
Effect of Ru Species on N2O Decomposition over Ru/Al2O3 Catalysts



Abstract
:1. Introduction
2. Results
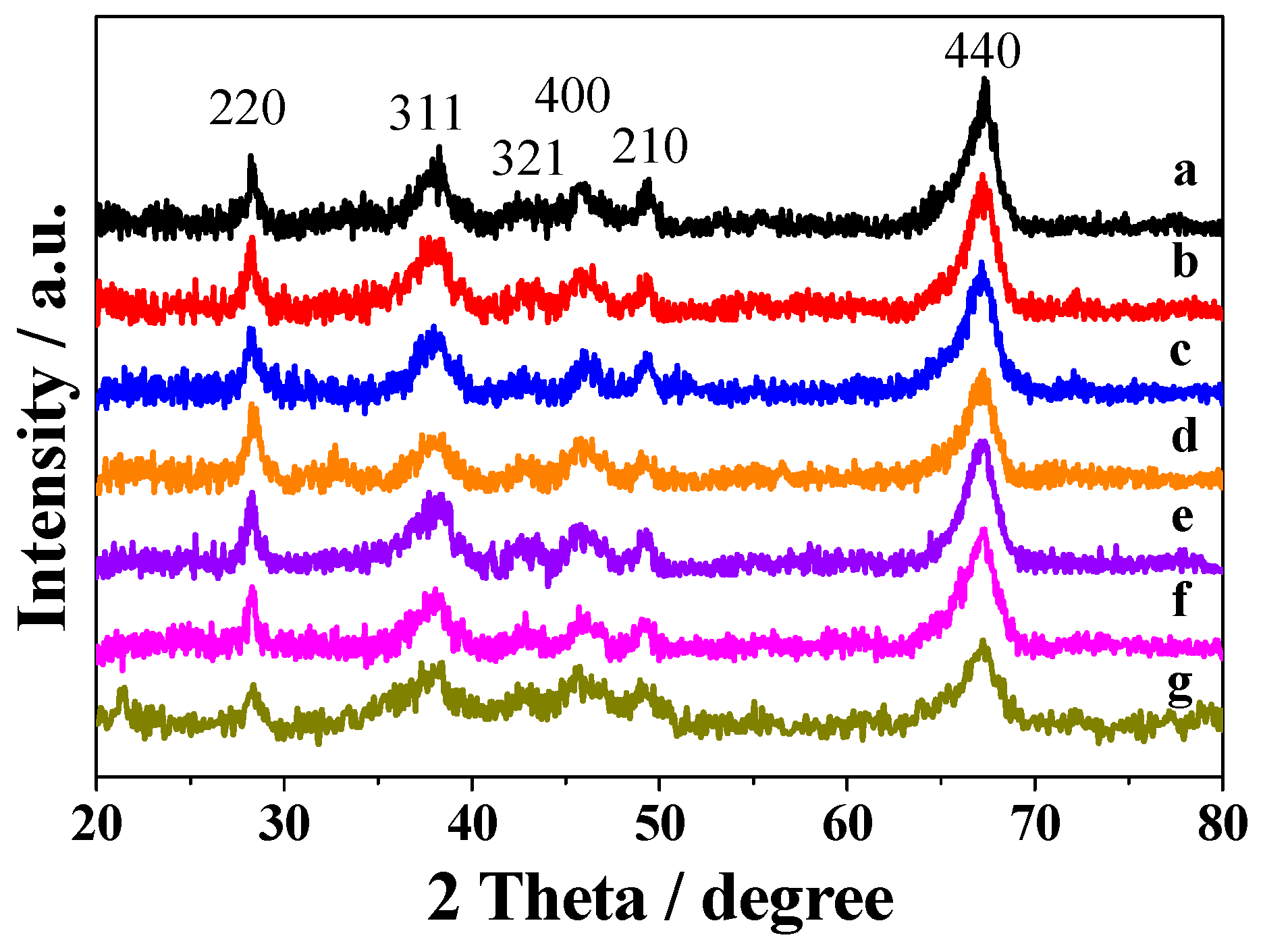
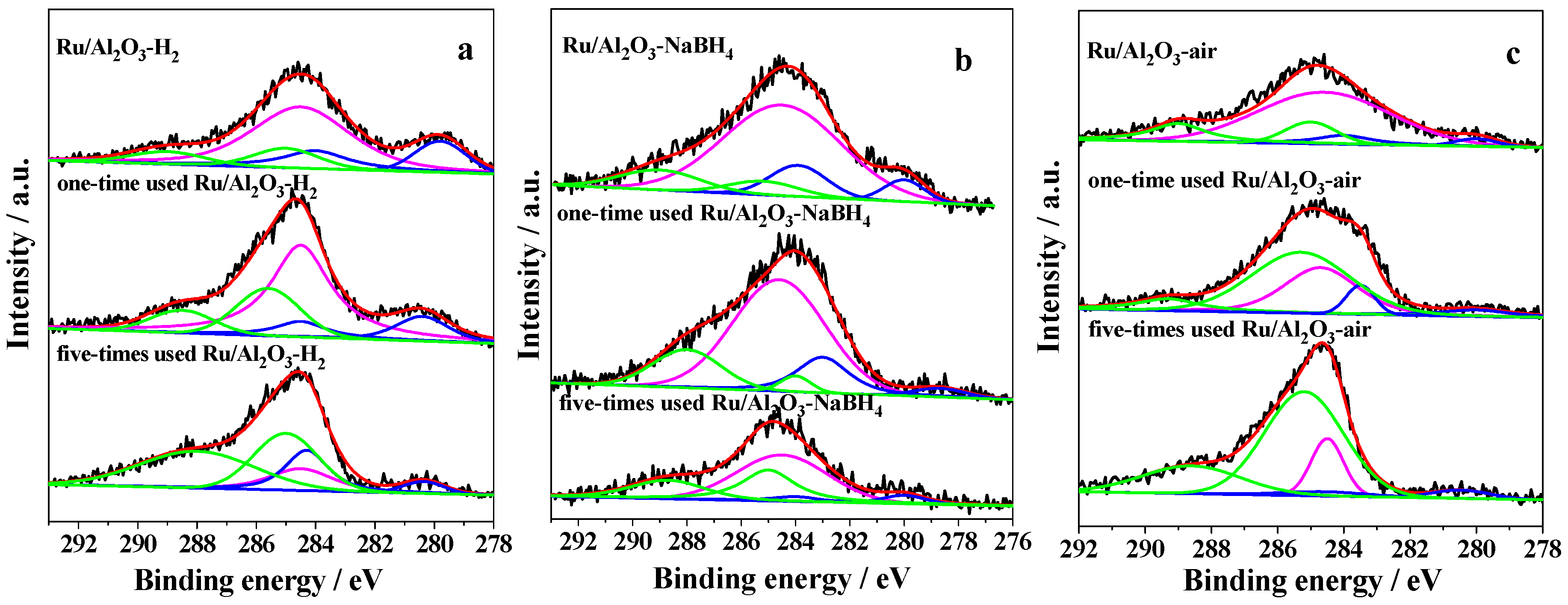
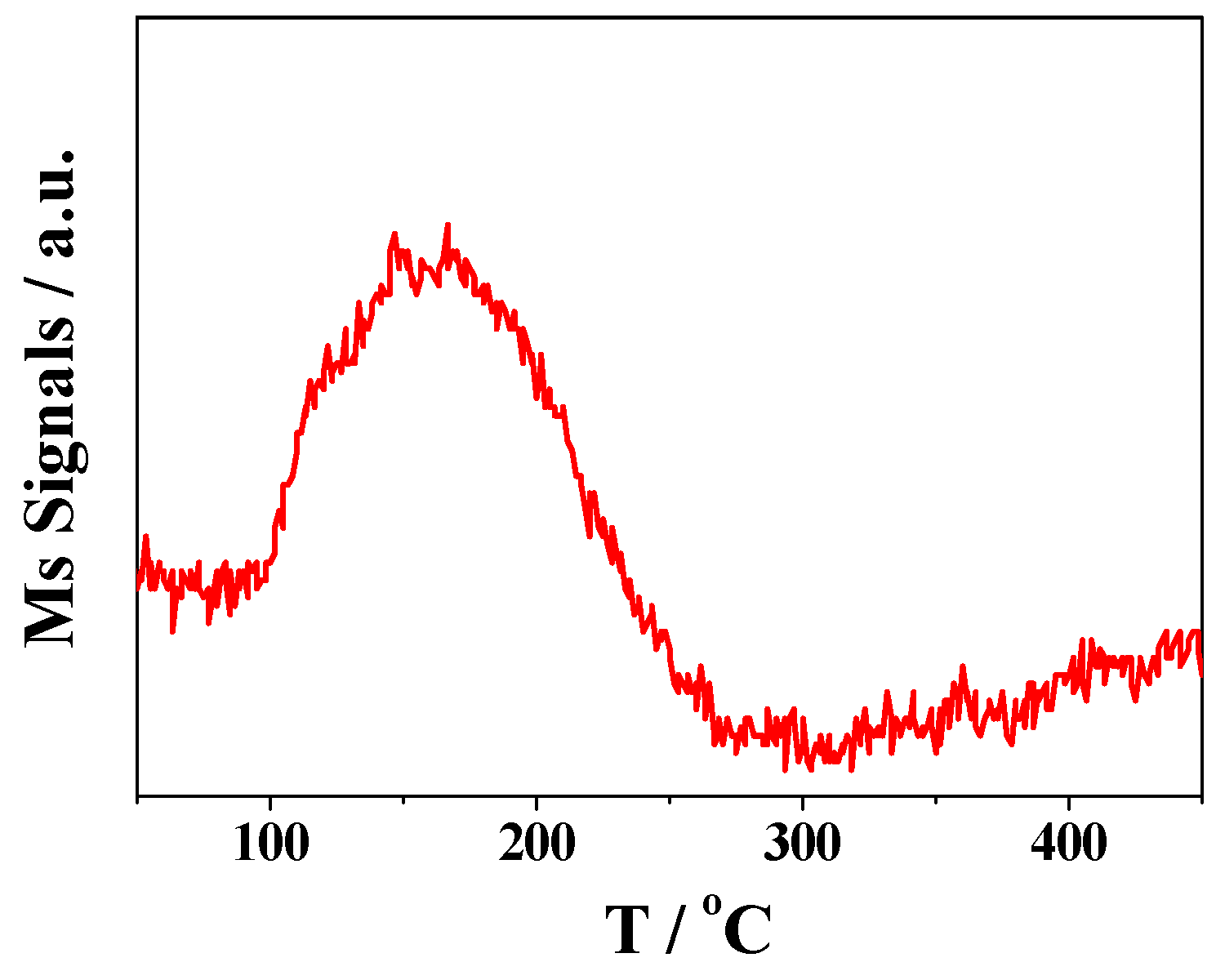
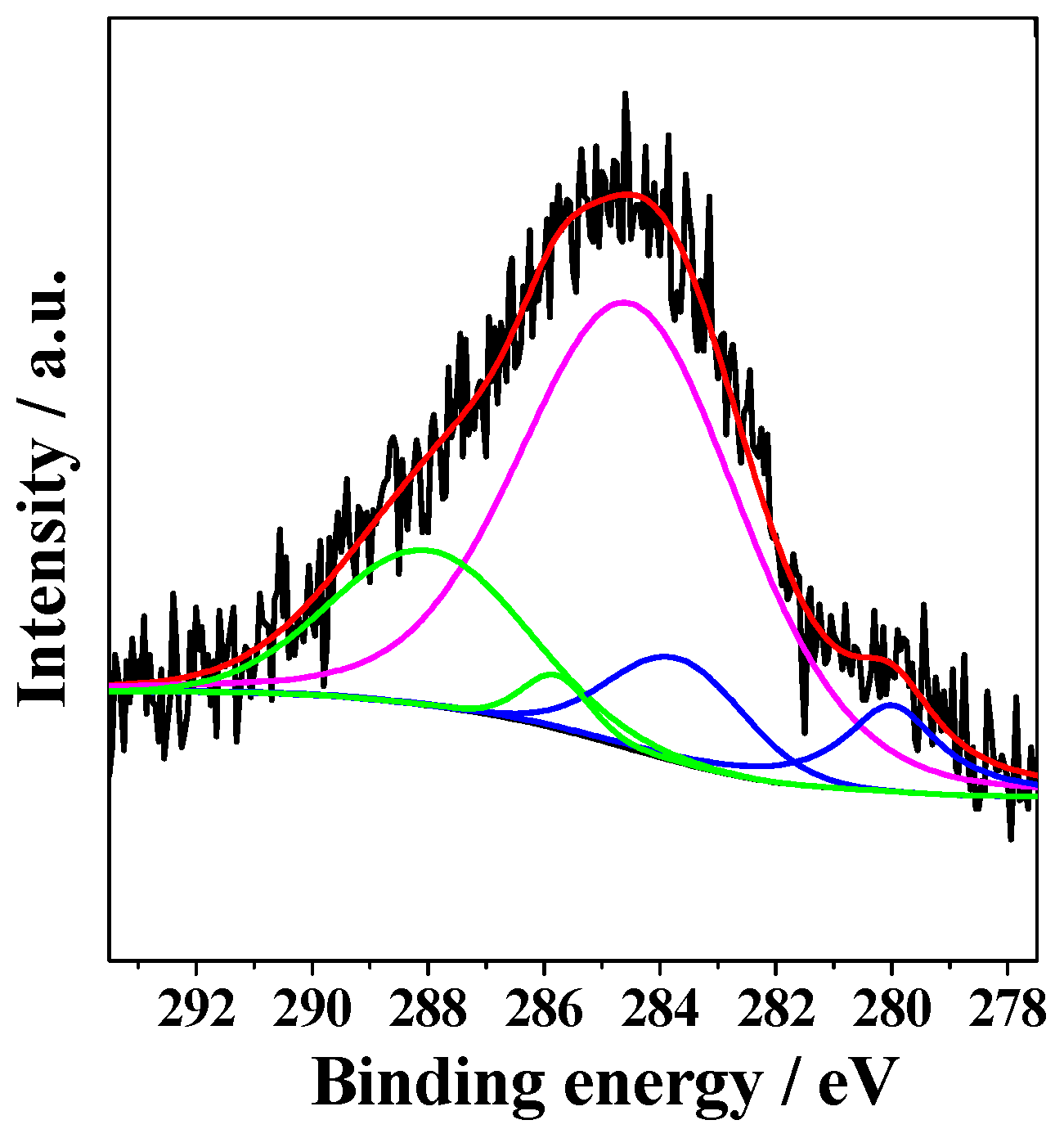
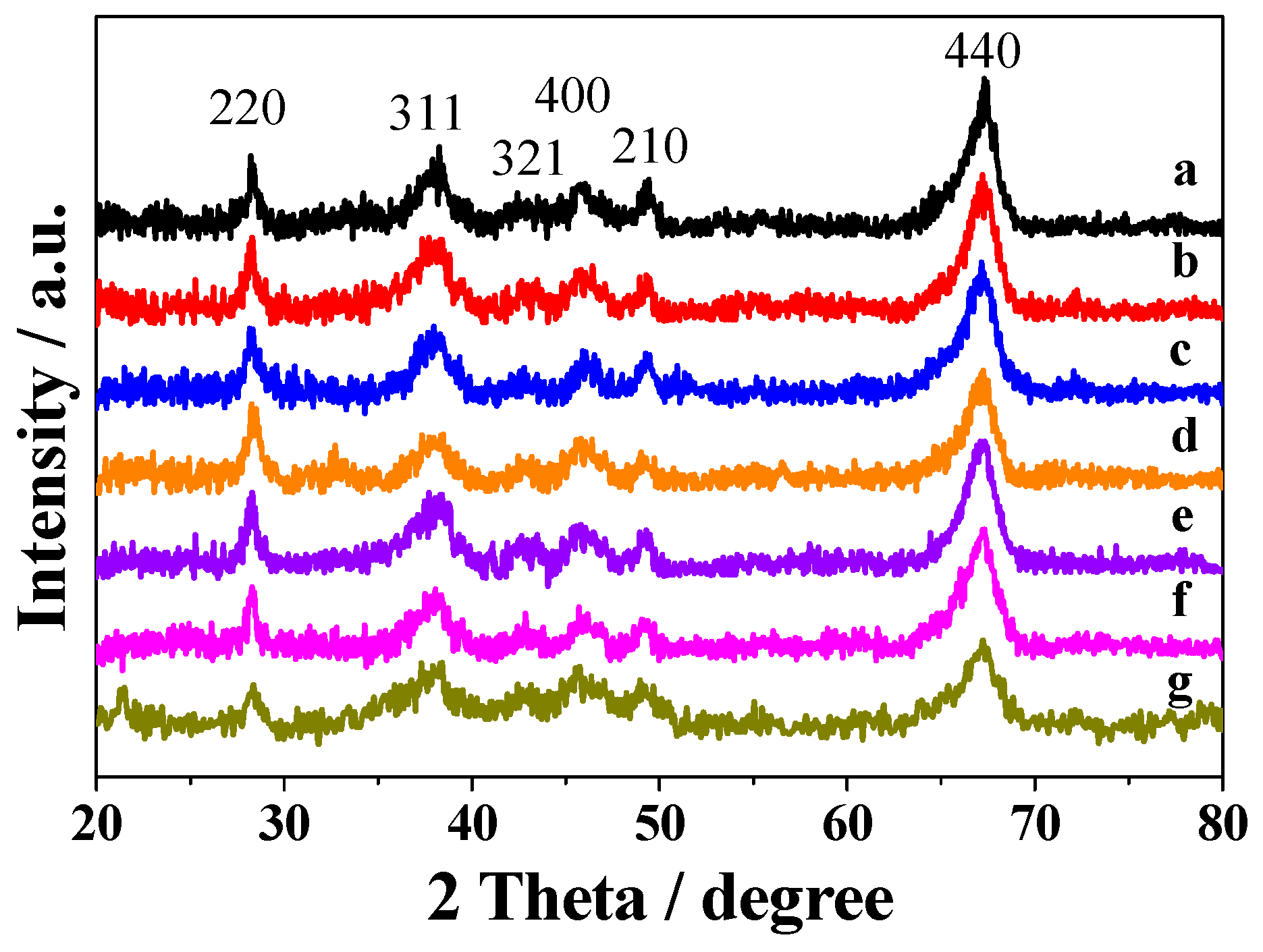
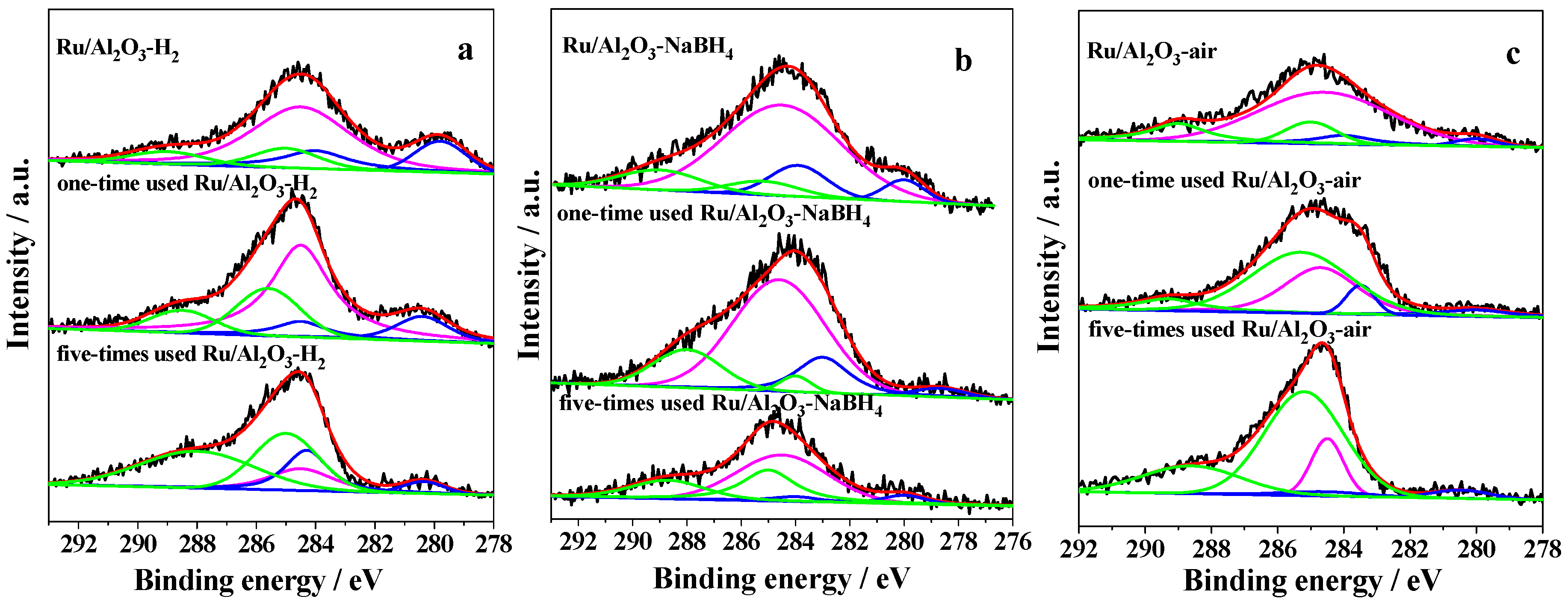
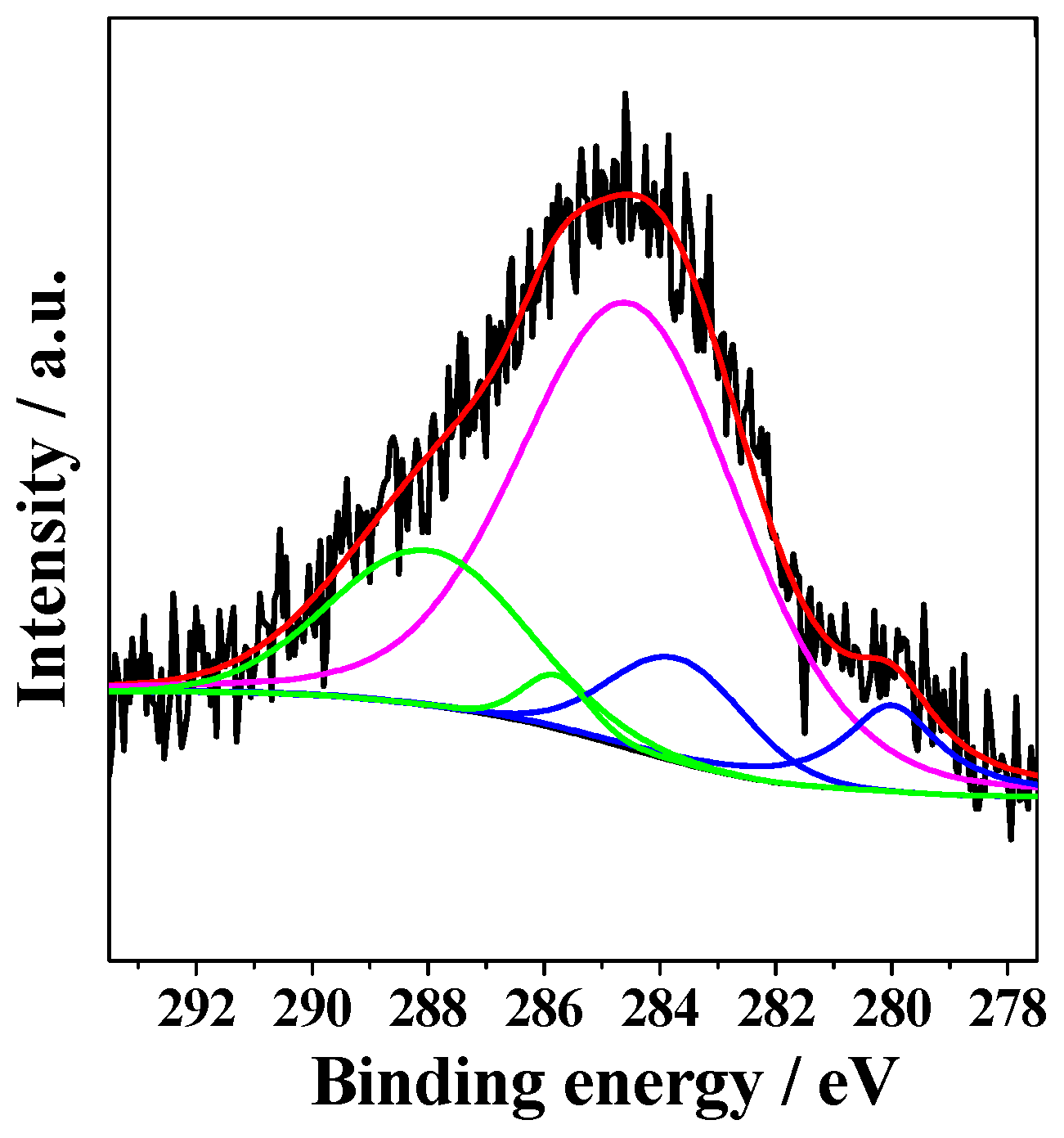
2.1. Characterization of Prepared Samples
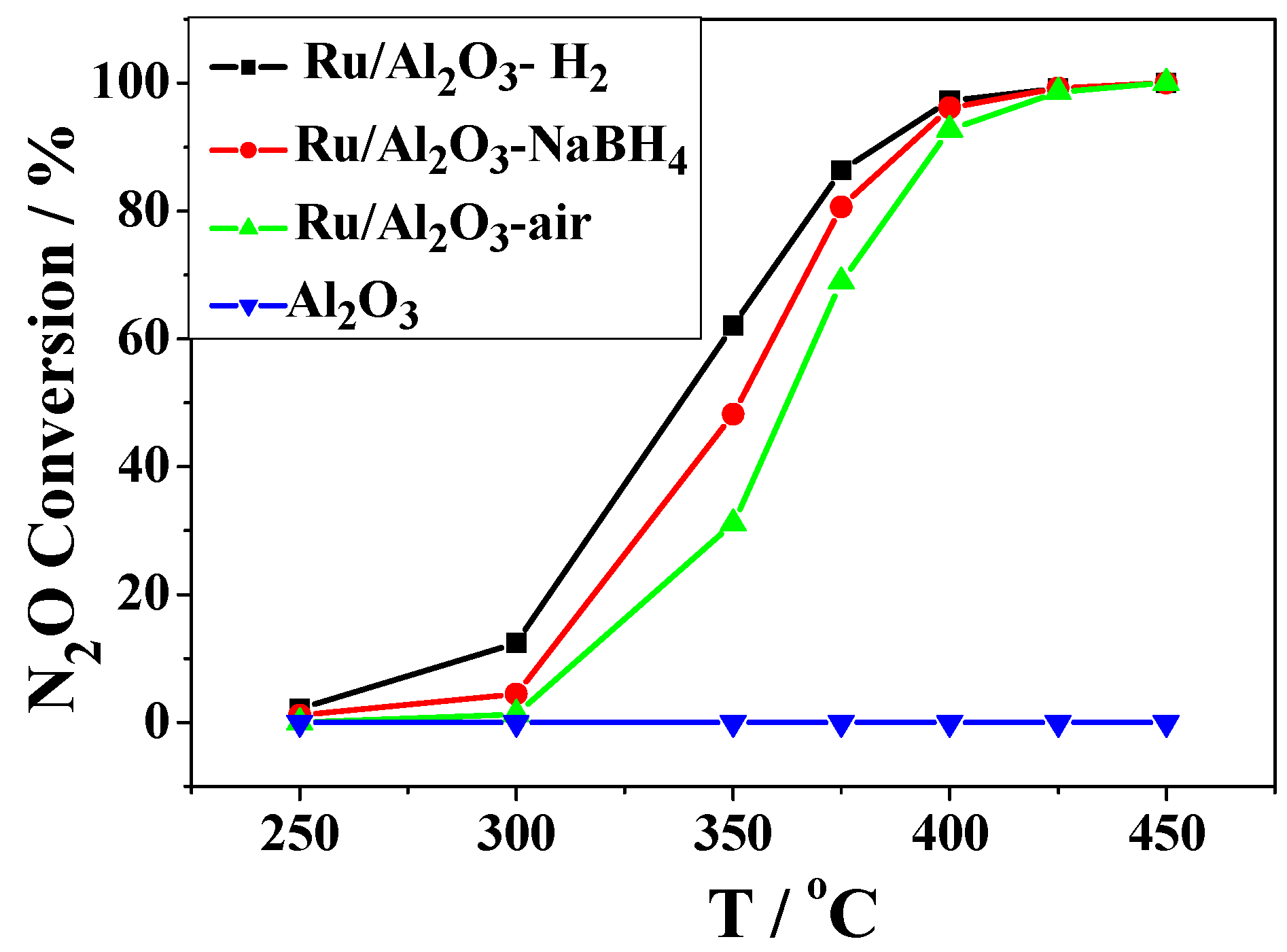
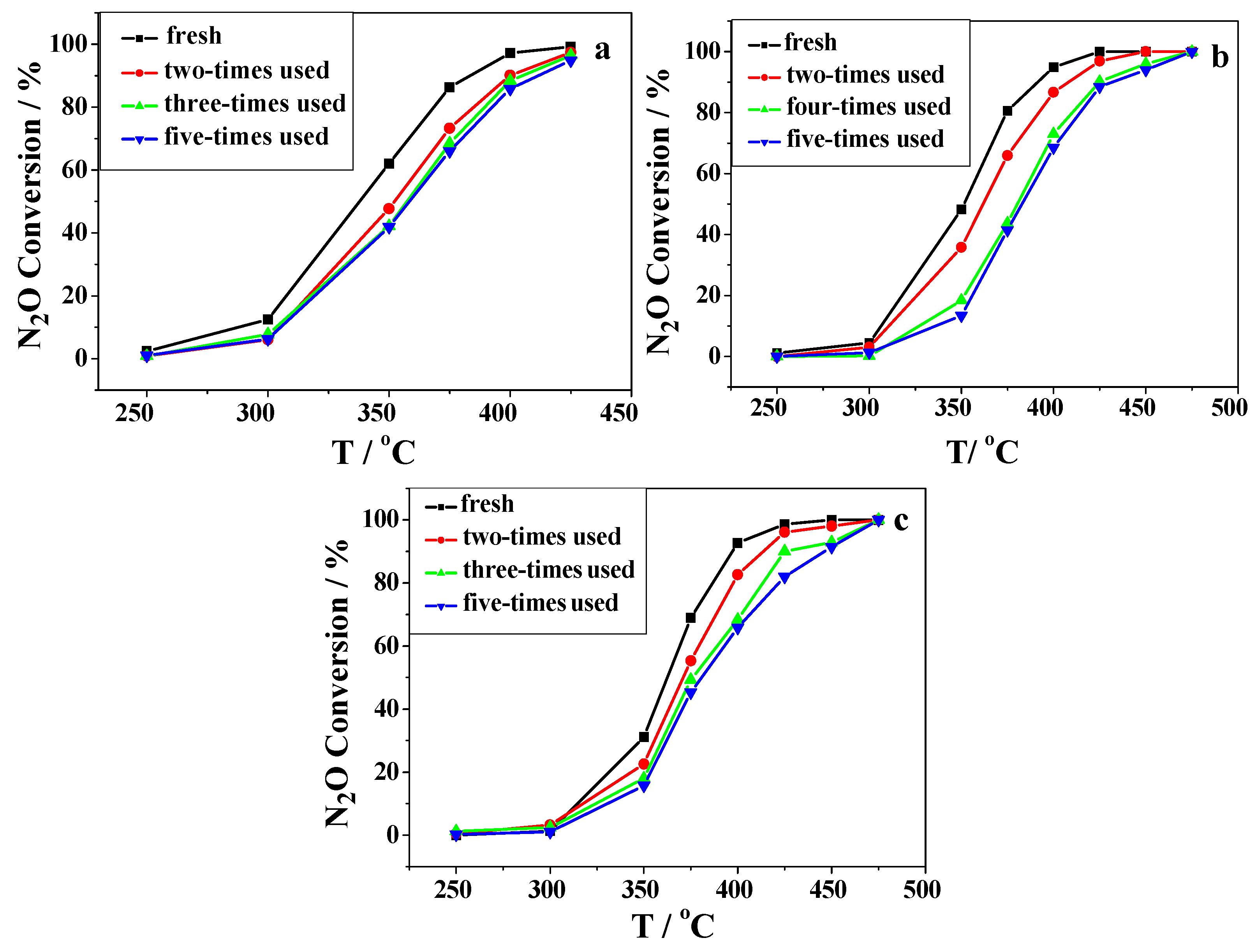
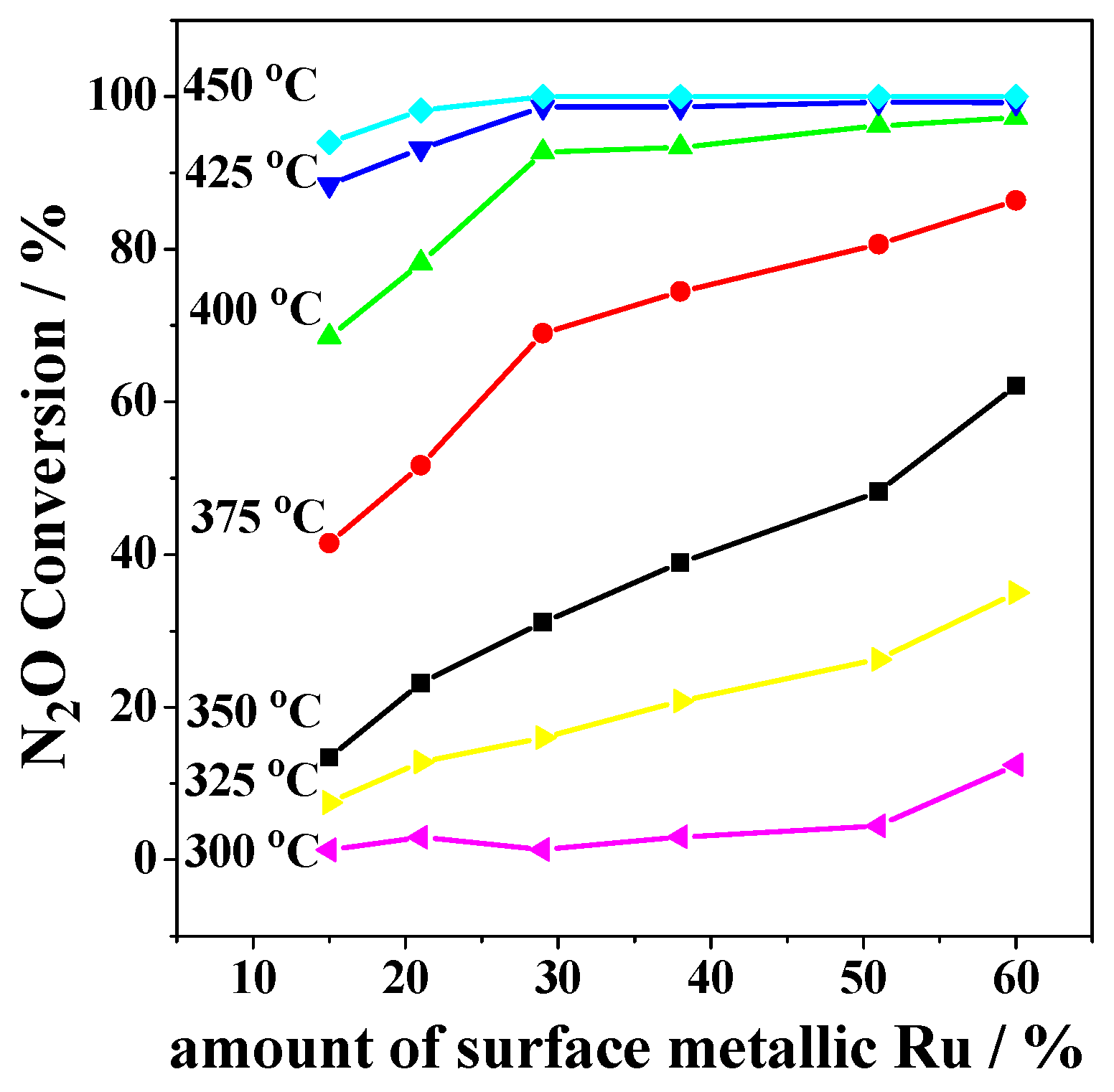
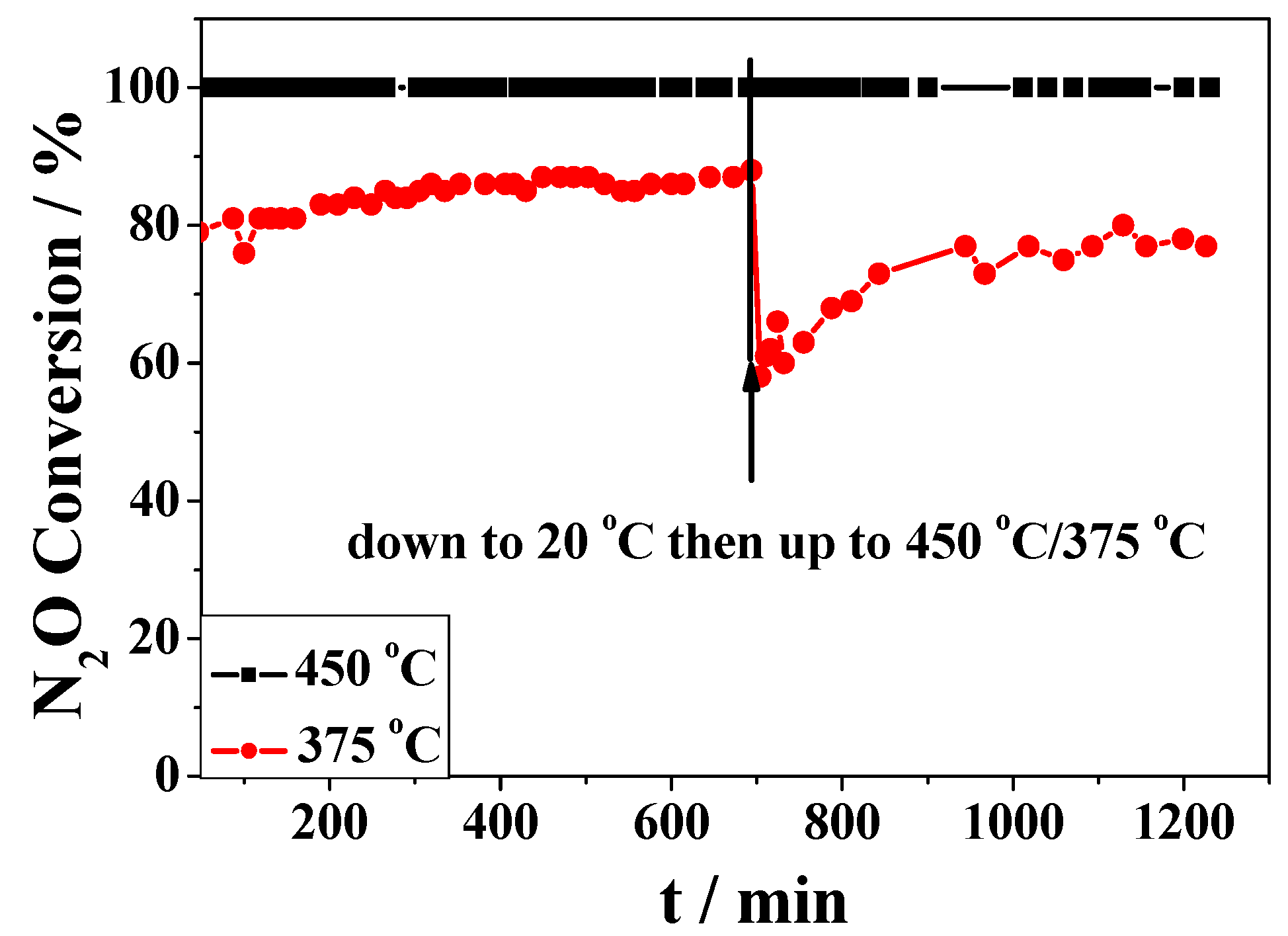
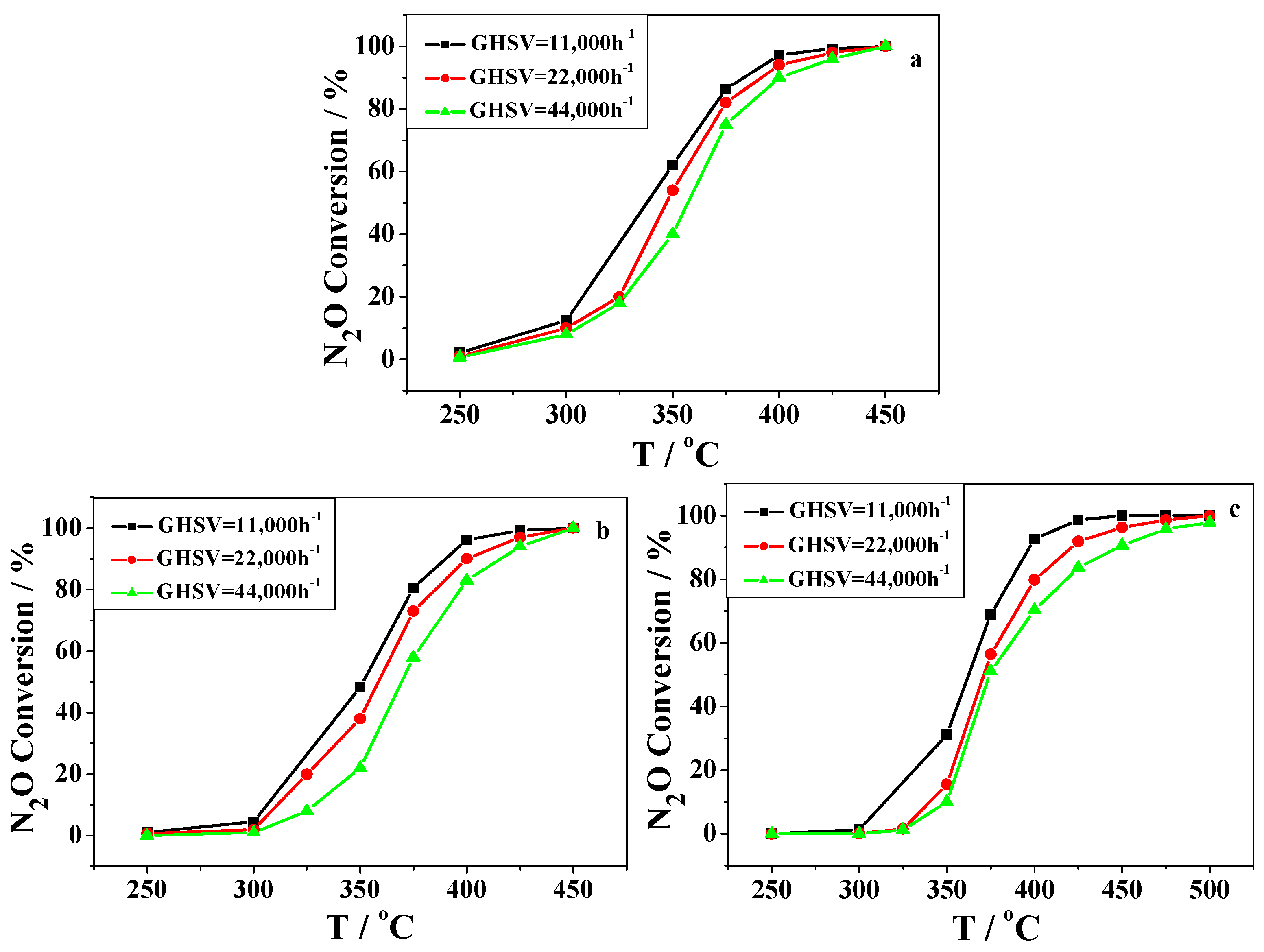
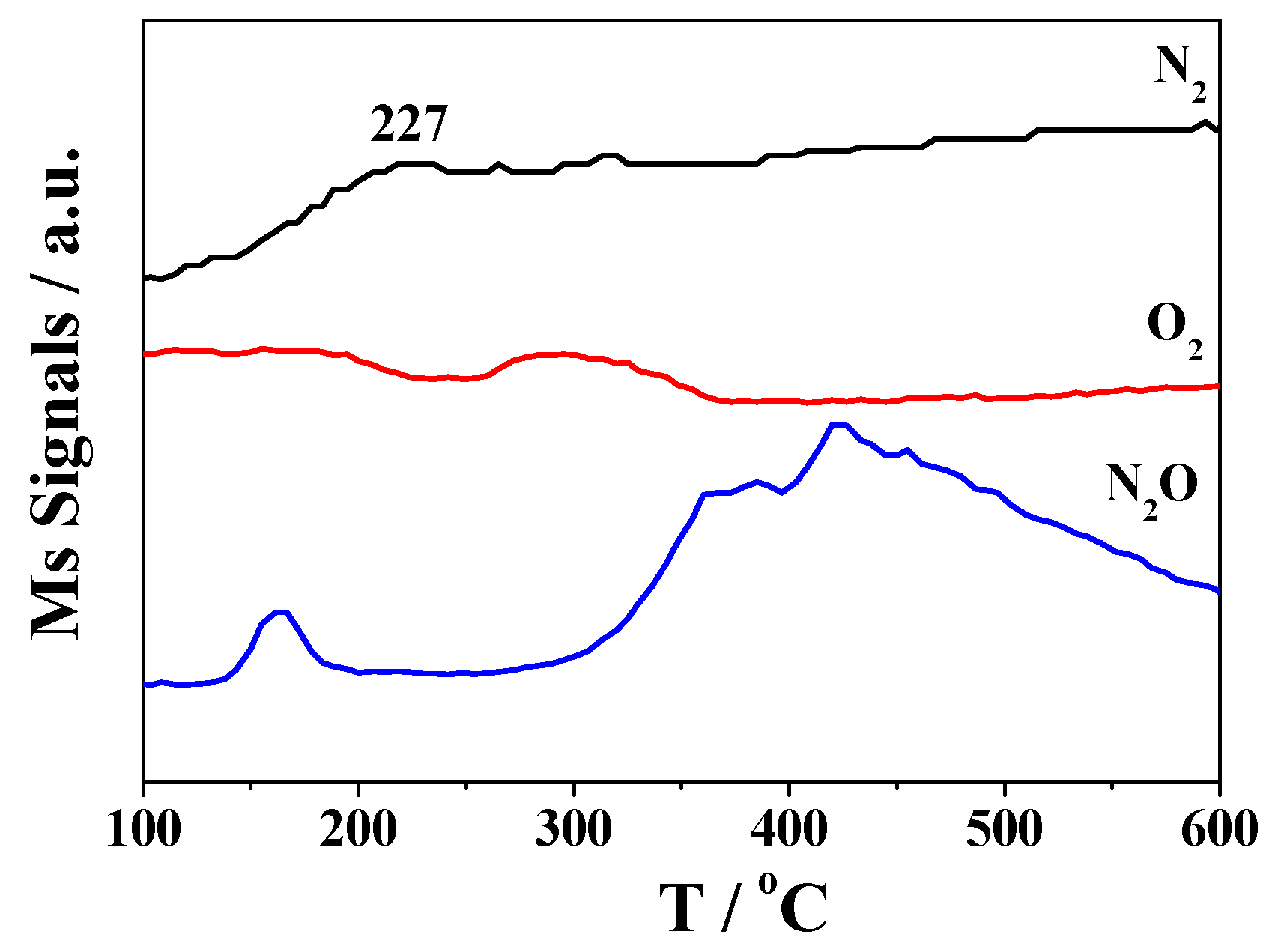
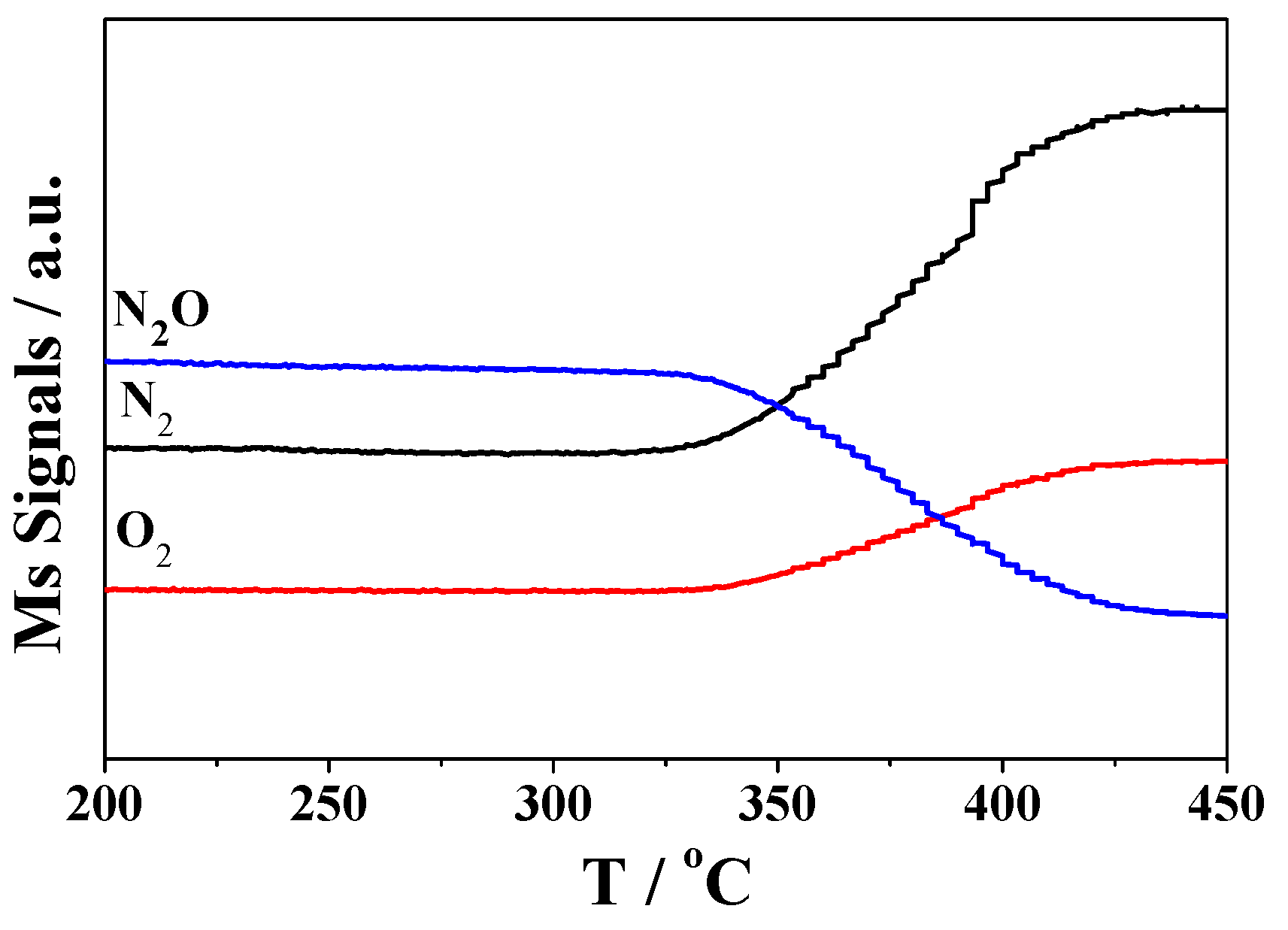
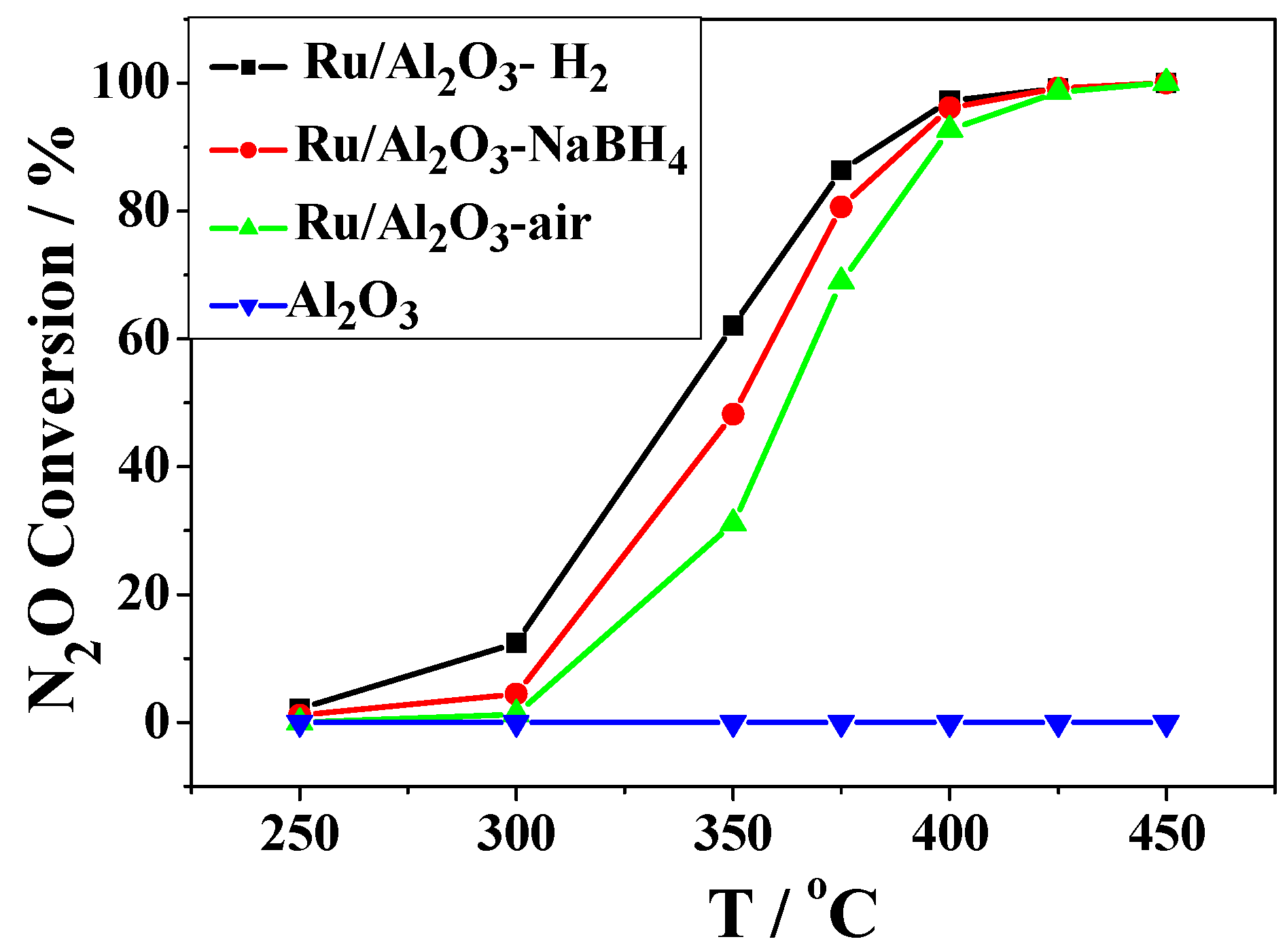
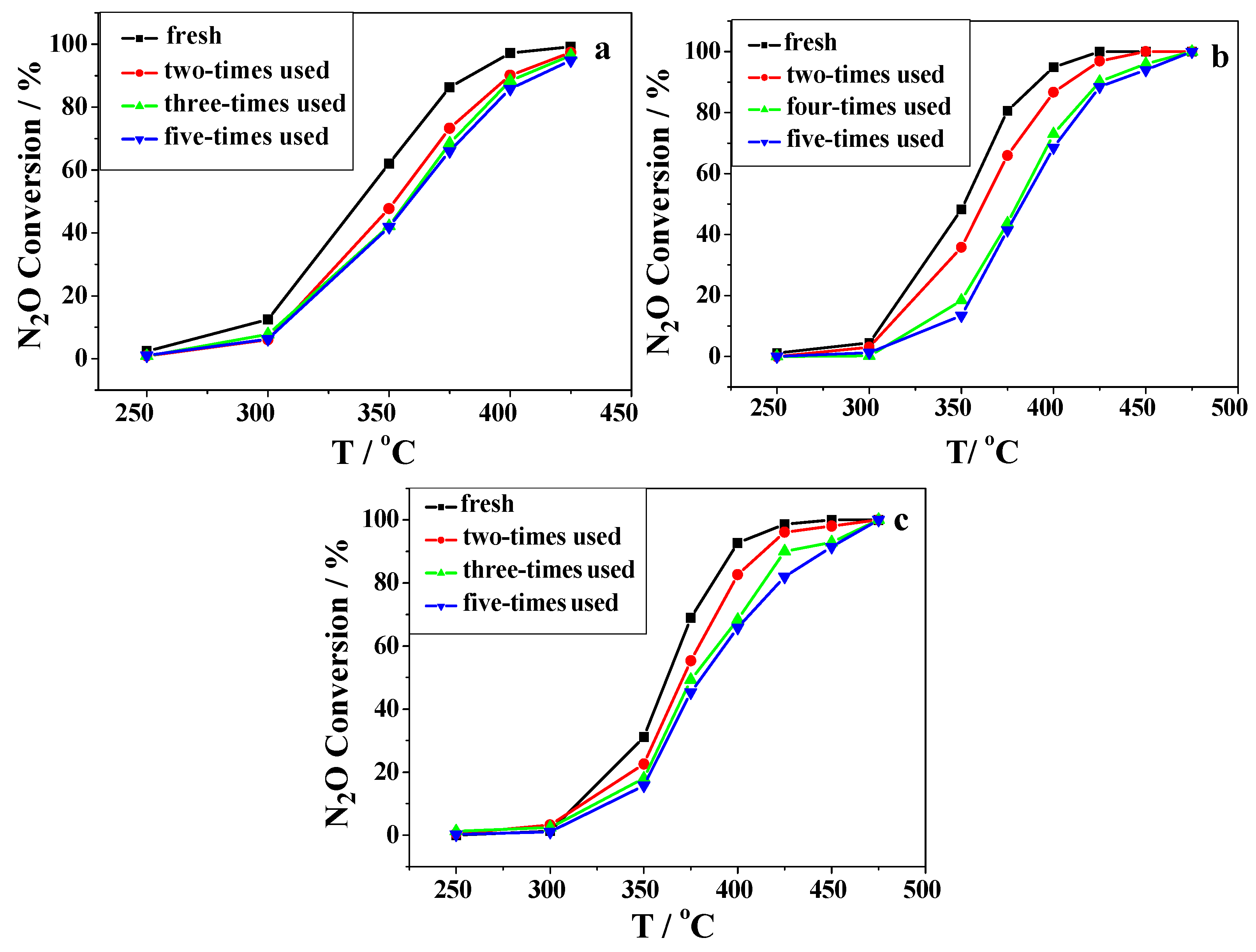
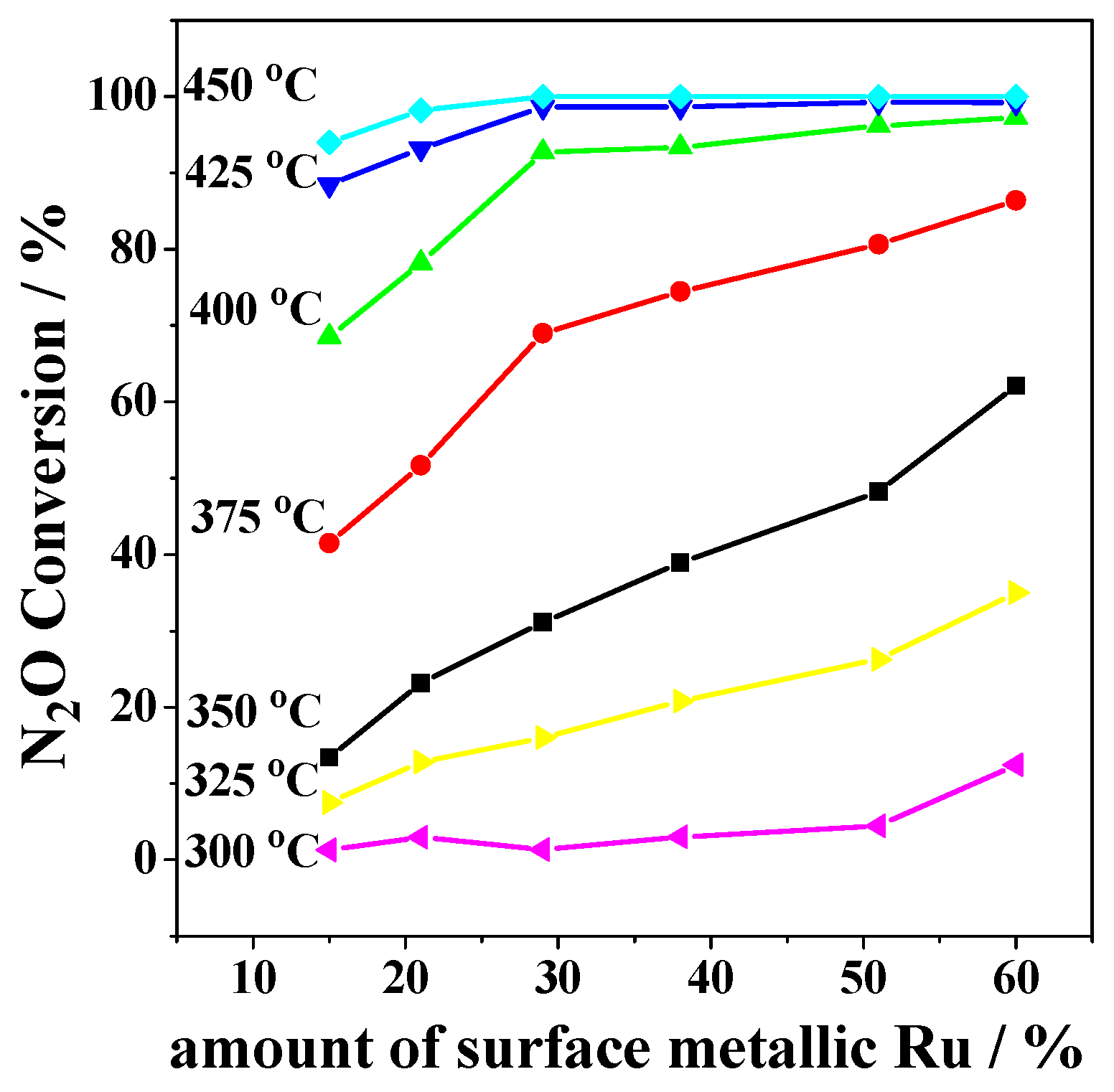
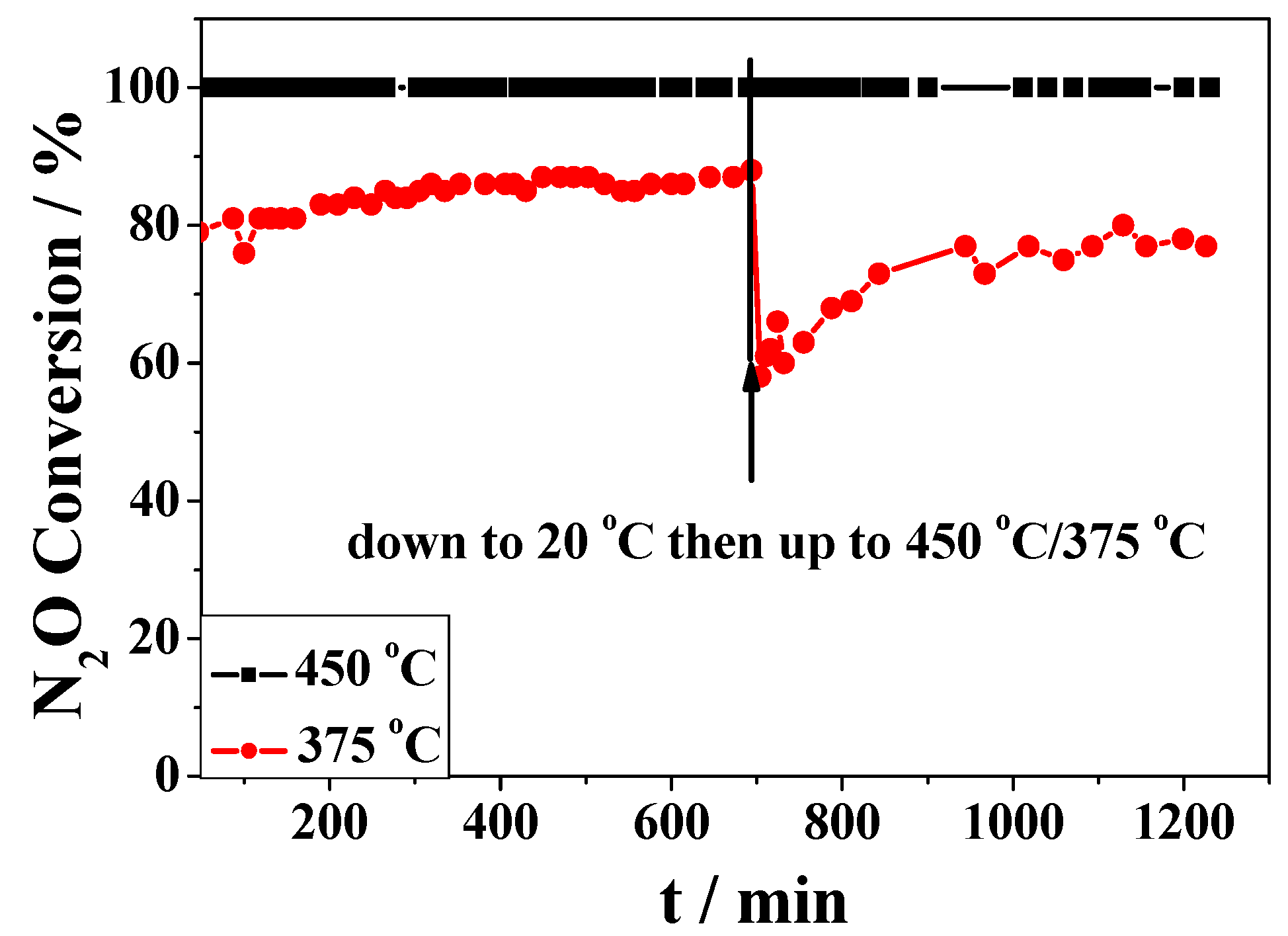
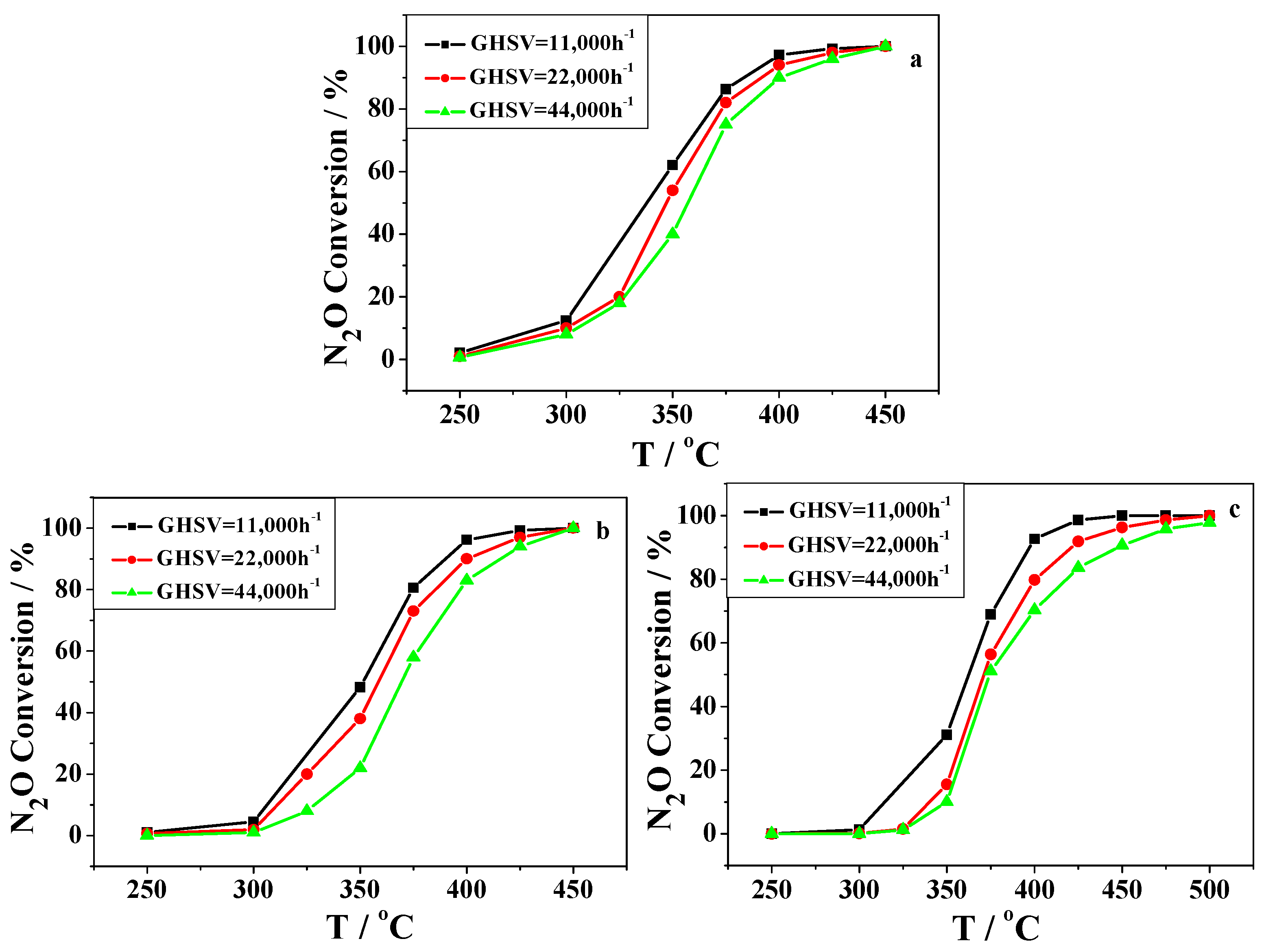
2.2. Catalytic Activity Studies
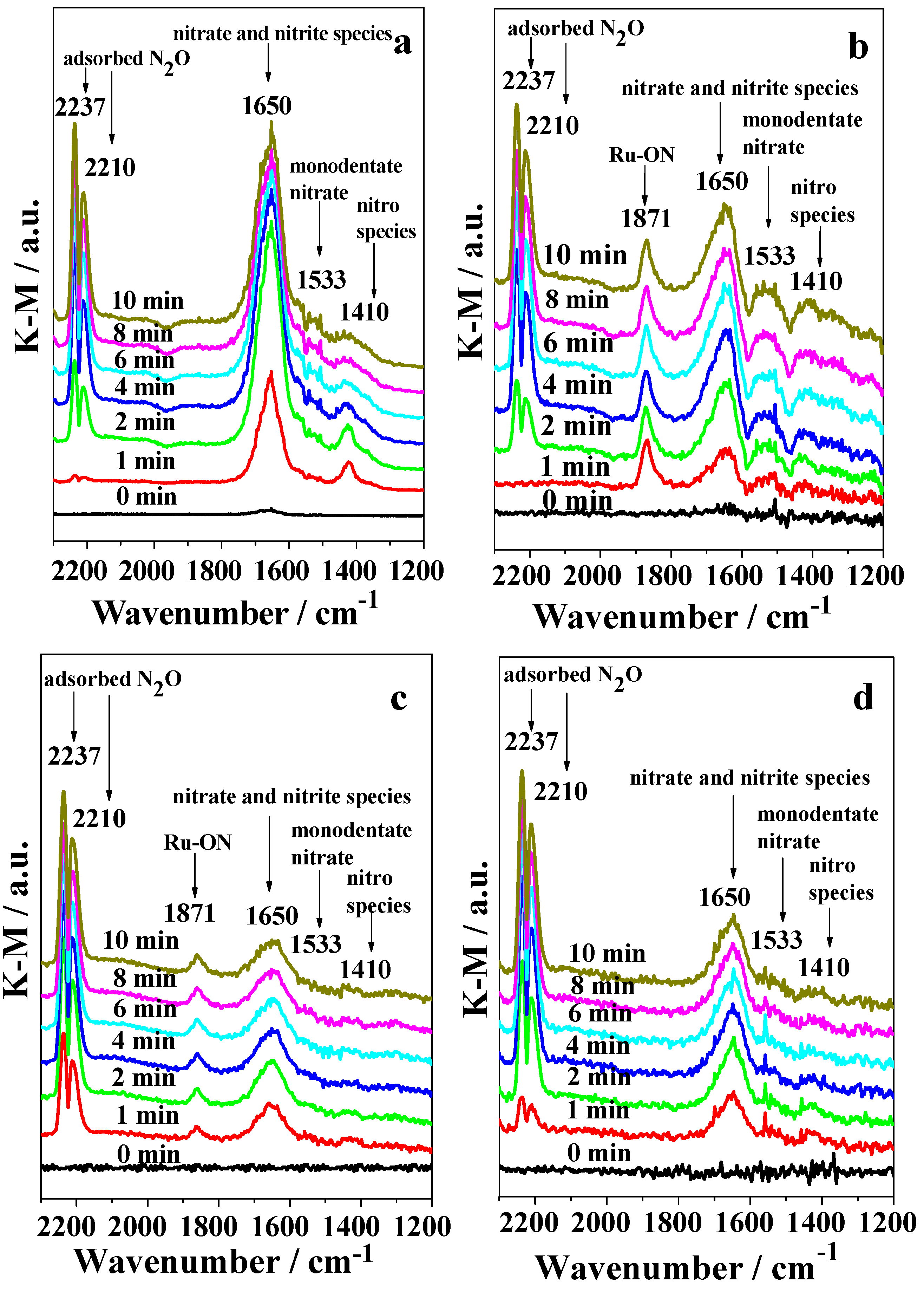
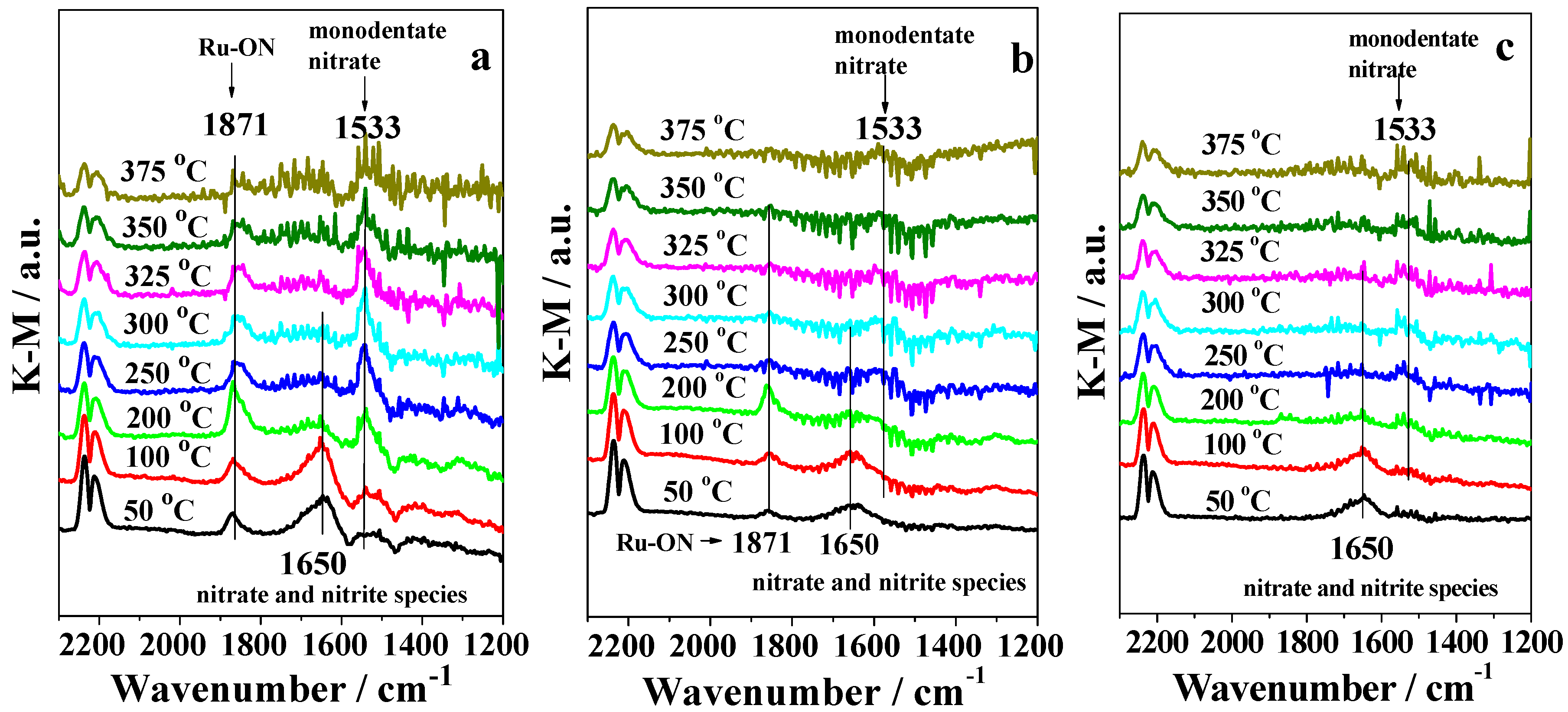
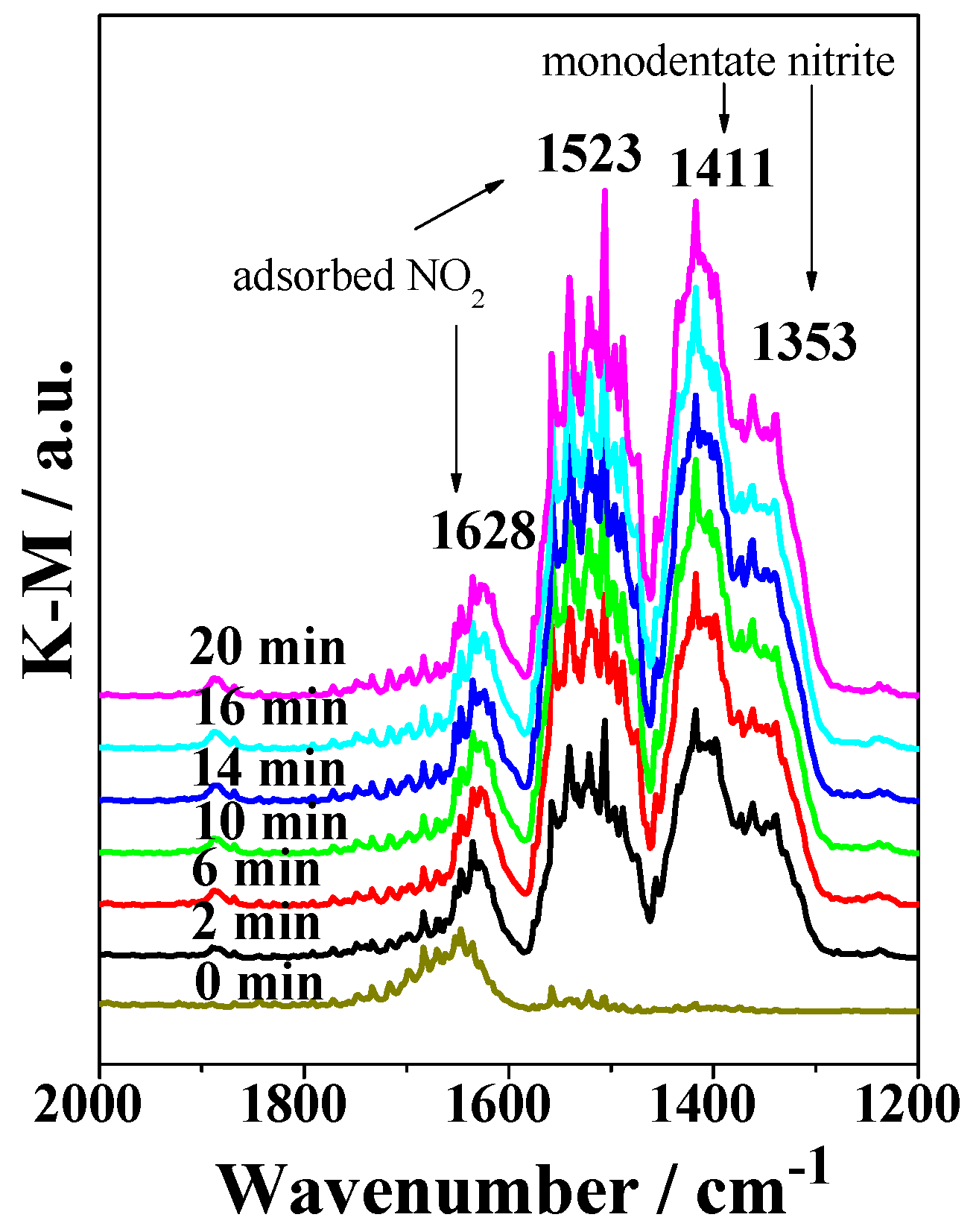
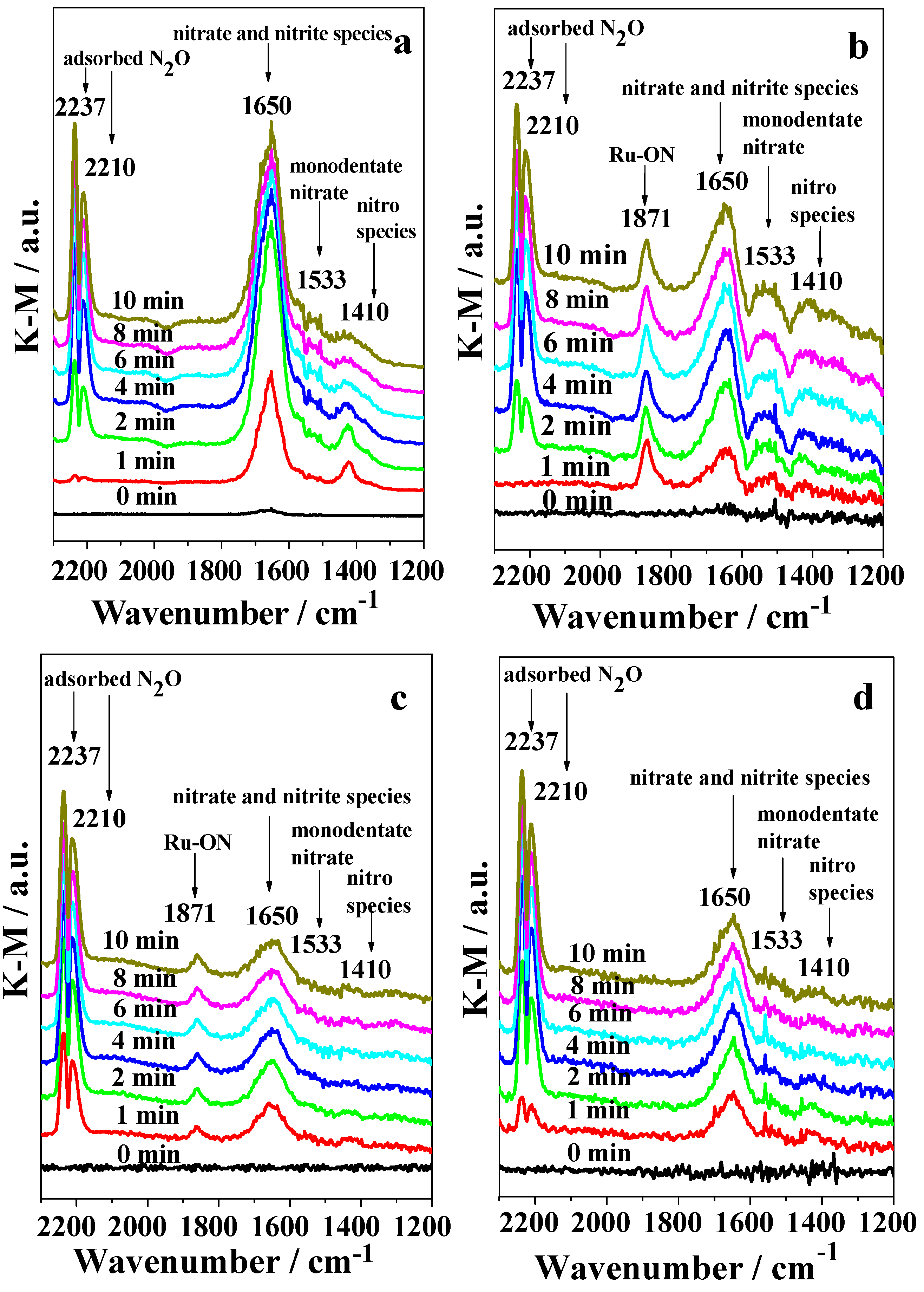
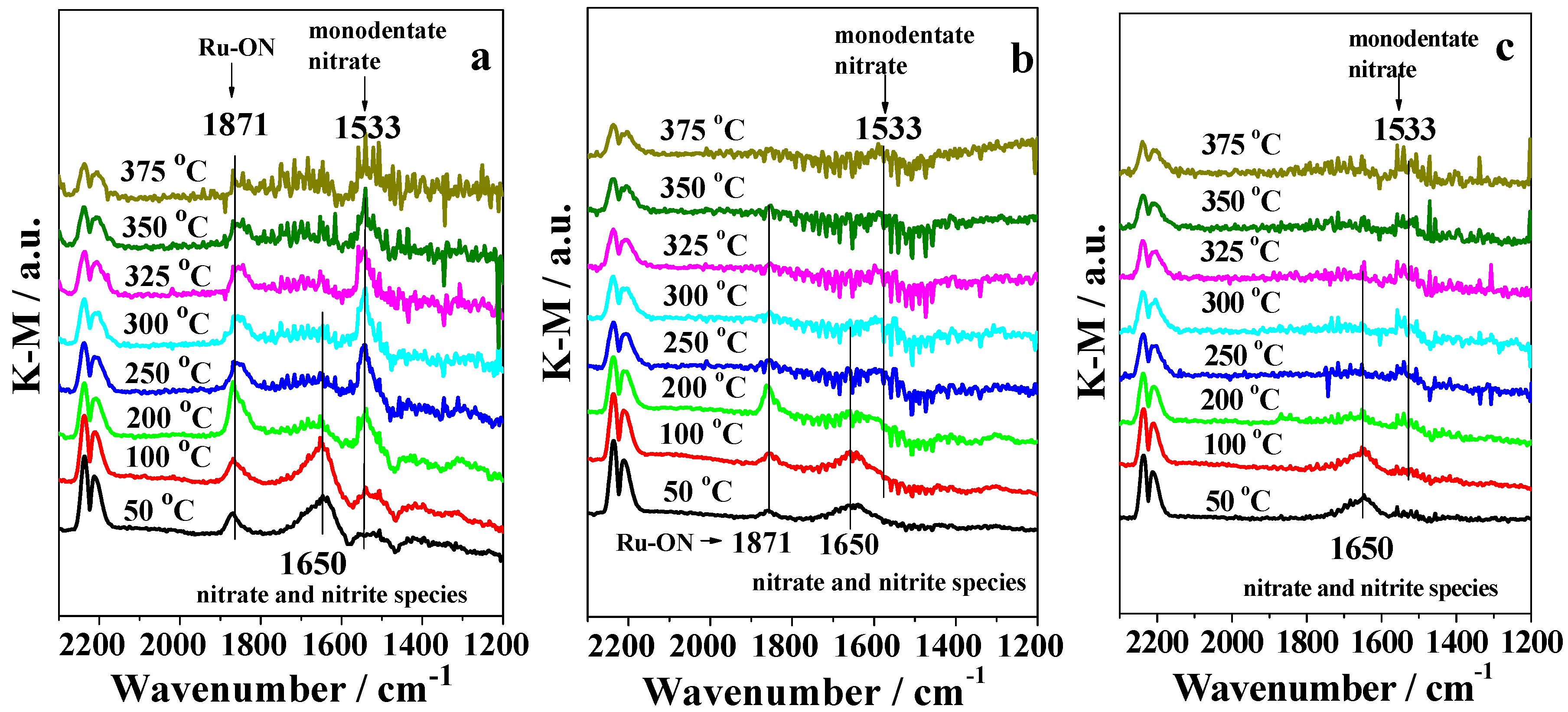
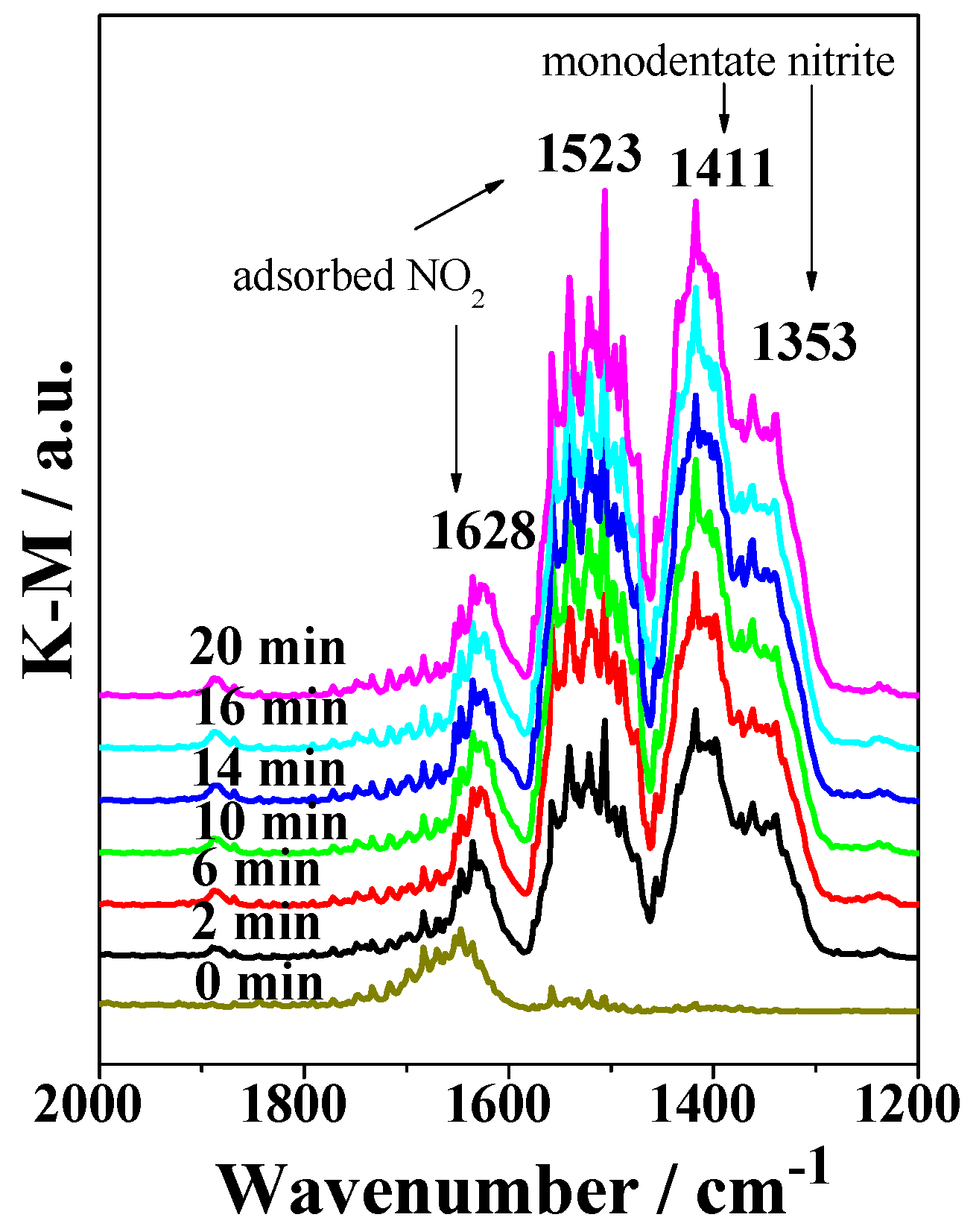
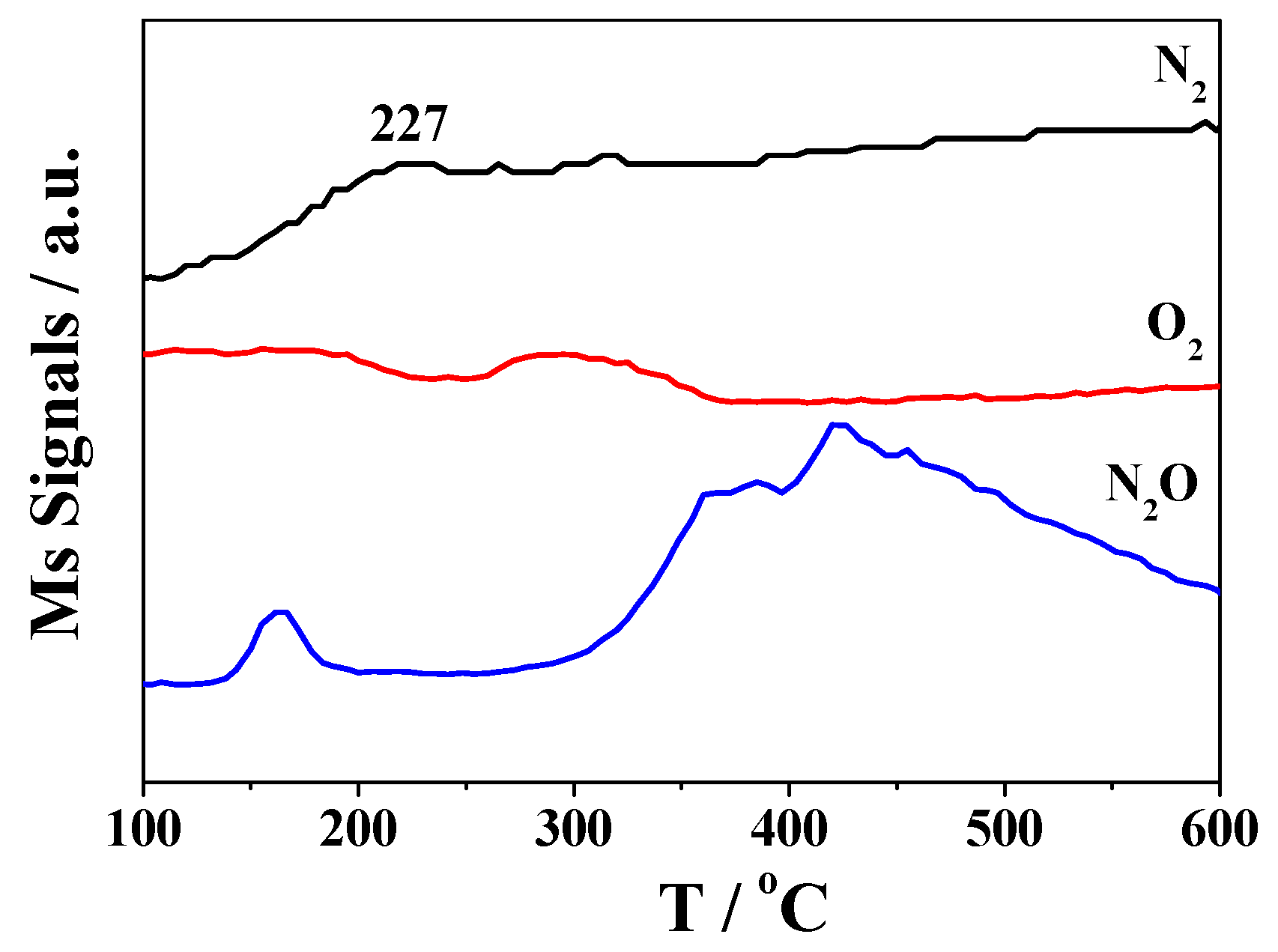
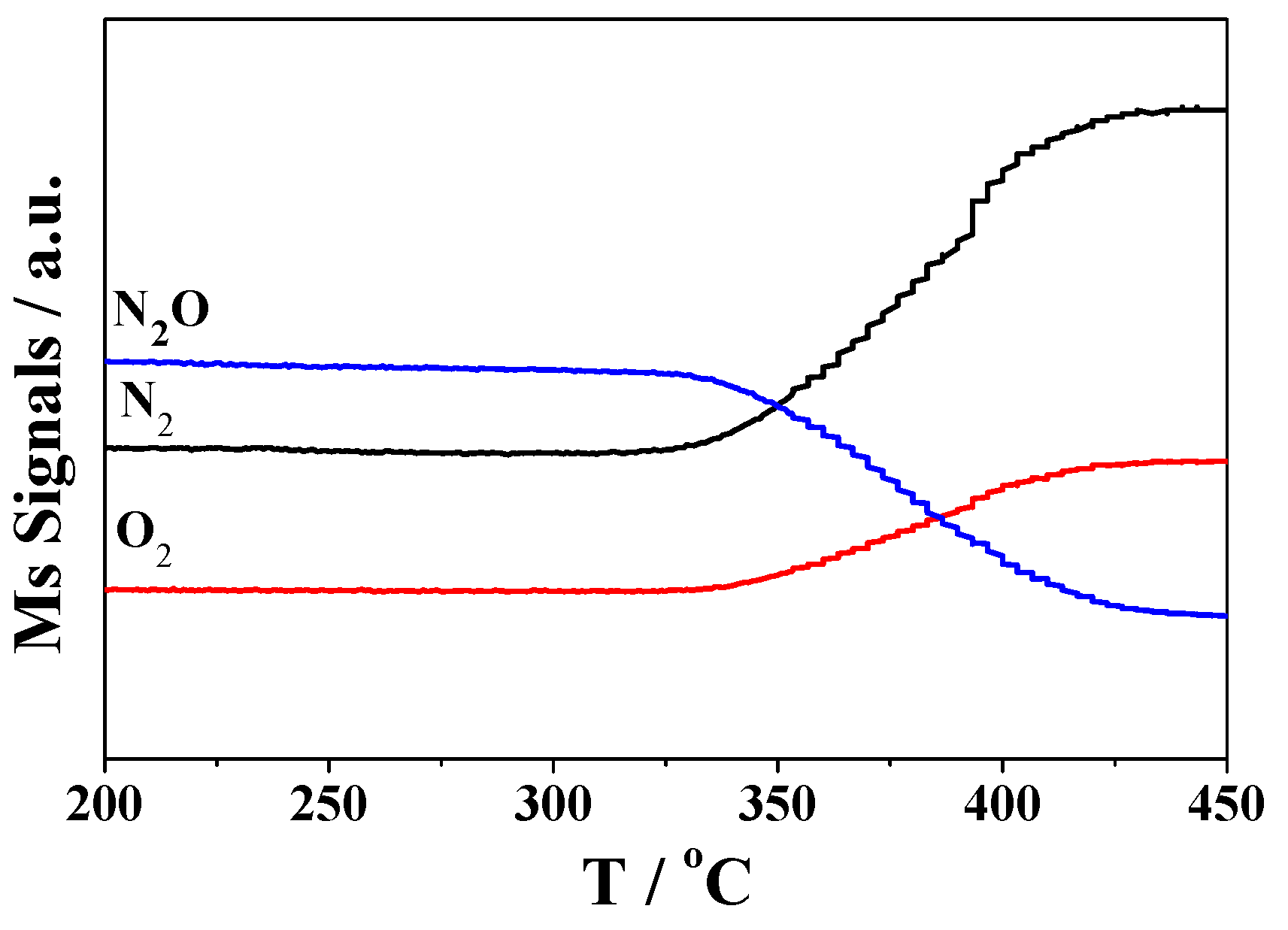
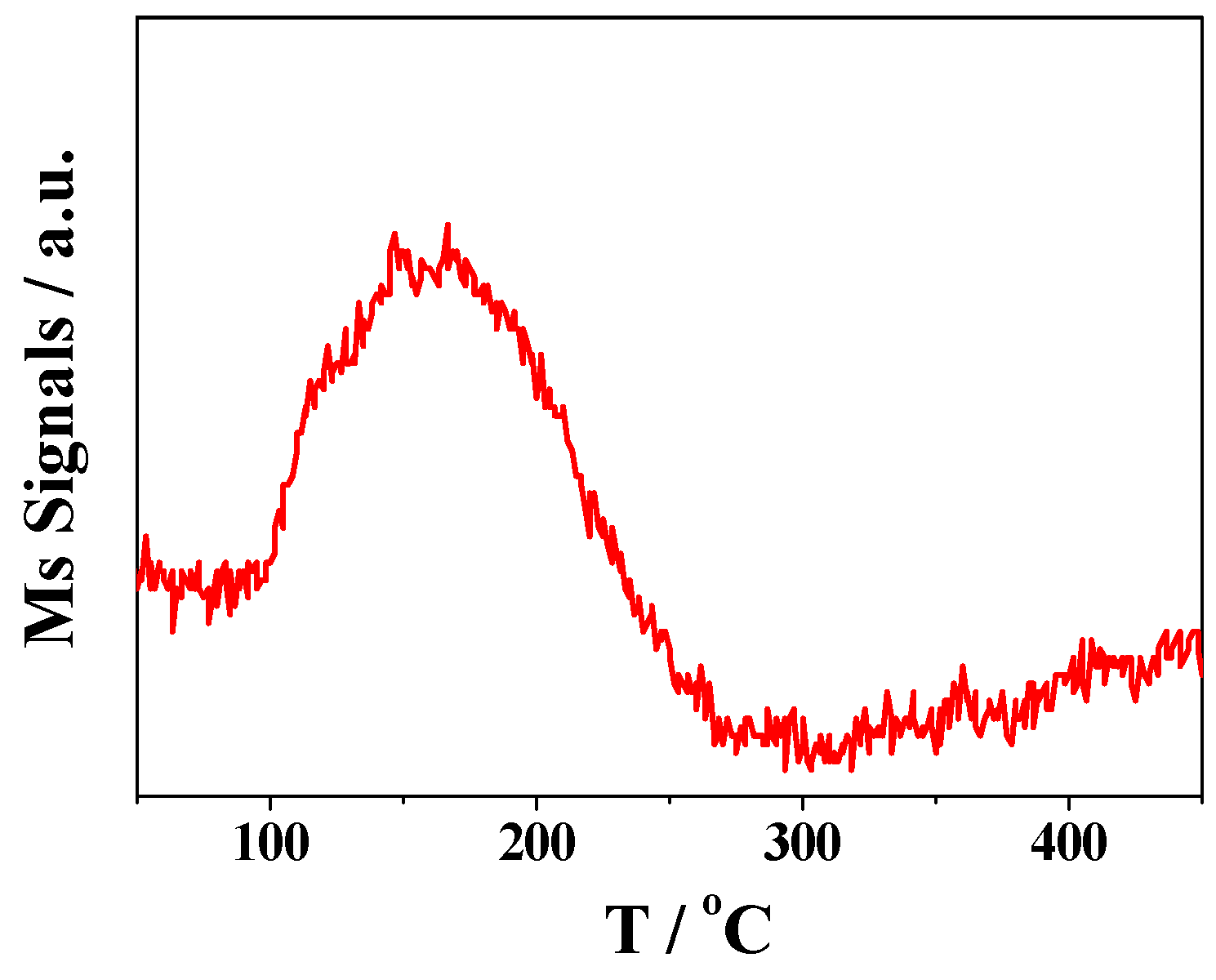
2.3. DRIFTS Studies
3. Discussion
4. Experimental Section
4.1. Materials
4.2. Catalyst Preparation
4.3. Catalyst Characterization
4.4. Catalytic Activity Test
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rodhe, H. A comparion of the contribution of various gases to the greenhouse effect. Science 1990, 248, 1217–1219. [Google Scholar] [CrossRef] [PubMed]
- Parks, J.E. Less costly catalysts for controlling engine emissions. Science 2010, 327, 1584–1585. [Google Scholar] [CrossRef] [PubMed]
- Kramlich, J.; Linak, W.P. Nitrous oxide behavior in the atmosphere, and in combustion and industrial systems. Prog. Energy Combust. Sci. 1994, 20, 149–202. [Google Scholar] [CrossRef]
- Wang, J.; Ji, Y.; He, Z.; Crocker, M.; Dearth, M.; McCabe, R.W. A non–NH3 pathway for NOx conversion in coupled LNT-SCR systems. Appl. Catal. B 2012, 111, 562–570. [Google Scholar] [CrossRef]
- Perez-Ramirez, J.; Kapteijn, F.; Schoffel, K.; Moulijn, J.A. Formation and control of N2O in nitric acid production: Where do we stand today? Appl. Catal. B 2003, 44, 117–151. [Google Scholar] [CrossRef]
- Perez-Ramirez, J. Prospects of N2O emission regulations in the European fertilizer industry. Appl. Catal. B 2007, 70, 31–35. [Google Scholar] [CrossRef]
- Kondratenko, E.V.; Kondratenko, V.A.; Santiago, M.; Pérez-Ramírez, J. Mechanistic origin of the different activity of Rh-ZSM-5 and Fe-ZSM-5 in N2O decomposition. J. Catal. 2008, 256, 248–258. [Google Scholar] [CrossRef]
- Wang, J.; Xia, H.; Ju, X.; Feng, Z.; Fan, F.; Li, C. Influence of extra-framework Al on the structure of the active iron sites in Fe/ZSM-35. J. Catal. 2013, 300, 251–259. [Google Scholar] [CrossRef]
- Konsolakis, M.; Drosou, C.; Yentekakis, I.V. Support mediated promotional effects of rare earth oxides (CeO2 and La2O3) on N2O decomposition and N2O reduction by CO or C3H6 over Pt/Al2O3 structured catalysts. Appl. Catal. B 2012, 123–124, 405–413. [Google Scholar] [CrossRef]
- Pietrogiacomi, D.; Campa, M.C.; Carbone, L.; Tuti, S.; Occhiuzzi, M. N2O decomposition on CoOx, CuOx, FeOx or MnOx supported on ZrO2: The effect of zirconia doping with sulfates or K+ on catalytic activity. Appl. Catal. B 2016, 187, 218–227. [Google Scholar] [CrossRef]
- Yu, H.; Tursun, M.; Wang, X.; Wu, X. Pb0.04Co catalyst for N2O decomposition in presence of impurity gases. Appl. Catal. B 2016, 185, 110–118. [Google Scholar] [CrossRef]
- Zabilskiy, M.; Djinović, P.; Tchernychova, E.; Tkachenko, O.P.; Kustov, L.M.; Pintar, A. Nanoshaped CuO/CeO2 materials: Effect of the exposed ceria surfaces on catalytic activity in N2O decomposition reaction. ACS Catal. 2015, 5, 5357–5365. [Google Scholar] [CrossRef]
- Komvokis, V.G.; Marti, M.; Delimitis, A.; Vasalos, I.A.; Triantafyllidis, K.S. Catalytic decomposition of N2O over highly active supported Ru nanoparticles (≤3 nm) prepared by chemical reduction with ethylene glycol. Appl. Catal. B 2011, 103, 62–71. [Google Scholar] [CrossRef]
- Benco, L. Compensation effect. A DFT study of the activation of N2O over M-CHA (M = Fe2+, Co2+, RuO2+, RuO+). J. Catal. 2013, 298, 122–129. [Google Scholar] [CrossRef]
- Liu, N.; Zhang, R.; Li, Y.; Chen, B. Local Electric Field Effect of TMI (Fe, Co, Cu)-BEA on N2O Direct Dissociation. J. Phys. Chem. C 2014, 118, 10944–10956. [Google Scholar] [CrossRef]
- Kaczmarczyk, J.; Zasada, F.; Janas, J.; Indyka, P.; Piskorz, W.; Kotarba, A.; Sojka, Z. Thermodynamic Stability, Redox Properties, and Reactivity of Mn3O4, Fe3O4, and Co3O4 Model Catalysts for N2O Decomposition: Resolving the Origins of Steady Turnover. ACS Catal. 2016, 6, 1235–1246. [Google Scholar] [CrossRef]
- Kim, B.; Li, Z.; Kay, B.D.; Dohnalek, Z.; Kim, Y.K. Unexpected Nondissociative Binding of N2O on Oxygen Vacancies on a Rutile TiO2 (110)-1 × 1. J. Phys. Chem. C 2012, 116, 1145–1150. [Google Scholar] [CrossRef]
- Pachatouridou, E.; Papista, E.; Delimitis, A.; Vasiliades, M.A.; Efstathiou, A.M.; Amiridis, M.D.; Alexeev, O.S.; Bloom, D.; Marnellos, G.E.; Konsolakis, M.; et al. N2O decomposition over ceria-promoted Ir/Al2O3 catalysts: The role of ceria. Appl. Catal. B 2016, 187, 259–268. [Google Scholar] [CrossRef]
- Amrousse, R.; Tsutsumi, A. Novel Rh-substituted hexaaluminate catalysts for N2O decomposition. Catal. Sci. Technol. 2016, 6, 438–441. [Google Scholar] [CrossRef]
- Jisa, K.; Novakova, J.; Schwarze, M.; Vondrova, A.; Sklenak, S.; Sobalik, Z. Role of the Fe-zeolite structure and iron state in the N2O decomposition: Comparison of Fe-FER, Fe-BEA, and Fe-MFI catalysts. J. Catal. 2009, 262, 27–34. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, Z.; Sui, C.; Yuan, F.; Niu, X.; Zhu, Y. Catalytic Decomposition of N2O over Co-Ti Oxide Catalysts: Interaction between Co and Ti Oxide. ChemCatChem 2016, 8, 2155–2164. [Google Scholar] [CrossRef]
- Konsolakis, M. Recent Advances on Nitrous Oxide (N2O) Decomposition over Non-Noble-Metal Oxide Catalysts: Catalytic Performance, Mechanistic Considerations, and Surface Chemistry Aspects. ACS Catal. 2015, 5, 6397–6421. [Google Scholar] [CrossRef]
- Li, Y.; Armor, J.N. Catalytic decomposition of nitrous oxide on metal exchanged zeolites. Appl. Catal. B 1992, 1, L21–L29. [Google Scholar] [CrossRef]
- Chang, Y.; McCarty, J.G.; Wachsman, E.D. Effect of ruthenium-loading on the catalytic activity of Ru-NaZSM-5 zeolites for nitrous oxide decomposition. Appl. Catal. B 1995, 6, 21–23. [Google Scholar] [CrossRef]
- Pinna, F.; Scarpa, M.; Strukul, G.; Guglielminotti, E.; Boccuzzi, F.; Manzoli, M. Ru/ZrO2 Catalysts: II. N2O Adsorption and Decomposition. J. Catal. 2000, 192, 158–162. [Google Scholar] [CrossRef]
- Marnellos, G.E.; Efthimiadis, E.A.; Vasalos, I.A. Effect of SO2 and H2O on the N2O decomposition in the presence of O2 over Ru/Al2O3. Appl. Catal. B 2003, 46, 523–539. [Google Scholar] [CrossRef]
- Lin, Q.; Huang, Y.; Wang, Y.; Lin, L.; Liu, X.Y.; Lv, F.; Wang, A.; Li, W.C.; Zhang, T. RuO2/rutile-TiO2: A superior catalyst for N2O decomposition. J. Mater. Chem. A 2014, 2, 5178–5181. [Google Scholar] [CrossRef]
- Kawi, S.; Liu, S.Y.; Shen, S.C. Catalytic decomposition and reduction of N2O on Ru/MCM-41 catalyst. Catal. Today 2001, 68, 237–244. [Google Scholar] [CrossRef]
- Beyer, H.; Emmerich, J.; Chatziapostolou, K.; Koehler, K. Decomposition of nitrous oxide by rhodium catalysts: Effect of rhodium particle size and metal oxide support. Appl. Catal. A 2011, 391, 411–416. [Google Scholar] [CrossRef]
- Zheng, J.; Meyer, S.; Köhler, K. Abatement of nitrous oxide by ruthenium catalysts: Influence of the support. Appl. Catal. A 2015, 505, 44–51. [Google Scholar] [CrossRef]
- Komvokis, V.G.; Marnellos, G.E.; Vasalos, I.A.; Triantafyllidis, K.S. Effect of pretreatment and regeneration conditions of Ru/γ-Al2O3 catalysts for N2O decomposition and/or reduction in O2-rich atmospheres and in the presence of NOX, SO2 and H2O. Appl. Catal. B 2009, 89, 627–634. [Google Scholar] [CrossRef]
- Boissel, V.; Tahir, S.; Koh, C.A. Catalytic decomposition of N2O over monolithic supported noble metal-transition metal oxides. Appl. Catal. B 2006, 64, 234–242. [Google Scholar] [CrossRef]
- Chen, X.; Delgado, J.J.; Gatica, J.M.; Zerrad, S.; Cies, J.M.; Bernal, S. Preferential oxidation of CO in the presence of excess of hydrogen on Ru/Al2O3 catalyst: Promoting effect of ceria-terbia mixed oxide. J. Catal. 2013, 299, 272–283. [Google Scholar] [CrossRef]
- Elmasides, C.; Kondarides, D.I.; Grunert, W.; Verykios, X.E. XPS and FTIR study of Ru/Al2O3 and Ru/TiO2 catalysts: Reduction characteristics and interaction with a methane-oxygen mixture. J. Phys. Chem. B 1999, 103, 5227–5239. [Google Scholar] [CrossRef]
- Sayan, S.; Suzer, S.; Uner, D.O. XPS and in-situ IR investigation of RuSiO2 catalyst. J. Mol. Strut. 1997, 410–411, 111–114. [Google Scholar] [CrossRef]
- Lin, J.; Li, L.; Pan, X.; Wang, X.; Cong, Y.; Zhang, T.; Zhu, S. Catalytic decomposition of propellant N2O over Ir/Al2O3 catalyst. AIChE J. 2016. [Google Scholar] [CrossRef]
- Pirngruber, G.D.; Frunz, L.; Pieterse, J.A.Z. The synergy between Fe and Ru in N2O decomposition over FeRu-FER catalysts: A mechanistic explanation. J. Catal. 2006, 243, 340–349. [Google Scholar] [CrossRef]
- Carl, P.J.; Larsen, S.C. Characterization of ruthenium-exchanged zeolites (beta, Y, and ZSM-5) by EPR spectroscopy. J. Catal. 2000, 196, 352–361. [Google Scholar] [CrossRef]
- Esclapez, S.P.; Illán-Gómez, M.J.; Lecea, C.S.; Bueno-López, A. On the importance of the catalyst redox properties in the N2O decomposition over alumina and ceria supported Rh, Pd and Pt. Appl. Catal. B 2010, 96, 370–378. [Google Scholar] [CrossRef]
- Hussain, M.; Fino, D.; Russo, N. N2O decomposition by mesoporous silica supported Rh catalysts. J. Hazard. Mater. 2012, 211–212, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Hadjiivanov, K.I. Identification of neutral and charged NxOy surface species by IR spectroscopy. Catal. Rev. Sci. Eng. 2000, 42, 71–144. [Google Scholar] [CrossRef]
- Oktar, N.; Mitome, J.; Holmgreen, E.M.; Ozkan, U.S. Catalytic reduction of N2O and NO2 with methane over sol–gel palladium-based catalysts. J. Mol. Catal. A 2006, 259, 171–182. [Google Scholar] [CrossRef]
- Wang, Y.; Lei, Z.; Chen, B.; Guo, Q.; Liu, N. Adsorption of NO and N2O on Fe-BEA and H-BEA zeolites. Appl. Surf. Sci. 2010, 256, 4042–4047. [Google Scholar] [CrossRef]
- Haq, S.; Hodgson, A. N2O adsorption and reaction at Pd (110). Surf. Sci. 2000, 463, 1–10. [Google Scholar] [CrossRef]
- Parres-Esclapez, S.; Such-Basanez, I.; Illan-Gomez, M.J.; Lecea, C.S.; Bueno-Lopez, A. Study by isotopic gases and in situ spectroscopies (DRIFTS, XPS and Raman) of the N2O decomposition mechanism on Rh/CeO2 and Rh/γ-Al2O3 catalysts. J. Catal. 2010, 276, 390–401. [Google Scholar] [CrossRef]
- Kondarides, D.I.; Chafik, T.; Verykios, X.E. Catalytic Reduction of NO by CO over Rhodium Catalysts: 2. Effect of Oxygen on the Nature, Population, and Reactivity of Surface Species Formed under Reaction Conditions. J. Catal. 2000, 191, 147–164. [Google Scholar] [CrossRef]
- Matsouka, V.; Konsolakis, M.; Lambert, R.M.; Yentekakis, I.V. In situ DRIFTS study of the effect of structure (CeO2–La2O3) and surface (Na) modifiers on the catalytic and surface behaviour of Pt/γ-Al2O3 catalyst under simulated exhaust conditions. Appl. Catal. B 2008, 84, 715–722. [Google Scholar] [CrossRef]
- Karakas, G.; Mitome-Watson, J.; Ozkan, U.S. In situ DRIFTS characterization of wet-impregnated and sol–gel Pd/TiO2 for NO reduction with CH4. Catal. Commun. 2002, 3, 199–206. [Google Scholar] [CrossRef]
- Baidya, T.; Bera, P.; Mukri, B.D.; Parida, S.K.; Kroher, O.; Elsener, M.; Hegde, M.S. DRIFTS studies on CO and NO adsorption and NO + CO reaction over Pd2+-substituted CeO2 and Ce0.75Sn0.25O2 catalysts. J. Catal. 2013, 303, 117–129. [Google Scholar] [CrossRef]
- Delabie, A.; Vinckier, C.; Flock, M.; Pierloot, K. Evaluating the activation barriers for transition metal N2O reactions. J. Phys. Chem. A 2001, 105, 5479–5485. [Google Scholar] [CrossRef]
- Miller, D.D.; Chuang, S.S.C. The effect of O2 on the NO-CO reaction over Ag-Pd/Al2O3: An in situ infrared study. Catal. Commun. 2009, 10, 1313–1318. [Google Scholar] [CrossRef]
- Kapteijn, F. Rodriguez-Mirasol, J. Moulijn, J.A. Heterogeneous catalytic decomposition of nitrous oxide. Appl. Catal. B 1996, 9, 25–64. [Google Scholar] [CrossRef]
- Liu, N.; Chen, B.; Li, Y.; Zhang, R.; Liang, X.; Li, Y.; Lei, Z. Charge Transfer Analysis on the Direct Decomposition of Nitrous Oxide over Fe-BEA Zeolite: An Experimental and Density Functional Study. J. Phys. Chem. C 2011, 115, 12883–12890. [Google Scholar] [CrossRef]
- Pérez-Ramírez, J.; Kapteijn, F.; Mul, G.; Moulijn, J.A. NO-assisted N2O decomposition over Fe-based catalysts: Effects of gas-phase composition and catalyst constitution. J. Catal. 2002, 208, 211–223. [Google Scholar] [CrossRef]
- Kapteijn, F.; Marban, G.; Rodriguez-Mirasol, J.; Moulijn, J.A. Kinetic analysis of the decomposition of nitrous oxide over ZSM-5 catalysts. J. Catal. 1997, 167, 256–265. [Google Scholar] [CrossRef]
- Lavrov, V.V.; Blagojevic, V.; Koyanagi, G.K.; Orlova, G.; Bohme, D.K. Gas-phase oxidation and nitration of first-, second-, and third-row atomic cations in reactions with nitrous oxide: Periodicities in reactivity. J. Phys. Chem. A 2004, 108, 5610–5624. [Google Scholar] [CrossRef]
- Fu, C.M.; Korchak, V.N.; Hall, W.K. Decomposition of nitrous oxide on FeY zeolite. J. Catal. 1981, 68, 166–171. [Google Scholar] [CrossRef]
- Wood, B.R.; Reimer, J.A.; Bell, A.T.; Janicke, M.T.; Ott, K.C. Nitrous oxide decomposition and surface oxygen formation on Fe-ZSM-5. J. Catal. 2004, 224, 148–155. [Google Scholar] [CrossRef]
- Heyden, A.; Keil, F.J.; Peters, B.; Bell, A.T. Comprehensive DFT study of nitrous oxide decomposition over Fe-ZSM-5. J. Phys. Chem. B 2005, 109, 1857–1873. [Google Scholar] [CrossRef] [PubMed]
- Kondratenko, E.V.; Pérez-Ramírez, J. Mechanis and kinetics of direct N2O decomposition over Fe-MFI zeolites with different iron speciation from temporal analysis of products. J. Phys. Chem. B 2006, 110, 22586–22595. [Google Scholar] [CrossRef] [PubMed]
- Ryder, J.A.; Chakraborty, A.K.; Bell, A.T. Density functional theory study of benzene oxidation over Fe-ZSM-5. J. Catal. 2003, 220, 84–91. [Google Scholar] [CrossRef]
- Chen, B.; Liu, N.; Liu, X.; Zhang, R.; Li, Y.; Li, Y.; Sun, X. Study on the direct decomposition of nitrous oxide over Fe-beta zeolites: From experiment to theory. Catal. Today 2011, 175, 245–255. [Google Scholar] [CrossRef]
- Zhang, X.; Shen, Q.; He, C.; Ma, C.; Cheng, J.; Li, L.; Hao, Z. Investigation of Selective Catalytic Reduction of N2O by NH3 over an Fe-Mordenite Catalyst: Reaction Mechanism and O2 Effect. ACS Catal. 2012, 2, 512–520. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Surface Area (m2/g) | Metallic Ru (eV) a | Ru4+ (eV) a | Metallic Ru/Ru4+ (%) b | ||
|---|---|---|---|---|---|---|
| 3d5/2 | 3d3/2 | 3d5/2 | 3d3/2 | |||
| Al2O3 | 204 | - | - | - | - | - |
| Ru/Al2O3-H2 | 199 | 279.8 | 284.0 | 285.0 | 289.0 | 60/40 |
| 1-time used Ru/Al2O3-H2 | 196 | 280.4 | 284.5 | 285.6 | 288.5 | 38/62 |
| 5-times used Ru/Al2O3-H2 | 196 | 280.4 | 284.3 | 285.0 | 288.0 | 26/74 |
| Ru/Al2O3-NaBH4 | 201 | 280.0 | 284.1 | 285.0 | 289.1 | 51/49 |
| 1-time used Ru/Al2O3-NaBH4 | 199 | 278.8 | 283.0 | 284.0 | 288.0 | 47/53 |
| 5-times used Ru/Al2O3-NaBH4 | 199 | 280.0 | 284.0 | 285.0 | 288.7 | 15/85 |
| Ru/Al2O3-air | 200 | 280.0 | 284.0 | 285.0 | 289.0 | 29/71 |
| 1-time used Ru/Al2O3-air | 196 | 280.0 | 283.5 | 285.3 | 289.3 | 21/80 |
| 5-times used Ru/Al2O3-air | 196 | 280.5 | 284.6 | 285.2 | 288.6 | 8/92 |
| used Ru/Al2O3-NaBH4 (cool under He) | - | 280.0 | 283.7 | 285.8 | 288.0 | 40/60 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sui, C.; Yuan, F.; Zhang, Z.; Zhang, C.; Niu, X.; Zhu, Y. Effect of Ru Species on N2O Decomposition over Ru/Al2O3 Catalysts. Catalysts 2016, 6, 173. https://doi.org/10.3390/catal6110173
Sui C, Yuan F, Zhang Z, Zhang C, Niu X, Zhu Y. Effect of Ru Species on N2O Decomposition over Ru/Al2O3 Catalysts. Catalysts. 2016; 6(11):173. https://doi.org/10.3390/catal6110173
Chicago/Turabian StyleSui, Chao, Fulong Yuan, Zhiping Zhang, Chi Zhang, Xiaoyu Niu, and Yujun Zhu. 2016. "Effect of Ru Species on N2O Decomposition over Ru/Al2O3 Catalysts" Catalysts 6, no. 11: 173. https://doi.org/10.3390/catal6110173
APA StyleSui, C., Yuan, F., Zhang, Z., Zhang, C., Niu, X., & Zhu, Y. (2016). Effect of Ru Species on N2O Decomposition over Ru/Al2O3 Catalysts. Catalysts, 6(11), 173. https://doi.org/10.3390/catal6110173