2.2. Catalytic Studies of Allyl Alcohol Epoxidation
Whilst previous studies of TS-1B have focused upon aromatic hydroxylation [
8,
9,
10], we chose to investigate the impact of NH
4HF
2 treatment on the epoxidation activity of TS-1. Allyl alcohol was chosen as a model HPPO substrate [
6]. Both TS-1 (
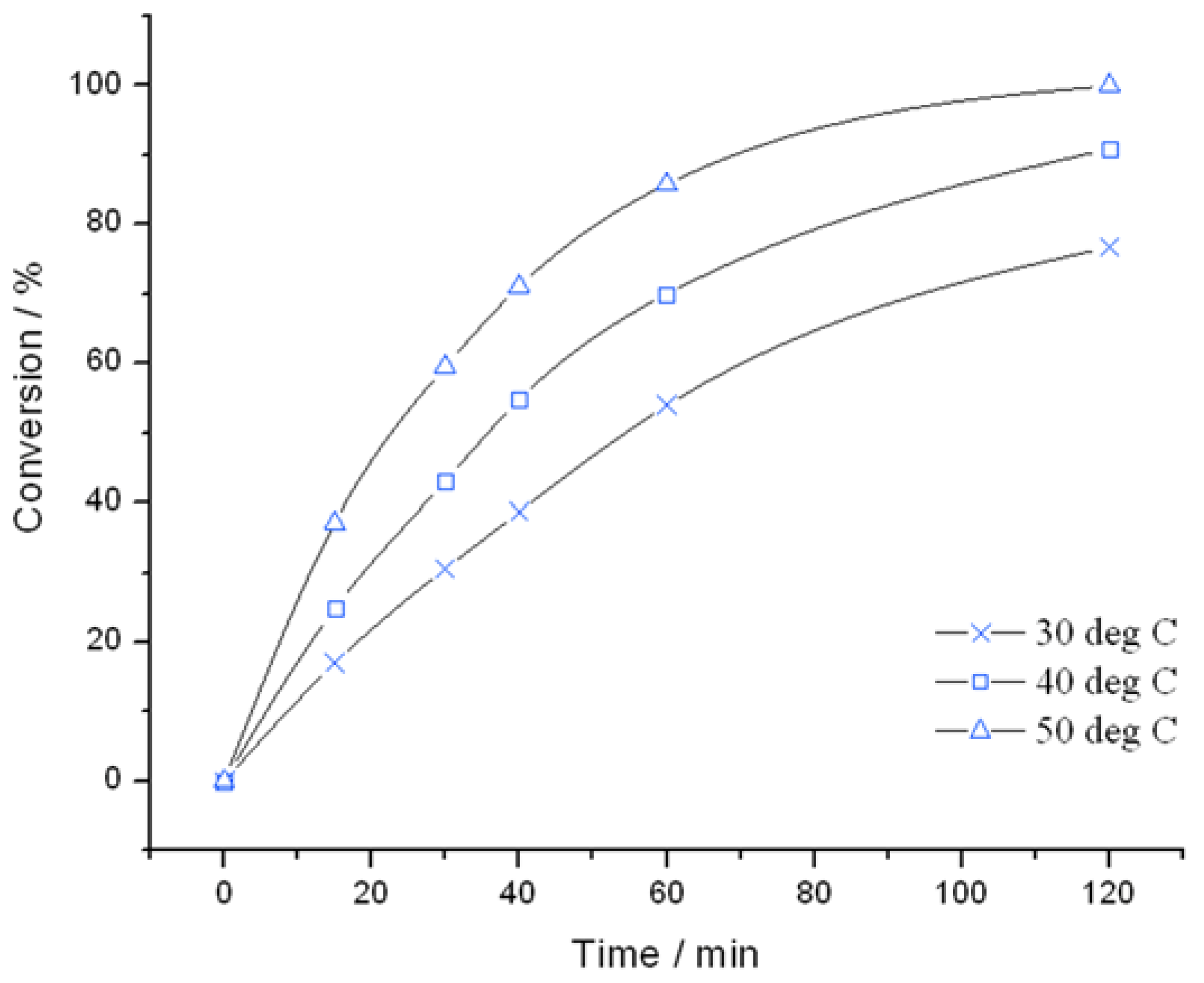
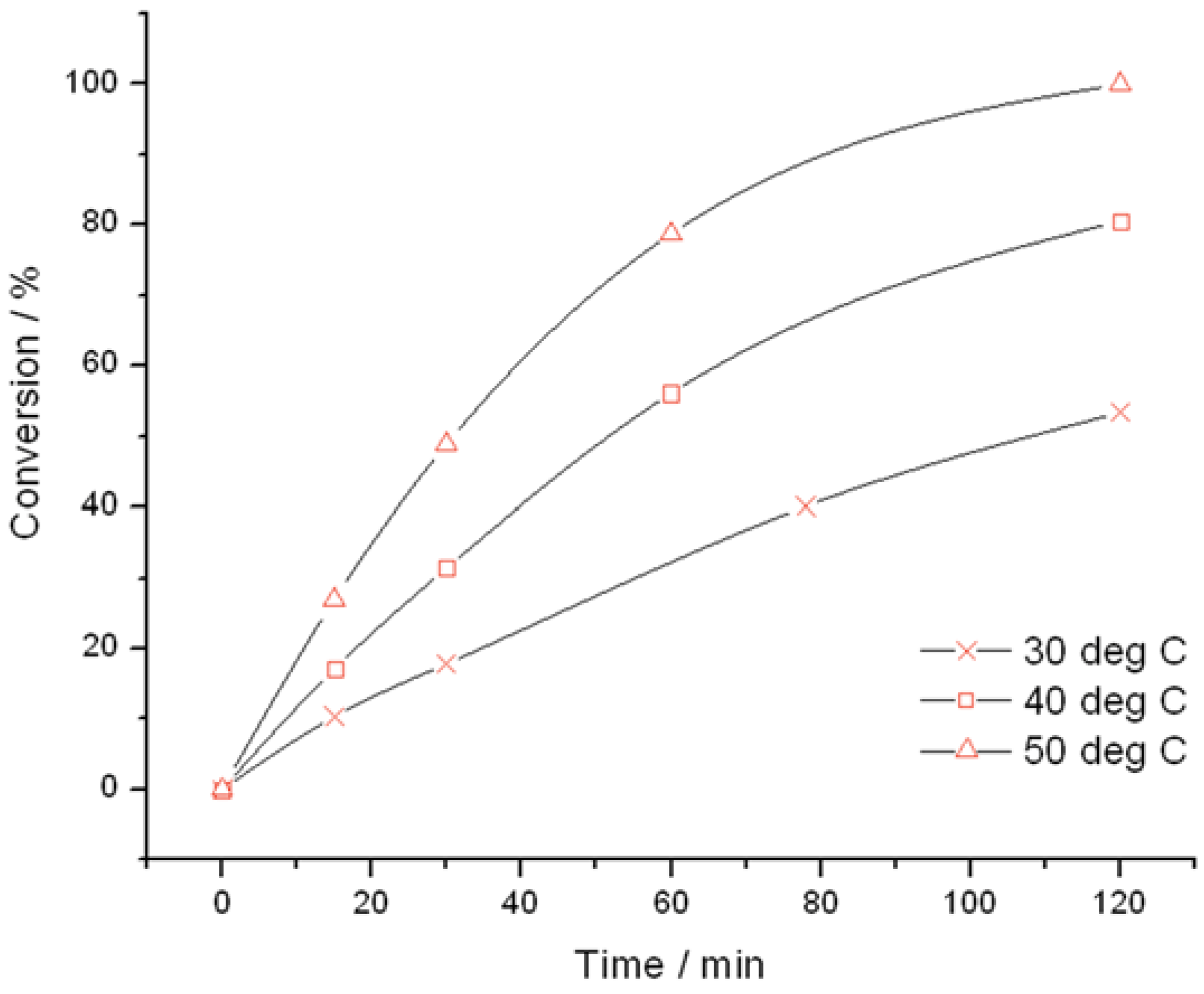
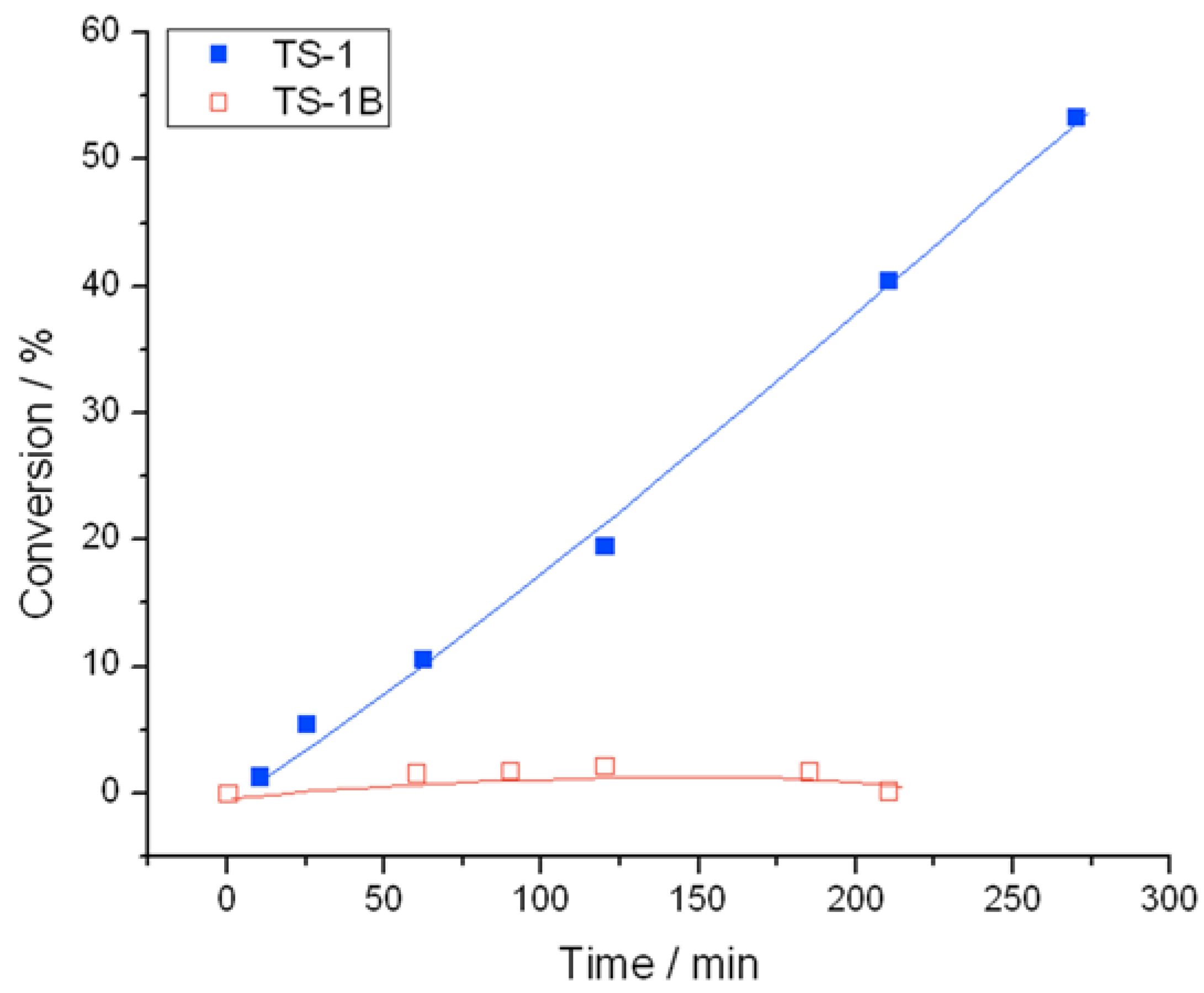
Figure 1) and TS-1B (
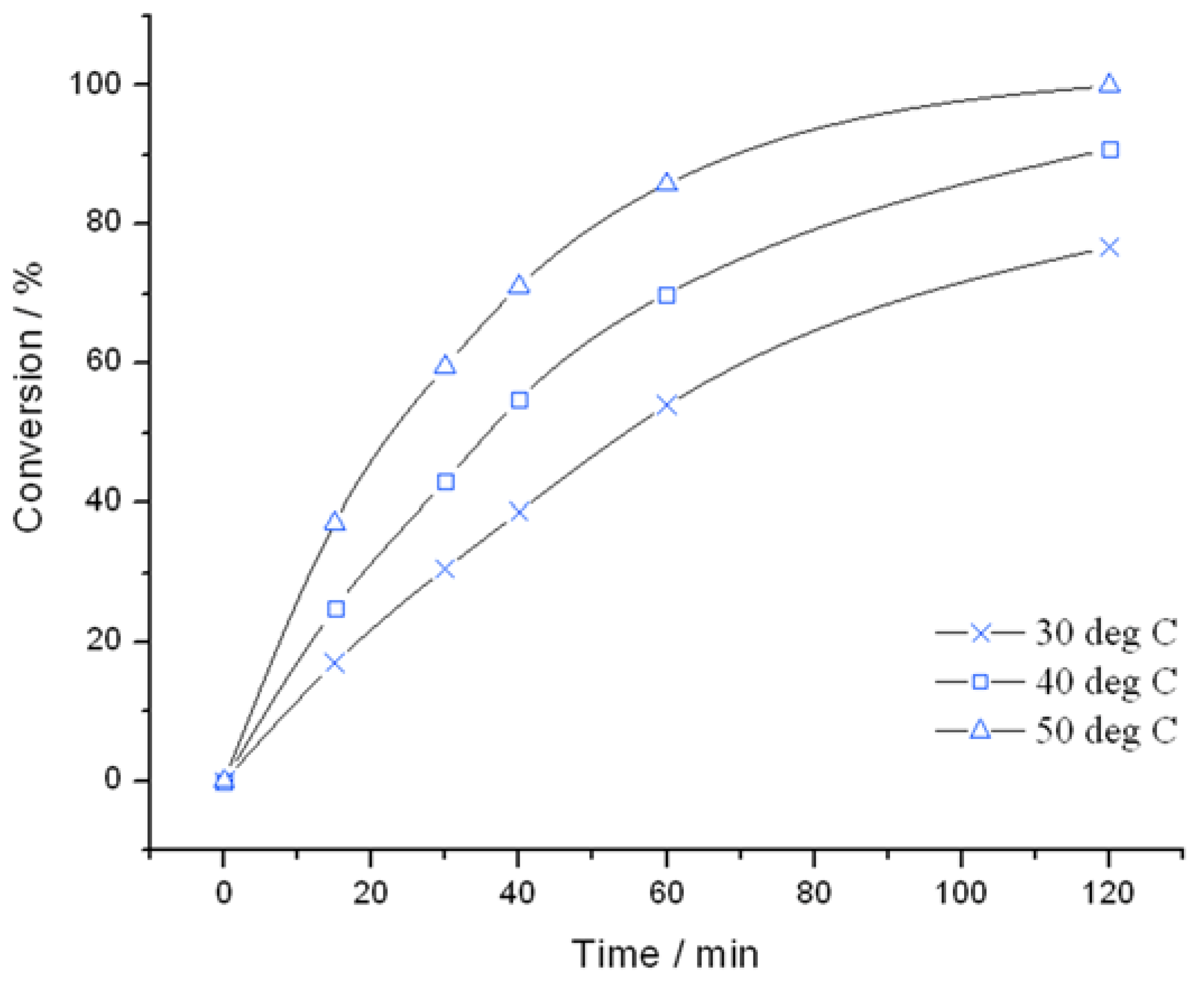
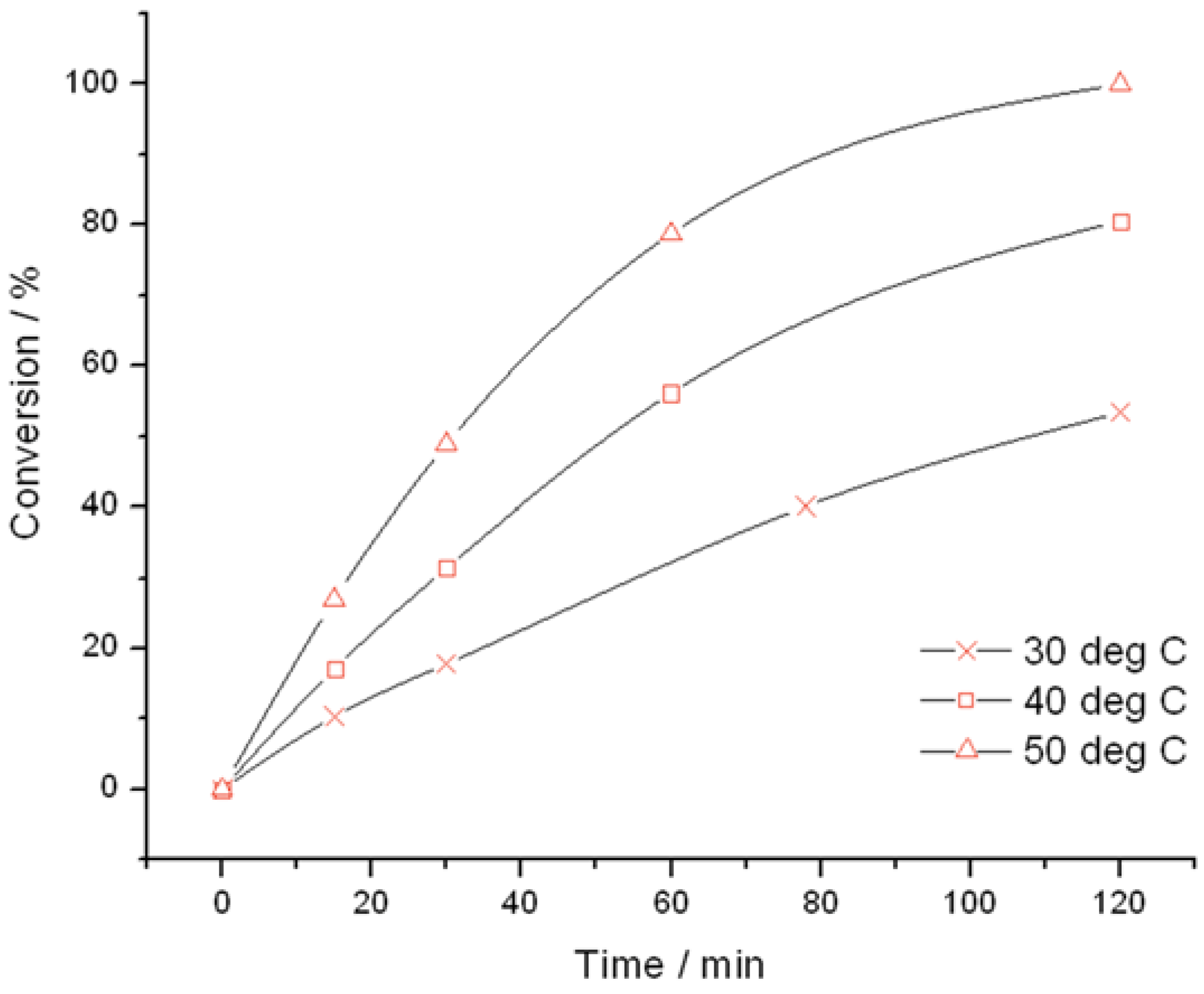
Figure 2) were found to be very active catalysts for this reaction. Both catalysts are able to reach full conversion in 120 min or less at the relatively mild reaction temperature of 50 °C. In both cases, glycidol is the main reaction product obtained with >95% selectivity, although 3-methoxy-1,2-propanediol (3M12PD) was also detected, almost fully accounting for the remaining carbon balance. Carbon balances of 95%–98% were always obtained, thus indicated that any other potential by-products below the detectability limit of our analytical protocols were not formed to a major extent. The observation of 3M12PD indicates that ring opening methanolysis of glycidol occurs across the least hindered end of the epoxide. Under a conventional acid catalysed mechanism, 2M13PD would be expected according to Markovnikov’s rules. This difference may be due to the active sites in TS-1 catalysing ring opening via a basic mechanism, or may alternatively be a consequence of shape selectivity favouring the linear methoxy-substituted diol over the branched. Although comparable in activity at 50 °C, further kinetic analysis at multiple temperatures reveals distinct differences in the activities of TS-1 and TS-1B.
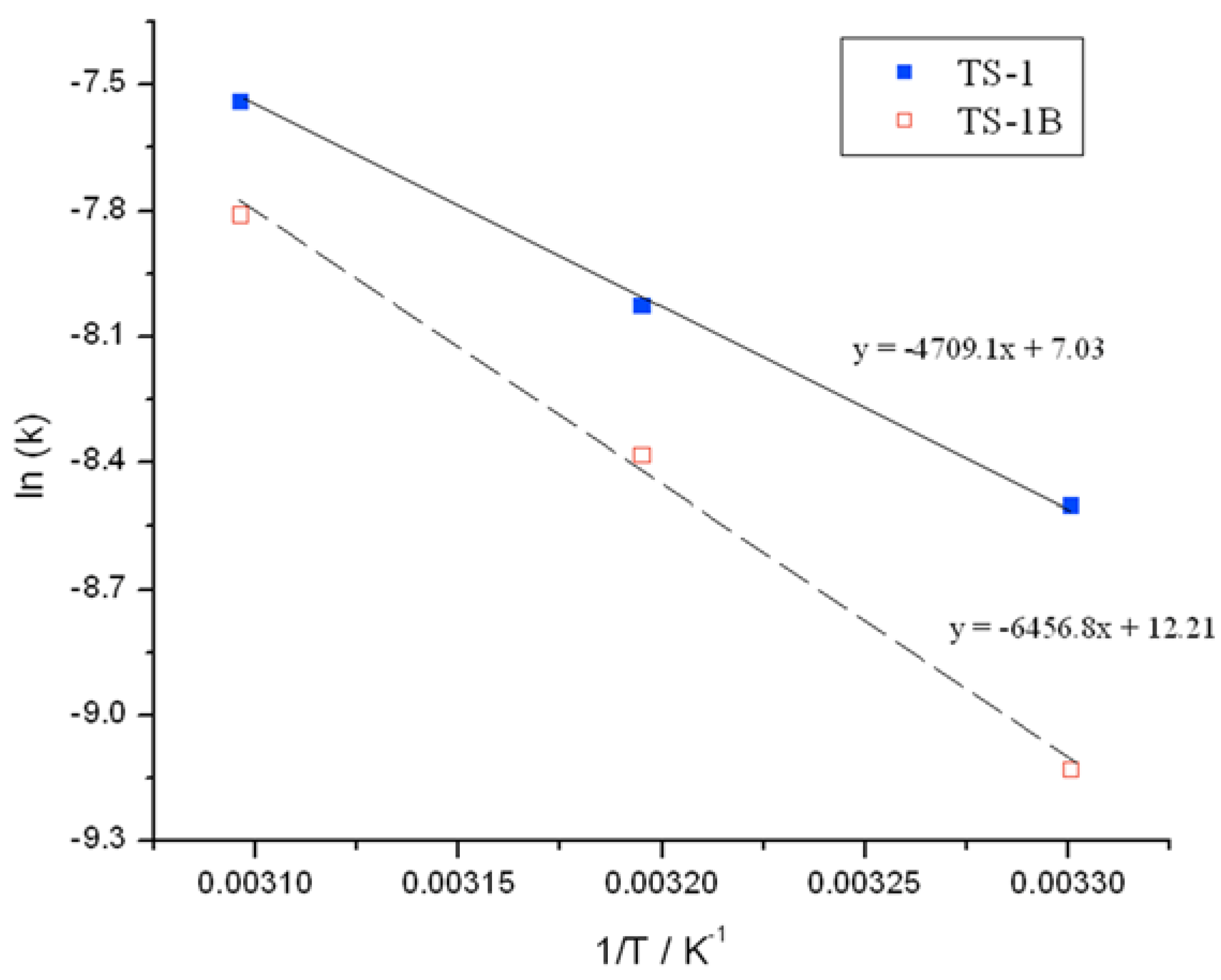
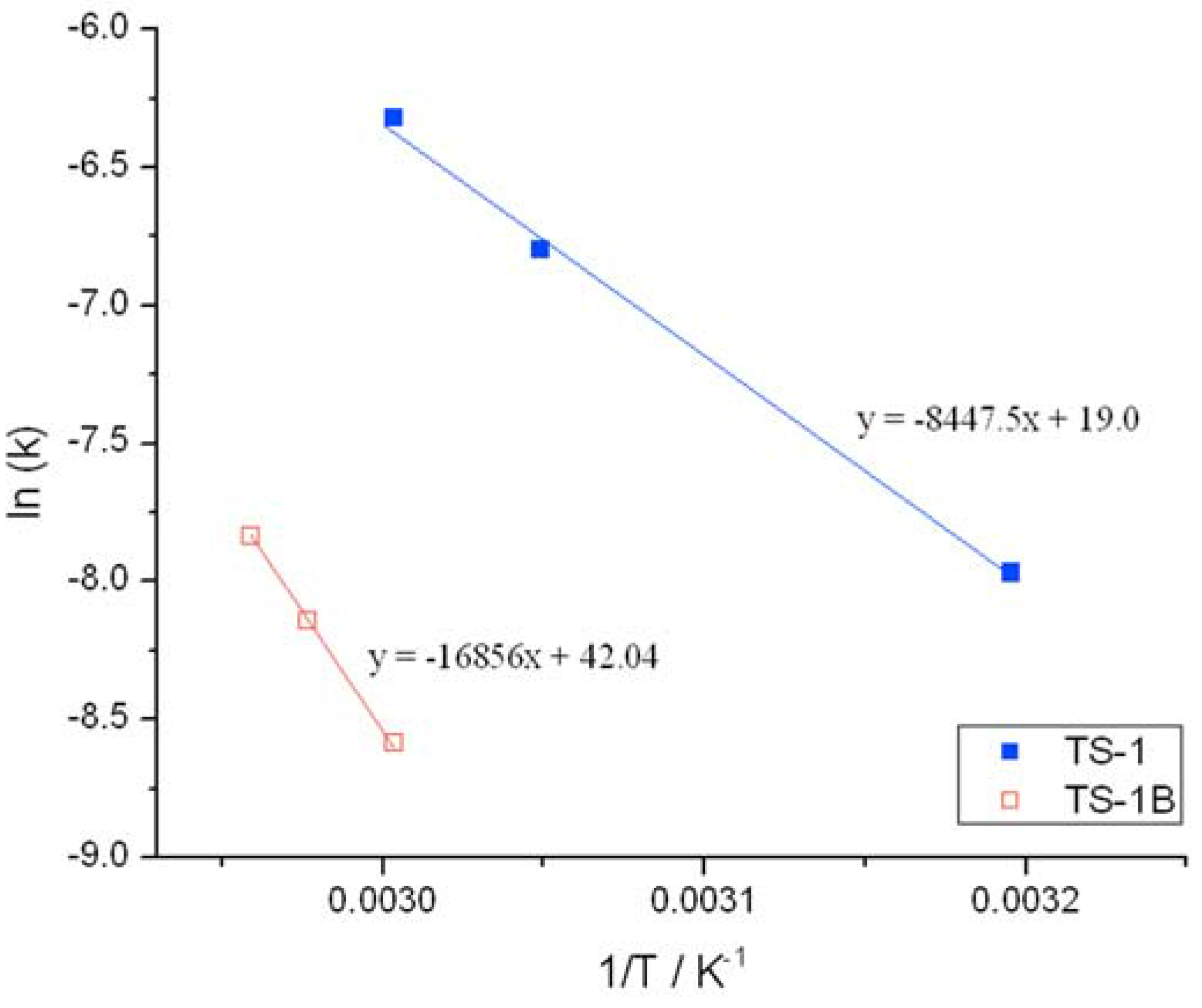
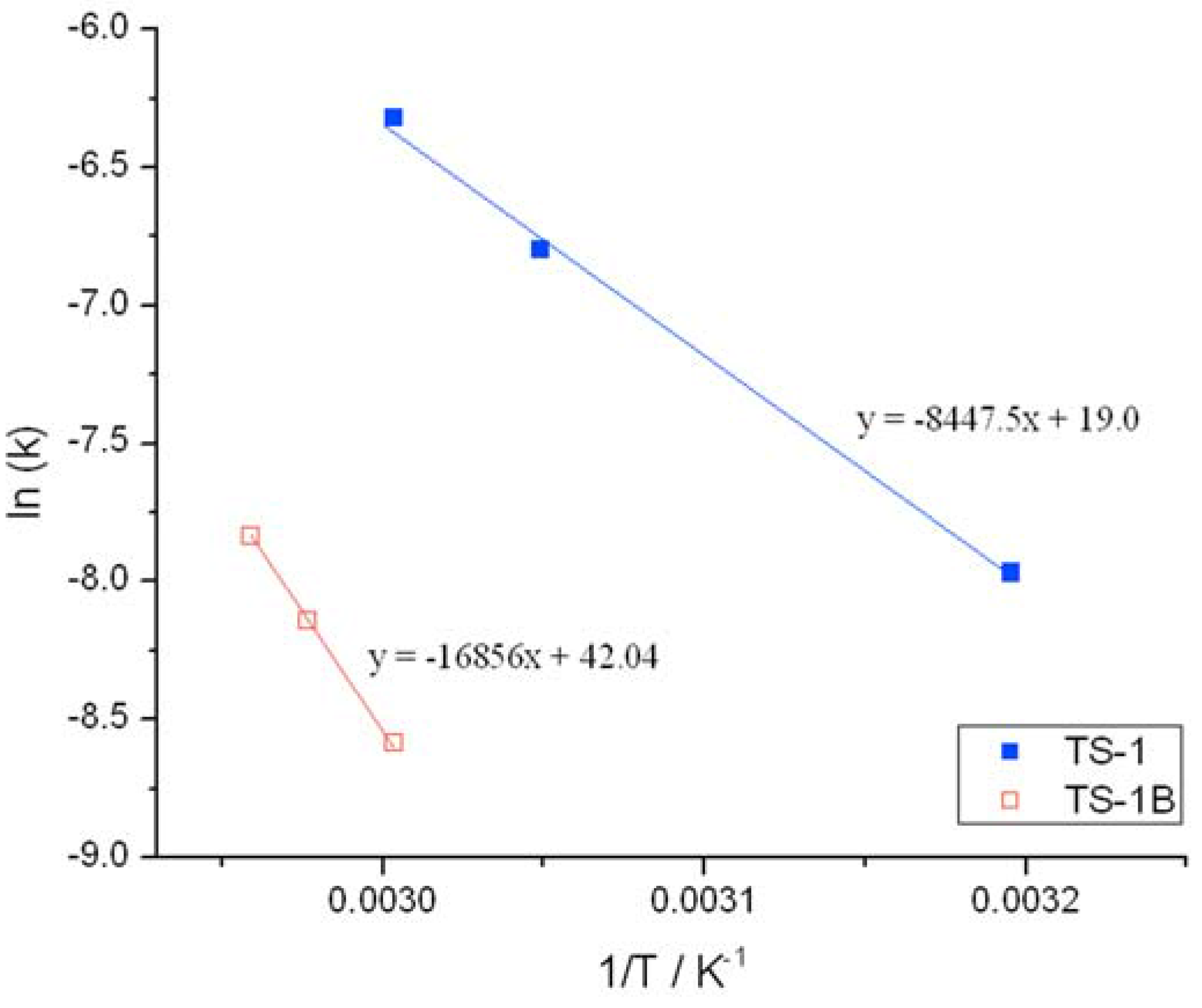
Initial rate analysis (ESI
Figures S4 and S5), calculated from the linear region of the rate plot, and the resulting Arrhenius plot (
Figure 3) clearly shows a change to both the temperature dependence and the pre-exponential factor following NH
4HF
2 treatment. The activation energy between 30 and 50 °C is found to increase from 39.1 kJ·mol
−1 for TS-1, to 53.7 kJ·mol
−1 for TS-1B. The Arrhenius barrier obtained for TS-1 is found to be in good agreement to previous theoretical studies [
12,
13]. Although this data indicates that TS-1B is a less active epoxidation catalyst—being less able to reduce the kinetic barrier—this decrease in activity is partially compensated for by an increase in the pre-exponential factor, which increases from 1 × 10
3 for TS-1, to 2 × 10
5 for TS-1B. These changes may indicate a modification to the rate-limiting step, and potentially some modifications to the epoxidation pathway.
Figure 1.
Time online analysis of allyl alcohol epoxidation with TS-1. Reaction conditions: [allyl alcohol] = 0.5 M, [H2O2] = 0.25 M, mass TS-1 = 50 mg, various temperatures.
Figure 1.
Time online analysis of allyl alcohol epoxidation with TS-1. Reaction conditions: [allyl alcohol] = 0.5 M, [H2O2] = 0.25 M, mass TS-1 = 50 mg, various temperatures.
Figure 2.
Time online analysis of allyl alcohol epoxidation with TS-1B. Reaction conditions: [allyl alcohol] = 0.5 M, [H
2O
2] = 0.25 M, mass TS-1B = 50 mg, various temperatures. Conversion is calculated according to the initial concentration of H
2O
2 (see
experimental section).
Figure 2.
Time online analysis of allyl alcohol epoxidation with TS-1B. Reaction conditions: [allyl alcohol] = 0.5 M, [H
2O
2] = 0.25 M, mass TS-1B = 50 mg, various temperatures. Conversion is calculated according to the initial concentration of H
2O
2 (see
experimental section).
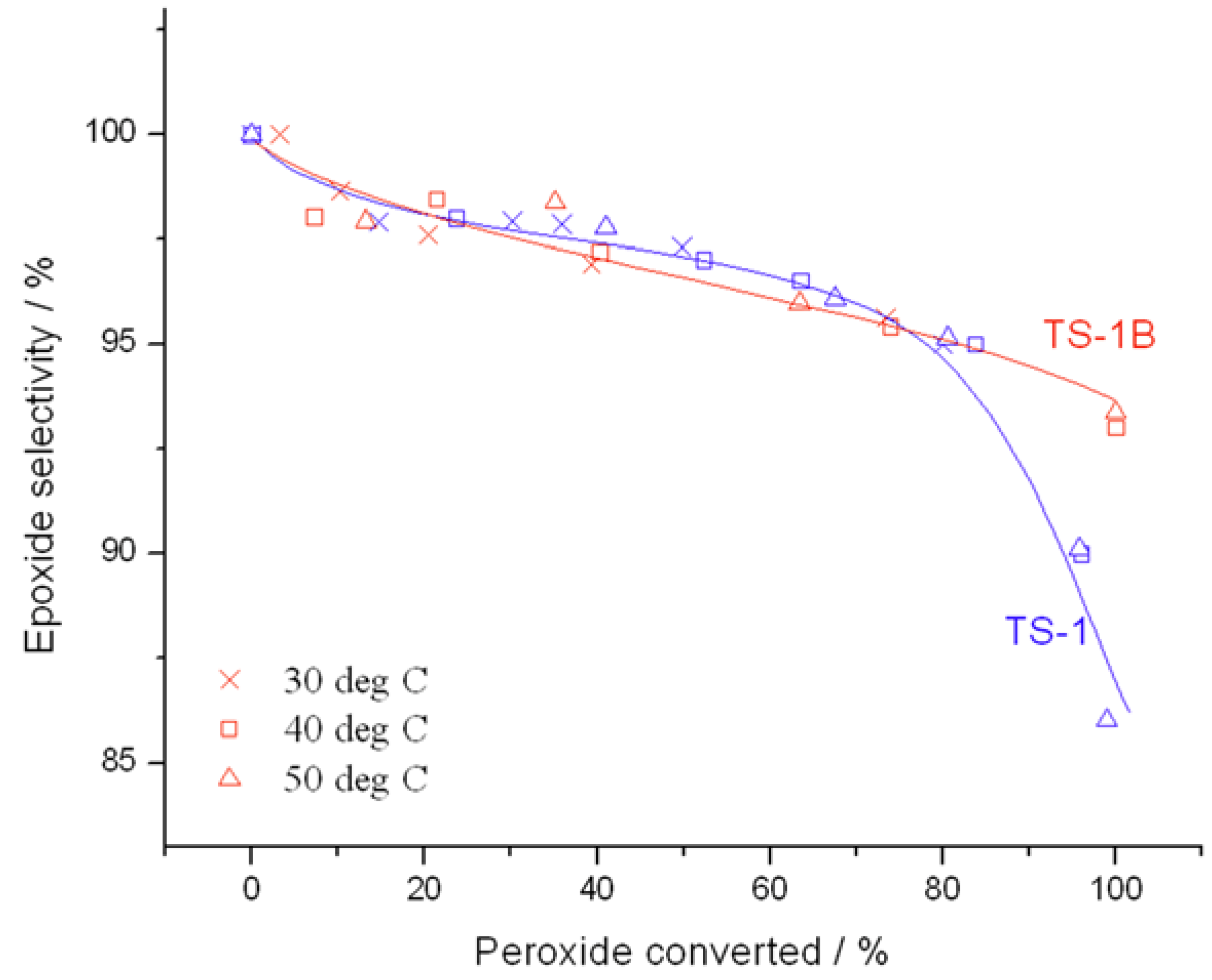
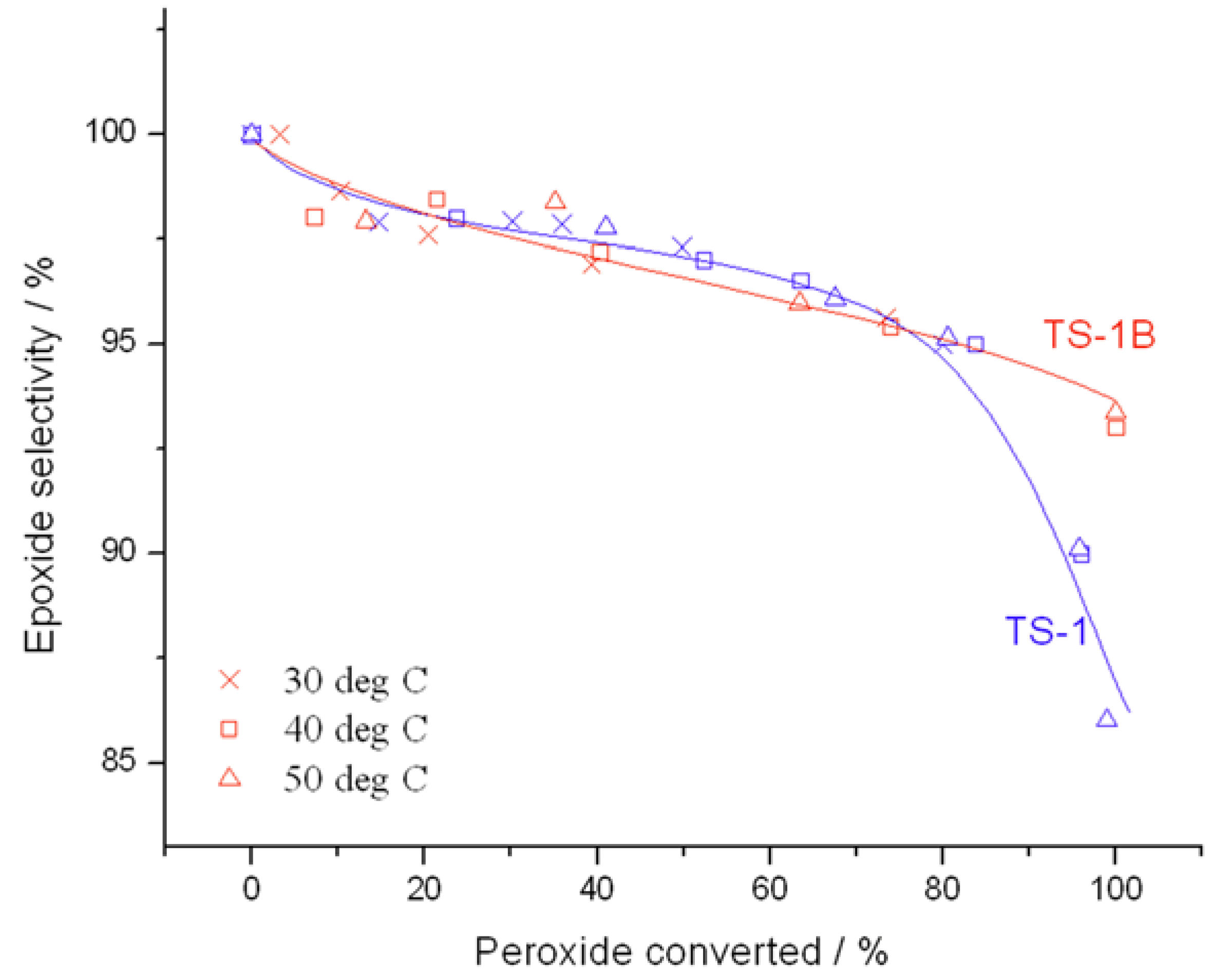
Further changes in catalytic performance may also be observed from the conversion
versus selectivity plots in
Figure 4, determined at maximum levels of conversion. Although both catalysts are almost exclusively selective to the main product, glycidol, up to a peroxide conversion level of 80%, it is clear that TS-1B is able to maintain excellent glycidol selectivity even at substantially higher levels of peroxide conversion. Indeed, whilst glycidol selectivity decreases rapidly to ±85% at 95% conversion with TS-1, glycidol selectivity remains well above 90% throughout the conversion range for TS-1B. This increase in selectivity is certainly non-trivial, and would allow the conversion to be held at higher levels. Not only does this avoid the formation of unwanted by-products, and hence decreases separation issues, but it also leads to higher space-time-yields and the avoidance of unnecessary recycles, which could improve the potential performance of the system on the whole. This improved selectivity may be related to a change in the acid/base character of the active sites, to a change in the way H
2O
2 is activated, or to the better availability of H
2O
2 at very high levels of conversion (See below), limiting the ability of the active sites to catalyse the consecutive reaction.
Figure 3.
Arrhenius plots obtained for (blue squares) TS-1 and (hollow red squares) TS-1B.
Figure 3.
Arrhenius plots obtained for (blue squares) TS-1 and (hollow red squares) TS-1B.
Figure 4.
Comparison of selectivity as a function of conversion for (blue) TS-1 and (red) TS-1B. Reaction conditions: [allyl alcohol] = 0.5 M, [H2O2] = 0.25 M, mass of catalyst = 50 mg, various temperatures. Conversion is calculated according to the initial concentration of H2O2, as a 2:1 olefin:H2O2 ratio was employed.
Figure 4.
Comparison of selectivity as a function of conversion for (blue) TS-1 and (red) TS-1B. Reaction conditions: [allyl alcohol] = 0.5 M, [H2O2] = 0.25 M, mass of catalyst = 50 mg, various temperatures. Conversion is calculated according to the initial concentration of H2O2, as a 2:1 olefin:H2O2 ratio was employed.
2.3. Catalytic Studies of H2O2 Decomposition
To further understand the impact of NH4HF2 treatment, we also investigated the ability of TS-1 and TS-1B to catalyse H2O2 decomposition. As described above, unwanted H2O2 decomposition is clearly disadvantageous, leading to an unwanted loss of expensive oxidant, and potentially increasing safety issues associated. To evaluate the catalytic decomposition of H2O2, we performed the epoxidation reaction without the olefin being present, i.e., solvent, catalyst and H2O2 concentrations were maintained, but the olefin substrate was absent. The loss of H2O2 with time could then be compared to control experiments in the absence of catalyst over the same time period, so as to rule out purely thermal losses (we note here that no thermal losses were observed at temperatures at or below 70 °C).
Both TS-1 and TS-1B are unsurprisingly less reactive for H
2O
2 decomposition than for epoxidation, and longer reaction times are required to induce substantial decomposition. Under reaction conditions identical to the epoxidation reaction,
i.e., at 50 °C, neither catalyst decomposes more than 6% of the initial H
2O
2 present in the first 90 min, even in the absence of the olefin. Despite this, clear differences between the decomposition activities of each catalyst can be observed following NH
4HF
2 treatment. Between 40 and 60 °C, TS-1 displays considerable decomposition activity, with conversion reaching a maximum of 53% over TS-1 after 270 min at 60 °C (
Figure 5). Whilst these values appear high, we note that decomposition in general is substantially retarded by the co-presence of the olefin, particularly under the reaction conditions employed for epoxidation. The presence of an epoxidisable double bond likely prohibits the Ti-(hydro)peroxo intermediate from liberating O
2 via decomposition or oxidation. Over TS-1, an activation barrier for H
2O
2 decomposition of 70.2 kJ·mol
−1 was obtained, in excellent agreement to the values found in the open literature (69–72 kJ·mol
−1) [
14,
15,
16].
Figure 5.
Conversion versus time for H2O2 decomposition at 60 °C over TS-1 and TS-1B.
Figure 5.
Conversion versus time for H2O2 decomposition at 60 °C over TS-1 and TS-1B.
In contrast, H
2O
2 decomposition activity is almost completely suppressed following NH
4HF
2 treatment. Indeed, even at 60 °C almost no decomposition is observed following the post-synthetic treatment, with a maximum conversion of 2.1% being observed under the same conditions. The suppression of H
2O
2 decomposition is further exemplified by the Arrhenius plot in
Figure 6, following calculation of the initial rate constants over the linear region of the kinetic plot (ESI
Figures S6 and S7). Due to the extremely low reactivity observed over TS-1B we were forced to work at slightly higher temperatures for experiments with this material. Nevertheless, an acceptable Arrhenius barrier of 140 kJ·mol
−1 was obtained over TS-1B, a two-fold increase in kinetic barrier.
Figure 6.
Arrhenius expression for H2O2 decomposition over (blue squares) TS-1 and (hollow red squares) TS-1B. Reaction conditions: [H2O2] = 0.25 M, mass of catalyst = 50 mg, various temperatures.
Figure 6.
Arrhenius expression for H2O2 decomposition over (blue squares) TS-1 and (hollow red squares) TS-1B. Reaction conditions: [H2O2] = 0.25 M, mass of catalyst = 50 mg, various temperatures.
2.4. Spectroscopic Studies with in Situ Raman
To account for these interesting effects, particularly the suppression of H
2O
2 decomposition, we considered several possibilities. Although the treatment of various zeolites with NH
4HF
2 has been found to induce some mesopores formation [
17], the impact of this was ruled out given the negligible changes observed in the porosimetry data (
Table S1). Similarly, changes to the potential acid/base character of TS-1—an important factor in H
2O
2 decomposition chemistry [
18]—were also ruled out, based on previous observations that NH
4HF
2 treatment does not change surface acidity (neither Lewis nor Brønsted) to any extent [
17]. Fluorine analysis of TS-1B revealed that no residual fluorine remained after treatment (
Table S2), and thus changes to the hydrophilicity or polarity of the catalyst based on fluorine are unlikely. Further indication of this was provided through FTIR spectroscopy: Although additional defect sites are indeed observed in TS-1B (ESI
Figure S8), these do not obviously change the observed dehydration/rehydration rate compared to TS-1. The potential role of defect sites can also be ruled out given that theoretical studies indicate these would decrease the activation barrier for epoxidation [
13], in contrast to our observed kinetics. Whilst ICP analysis also revealed a decrease in trace Fe content following etching (Fe content decreased from 326 to 254 ppm), control measurements investigating the ability of Fe-silicalite-1 to decompose H
2O
2 revealed that the impurity Fe content was not responsible for H
2O
2 decomposition (ESI
Table S3). The inability of this MFI material to catalyse H
2O
2 decomposition at a similar Fe loading to that found in TS-1 as impurities clearly indicates that Ti is required for decomposition to proceed, in good agreement with previous studies [
18].
Accordingly, we re-focused our attention on spectroscopic study of the Ti active sites with more sensitive spectroscopic techniques. UV-resonance enhanced Raman (UVR) spectroscopy is a particularly promising technique, given its ability to directly probe the Ti active site speciation of each catalyst, improve sensitivity, minimise fluorescence, and selectively probe the different active sites present through the resonance Raman effect [
19,
20,
21]. The tremendous insight offered by UVR is illustrated in
Figure 7. Irradiating TS-1 with a progressively higher energy excitation wavelength allows the LMCT bands of the Ti
IV species to be probed, and leads to the identification of Ti
IV-specific vibrations more sensitively and selectively. Furthermore, vibrations that are totally undetectable by conventional Raman spectroscopy with IR (
Figure 7a) and visible (
Figure 7b) sources are also observed due to resonance enhancement effects, although it should be added that species that do not undergo resonance enhancement will not experience improved detectability.
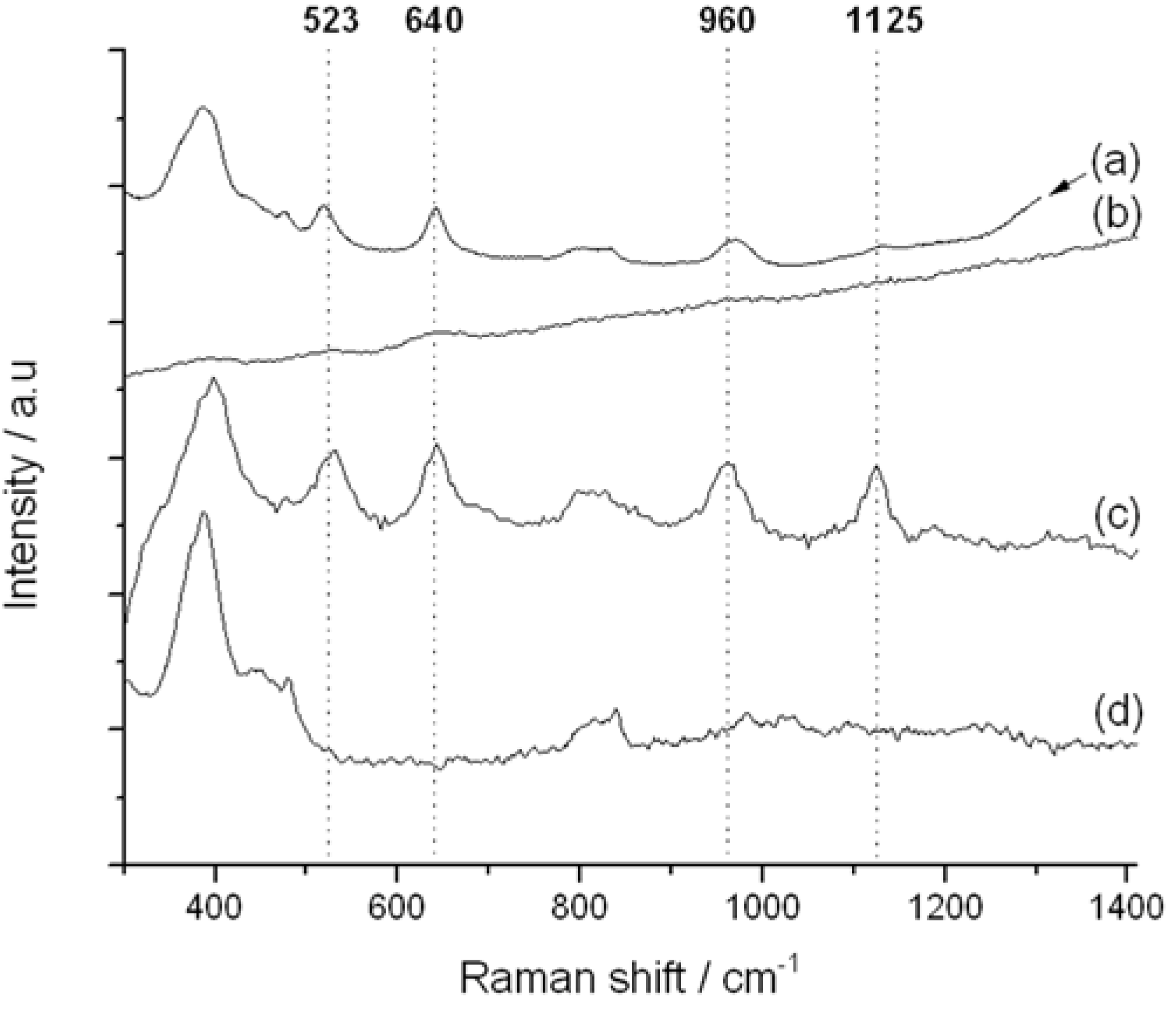
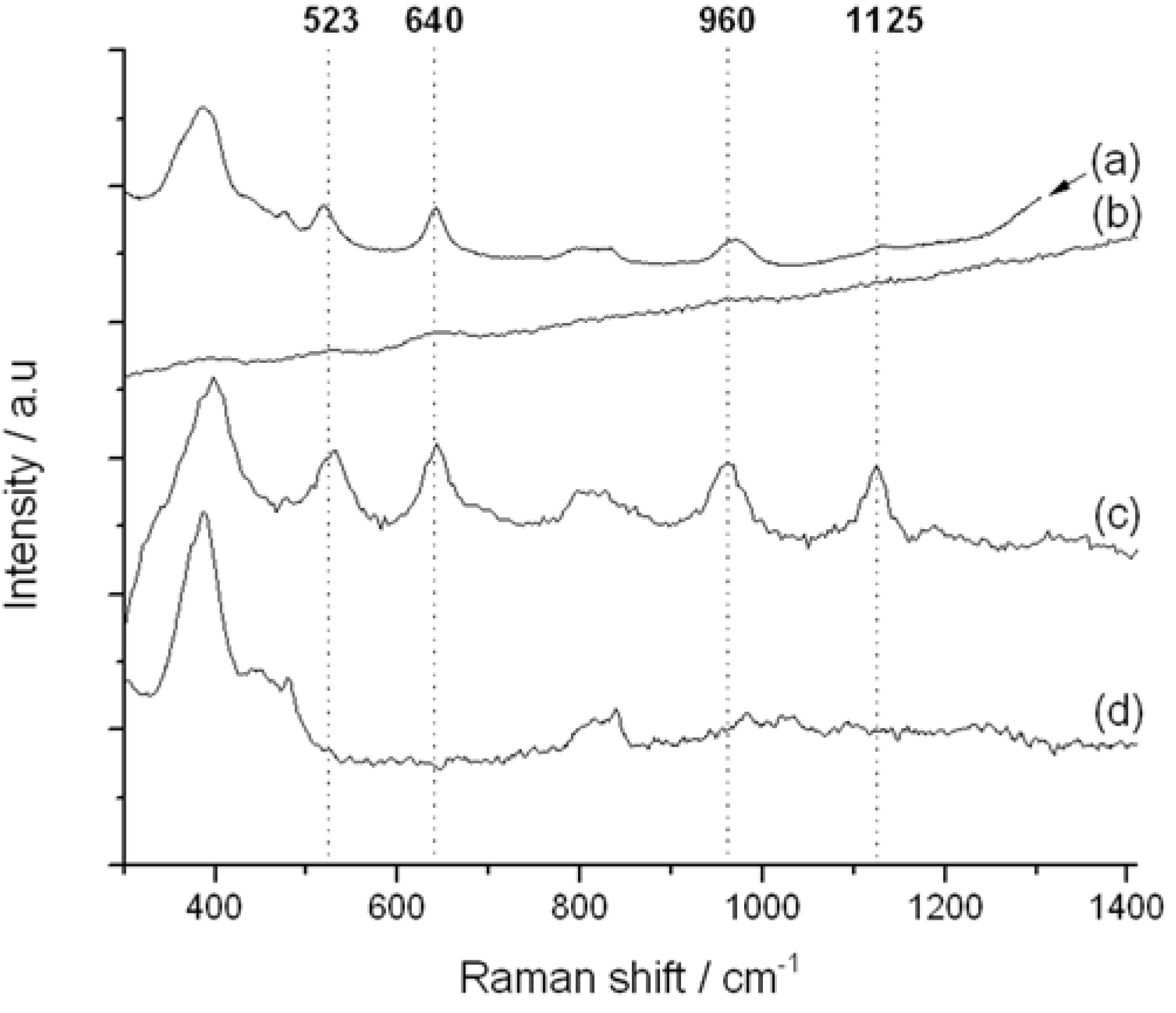
Figure 7.
Impact of Raman excitation laser wavelength on Raman spectrum of TS-1. (a) TS-1 @785 nm excitation; (b) TS-1 @514 nm excitation; (c) TS-1 @325 nm excitation; (d) silicalite-1 @325 nm excitation.
Figure 7.
Impact of Raman excitation laser wavelength on Raman spectrum of TS-1. (a) TS-1 @785 nm excitation; (b) TS-1 @514 nm excitation; (c) TS-1 @325 nm excitation; (d) silicalite-1 @325 nm excitation.
The 325 nm UV-Raman spectrum of TS-1 is presented in
Figure 7c. In addition to the two vibrations related to the zeolite framework at 380 and 800 cm
−1 (also present in Ti-free silicalite-1,
Figure 7d), four vibrations associated with the Ti
IV active sites are visible at 523, 640, 960 and 1125 cm
−1. The first band (532 cm
−1) has previously been assigned to the symmetric stretching vibration of framework Ti–O–Si species [
22,
23]. Both the 960 and 1125 cm
−1 bands have also been attributed to the Ti
IV active sites of TS-1. These arise from the combination of three asymmetric stretching modes of a tetrahedral [Ti(OSi)
4] unit (960 cm
−1), and a totally symmetric stretching mode,
i.e., breathing mode, of the same unit (1125 cm
−1), respectively [
11]. Accordingly, these final two stretches are directly related to the tetrahedrally-coordinated Ti atom present in the [Ti(OSi)
4] active sites. We note that 1125 cm
−1 band is also known to undergo significant resonance enhancement as the laser is progressively tuned into the LMCT modes, in good agreement with our data (
Figure 10) [
11]. Accordingly, the 530, 960 and 1125 cm
−1 UV-Raman bands provide a unique opportunity to monitor the Ti site speciation in TS-1 and TS-1B as a function of post-synthetic modification.
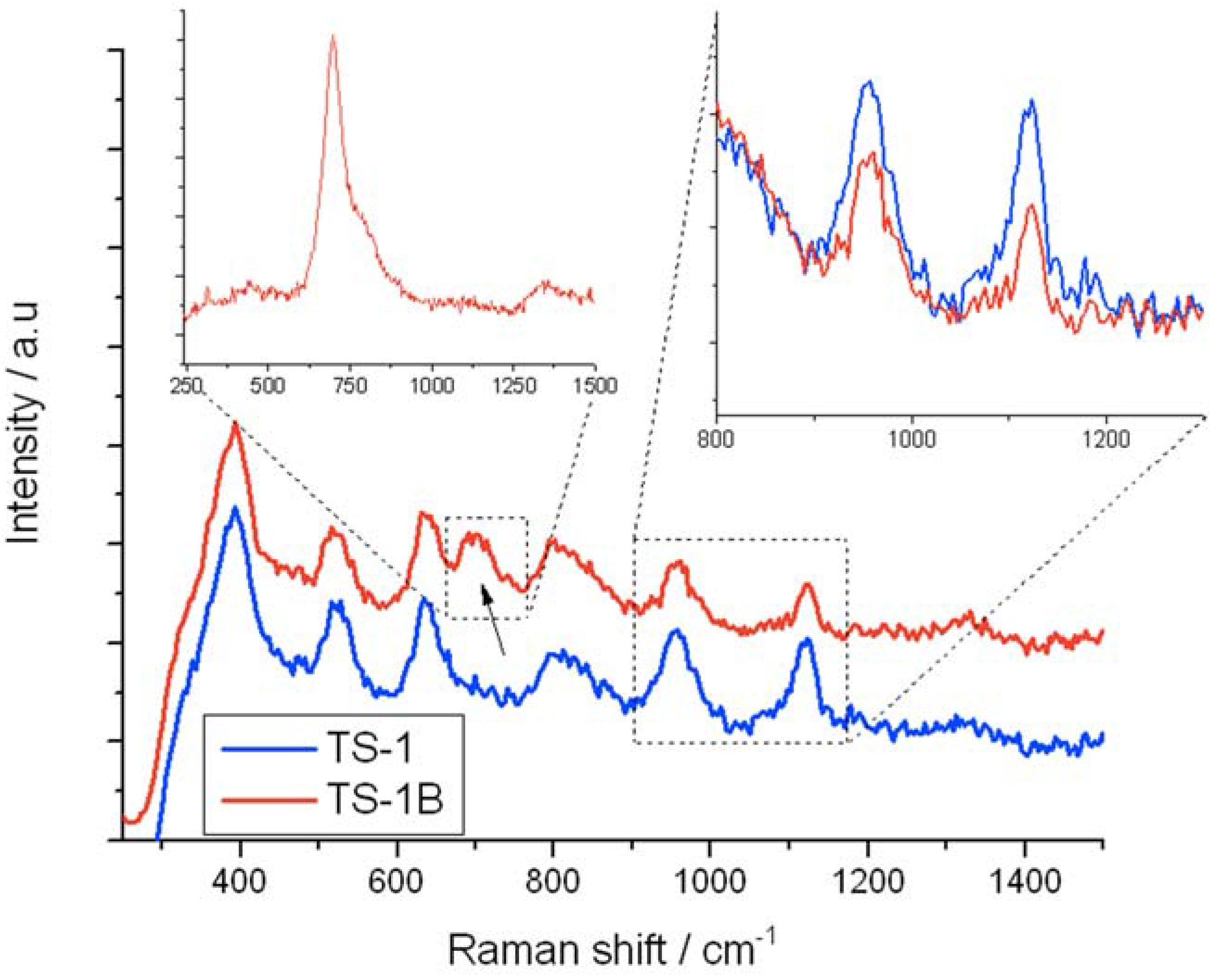
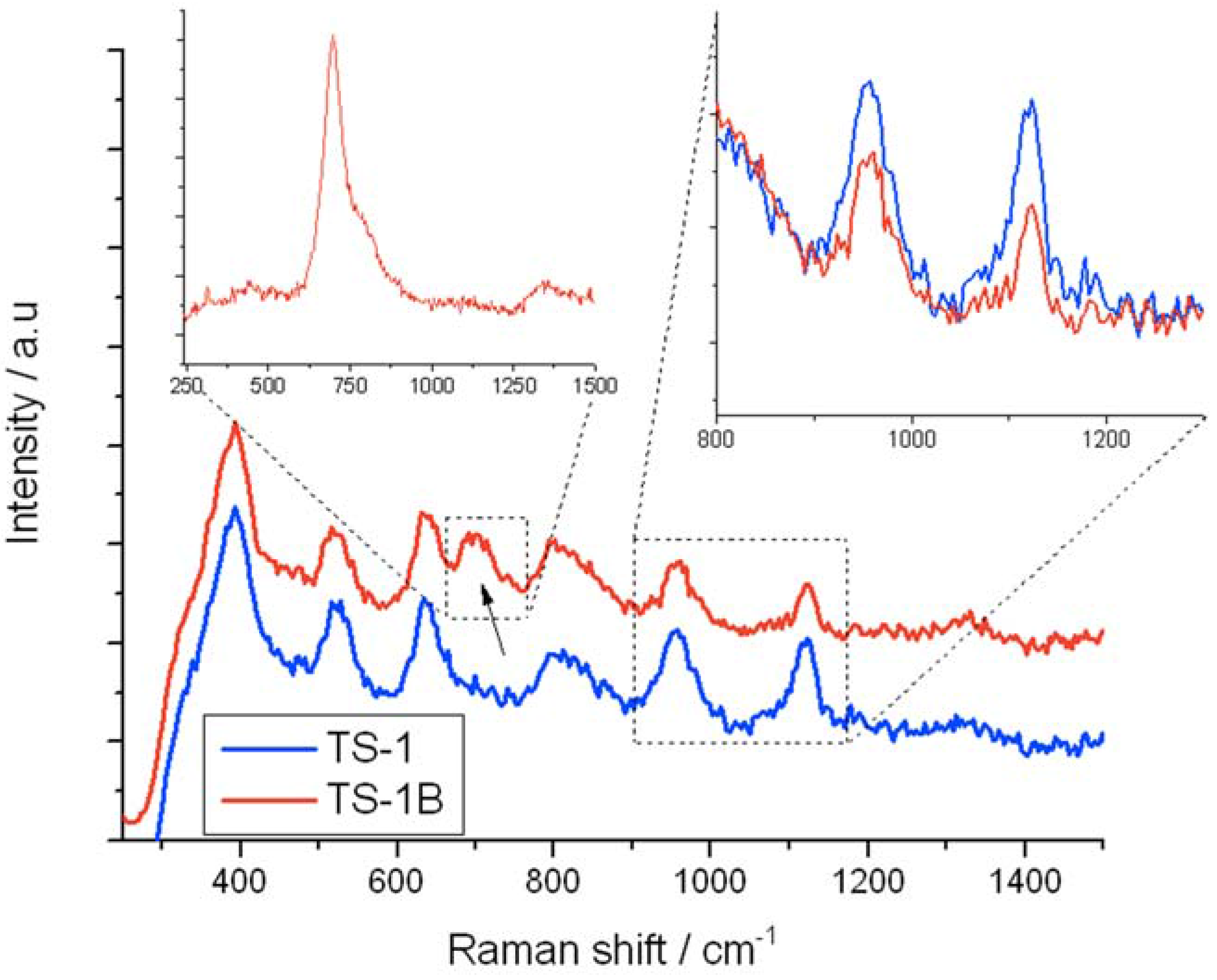
Figure 8 displays the UV-Raman (325 nm) spectra of TS-1 (blue) and TS-1B (red), with the right inset showing the relative intensities of the 960 and 1125 cm
−1 bands. As can be seen, NH
4HF
2 treatment of TS-1 leads to significant changes in the UV Raman spectrum. The 1125 (inset), 960 (inset), and to a lesser extent the 530 cm
−1 band (not shown) of TS-1 experience a significant decrease in relative intensity upon NH
4HF
2 treatment. Since the 960 and 1125 cm
−1 bands have been assigned to stretching modes of a purely tetrahedral [Ti(OSi)
4] unit [
11], it can be proposed that a general decrease in tetrahedral, framework [Ti(OSi)
4] atoms occurs upon post-synthetic modification,
i.e., extraction of framework Ti occurs. This is in line with the decrease in the intensity of the 210 nm band in the UV-Vis spectra (ESI
Figure S2). Based on the relative intensities of the 960 and 1125 cm
−1 bands before and after treatment, we estimate the concentration of the original [Ti(OSi)
4] atoms have decreased by approximately 25%–50% following NH
4HF
2 treatment, in further agreement to the decreased intensity of the Si–O–Ti band in the FTIR spectra (ESI
Figure S3).
Figure 8.
UV-Raman (325 nm) spectra of (blue/bottom) TS-1 and (red/top) TS-1B. The right inset shows the relative intensities of the 960 and 1125 cm−1 bands, whilst the left inset shows the intensity of the 695 cm−1 band when TS-1B is excited with a 266 nm laser.
Figure 8.
UV-Raman (325 nm) spectra of (blue/bottom) TS-1 and (red/top) TS-1B. The right inset shows the relative intensities of the 960 and 1125 cm−1 bands, whilst the left inset shows the intensity of the 695 cm−1 band when TS-1B is excited with a 266 nm laser.
Concurrently, a new Raman stretch at 695 cm
−1 is observed. The generation of a new Raman band is in good agreement to the original UV-Vis data, which demonstrated that a new LMCT band was formed (270 nm) following NH
4HF
2 treatment. Given that this Raman stretch is in the framework,
i.e., fingerprint, region of the Raman spectrum, it is likely associated with the Ti
IV active sites. However, to verify if this new species was indeed responsible of the absorption band at 270 nm (ESI
Figure S2), and is therefore related to the extraction of the [Ti(OSi)
4] units, an additional Raman experiment at 266 nm excitation wavelength was performed (
Figure 8, left inset). As can be seen, irradiating TS-1B with a 266 nm laser significantly increases the intensity of this stretch by several orders of magnitude. The 266 nm excitation laser overlaps perfectly with the 270 nm absorbance band, thus allowing true resonance conditions to be met, strongly indicating that this Raman stretch at 695 cm
−1 is associated with the 270 nm UV-Vis band, and is hence directly proportional to the decrease in [Ti(OSi)
4] atoms. It appears, therefore, that treatment of TS-1 with NH
4HF
2 leads to a general extraction of framework [Ti(OSi)
4] atoms, and the concurrent generation of new Ti sites, which exhibit LMCT bands at ±270 nm, and a Raman stretch at 695 cm
−1.
To gain further insight into the active sites present in both TS-1 and TS-1B, we also performed
in situ UV-Raman spectroscopy (
Figure 9), where both TS-1 and TS-1B were treated with H
2O
2. In good agreement to previous
in situ Raman studies of TS-1, complete elimination of the 960 and 1125 cm
−1 bands are observed upon treatment of TS-1 with H
2O
2 [
24,
25]. Given that these bands are associated with various stretching and breathing modes of the [Ti(OSi)
4] tetrahedron, the total loss of these stretches can be attributed to the change in geometry (from tetrahedral to octahedral) that occurs upon the formation of the relevant Ti-(hydro)peroxo species. We note that the original spectrum can be restored by a heat treatment at 90 °C, and may even partially be restored by extended scanning under the Raman beam.
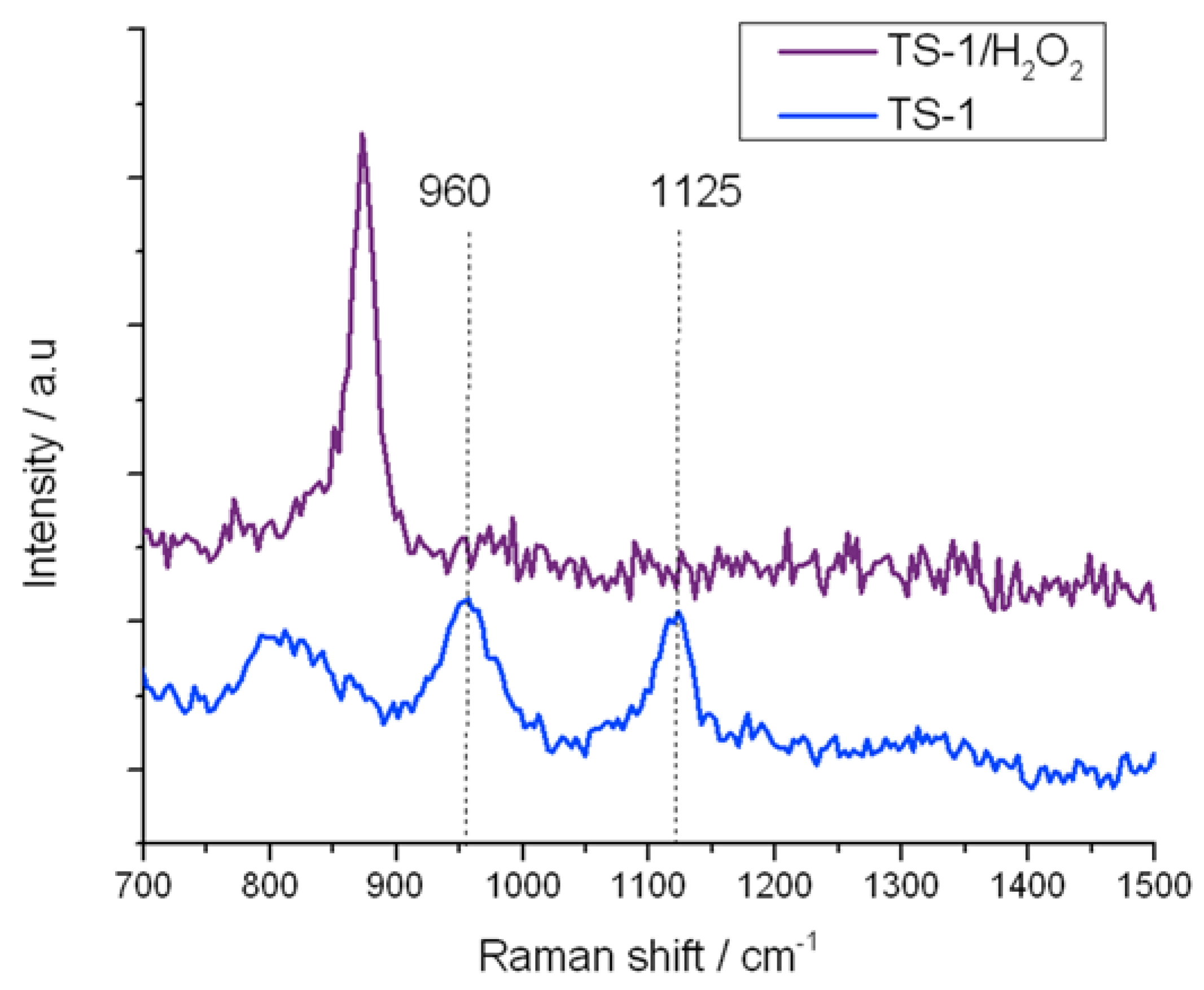
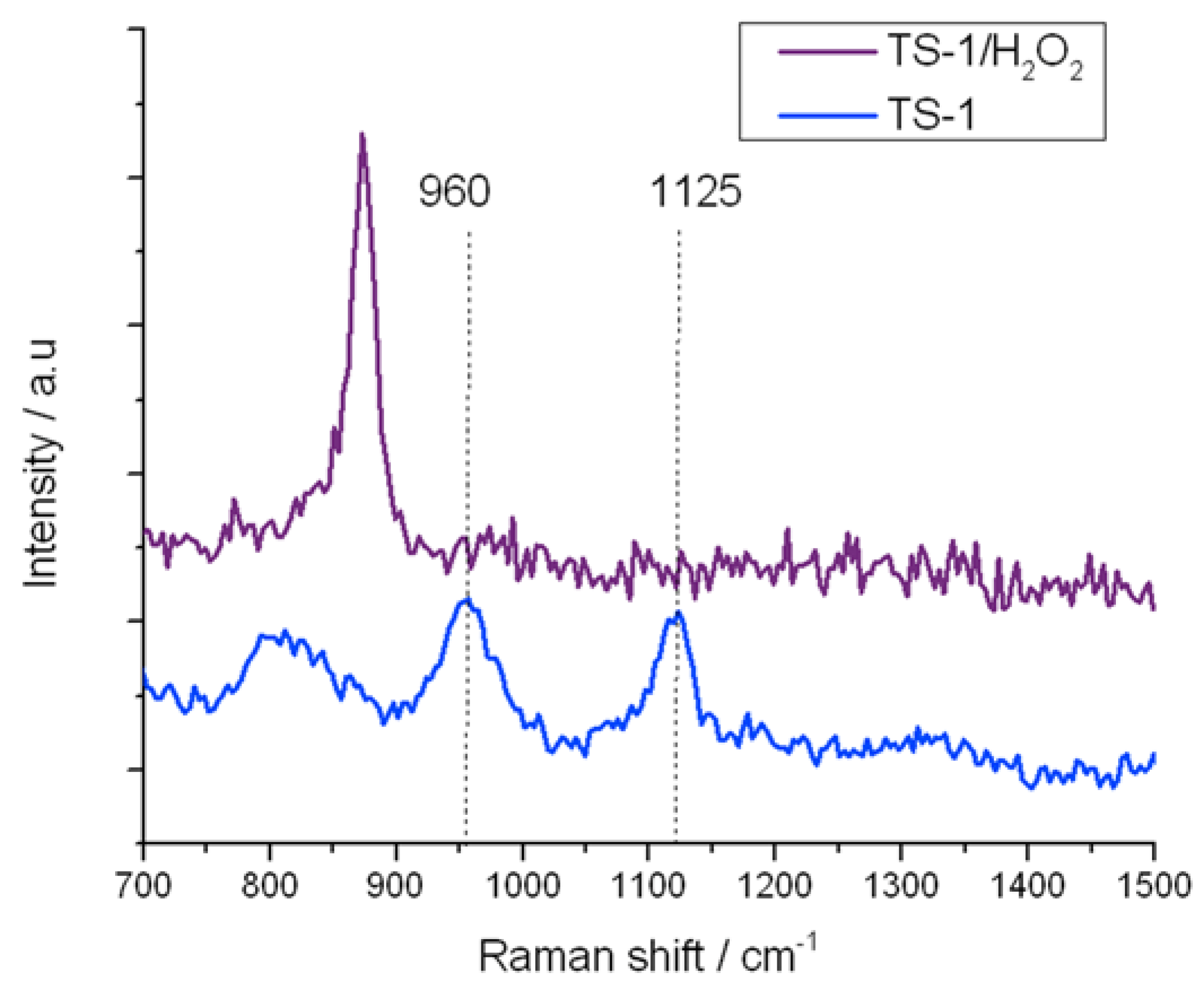
Figure 9.
In situ UV-Raman (325 nm) spectra of (blue) TS-1 and (purple) TS-1 treated with H2O2.
Figure 9.
In situ UV-Raman (325 nm) spectra of (blue) TS-1 and (purple) TS-1 treated with H2O2.
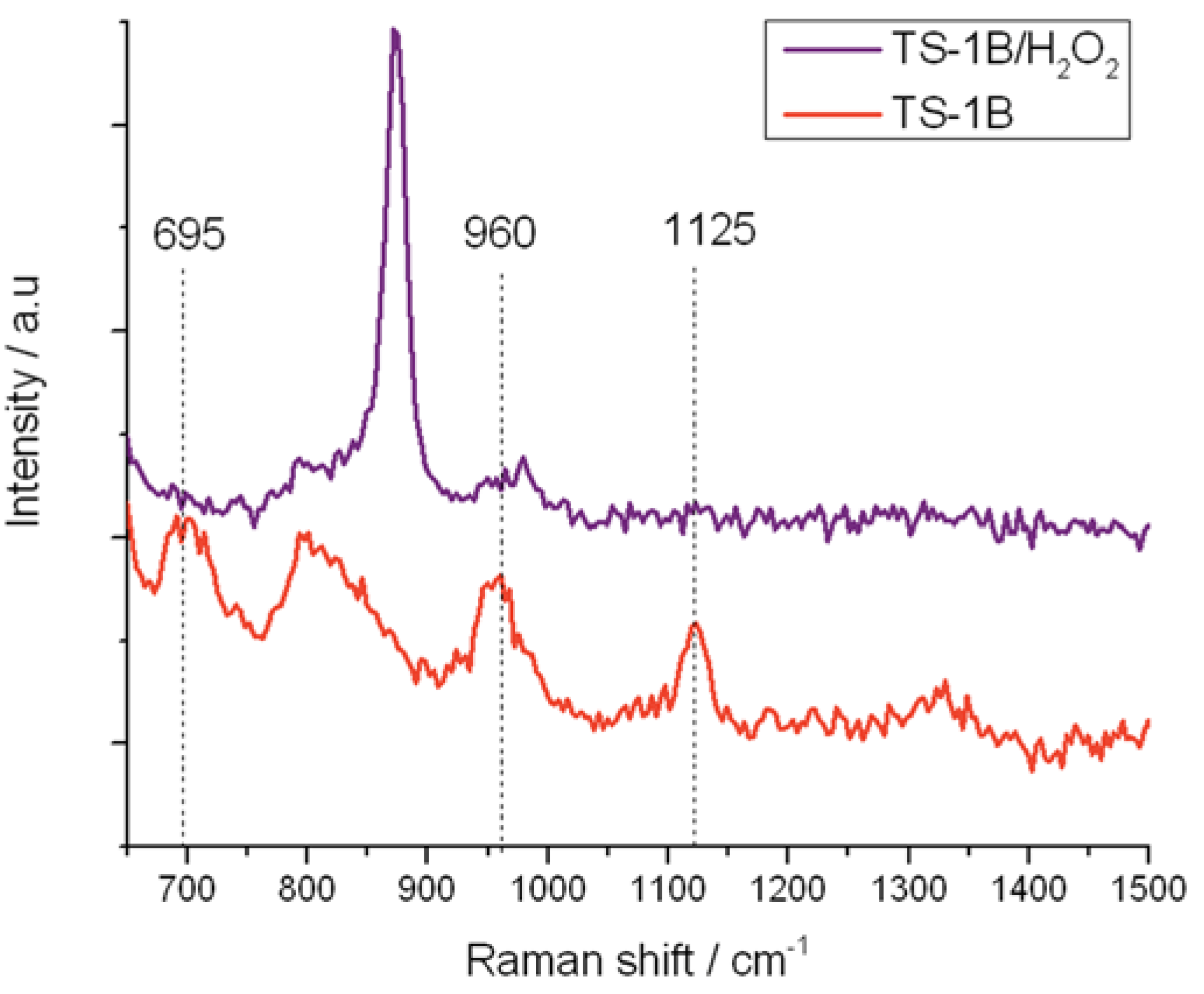
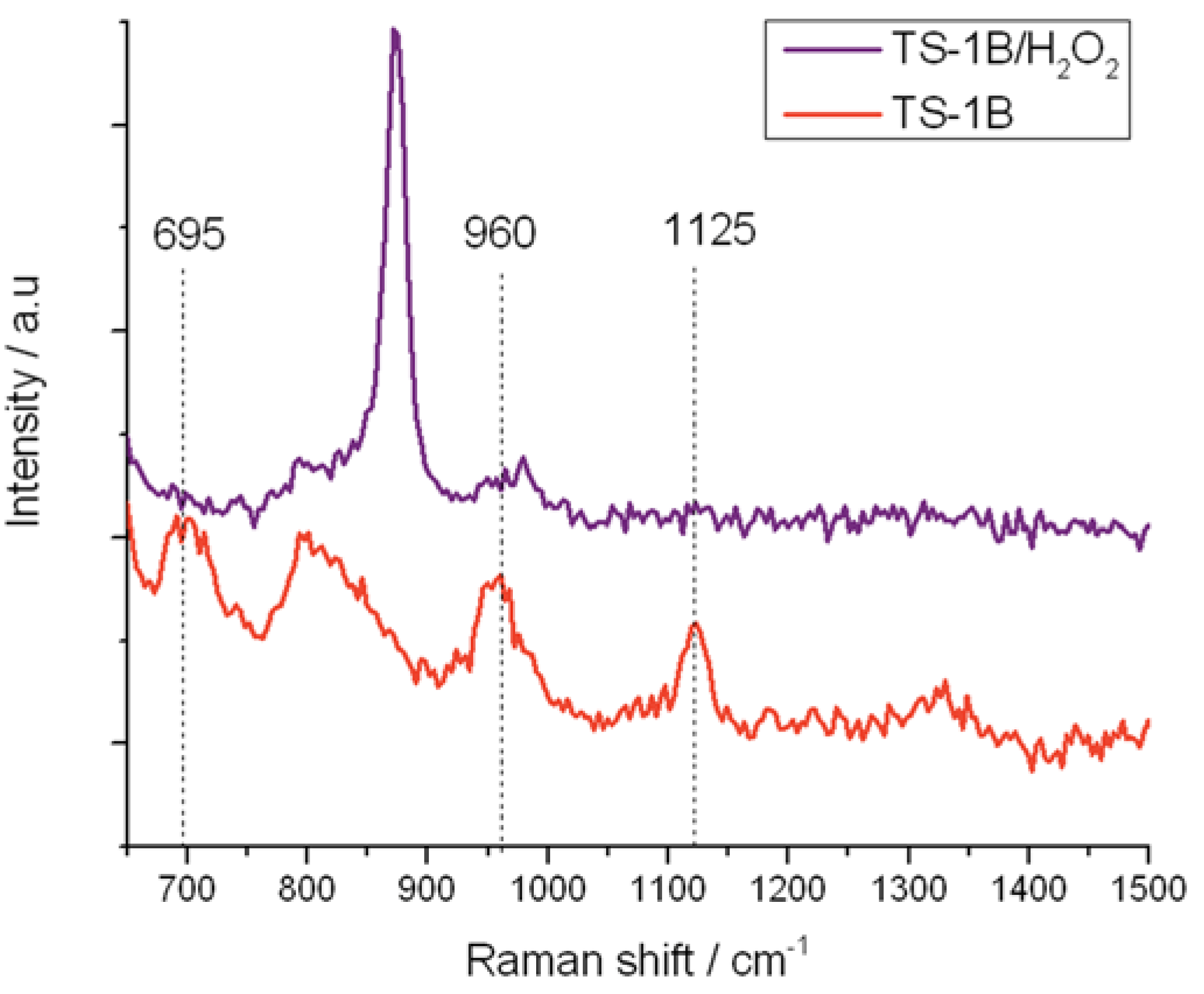
Similarly, the residual 960 and 1125 cm−1 stretches in TS-1B are also eliminated upon treatment with H2O2, confirming that at least some of the original active site remains following NH4HF2 treatment. However, it is also notable that the 695 cm−1 band, which has been attributed to the new 270 nm UV-Vis absorbance band, is also eliminated from the Raman spectrum upon coordination of H2O2. The total loss of this Raman band confirms (i) that this Raman mode arises from a species whose vibrational signature is destroyed upon coordination of H2O2; and (ii) that the new active site formed following NH4HF2 treatment reacts with H2O2, indicating that this species also provides a route toward H2O2 activation. From the characterization data obtained thus far, we assign this change in Ti speciation to the extraction of framework [Ti(OSi)4] atoms, and to the concurrent generation of new active sites. Given that a redistribution of Ti from one T-site to another would not change the intensity of the breathing mode of the [Ti(OSi)4] atoms at 1125 cm−1, it is likely that the new active sites are located in extra-framework positions of the zeolite.
At this time, we are not able to conclusively attribute the modified kinetic behaviour to the generation of extra-framework Ti species. Yet, the
in situ UV-Raman data and the absence of any other contributing factors (see above) strongly suggest that these factors are related. We consider there to be two potential ways that the extraction of framework Ti could lead to such pronounced changes. The first possibility is that NH
4HF
2 treatment removes a particularly non-selective framework [Ti(OSi)
4] site, responsible for both epoxidation and undesirable decomposition. Alternatively, the extraction of framework Ti could result in the formation of active, extra-framework Ti complexes, which may provide an alternative pathway for H
2O
2 activation. In light of the
in situ UV-Raman analysis (
Figure 10), we favour the latter. Given the narrow FWHM observed in the UV-Vis spectrum of TS-1B (ESI
Figure S2), and the generation of only one new Raman active mode, we hypothesise that any extra-framework complex must be either mononuclear, or of low nuclearity (
cf. dimeric). Although reports suggest mononuclear, extra-framework Ti to be inactive for epoxidation [
26], several recent reports have highlighted the potential for two concerted Ti sites to catalyse epoxidation reactions: Kortz
et al. demonstrated that a polyoxometallate containing a unique di-titanium centre ([Ti
2(OH)
2As
2W
19O
67(H
2O)] [
8]) is able to activate H
2O
2 for epoxidation catalysis [
27], and the barrier calculated for H
2O
2 activation over a model Ti-dimer complex was found to be 53.5 kJ·mol
−1 by Lundin
et al. [
28] This value is especially close to the experimental barrier of TS-1B. Definitive identification of the new active sites, and their potential role in the epoxidation/decomposition process, will require further (
in situ) spectroscopic study with complimentary techniques, such as X-ray Absorption spectroscopy and computational methodologies. These additional spectroscopic, computational and kinetic studies remain the focus of our on-going work.
Figure 10.
In situ UV-Raman (325 nm) spectra of (red) TS-1B and (purple) TS-1B treated with H2O2.
Figure 10.
In situ UV-Raman (325 nm) spectra of (red) TS-1B and (purple) TS-1B treated with H2O2.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}