1. Introduction
In a gas-to-liquid (GTL) plant the high H2/CO ratio obtained from reforming of natural gas to synthesis gas (syngas) obviates the need for shifting CO with steam to yield more hydrogen (and CO2) for the FT unit. This is one main reason for using a cobalt catalyst instead of the much cheaper iron alternative as catalytic metal for the FT reaction. In addition, the cobalt catalyst is more active and has a simpler product slate of mainly paraffins and some α–olefins. However, both cobalt metal in itself, precious metal promoters as well as advanced overall formulations, make the catalyst inherently costly. Further, Co catalysts typically lose about half their activity within a few months. Assuming an economically acceptable catalyst lifetime of 2–3 years, this means that the catalyst cost will add several USD to the price per bbl of produced synthetic crude. Therefore, improving catalyst stability is a major focus among technology providers and plant operators. It follows logically that a basic understanding of the mechanisms involved in the deactivation process is vital to improving catalyst stability. Fortunately, it appears that, at least for most commercial Co catalysts, rejuvenation of catalyst activity is possible.
A comprehensive review of deactivation of Co FT catalysts appeared a few years ago [
1]. This review discusses a wide variety of deactivation mechanisms comprising sintering; re-oxidation of cobalt, including surface oxidation; formation of stable compounds between cobalt and the support, e.g., cobalt aluminate; surface reconstruction; formation of carbon species on the cobalt surface; carbiding; and poisoning. However, less focus is given in the review to long-term deactivation under commercial conditions.
Historically, details of deactivation mechanisms and rates have been scarce particularly as only a few plants are operated commercially. Nevertheless, some data can be found in the patent literature, mainly based on operation of pilot or demonstration plants, and in conference presentations. A complicating factor is that an industrial process is typically operated at constant global production,
i.e., deactivation is counteracted by a steady increase in operating temperature. Only a few reports on deactivation under these most relevant conditions could be found. Fortunately, Sasol and its collaborator, Eindhoven University of Technology, have published extensive data on mechanisms and rates of deactivation at industrial conditions for their Co/Pt/alumina catalyst. Several of their papers have focused on long term deactivation due to polycarbon deposition [
2].
We would also like to draw attention to an extensive report to US-DOE where long term experiments are reported focusing mainly on the effect of water [
3]. Catalysts with the formulations Co(15 wt.%)/Re(0.2; 0.5; 1.0 wt.%)/γ-Al
2O
3, Co(10 wt.%)/Ru(0.2 wt.%)/TiO
2, Co(15 wt.%)/Pt(0;0.5 wt.%)/γ-Al
2O
3, Co(15 wt.%)/Ru(0.2;0.5;1.0 wt.%)/ γ-Al
2O
3 and Co(12.4 wt.%)/SiO
2 were tested for up to 3500 h in a 1L autoclave (CSTR-reactor). The authors claim that carbon deposition may be minimized by careful temperature control, and that deactivation caused by sintering and oxidation are the major concerns. These conclusions are controversial and have been disputed by several investigations; see later in this review and in our previous review [
1].
Argyle
et al. have fitted previously published activity
versus time data to first or second order general power law rate expressions incorporating a limiting activity and have shown how parallel routes, e.g., sintering and carbon deposition deactivation, can be modeled separately. For example, their model predicts that during a 60 day run under typical FTS conditions a commercial Co catalyst loses about 30% activity within 10–15 days due to rapid sintering, while an additional 30% activity is lost gradually over the 60 day period due to carbon [
4].
Causes of deactivation may depend on catalyst material and properties, e.g., support, promoters, dispersion, extent of reduction, etc.; reactor type; and especially operating conditions. It appears that after an initial break-in period during which cobalt is equilibrated with its reactor environment in terms of crystallite size, possibly crystal structure, and degree of reduction, a slow long term deactivation is observed. The origin of this latter deactivation period is discussed in terms of carbon formation and/or re-oxidation of the metal.
2. Catalyst Activity
To understand catalyst deactivation, it is first necessary to understand the factors that determine initial catalyst activity. Activity is largely dependent on the degree of reduction of the cobalt metal precursor and the average size of the cobalt crystallites, which together determine the surface density of catalytically active sites,
i.e., the dispersion of the metal. It has been verified that the turn-over-frequency (TOF) is rather constant for Co crystallites larger than 6–8 nm [
5]. As activity falls off rapidly below this threshold, methods for making very high dispersion catalysts have limited relevance for FT-synthesis. Cobalt crystallite size and degree of reduction depend on several factors, including cobalt precursor, support material and its pretreatment; pore diameter, pore volume and available total surface area; method of impregnation or deposition; drying and calcination conditions; reduction conditions;
etc. It is especially important to calcine and reduce the catalyst at optimum conditions,
i.e., optimal gas flow rates, temperature ramp and final temperature [
6]. For example, overly high calcination and reduction temperatures result in large cobalt oxide and cobalt metal crystallites and, therefore, undesirably low dispersion due to over-sintering. For a given degree of reduction and crystallite size, the activity per kg catalyst is proportional to the cobalt loading. The loading of a commercial type catalyst may vary from 12 to 30 wt.% and is a compromise between several catalyst properties. For instance, a lower surface area and pore volume support will be able to accommodate less cobalt, but might be considerably more attrition resistant.
To optimize cobalt crystallite size is not particularly challenging as long as one is able to control preparation conditions. It is industrial practice to add a metal promoter that enhances the degree of reduction and maintains a targeted dispersion [
7,
8]. The literature provides no solid evidence that such metal promoters are able to enhance reaction rates or surface concentration of intermediates. Promoters used at a commercial or demo scale include platinum, rhenium and ruthenium. The promoter will add significantly to the cost of the catalyst; catalyst grade Re is today priced at
ca. 3000 USD/kg and Pt at 45,000 USD/kg [
9]. Typical loadings are up to 0.5 and 0.05 wt.% for Re and Pt, respectively. It can also be mentioned that a possible effect of cobalt being in the
fcc or
hcp crystallographic phase, with the latter being more active, has been reported [
10]. However, studies on the actual configuration of an active cobalt crystal are needed to be able to correlate activity with atomic arrangement on the surface of a working catalyst. It is well documented that the catalyst support has a strong influence on the selectivity to C
5+ of the process, but as long as known impurities like alkali, alkaline earth metals and sulfur are eliminated it is less clear how support chemistry and pore structure influence activity of a catalyst for a given cobalt crystallite size. However, different supports have varying interactions with cobalt oxide and therefore influence reducibility.
It is well known that reaction rate greatly depends on process conditions. Generally, rate increases with increasing temperature and overall pressure. Furthermore, indications are that the rate increases with H
2/CO ratio, possibly due to higher methane make, and decreases with increasing conversion as the partial pressure of syngas is reduced and a high level of product water may block active sites [
11].
Of the CH
x monomers generated on the surface of a catalyst, CH
2 is probably the most abundant intermediate and is probably readily incorporated in the chain during polymerization. A smaller portion of the monomer will be hydrogenated all the way to methane. The growing chain can terminate by β–hydrogen abstraction and leave the surface as an olefin or be hydrogenated to an alkane. Olefins can also be hydrogenated in a secondary reaction. There is evidence from experiments at low conversion and small catalyst particle size that the primary product is dominated by olefins, but for practical purposes the olefin to paraffin ratio is well above two for C
3 and then diminishes rapidly with chain length [
12].
3. Fischer-Tropsch Reactors
Apart from the type of FT-catalyst, selection of the FT-reactor, as well as how it is operated and incorporation in the XTL flowsheet, is the principal factor influencing catalyst deactivation. Comparison of properties of the main reactor types for low-temperature Fischer-Tropsch synthesis is given in
Table 1. Conversion per path will vary, mainly with the propensity for heat removal and temperature control, whereas the temperature typically is between 200 and 250 °C and the pressure will be in the range 15–30 bar. H
2/CO ratio in the make-up gas from the syngas generator will be slightly below 2, whereas the outlet ratio of the FT-reactor will be considerable lower; down to 1.2, in some cases even below 1.0. Evidently, high temperature and low H
2/CO ratio are expected to promote deactivation, but reports on these effects are not available.
Table 1.
Properties for different reactor types for Low-Temperature Fischer-Tropsch synthesis *.
Table 1.
Properties for different reactor types for Low-Temperature Fischer-Tropsch synthesis *.
| Reactor | Conversionper path(%) | Capacity per reactor (bbl/day) | Characteristics |
|---|
| Tubular fixed-bed | 30–35 | ≤6000 | ≤ 30,000 tubes with catalysts pellets or extrudates. |
| Slurry bubble column | 55–65 | ≤25,000 | Internal heat exchanger and optional product filter |
| Micro-channel | 65–75 | ≤1000 | Metal block with < 2mm diameter channels |
In a comparative study of reactor types for LTFT synthesis Guettel and Turek conclude that the productivity per reactor volume of a slurry bubble-column reactor or monolith reactor is up to one order of magnitude higher than for a fixed-bed or micro-channel reactor [
15]. However, this conclusion is at variance with other work, reported for micro-channel reactors for which superior productivities have been reported [
16].
Fixed-bed reactor. Due to the necessity of controlling the heat evolved during reaction, the design of a fixed-bed FT reactor is based on a multi-tube heat-exchange type of reactor where catalyst pellets are loaded into the tube bundles and the shell contains evaporating water. In this type of reactor axial temperature typically increases through a maximum of 5–20°°C, and it is imperative to avoid hot-spots which cause sintering. In order to minimize deactivation due to temperature effects several measures are taken. Once-through CO conversion is limited to 30–35%. Tube diameter is typically 2.5–5 cm and the size of the catalyst pellets or extrudates is relatively small, in the range of 1–3 mm. Small extrudates size is also important to secure good radial mixing and minimize diffusion limitations, thus maintaining high selectivity to liquids. It has been shown that above
ca. 200 µm particle size the higher effective H
2/CO ratio in the inner part of the pellets significantly reduces C
5+ yield [
12]. Therefore, an egg-shell catalyst design is preferred where only the outer parts are impregnated with active metal. Another factor limiting the applicable superficial gas velocity and the global rate is pressure drop. In general, a challenge related to deactivation in a fixed-bed reactor is variations in the partial pressures of reactants and products along the tube length and within the catalyst particles.
H2/CO ratio depends on several factors, but the make-up gas typically has a ratio slightly below 2 for maximizing C5+ selectivity. With recycle of the product gas to the FT-section the feed ratio to the reactor may be significantly lower, possibly 1.6–1.7, and there may be a gradual reduction from the inlet to around 1.4–1.5 at the outlet. Compared to a slurry reactor, the average gas composition may be richer in hydrogen and due to less efficient temperature control, the overall operating temperature usually lower. A consequence is lower reaction rates, but this is at least in part compensated for by a lower average partial pressure of product water and thereby a higher syngas pressure. The overall effect on deactivation, in particular carbon deposition, is complex and challenging to predict.
A distinct advantage of the tubular fixed-bed reactor is a well proven commercial design. Several tens of thousands of tubes can be incorporated within the reactor shell. Scale-up is comparatively easy, and optimization can be done in a single tube laboratory reactor. Operational experience with catalyst fouling or attrition and resulting difficulties with loading and unloading tubes are trade secrets, but it can be expected that an experienced operator is able to control these factors. Minimizing catalyst deactivation or being able to perform in situ regeneration is critical in order to reduce catalyst consumption and avoid an extensive unloading-reloading sequence. The liquid product is inherently separated from the catalyst and any need for removing residual particles and metal components will be low.
Slurry bubble-column reactor (SBCR). Catalyst particles are suspended in the liquid hydrocarbon product of the FT process and synthesis gas is bubbled through the slurry. Depending on the density of the catalysts particles, their diameter and the superficial gas velocity, there is a profile of solid concentration diminishing from bottom to top of the reactor. Gaseous components leave from the top of the reactor. Higher boiling products have to be removed from the reactor as liquid, and separated from the catalyst. Several methods for this purpose have been patented, both
in situ and
ex situ techniques. Broadly they can be classified as employment of filters, settling devices, magnetic separation and hydrocyclones. Sasol uses internal filters combined with secondary polishing filters of the product [
14].
Settling of the catalyst should be avoided as overheating and consequently catalyst deactivation will occur. Particularly critical are the gas distribution system and, depending on design measures to prevent particle settling in stagnant regions below the nozzles. One way to improve overall liquid circulation, and thereby avoid settling, is to install so-called internal down-comers. A serious threat to the catalyst in a slurry operation is any upset in production, like a sudden reduction in syngas flow. Without adequate back-up systems such events will lead to settling and serious overheating in the catalyst mass due to continuous FT-synthesis with residual syngas. Similar conditions may occur in slurry separation devices like filters, but no public information is available on any effects on catalyst deactivation in these devices.
An SBCR operates preferentially in the churn turbulent flow regime for best distribution of catalyst particles as well as minimizing mass and heat transport restrictions. In the churn turbulent flow regime there is a mixture of smaller and larger bubbles that undergo frequent beak-up and coalescence. This mechanism prevents serious film transport restrictions on the catalyst slurry interphase, and with catalyst particles below 200 μm the H
2/CO ratio as well as water vapor pressure can be assumed constant over the entire reactor volume. Thus, deactivation should be more easily controlled compared to other reactor configurations. Further details on operation of SBCRs can be found in the book on Fischer-Tropsch technology by Steinberg and Dry [
17].
Reasons for selection of a slurry bubble column reactor for Fischer-Tropsch synthesis include: (1) a comparatively simple construction; (2) high space-time-yield and catalyst efficiencies; (3) high heat transfer coefficients; and (4) isothermal conditions. Continuous catalyst regeneration of a slip stream is a viable option. Challenges include minimizing catalyst particle attrition and efficiently separating catalyst from the products. Efficient liquid and gas back-mixing and a high exit water concentration lead to high selectivity; the high exit water concentration is beneficial in reducing coke deposition. On the other hand reactant concentrations are lower than the average of a fixed-bed reactor resulting in comparatively lower global rates. Single pass conversion is typically in the range of 55–65%, significantly higher than for fixed-bed. Conversion is limited by the feasible height of the reactor, but there is also an upper conversion limit above about 75–80% for which the water-gas-shift activity will lead to possible catalyst oxidation and a steep increase in CO
2 yield [
18]. Naturally, extensive recycle of syngas in the FT-section of the plant is necessary to obtain a very high overall CO conversion.
Micro-channel reactor. Micro-channel reactor technology holds great promise for process intensification due to outstanding heat and mass transfer rates [
19,
20]. Combined with highly active and stable catalysts, micro-channel reactors can achieve high volume based productivities. In some cases very high conversions (~ 90%) can be obtained while maintaining high C
5+ selectivity. Detailed studies of flow and temperature behavior have shown that a micro-channel reactor can operate isothermally and with very low pressure drop [
19]. Except for the normal initial deactivation, the catalyst in the micro-channel reactor is remarkably stable even at very high conversions [
21]. Observed rates of deactivation appear to be lower in the micro-channel reactor compared with the fixed bed laboratory reactor at similar conditions. Velocys and CompactGTL are operating microchannel demonstration plants [
20].
3. Commercial Catalyst Formulations
Scale-up to commercial catalyst production is demanding, and little public information on the industrial manufacturing processes is available. Great care must be taken to obtain a homogeneous catalyst material, but the targeted distribution of cobalt on the support depends on the actual process. For slurry catalysts with diameters typically in the range 40–120 µm pore diffusion resistance of the syngas is negligible ensuring full utilization of the available surface area [
12]. For micro-channel reactors the catalyst either will resemble a slurry catalyst or be impregnated onto special trays that are inserted into the channels. On the other hand, a fixed-bed catalyst is typically designed as an egg-shell or rim catalyst in which only the outer few hundred micrometers contain the active phase. As to the degree of reduction it has been shown by Sasol that the initial value is not critical, as the syngas will reduce the catalyst further under the first months of operation, thereby increasing CO conversion [
2].
Cobalt FT catalysts can be classified according to supports and promoters used.
Table 2 lists several commercial type catalysts. Precious metal promoters like Pt and Ru may facilitate hydrogenation activity and hence reduce propensity for carbon deposition as indicated, for example, by Exxon Mobil for ruthenium.
Some relative activities have been estimated and are included in the table. The values are based on fixed-bed data reported in the patent literature. As the process conditions vary considerably, an attempt to normalize the activities is made by using a simplified kinetic expression with activation energy of 110 kJ/mol, and partial pressures are average pressures in the reactor [
22]. It is recognized that this comparison is only approximate, but still a guide. Activities are generally lower for a given catalyst operated in slurry compared to fixed-bed in spite of limited apparent diffusion limitations. The origin of this effect is not understood. Particularly low activities have been reported by Nippon and ENI/IFP, perhaps an indication of large cobalt crystals in the working catalyst.
It appears that Shell favors a titania based support over their previous zirconia modified silica system. Promoters are either Mn or V, the latter claimed to lower CO
2 make [
23]. Titania has relatively large pores and moderately low surface areas, but is known to facilitate high selectivities to C
5+ products. Fixed-bed catalysts contain modifiers like citric acid added prior to the forming step. In addition to Shell, it should be mentioned that BP is promoting its fixed-bed technology and claims that a CO
2 resistant support is vital [
24].
Velocys/Oxford Catalyst and Compact GTL are offering micro-channel fixed-bed technologies with catalyst diameter or thickness of 0.1–0.5 mm. Velocys’ carbon combustion preparation technique may very well take the edge off initial sintering during FT synthesis by optimizing cobalt crystallite size already at the catalyst manufacturing stage. Loading and off-loading the catalyst can be particular challenge for these systems, but efficient methods for this purpose are claimed by these companies.
Table 2.
Formulation of commercial type cobalt catalysts and their application *.
Table 2.
Formulation of commercial type cobalt catalysts and their application *.
| Technology provider | Support/ modifier | Promoter | Reactor type | Reactor scale (bbl/d) | Relative activity | Reference |
|---|
| Sasol | γ-Alumina/ Si ** | Pt | Slurry | 16,000 | | [25] |
| Shell | Titania | Mn; V | Fixed | 6000 | 0.3 | [26] |
| GTL.F1 | Ni-aluminate/ α-Alumina | Re | Slurry | 1000 | 0.9 | [27] |
| ENI/IFP/ Axens | γ-alumina/SiO2;spinel | ? | Slurry | 20 | 0.19 | [28] |
| Nippon Oil | Silica/ Zirconia | Ru | Slurry | 500 | 0.16 | [29] |
| Syntroleum | γ-Alumina/ Si **; La | Ru | Slurry | 80 | | [30] |
| BP | ZnO | ? | Fixed | | | [24] |
| Exxon Mobil | Titania/ γ-Alumina | Re | Slurry | 200 | | [31] |
| Conoco-Phillips | γ-Alumina/ Boron | Ru/Pt/Re | Slurry | 400 | 0.68 | [32] |
| Compact GTL | Alumina? | Re? | Micro | 500 | | [33] |
| Oxford cat. /Velocys | Titania-silica | Re | Micro | 1000 | | [34] |
The platinum promoted Sasol catalyst is prepared on γ-alumina (Puralox SCCa-2/150 or -5/150: pore volume 0.5 mL/g; surface area 150 m2/g) and stabilized by impregnation with tetra-ethoxy-silane (TEOS) followed by calcination to give a surface concentration of ca. 2.5 Si atoms/nm2. This procedure apparently modifies the surface so that the support becomes less prone to dissolution in the acidic product water. The GTL.F1/Statoil catalyst is based on a larger pore diameter γ-alumina modified with nickel and fired at high temperature to produce a nickel-aluminate (spinel)/α-alumina mixture. The pore properties resemble titania-based supports, but with very high attrition resistance. Also the ENI/IFP catalyst is supported on Si-modified γ-alumina, but probably strengthened by silanation and calcination giving a final SiO2 content of 6–7 wt.%. Other support modification methods have been described by ENI/IFP in earlier patents, including formation of spinel compounds. It is unclear whether the catalyst formulation contains a reduction or other type of promoter. In their slurry catalyst development, Nippon has apparently adopted a silica based support formulation similar to that of Shell’s previous fixed-bed catalyst, but using ruthenium as a promoter. No information on attrition resistance has been revealed, as is the case for most other slurry catalysts as well. Syntroleum used a catalyst similar to Sasol’s, but also with a ruthenium promoter. Exxon Mobil, that pioneered titania as a support with an alumina binder, BP, Conoco-Phillips and Syntroleum have terminated their developments in FT-technology. However, BP is still licensing their FT technology and Exxon Mobil has announced that the technology is ready for commercialization should the right project be prioritized.
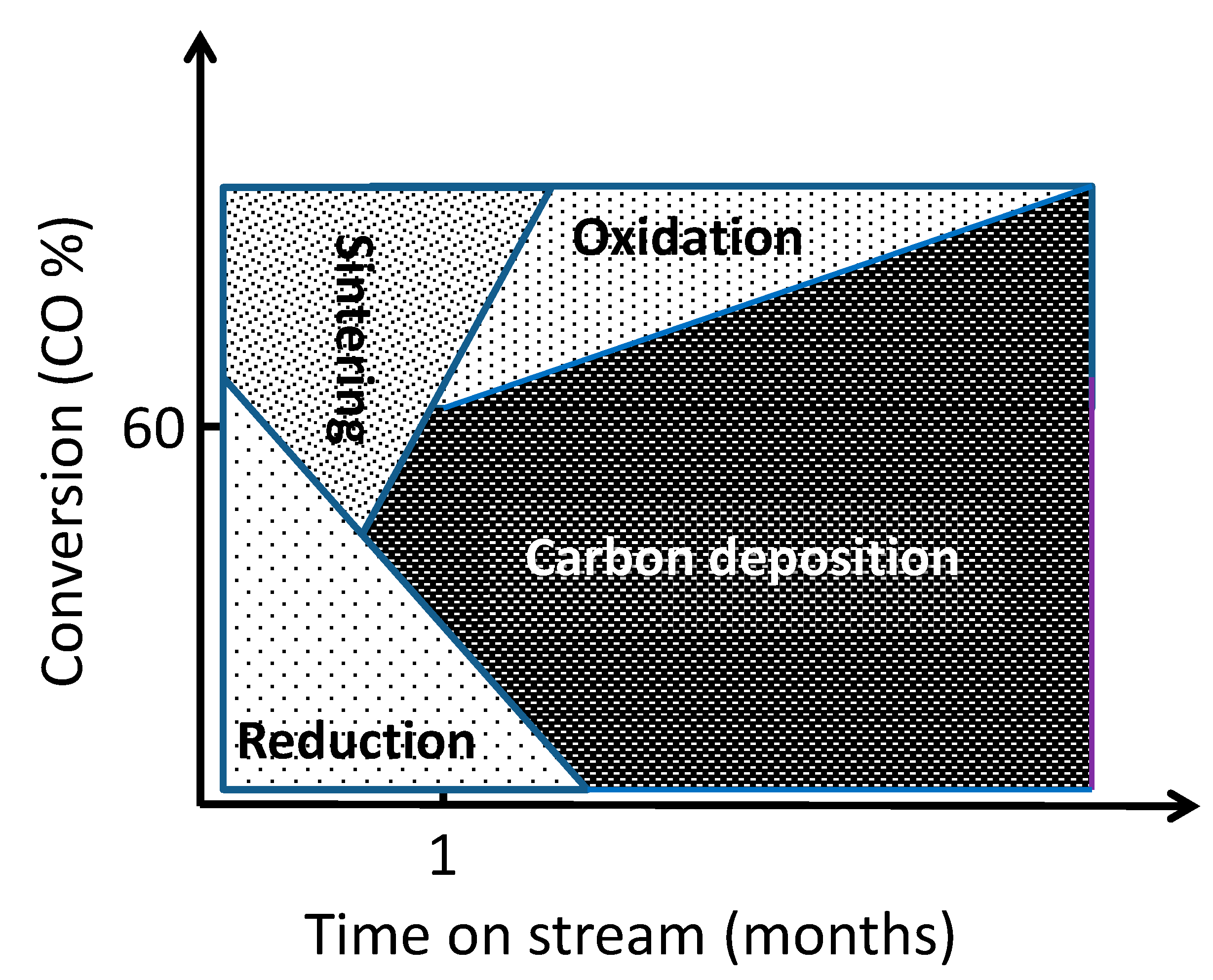
4. Causes of Deactivation
From our previous review on deactivation during LTFT synthesis the main causes of deactivation are sintering, re-oxidation of cobalt, formation of stable compounds between cobalt and the support, surface reconstruction, formation of carbon species on the cobalt surface, carbiding and poisoning [
1]. In addition there can be a loss of catalyst material from the reaction zone due to attrition. The chemical environment is challenging with a number of reactive chemical species generated including significant amounts of water. In addition, the exothermicity of the reaction may lead to hot spots in the catalyst.
There appear to be two main “schools” for describing long-term deactivation mechanisms based on demo slurry operations, one favoring re-oxidation [
35,
36], and one poly-carbon formation on the surface [
2]. It should be realized that both catalyst system and process conditions can affect the results. In addition, an initial sintering stage may be observed if the fresh catalyst contains crystallites in the range of 6–12 nm. The consequences of severe deactivation can be a significant decline in the activity over a typical design period for catalyst life of two years to an estimated 25–30% of the initial value. In addition, slurry catalyst loss due to attrition can be significant. All previous experience considered catalyst replacement due to deactivation will contribute significantly to the operational costs of an FT plant.
In a study on the effect of impurities it was found [
37], by impregnating 400 ppm of the impurity element from nitrate precursors, a poisoning effect which decreases in the following order:
Mn, Fe and Cl showing minimal effects. The latter is surprising as chlorine causes a 25% reduction in hydrogen chemisorption. Even alkali concentrations of less than 100 ppm have a large effect on the rate (site time yield) [
38]. However, no effect of alkali on the hydrogen chemisorption was observed. The impact of alkali and alkaline earth elements is far stronger than any stoichiometric blocking of surface sites, and might be related to the strong electronegativity of the elements leading to blocking of steps on cobalt thus preventing CO dissociation [
39]. Particular care must be taken to avoid alkali and alkaline earth elements in impregnation fluids and washing water as well as contaminants in the catalyst support. Sulfur as H
2S or (CH
3)
2S added to the syngas gives a deactivation consistent with stoichiometric blocking of cobalt surface sites as
in situ measurement of cobalt dispersion by H
2S is consistent with hydrogen derived dispersion of a fresh catalyst. The effect of ammonia appears to be strongly catalyst dependent, and reports vary from negligible influence to strong negative consequences.
Contributions to attrition of catalysts for three-phase slurry bubble column operation include mechanical abrasion and breakage of catalyst particles; chemical dissolution; and synergisms between these mechanisms. It appears that Sasol focuses more on avoiding chemical attack on their γ-alumina based support [
40], whereas Statoil/GTL.F1 [
41], IFP/ENI/Axens [
28], and probably Exxon [
42], have developed more mechanically robust slurry catalysts.
Sasol has reported deactivation profiles in several publications for their Co/Pt/modified γ-alumina catalyst [
2]. We refer to the section below on carbon deposition and to the previous review for further details on the Sasol work and their extensive documentation of carbon deposits [
1].
Statoil/GTL.F1 have disclosed deactivation profiles at several conferences for their attrition resistant catalyst of Co/Re/aluminate spinel catalyst. In a CSTR slurry reactor test over 3000 h the temperature was adjusted regularly, typically in 2–3 weeks intervals, to keep conversion reasonable constant [
43]. Somewhat surprisingly, temperature was
decreased from 222 °C to 215 °C during the operation meaning that the catalyst activity increased regularly. This is in line with the reported enhanced reduction during first months of slurry FT-synthesis. More surprisingly, the C
5+ selectivity increased simultaneously by
ca. 5%, significantly more than expected given the reduction in temperature. In another presentation on CSTR results, the rate of hydrocarbon formation increased up to 800 h time-on-stream (TOS) followed by decline towards end of test at 1600 h [
44]. Characterization of a commercial catalyst after
ca. one month operation in a commercial scale slurry-bubble column confirms an increase in degree of reduction [
45]. This is concurrent with sintering probably facilitated by a high steam partial pressure [
46].
5. Deactivation by Carbon Deposition
Higher hydrocarbons (waxes) are desired products from LTFT synthesis on Co catalysts. The hydrocarbons can accumulate on the surface and can slowly be converted to carbon or coke that blocks the active sites. By using TPR Lee
et al. [
47] could distinguish between several forms of carbon on the catalyst surface from CO disproportionation; they suggested that carbon was present in two forms: atomic and polymeric carbon. Support for stabilization of graphitic carbon on an
fcc-Co(111) surface has been obtained by quantum-mechanical calculations [
48]. DFT data indicate chemical bonding between graphene and cobalt, as also supported by other studies [
49]. Direct STM evidence for formation of graphene on Co(0001) has been demonstrated through decomposition of ethylene [
50]. It was found that carbon on the surface induces cobalt reconstruction and weakens CO and H
2 adsorptions.
Thus, there are ample investigations showing that carbon in different forms can interact with and block cobalt surfaces. Support for this deactivation mechanism comes from a few long term studies using commercial catalysts in pilot reactors. Sasol studied catalyst deactivation by periodically removing samples from a pilot slurry bubble column reactor operated for 6 months [
2]. Wax was removed by inert solvent extraction before the catalyst samples were characterized by temperature programmed hydrogenation and oxidation, chemisorption, TEM and LEIS. Polymeric carbon was found both on the alumina support and on cobalt. This carbon is resistant to hydrogen treatment at temperatures above the FT synthesis temperature. The amount of polymeric carbon correlated well with observed long term deactivation. From XANES data they ruled out oxidation of cobalt during the run, but there was significant sintering taking place during the first 10–15 days on stream. Moodley
et al. [
2] concluded that accumulation of polymeric carbon was responsible for at least a part of long term catalyst deactivation.
Build-up of graphitic or polymeric carbon as deactivation mechanism was recognized by Syntroleum [
51]. By TGA-MS they estimated that about 1% carbon was deposited on the catalyst. Carbon deposition on cobalt/ZnO has also been proposed by BP based on results from a demonstration plant and laboratory studies [
52].
6. Deactivation by Re-Oxidation
Schanke
et al. investigated the influence of water on deactivation of unpromoted or Re promoted alumina supported cobalt catalysts. Adding 20–28% steam to 50% syngas in the feed of a lab-scale fixed-bed reactor resulted in significant deactivation due to oxidation of highly dispersed cobalt crystals and surface cobalt atoms [
53]. Although these experiments clearly show oxidation of cobalt, the conditions represent very high conversion levels (> 80%). Oxidation takes place within the stability range for bulk cobalt metal, and is presumed to be a consequence of surface reactivity of small crystallites. It is also evident that the effect of water depends critically on the support material used, e.g., samples of γ–alumina from different sources behave very differently. Even a positive effect of water on activity has been reported; see [
1,
3] for further discussion. From activity tests over a range of conversions in a slurry CSTR reactor Co/Re/γ-alumina catalyst activity is observed to slightly increase with conversion up to 85%; above 85% conversion it drops rapidly [
18]. At high conversions high partial pressures of H
2O may oxidize small cobalt crystallites and promote aluminate formation thus enhancing WGS activity as shown by a significant CO
2 make. Similar results were found for a Pt promoted catalyst [
54].
Very recently, the group of A.Y. Kodakov in cooperation with Total published evidence for surface oxidation of cobalt nano-crystallites in alumina supported, Pt promoted catalysts during FT synthesis [
55]. However, surface oxidation was only clearly evident by STEM-EELS after an excursion to 340 °C and 100% CO conversion. B.H. Davis and coworkers exposed a freshly reduced catalyst directly to a water vapor pressure equivalent to 50% conversion [
56]. They observed oxidation of a fraction of the smaller cobalt crystallites when supported on alumina or activated carbon, and recommended that careful crystallite size management is required for a commercial catalyst. Kliewer
et al. studied redox transformations of cobalt catalysts by TEM in terms of agglomeration of the metal, mixed-oxide formation with the support and reversible oxidation followed by reduction under mild hydrogen treatment [
57]. They claim that reactor and TEM studies show that nanoscale Co crystallites can oxidize to CoO during commercially relevant FT synthesis conditions in spite of bulk thermodynamic data that suggest otherwise [
58]. The propensity for oxidation is enhanced by small Co crystallites and high CO conversion with attendant high water vapor pressure and a high H
2O to CO ratio in the reactor. The oxide crystallites thus formed can be fully reduced by hydrogen at standard FT temperature and pressure. Reported TEM images from Exxon show that the oxidized cobalt metal wets the support surface and thereby facilitates contact between nearby crystallites. This has also been illustrated in a presentation from Statoil in
Figure 1 [
59]. Both images show a thickness of a cobalt oxide crystallite of
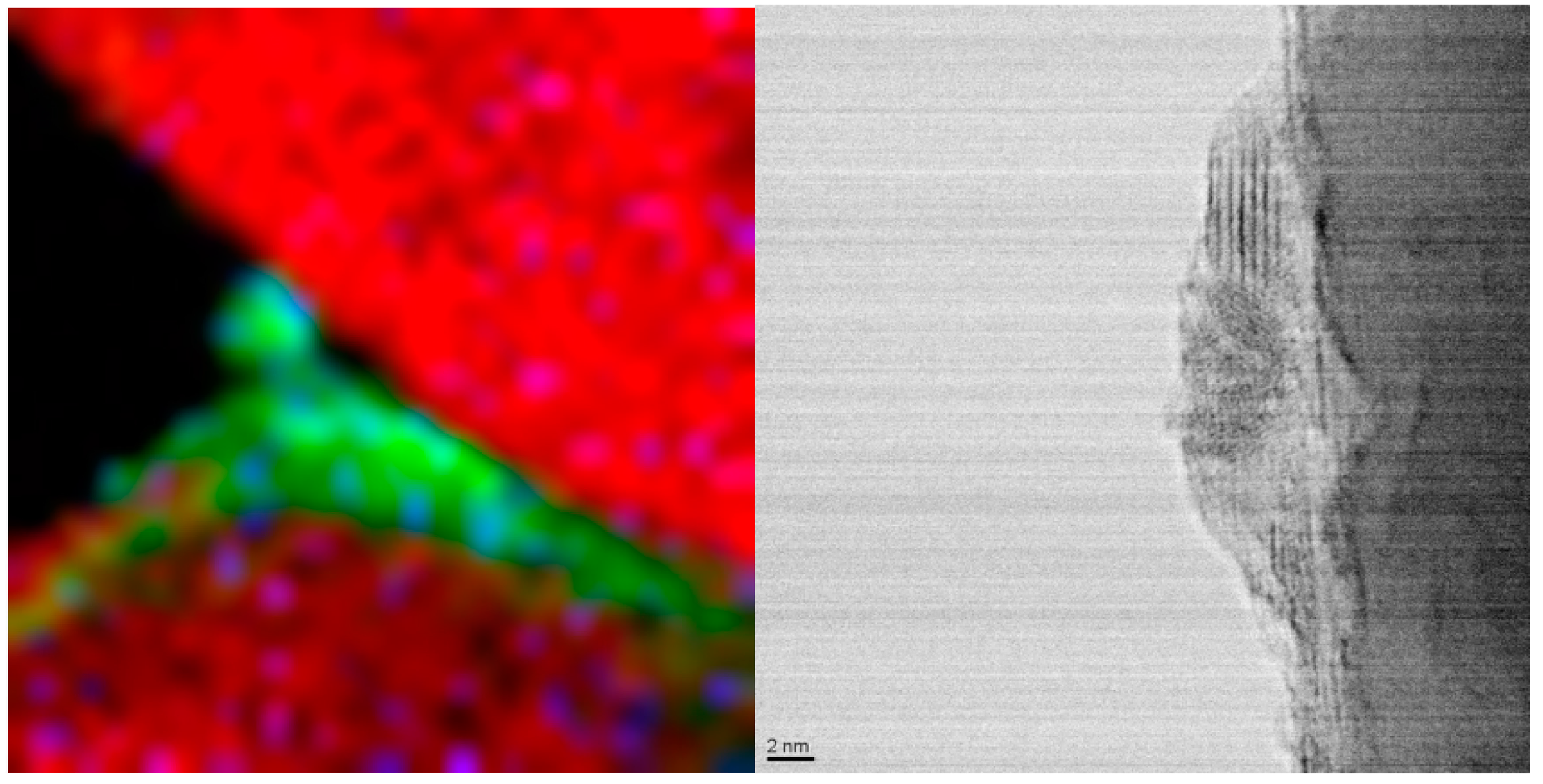
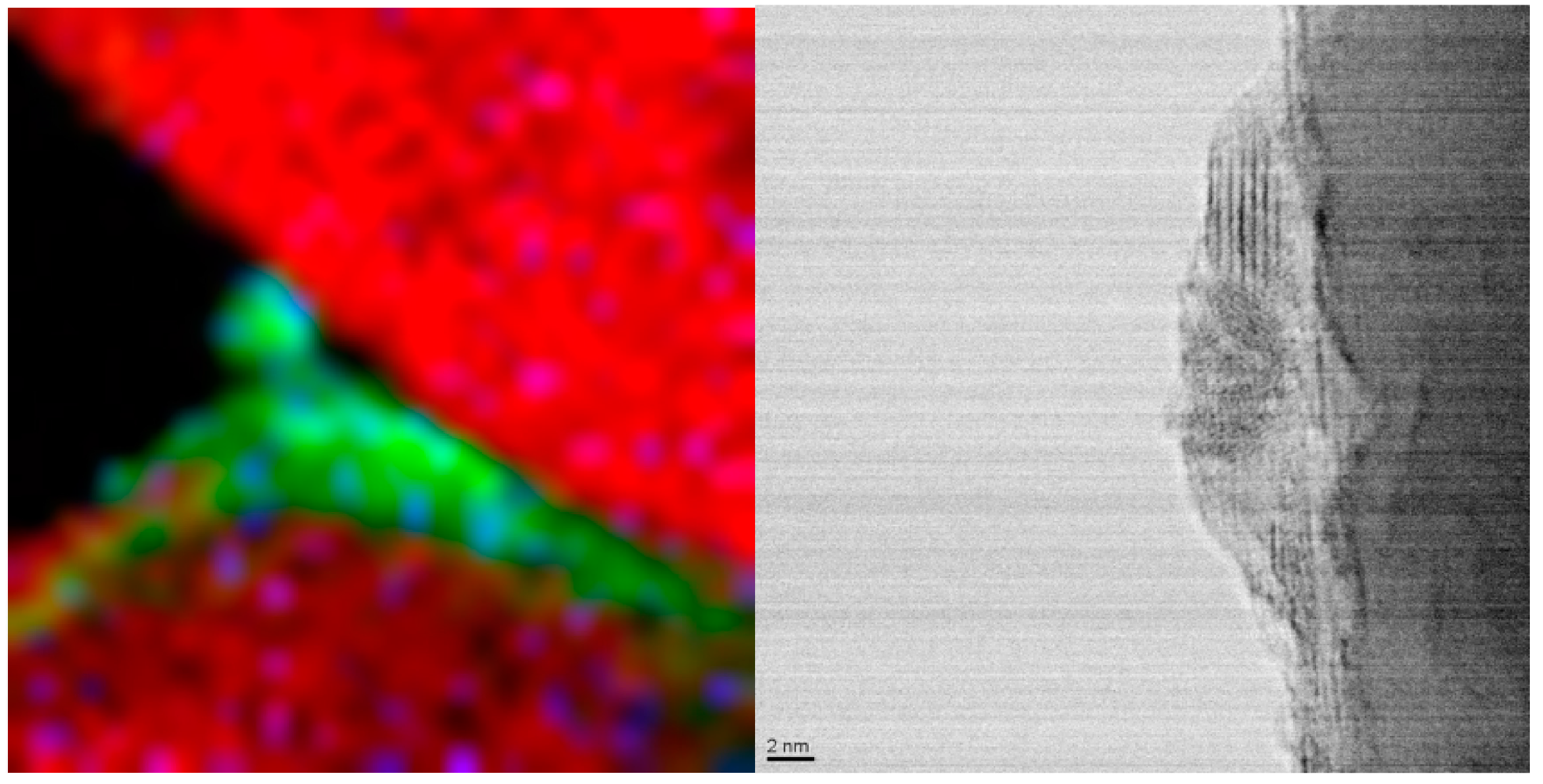
ca. 5 nm wetting the surface. Further, the TEM image indicates mixed orientation of several cobalt oxide crystallites and an amorphous layer at the support interphase. Upon re-reduction metal crystallites may agglomerate depending on the initial spatial distribution on the catalyst support.
Figure 1.
Wetting of cobalt oxide on a support. Left: EELS spectra; alumina: red; cobalt: green. Right: TEM.
Figure 1.
Wetting of cobalt oxide on a support. Left: EELS spectra; alumina: red; cobalt: green. Right: TEM.
That cobalt distribution can vary significantly with catalyst preparation procedure is illustrated in
Figure 2 for a Co/Re/γ-alumina catalyst [
43]. Cobalt dispersions are comparable, but we see that the distribution of clusters of the oxide varies significantly. Although it can be imagined that small well dispersed clusters are less prone to deactivation, this needs to be verified.
Figure 2.
Cobalt oxide clusters on a γ-alumina. Bar shows 1 µm for all images.
Figure 2.
Cobalt oxide clusters on a γ-alumina. Bar shows 1 µm for all images.
It is well known that mixed-oxides can be formed between cobalt and silica, alumina and titania. In part, a surface layer of mixed-oxide is formed during catalyst preparation from water solution followed by drying and calcination. It has also been claimed that enhanced mixed oxide formation takes place during FT-reaction at high conversion levels (> 70%) concurrent with oxidation of cobalt to Co
2+. In the case of silica supported catalysts, well defined crystalline needles of cobalt silicate are rapidly formed at higher conversions [
60].
Redox reactions of cobalt have been used deliberately to enhance the catalytic properties of FT catalysts by employing a reduction-oxidation-reduction (ROR) procedure [
61,
62]. An ROR treatment can increase Co dispersion presumably by forming hollow oxide domes during controlled oxidation that break up into smaller Co crystallites during re-reduction [
63]. Whether the resulting Co crystals that will be in close proximity with each other experience agglomeration during the first months of commercial FT synthesis has apparently not been reported.
7. Catalyst Rejuvenation and Regeneration
Regeneration of cobalt LTFT synthesis catalysts is largely described in the patent literature. The options involve treatment of the catalyst with air (oxygen), hydrogen and/or CO and variations thereof in addition to procedures for removing produced wax. Therefore, regeneration addresses reversing the main deactivation processes of carbon deposition, metal oxidation and sintering by combustion, reduction and re-dispersion, respectively. A review of early reports on regeneration covering 1930–1952 has been presented [
64]. It was concluded that there is no universal process for regenerating Co FT-catalysts; the art over this relatively short time period comprising conflicting results with patents covering a wide range of processes involving oxidation, reduction, combined oxidation-reduction, steam-reduction, operating at elevated temperatures and solvent extraction.
Commercial regeneration processes are either in situ in the FT-reactor itself or ex situ after removal of part of or the entire catalyst inventory. Indications are that Shell successfully regenerates their catalyst regularly, but it is undisclosed whether this takes place inside the tubes of the fixed-bed reactor or in a separate unit, whereas Sasol apparently removes part of the slurry from the reactor continuously for regeneration and re-deployment of the catalyst into the reactor. The latter approach allows continuous operation of the LTFT synthesis. Micro-channel reactors pose special challenges depending on the catalyst configuration. In situ regeneration is an option, or the catalyst can be removed for external treatment either by unloading the catalyst particles or removing multi-channel trays with catalyst attached.
A summary of regeneration procedures from some of the main industrial companies that are or recently have been involved in Fischer-Tropsch technology development is given in
Table 3. The table is representative of available information but should not be assumed to be complete or up-to-date; nevertheless it illustrates what is probably the preferred approach of each individual company. In the first column the type of commercial regeneration process and primary FT-reactor type are listed and whether the regeneration is intermittent or continuous, while data in the other columns refer to the actual test protocol and results described in the patent literature. Note that reactor type can be different in columns one and three.
It is clearly possible to regenerate a deactivated catalyst to a level close to the original activity by different combinations of wax removal, hydrogenation and combustion of carbonaceous deposits; followed by re-reduction if needed. There is unfortunately little information available on the long-term performance of regenerated catalysts.
Sasol has published a brief summary of their procedure for removing most of the wax, followed by combustion of the remaining carbonaceous species and complete reactivation [
65]. Specifically, Sasol describes dewaxing by hydrogenolysis with pure hydrogen for 2 h at 220 °C and reduction for 2 h at 350 °C [
66]. After passivation with CO
2, the catalyst is subjected to oxidation with air in a fluidized-bed calcination unit at 250 °C for 6 h under a pressure preferably of
ca. 10 bar. Re-reduction is performed at 425 °C and 98% of the original activity is regained. In cooperation with Eindhoven University, Sasol has investigated mechanisms of deactivation and regeneration for both model and commercial catalysts [
67]. After a heptane wash and reoxidation, several FT cycles were demonstrated with no apparent permanent loss in activity. The oxidation step is described in terms of the Kirkendall effect involving spreading of a Co oxide film during oxidation followed by breaking up of the film to form small re-dispersed Co crystallites.
Table 3.
Summary of regeneration concepts and procedures based on patents and presentations from LTFT technology companies.
Table 3.
Summary of regeneration concepts and procedures based on patents and presentations from LTFT technology companies.
| Technology owner Regeneration configuration* | Catalyst TOS | FT test reactor | Wax removal step | Primary hydrogenation | Calcination/ Oxidation/ Re-dispersion | Regeneration effect (activity) |
|---|
| Sasol [66,68] Slurry; continuous Ex situ | Co/Pt/ alumina - | Slurry** | H2 strip at 220 °C or xylene wash | H2 at 350 °C | Air in fluid-bed | 98% |
| Shell [69] Fixed-bed; intermittent in situ | Co/Mn/ Titania- Years | Fixed-bed Full scale | Gas oil wash | Diluted H2 | Diluted O2 at 270 °C; NH3/CO2 | > 100% < 100% (800 h TOS) |
| GTL.F1 [70] Slurry; continuous Ex situ | Co/Re/ aluminate - | Slurry** | Draining or cyclohexane wash | No | Diluted air in fluid-bed | 98–115% |
| ExxonMobil [71] Slurry; continuous Ex situ | Co/Re/titania - A few days | Fixed-bed (lab) | Filtration + H2 strip | No | Diluted O2 in fixed-bed | ~ 100% |
| Nippon Oil [72] Slurry; continuous Ex situ | Co/ zirconia-silica - | Slurry (lab) | “de-oiling” | No | Steaming at 200 °C; 25 barin fixed-bed | 95% |
| ConocoPhillips [73] Slurry; continuous Ex situ | Co/Ru or Re/ (F-)alumina - 1014 h | Fixed-bed (lab) or slurry.** | No | 7% H2/steam at 300 °C/ 3 bar | No | 80–95% |
| Syntroleum [74] Slurry; continuous Ex situ | Co/alumina - 4000 h | Slurry** | N2 at 316–343 °C | No | Diluted O2 in fluid-bed | “Good performance” |
| Oxford Catalyst/ Velocys [75] Micro-channel; in situ intermittent | Co/Re/ titania-silica? | Micro-channel | N2 flow? | H2in situ | O2in situ | ~ 100% |
It appears that Shell is aiming at
in situ regeneration in the tubes of their fixed-bed FT reactor, although an external post FT-reaction step is part of their described procedure [
69]. Regeneration is based on a procedure that most likely comes from their Bintulu plant in Malaysia. After wax removal a mild hydrogenation and oxidation is conducted. The catalyst is then taken out of the FT-reactor, treated with a concentrated aqueous ammonia solution and subsequently with CO
2, giving Co ammonium carbonate. Most likely the latter procedure gives cobalt ammine carbonate complexes suitable for re-dispersing cobalt [
76].
Regeneration may re-disperse cobalt, possibly even to a level higher than for a freshly reduced catalyst [
70]. A promoted cobalt catalyst on a spinel support was subjected to an extended run in a slurry-bubble column reactor as described by Schanke
et al. [
14]
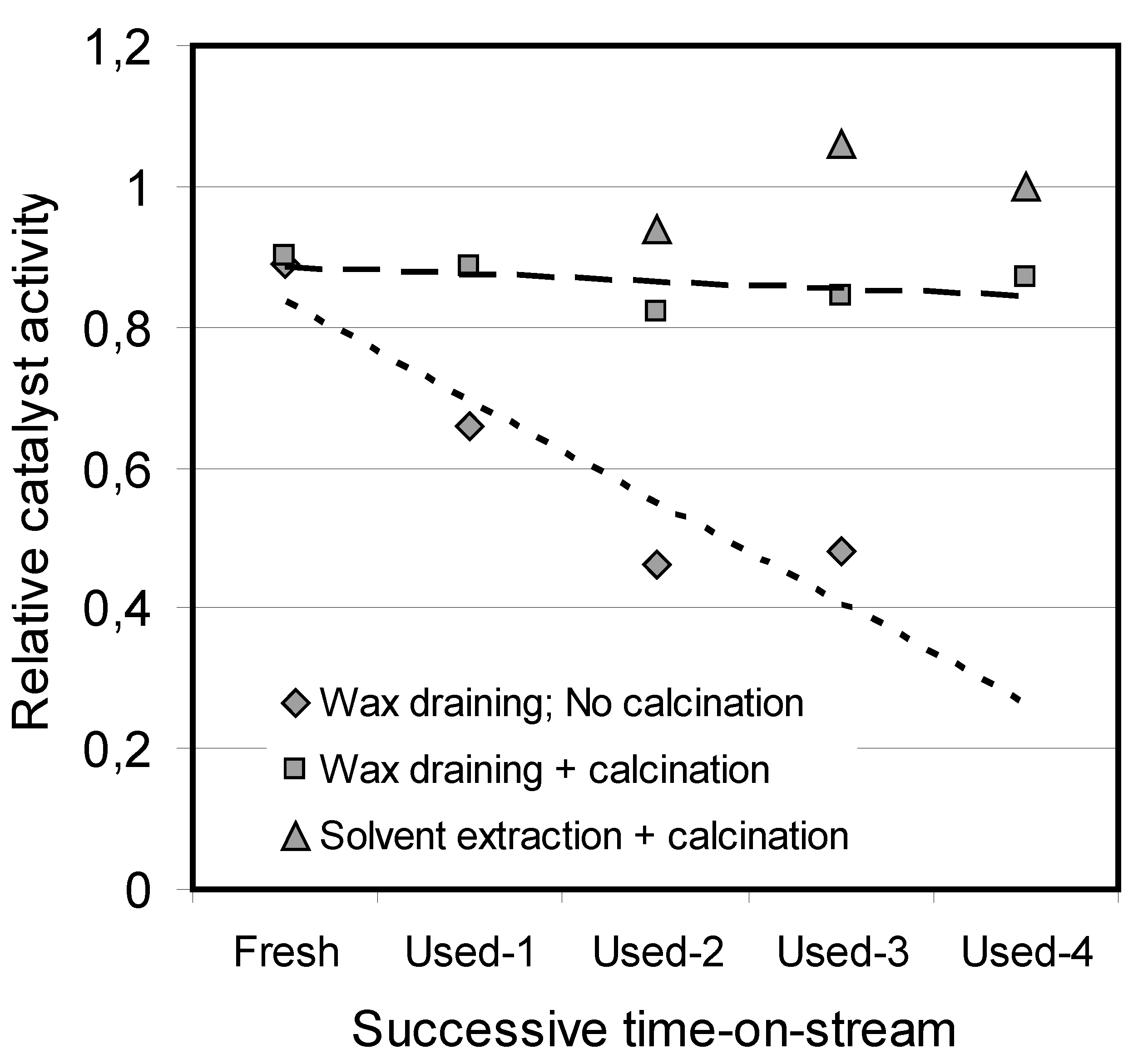
. Actual time on stream was not reported, but samples from four different TOS’s were analyzed and regenerated. Data from the patent have been plotted in
Figure 3. All activity data are from standard runs in a laboratory fixed-bed reactor at 210 °C, 20 bar pressure and H
2/CO ratio of 2:1, and after a conventional reduction protocol with hydrogen at 350 °C. Fresh and used catalysts, all embedded in wax, were drained to remove excessive wax at 85 °C before activity testing. A successive reduction in activity to
ca. 50% was experienced. If the spent catalyst was calcined at 300 °C to burn off excessive wax and carbon deposits before testing, the activity is only slightly reduced compared to the fresh catalyst. Interestingly, even an enhanced activity was experienced if the wax was removed by cyclohexane/n-heptane solvent extraction before calcination. Pore volumes and surface areas of freshly calcined catalysts and used catalysts subject to wax draining and oxidation are unchanged. From these data it appears that hydrogenation by itself has moderate or no effect on regeneration, but that burning off coke deposits completely rejuvenates the catalyst.
Figure 3.
Catalyst activity before and after oxidation of carbon deposits of a catalyst used in a slurry bubble column reactor (data from ref. [
70]).
Figure 3.
Catalyst activity before and after oxidation of carbon deposits of a catalyst used in a slurry bubble column reactor (data from ref. [
70]).
In an early patent, Exxon researchers describe successful rejuvenation by atmospheric hydrogen treatment at typical FT synthesis temperatures of 200–230 °C of spent Co/Ru/titania catalysts [
77]. The promoter is needed to facilitate the rejuvenation and is claimed also to inhibit carbon deposits [
78]. However, examples are only for FT runs of 10–30 days at 50–60% CO conversion in lab-scale, fixed-bed reactors, and therefore may only addresses hydrogenolysis of very heavy waxes that partially block pores and possibly hydrogenation of oligomeric carbons on the surface of the catalyst. In a later patent, as shown in the table above, Exxon Mobil has demonstrated that reduction with hydrogen after oxidative regeneration can be performed in the slurry FT-reactor itself at mild process conditions of 200 °C and 20 bar, thus resembling the FT synthesis conditions. The company has a number of patents describing regeneration procedures, including adding more active metal after combustion of carbon deposits [
79].
As pointed out above, - deposits of heavy hydrocarbon waxes reside in the pores of a used catalyst that should be largely removed before regeneration. Hydrogen treatment may reduce part of the wax through hydrogenation, but may also leave residual components at the surface. ConocoPhillips claims to have designed a suitable reactivation procedure that both removes heavy hydrocarbons and reduces the active metal [
73]. For a 19 wt.% Co/0.1 wt.% Ru on alumina catalyst run for 40 days at standard FT-conditions, best regeneration results were obtained in a fixed-bed using a mixture of 93% steam/7% H
2 at 300 °C and 3 bar where 95% of initial activity was regained. Similar experiments for a catalyst composition of 19 wt.% Co/0.1 wt.% Re on fluorinated alumina in a slurry reactor were less successful as 71% of initial activity was obtained, up from 33% after the FT period.
In their continuous regeneration of a spent slurry catalyst, Syntroleum focuses on removing as much wax as possible by nitrogen stripping at 300–350 °C and 3 bar, followed by calcination. Improved cobalt dispersion and reducibility are claimed, but no actual performance data are reported for the regenerated catalyst [
74]. Nippon Oil has followed an alternative approach where they hydrothermally treat the spent catalyst with steam at elevated pressure [
72]. Much of the activity is regained, although it appears that the TOS between regenerations is relatively short in view of the high activity level of
ca. 75% before regeneration, thus moderating build-up of polymeric carbonaceous deposits.
Oxford Catalyst/Velocys have presented interesting long-term operation and regeneration performances for their micro-channel reactor [
75]. Velocys’ catalyst and reactor operate at much higher space velocities and productivities than conventional catalyst/large-scale FT reactors. To compensate for deactivation, temperature is increased from 205 °C to 232 °C after
ca. 650 days TOS, accompanied by a slight increase in CO conversion from 71.2 to 73.6%. Naturally, the temperature increase is accompanied by a reduction in C
5+ selectivity, in this case by as much as from 87.9% to 75.8%. Both activity and selectivity are fully restored after
in situ hydrogen treatment followed by calcination with oxygen and re-reduction. The first step of hydrogenation probably re-reduces smaller cobalt crystallites and partly removes some wax and deposits. The hydrogenation step can be carried out under conditions resembling FT-synthesis and can thus be carried out at the plant location and with the catalyst loaded in the reactor. However, burning off the carbonaceous deposits is far more demanding and probably requires removing the catalyst from the reactor. These results are qualitatively in agreement with what can be expected from large scale fixed-bed and slurry bubble column operations although the rate of deactivation for the Velocys’ catalyst is quantitatively smaller than those reported in available literature for large scale reactors. The alternative micro-channel LTFT provider, CompactGTL, appears to have only a regeneration patent directed at removing accumulated ammonia deposits [
80].


{kind=link}
{kind=link}
{kind=link}
{kind=link}