–49. [Google Scholar] [CrossRef]
- Wei, J.M.; Iglesia, E. Structural requirements and reaction pathways in methane activation and chemical conversion catalyzed by rhodium. J. Catal. 2004, 225, 116–127. [Google Scholar] [CrossRef]
- Wei, J.M.; Iglesia, E. Isotopic and kinetic assessment of the mechanism of methane reforming and decomposition reactions on supported iridium catalysts. Phys. Chem. Chem. Phys. 2004, 6, 3754–3759. [Google Scholar] [CrossRef]
- Wei, J.M.; Iglesia, E. Structural and mechanistic requirements for methane activation and chemical conversion on supported iridium clusters. Angew. Chem. Int. Ed. 2004, 43, 3685–3688. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.M.; Iglesia, E. Mechanism and site requirements for activation and chemical conversion of methane on supported Pt clusters and turnover rate comparisons among noble metals. J. Phys. Chem. B 2004, 108, 4094–4103. [Google Scholar] [CrossRef]
- Sehested, J. Four challenges for nickel steam-reforming catalysts. Catal. Today 2006, 111, 103–110. [Google Scholar] [CrossRef]
- Murakhtina, T.; Site, L.D.; Sebastiani, D. Vibrational frequencies of water adsorbed on (111) and (221) nickel surfaces from first principle calculations. Chem. Phys. Chem. 2006, 7, 1215–1219. [Google Scholar] [CrossRef] [PubMed]
- Catapan, R.C.; Oliveira, A.A.M.; Chen, Y.; Vlachos, D.G. DFT study of the water–gas shift reaction and coke formation on Ni(111) and Ni(211) surfaces. J. Phys. Chem. C 2012, 116, 20281–20291. [Google Scholar] [CrossRef]
- Lindström, B.; Pettersson, L.J. Hydrogen generation by steam reforming of methanol over copper-based catalysts for fuel cell applications. Int. J. Hydrogen Energy 2001, 26, 923–933. [Google Scholar] [CrossRef]
- Andersson, K.; Ketteler, G.; Bluhm, H.; Yamamoto, S.; Ogasawara, H.; Pettersson, L.G.M.; Salmeron, M.; Nilsson, A. Autocatalytic water dissociation on Cu(110) at near ambient conditions. J. Am. Chem. Soc. 2008, 130, 2793–2797. [Google Scholar] [CrossRef] [PubMed]
- Llorca, J.; Homs, N.; Sales, J.; de la Piscina, P.R. Efficient production of hydrogen over supported cobalt catalysts from ethanol steam reforming. J. Catal. 2002, 209, 306–317. [Google Scholar] [CrossRef]
- Xu, L.S.; Ma, Y.S.; Zhang, Y.L.; Chen, B.H.; Wu, Z.F.; Jiang, Z.Q.; Huang, W.X. Water adsorption on a Co(0001) surface. J. Phys. Chem. C 2010, 114, 17023–17029. [Google Scholar] [CrossRef]
- Murata, K.; Wang, L.S.; Saito, M.; Inaba, M.; Takahara, I.; Mimura, N. Hydrogen production from steam reforming of hydrocarbons over alkaline-earth metal-modified Fe- or Ni-based catalysts. Energy Fuels 2004, 18, 122–126. [Google Scholar] [CrossRef]
- Polychronopoulou, K.; Bakandritsos, A.; Tzitzios, V.; Fierro, J.L.G.; Efstathiou, A.M. Absorption-enhanced reforming of phenol by steam over supported Fe catalysts. J. Catal. 2006, 241, 132–148. [Google Scholar] [CrossRef]
- Liang, C.H.; Ma, Z.Q.; Lin, H.Y.; Ding, L.; Qiu, J.S.; Frandsen, W.; Su, D.S. Template preparation of nanoscale CexFe1−xO2 solid solutions and their catalytic properties for ethanol steam reforming. J. Mater. Chem. 2009, 19, 1417–1424. [Google Scholar] [CrossRef]
- Noichi, H.; Uddin, A.; Sasaoka, E. Steam reforming of naphthalene as model biomass tar over iron-aluminum and iron-zirconium oxide catalyst catalysts. Fuel Process. Technol. 2010, 91, 1609–1616. [Google Scholar] [CrossRef]
- Mawdsley, J.R.; Krause, T.R. Rare earth-first-row transition metal perovskites as catalysts for the autothermal reforming of hydrocarbon fuels to generate hydrogen. Appl. Catal. A 2008, 334, 311–320. [Google Scholar] [CrossRef]
- Sato, K.; Nagaoka, K.; Nishiguchi, H.; Takita, Y. n-C4H10 autothermal reforming over MgO-supported base metal catalysts. Int. J. Hydrogen Energy 2009, 34, 333–342. [Google Scholar] [CrossRef]
- Anderson, A.B. Reactions and structures of water on clean and oxygen covered Pt(111) and Fe(100). Surf. Sci. 1981, 105, 159–176. [Google Scholar] [CrossRef]
- Baró1, A.M.; Erley1, W. The adsorption of H2O on Fe(100) studied by EELS. J. Vac. Sci. Technol. 1982, 20, 580–583. [Google Scholar] [CrossRef]
- Lu, J.P.; Albert, M.R.; Bernasek, S.L. The adsorption of oxygen on the Fe(100) surface. Surf. Sci. 1989, 215, 348–362. [Google Scholar] [CrossRef]
- Hung, W.H.; Schwartz, J.; Bernasek, S.L. Sequential oxidation of Fe(100) by water adsorption: formation of an ordered hydroxylated surface. Surf. Sci. 1991, 248, 332–342. [Google Scholar] [CrossRef]
- Freitas, R.R.Q.; Rivelino, R.; de Brito Mota, F.; de Castilho, C.M.C. Dissociative adsorption and aggregation of water on the Fe(100) surface: A DFT study. J. Phys. Chem. C 2012, 116, 20306–20314. [Google Scholar] [CrossRef]
- Eder, M.; Terakura, K. Initial stages of oxidation of (100) and (110) surfaces of iron caused by water. Phys. Rev. B 2001, 64, 115426:1–115426:7. [Google Scholar] [CrossRef]
- Jung, S.C.; Kang, M.H. Adsorption of a water molecule on Fe(100): Density-functional calculations. Phys. Rev. B 2010, 81, 115460:1–115460:7. [Google Scholar] [CrossRef]
- Govender, A.; Ferré, D.C.; Niemantsverdriet, J.W. The surface chemistry of water on Fe(100): A density functional theory study. Chem. Phys. Chem. 2012, 13, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Lazar, P.; Otyepka, M. Dissociation of water at iron surfaces: Generalized gradient functional and range-separated hybrid functional study. J. Phys. Chem. C 2012, 116, 25470–25477. [Google Scholar] [CrossRef]
- Liu, S.; Tian, X.; Wang, T.; Wen, X.; Li, Y.W.; Wang, J.; Jiao, H. High coverage water aggregation and dissociation on Fe(100): A computational analysis. J. Phys. Chem. C 2014, 118, 26139–26154. [Google Scholar] [CrossRef]
- Michaelides, A.; Ranea, V.A.; de Andres, P.L.; King, D.A. General model for water monomer adsorption on close-packed transition and noble metal surfaces. Phys. Rev. Lett. 2003, 90, 216102:1–216102:4. [Google Scholar] [CrossRef] [PubMed]
- Michaelides, A.; Ranea, V.A.; de Andres, P.L.; King, D.A. First-principles study of H2O diffusion on a metal surface: H2O on Al{100}. Phys. Rev. B 2004, 69, 075409:1–075409:4. [Google Scholar] [CrossRef]
- Wang, S.; Cao, Y.; Rikvold, P.A. First-principles calculations for the adsorption of water molecules on the Cu(100) surface. Phys. Rev. B 2004, 70, 205410:1–205410:4. [Google Scholar] [CrossRef]
- Li, J.B.; Zhu, S.L.; Li, Y.; Wang, F.H. Water adsorption on Pd {100} from first principles. Phys. Rev. B 2007, 76, 235433:1–235433:8. [Google Scholar] [CrossRef]
- Thiel, P.A.; Madey, T.E. The interaction of water with solid surfaces: fundamental aspects. Surf. Sci. Rep. 1987, 7, 211–385. [Google Scholar] [CrossRef]
- Henderson, M.A. The interaction of water with solid surfaces: fundamental aspects revisited. Surf. Sci. Rep. 2002, 46, 1–308. [Google Scholar] [CrossRef]
- Heras, J.M.; Estiu, G.; Viscido, L. The interaction of water with clean palladium films: A thermal desorption and work function study. Appl. Surf. Sci. 1997, 108, 455–464. [Google Scholar] [CrossRef]
- Nyberg, C.; Tengstal, C.G. Adsorption and reaction of water, oxygen, and hydrogen on Pd(100): Identification of adsorbed hydroxyl and implications for the catalytic H2–O2 reaction. J. Chem. Phys. 1984, 80, 3463–3468. [Google Scholar] [CrossRef]
- Wolf, M.; Nettesheim, S.; White, J.M.; Hasselbrink, E.; Ertl, G. Ultraviolet-laser induced dissociation and desorption of water adsorbed on Pd(111). J. Chem. Phys. 1990, 92, 1509–1510. [Google Scholar] [CrossRef]
- Wolf, M.; Nettesheim, S.; White, J.M.; Hasselbrink, E.; Ertl, G. Dynamics of the ultraviolet photochemistry of water adsorbed on Pd(111). J. Chem. Phys. 1991, 94, 4609–4619. [Google Scholar] [CrossRef]
- Zhu, X.Y.; White, J.M.; Wolf, M.; Hasselbrink, E.; Ertl, G. Photochemical pathways of water on palladium (111) at 6.4 eV. J. Phys. Chem. 1991, 95, 8393–8402. [Google Scholar] [CrossRef]
- Cao, Y.L.; Chen, Z.X. Theoretical studies on the adsorption and decomposition of H2O on Pd(111) surface. Surf. Sci. 2006, 600, 4572–4583. [Google Scholar] [CrossRef]
- Shavorskiy, A.; Eralp, T.; Gladys, M.J.; Held, G. A stable pure hydroxyl layer on Pt{110}-(1×2). J. Phys. Chem. C 2009, 113, 21755–21764. [Google Scholar] [CrossRef]
- Liu, S.L.; Tian, X.X.; Wang, T.; Wen, X.D.; Li, Y.W.; Wang, J.G.; Jiao, H.J. Coverage dependent water dissociative adsorption on the cleanand O-precovered Fe(111) surfaces. J. Phys. Chem. C 2015, 119, 11714–11724. [Google Scholar] [CrossRef]
- Hung, W.H.; Schwartz, J.; Bernasek, S.L. Adsorption of H2O on oxidized Fe(100) surfaces: comparison between the oxidation of iron by H2O and O2. Surf. Sci. 1993, 294, 21–32. [Google Scholar] [CrossRef]
- Pozzo, M.; Carlini, G.; Rosei, R.; Alfè, D. Comparative study of water dissociation on Rh(111) and Ni(111) studied with first principles calculations. J. Chem. Phys. 2007, 126, 164706:1–164706:11. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.J.; Wang, G.P. A theoretical study of water adsorption and dissociation on Ni(111) surface during oxidative steam reforming and water gas shift processes. J. Energy Inst. 2015, 88, 112–117. [Google Scholar] [CrossRef]
- Mohsenzadeh, A.; Bolton, K.; Richards, T. DFT study of the adsorption and dissociation of water on Ni(111), Ni(110) and Ni(100) surfaces. Surf. Sci. 2014, 627, 1–10. [Google Scholar] [CrossRef]
- Tang, Q.L.; Chen, Z.X.; He, X. A theoretical study of the water gas shift reaction mechanism on Cu(111) model system. Surf. Sci. 2009, 603, 2138–2144. [Google Scholar] [CrossRef]
- Wang, W.J.; Wang, G.P. Theoretical study of direct versus oxygen-assisted water dissociation on the Cu(110) surface. Appl. Surf. Sci. 2015, 351, 846–852. [Google Scholar] [CrossRef]
- Fajín, J.L.C.; Cordeiro, M.N.D.S.; Illas, F.; Gomes, J.R.B. Descriptors controlling the catalytic activity of metallic surfaces towardwater splitting. J. Catal. 2010, 276, 92–100. [Google Scholar] [CrossRef]
- Fajín, J.L.C.; Cordeiro, M.N.D.S.; Illas, F.; Gomes, J.R.B. Generalized Brønsted-Evans-Polanyi relationships and descriptors for O–H bond cleavage of organic molecules on transition metal surfaces. J. Catal. 2014, 313, 24–33. [Google Scholar] [CrossRef]
- Błoński, P.; Kiejna, A.; Hafner, J. Theoretical study of oxygen adsorption at the Fe(110) and (100) surfaces. Surf. Sci. 2005, 590, 88–100. [Google Scholar] [CrossRef]
- Sorescu, D.C. First principles calculations of the adsorption and diffusion of hydrogen on Fe(100) surface and in the bulk. Catal. Today 2005, 105, 44–65. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Yan, L.F.; Wang, G.C. Oxygen-assisted water partial dissociation on copper: a model study. Phys. Chem. Chem. Phys. 2015, 17, 8231–8238. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Li, M.M.; Yan, T.; Fang, T. Decomposition of H2O on clean and oxygen-covered Au(100) surface: A DFT study. Appl. Surf. Sci. 2014, 315, 16–21. [Google Scholar] [CrossRef]
- Cao, Y.; Chen, Z. Slab model studies of water adsorption and decomposition on clean and X- (X = C, N and O) contaminated Pd(111) surfaces. Phys. Chem. Chem. Phys. 2007, 9, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Kurth, S.; Perdew, J.P.; Blaha, P. Molecular and solid-state tests of density functional approximations: LSD, GGAs, and meta-GGAs. Int. J. Quantum Chem. 1999, 75, 889–909. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Kittel, C. Introduction to Solid State Physics, 8th ed.; Wiley: New York, NY, USA, 2005. [Google Scholar]
- Kohlhaas, R.; Donner, P.; Schmitz-Pranghe, N. The temperature-dependence of the lattice parameters of iron, cobalt, and nickel in the high temperature range. Z. Angew. Phys. 1967, 23, 245. [Google Scholar]
- Kittel, C. Introduction to Solid State Physics, 7th ed.; Wiley: New York, NY, USA, 1996. [Google Scholar]
- Jiang, D.E.; Carter, E.A. Diffusion of interstitial hydrogen into and through bcc Fe from first principles. Phys. Rev. B 2004, 70, 064102:1–064102:9. [Google Scholar] [CrossRef]
- Halgren, T.A. The synchronous-transit method for determining reaction pathways and locating molecular transition states. Chem. Phys. Lett. 1977, 49, 225–232. [Google Scholar] [CrossRef]
- Govind, N.; Petersen, M.; Fitzgerald, G.; King-Smith, D.; Andzelm, J. A generalized synchronous transit method for transition state location. Comp. Mater. Sci. 2003, 28, 250–258. [Google Scholar] [CrossRef]
- Cococcioni, M.; de Gironcoli, S. Linear response approach to the calculation of the effective interaction parameters in the LDA+U method. Phys. Rev. B 2005, 71, 035105. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).