
Theoretical Insights and Design Strategies of Metal–Nitrogen–Carbon Catalysts for Electrochemical Nitrogen Reduction Reaction


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Fundamentals of Electrochemical NRR
2.1. Mechanisms of NRR
2.1.1. Dissociative Mechanism
2.1.2. Associative Mechanism
2.1.3. Other Mechanisms
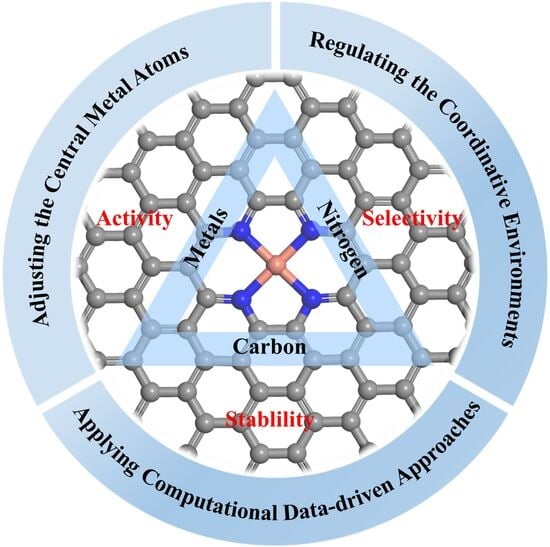
2.2. Performance of M–N–C for NRR
- Activity. The catalytic activity of M–N–C is widely attributed to modifications in the electronic properties of the active sites. When metal atoms interact with a substrate, the orbital hybridization occurs between the metal and nitrogen atoms [53]. The doped nitrogen atoms and carbon substrates can serve as electron donors or acceptors, inducing the redistribution of electrons around the metal active sites [77]. Normally, the electrons of the metal atoms are transferred to more electronegative nitrogen atoms, which modulates the d-band center of the metals [78]. In recent years, several studies have revealed that electrons of the metal active sites in M–N–C can be transferred to the adsorbed N2 molecules, which play a key role in NRR [79]. Therefore, adjusting the electronic MSI of M–N–C is critical for optimizing the catalytic activity of M–N–C.
- Selectivity. M–N–C materials have well-defined active sites, which help to improve the selectivity of NRR. M–N–C can inhibit HER through geometric and electronic effects [80,81,82,83]. The geometric effects of M–N–C restrict the adsorption to the top site of the active metal centers. The preferential adsorption of N2 at the active sites will inhibit the adsorption of H atoms on M–N–C. Additionally, owing to the MSI of M–N–C, the electrons in the metal atoms will be transferred to the substrates, leading to a positive charge of metal atoms. The charge repulsion between the positively charged metal and the H further prevents the adsorption of H atoms at the active sites, thereby enhancing the NRR selectivity with M–N–C.
- Stability. The stability of M–N–C materials is primarily attributed to the strong interaction between the metal active sites and the substrates. The non-metallic nitrogen ligands generate highly localized acceptor-like state near the Fermi level, resulting in strong interactions with the metal atoms [53,84]. This interaction suppresses atomic migration and prevents the agglomeration of metal atoms. Owing to the chemical bonding properties of the metal atoms on the N-doped carbon substrates, M–N–C exhibits satisfactory stability for NRR.
2.3. Computational Models of NRR
2.3.1. Electrode Potential Models
2.3.2. Solvation Models
2.3.3. Reaction Kinetics
2.3.4. Molecular Dynamics
3. Design Strategies of M–N–C Catalysts
3.1. Adjusting the Central Metal Atoms
3.2. Regulating the Coordinative Environments
3.3. Applying Computational Data-Driven Approaches
4. Summary and Outlook
- Accuracy of theoretical calculation. Although various synthetic methods have been employed to prepare M–N–C materials, the trial-and-error approach of experiments is both time-consuming and costly. DFT calculations have proven to be a powerful tool for guiding the selection of elements and the structural design of M–N–C catalysts. However, the current calculation models and reaction conditions are often highly idealized, neglecting factors such as temperature, pressure, and solvent effects. This creates a significant gap between theoretical research and practical experiments. It is urgent to develop more advanced computational techniques and strategies that can simulate catalytic activity to get closer to the real conditions. Solvation models and complex molecular dynamics have attracted increasing attention in this context [7,154,155]. The combined application of these advanced theoretical techniques will provide a robust tool for more comprehensive theoretical research. They can reduce the deviation between theoretical and experimental results and enhance the accuracy and adaptability of theoretical calculation results.
- Development of characterization techniques. Accurately characterizing the electronic state changes during the NRR process remains a significant challenge. Current characterization techniques are proficient in identifying the features of atomically dispersed metal active sites. However, many studies have demonstrated that the variations in nitrogen and carbon coordination numbers around the CMAs significantly influence catalytic performance. However, the existing characterization techniques (e.g., HAADF-STEM, AC-TEM, and XAS) are only for local information and average statistics, which are insufficient for precisely judging the local N-doped carbon structures near the CMAs [46,156]. Therefore, developing more advanced characterization techniques that can accurately determine the coordination structure remains a critical challenge for future research.
- Design of catalytic descriptors. Current catalyst screening efforts based on DFT calculations are often hindered by different algorithms and inconsistencies in calculation accuracy, which restrict their ability to offer comprehensive and systematic design guidance. Developing universal catalytic descriptors that transcend the limitations of LSR represents a crucial approach in advancing catalyst design [157]. These descriptors serve as essential tools for theoretical screening, enabling the identification of promising catalysts. By constructing rational descriptors derived from simple parameters, such as bond length and valence electron number, researchers can provide clear and actionable guidance for the design of M–N–C catalysts [158,159]. Furthermore, the well-defined active sites in M–N–C catalysts offer an ideal platform for the development of accurate descriptors, facilitating a deeper understanding of the structure–property relationship and advancing the rational design of catalytic systems.
- AI-assisted catalyst discovery. The rapidly expanding chemical and structural space of M–N–C catalysts makes traditional trial-and-error screening increasingly inefficient. Artificial intelligence (AI) offers powerful tools for accelerating catalyst discovery by identifying structure–property relationships from DFT-generated and literature-derived datasets. For example, Kim et al. developed a slab graph convolutional neural network to predict catalytic properties and accelerate the discovery of N2 electroreduction catalysts [160]. More recently, He et al. proposed an ML-driven four-step screening strategy to shorten the screening process for high-performance NRR electrocatalysts [148]. These studies show that AI methods can efficiently predict key catalytic parameters, including adsorption energies, UL, reaction selectivity, and catalyst stability, thereby reducing the need for exhaustive DFT calculations. To further improve the reliability and applicability of AI-assisted catalyst design, future studies should establish standardized NRR databases covering catalyst structures, calculation settings, solvation models, electrode potentials, key intermediates, and experimental validation. The integration of high-throughput DFT, interpretable AI models, and experimental feedback will provide an important pathway for advancing M–N–C catalyst design from empirical screening toward closed-loop intelligent discovery.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Shi, Y.; Zhao, Z.; Yang, D.; Tan, J.; Xin, X.; Liu, Y.; Jiang, Z. Engineering photocatalytic ammonia synthesis. Chem. Soc. Rev. 2023, 52, 6938–6956. [Google Scholar] [CrossRef] [PubMed]
- John, J.; MacFarlane, D.R.; Simonov, A.N. The why and how of NOx electroreduction to ammonia. Nat. Catal. 2023, 6, 1125–1130. [Google Scholar] [CrossRef]
- Qing, G.; Ghazfar, R.; Jackowski, S.T.; Habibzadeh, F.; Ashtiani, M.M.; Chen, C.-P.; Smith, M.R., III; Hamann, T.W. Recent Advances and Challenges of Electrocatalytic N2 Reduction to Ammonia. Chem. Rev. 2020, 120, 5437–5516. [Google Scholar] [CrossRef]
- Xu, X.; Hu, L.; Li, Z.; Xie, L.; Sun, S.; Zhang, L.; Li, J.; Luo, Y.; Yan, X.; Hamdy, M.S.; et al. Oxygen vacancies in Co3O4 nanoarrays promote nitrate electroreduction for ammonia synthesis. Sustain. Energy Fuels 2022, 6, 4130–4136. [Google Scholar] [CrossRef]
- Choudhary, A.; Halder, A.; Aggarwal, P.; Govind Rao, V. Plasmonic chemistry for sustainable ammonia production. Commun. Mater. 2024, 5, 69. [Google Scholar] [CrossRef]
- Dai, T.-Y.; Yang, C.-C.; Jiang, Q. Recent progress on catalyst design of nitrogen reduction reaction by density functional theory. Sci. China Mater. 2024, 67, 1101–1123. [Google Scholar] [CrossRef]
- Yao, X.; Halpren, E.; Liu, Y.Z.; Shan, C.H.; Chen, Z.W.; Chen, L.X.; Singh, C.V. Intrinsic and external active sites of single-atom catalysts. iScience 2023, 26, 107275. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Chen, Z.; Kou, S.; Lu, G.; Chen, D.; Liu, Z. Carbon-supported catalysts with atomically dispersed metal sites for oxygen electroreduction: Present and future perspectives. J. Mater. Chem. A 2021, 9, 15919–15936. [Google Scholar] [CrossRef]
- Kojima, Y. Hydrogen storage materials for hydrogen and energy carriers. Int. J. Hydrogen Energy 2019, 44, 18179–18192. [Google Scholar] [CrossRef]
- MacFarlane, D.R.; Cherepanov, P.V.; Choi, J.; Suryanto, B.H.R.; Hodgetts, R.Y.; Bakker, J.M.; Ferrero Vallana, F.M.; Simonov, A.N. A Roadmap to the Ammonia Economy. Joule 2020, 4, 1186–1205. [Google Scholar] [CrossRef]
- Li, S.; Fu, X.; Nørskov, J.K.; Chorkendorff, I. Towards sustainable metal-mediated ammonia electrosynthesis. Nat. Energy 2024, 9, 1344–1349. [Google Scholar] [CrossRef]
- Kyriakou, V.; Garagounis, I.; Vourros, A.; Vasileiou, E.; Stoukides, M. An Electrochemical Haber–Bosch Process. Joule 2020, 4, 142–158. [Google Scholar] [CrossRef]
- Jabarivelisdeh, B.; Jin, E.; Christopher, P.; Masanet, E. Model-Based Analysis of Ammonia Production Processes for Quantifying Energy Use, Emissions, and Reduction Potentials. ACS Sustain. Chem. Eng. 2022, 10, 16280–16289. [Google Scholar] [CrossRef]
- Smith, C.; Hill, A.K.; Torrente-Murciano, L. Current and future role of Haber–Bosch ammonia in a carbon-free energy landscape. Energy Environ. Sci. 2020, 13, 331–344. [Google Scholar] [CrossRef]
- IEA. Ammonia Technology Roadmap: Towards More Sustainable Nitrogen Fertiliser Production; IEA: Paris, France, 2021. [Google Scholar]
- Wang, L.; Wang, D.; Li, Y. Single-atom catalysis for carbon neutrality. Carbon Energy 2022, 4, 1021–1079. [Google Scholar] [CrossRef]
- Chen, J.; Cheng, H.; Ding, L.-X.; Wang, H. Competing hydrogen evolution reaction: A challenge in electrocatalytic nitrogen fixation. Mater. Chem. Front. 2021, 5, 5954–5969. [Google Scholar] [CrossRef]
- Zhao, X.; Hu, G.; Chen, G.F.; Zhang, H.; Zhang, S.; Wang, H. Comprehensive Understanding of the Thriving Ambient Electrochemical Nitrogen Reduction Reaction. Adv. Mater. 2021, 33, 2007650. [Google Scholar] [CrossRef]
- Shen, H.; Choi, C.; Masa, J.; Li, X.; Qiu, J.; Jung, Y.; Sun, Z. Electrochemical ammonia synthesis: Mechanistic understanding and catalyst design. Chem 2021, 7, 1708–1754. [Google Scholar] [CrossRef]
- Ren, Y.; Yu, C.; Wang, L.; Tan, X.; Wang, Z.; Wei, Q.; Zhang, Y.; Qiu, J. Microscopic-Level Insights into the Mechanism of Enhanced NH3 Synthesis in Plasma-Enabled Cascade N2 Oxidation-Electroreduction System. J. Am. Chem. Soc. 2022, 144, 10193–10200. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, W.; Tan, X.; Jiang, K.; Zhai, S.; Li, Z. Atomic-level reactive sites for electrocatalytic nitrogen reduction to ammonia under ambient conditions. Coord. Chem. Rev. 2023, 489, 215196. [Google Scholar] [CrossRef]
- Peramaiah, K.; Ramalingam, V.; Fu, H.-C.; Alsabban, M.M.; Ahmad, R.; Cavallo, L.; Tung, V.; Huang, K.-W.; He, J.-H. Optically and Electrocatalytically Decoupled Si Photocathodes with a Porous Carbon Nitride Catalyst for Nitrogen Reduction with Over 61.8% Faradaic Efficiency. Adv. Mater. 2021, 33, 2100812. [Google Scholar] [CrossRef]
- Schiffer, Z.J.; Manthiram, K. Electrification and Decarbonization of the Chemical Industry. Joule 2017, 1, 10–14. [Google Scholar] [CrossRef]
- Fu, X.; Pedersen, J.B.; Zhou, Y.; Saccoccio, M.; Li, S.; Sažinas, R.; Li, K.; Andersen, S.Z.; Xu, A.; Deissler, N.H.; et al. Continuous-flow electrosynthesis of ammonia by nitrogen reduction and hydrogen oxidation. Science 2023, 379, 707–712. [Google Scholar] [CrossRef]
- Moon, Y.H.; Kim, N.Y.; Kim, S.M.; Jang, Y.J. Recent Advances in Electrochemical Nitrogen Reduction Reaction to Ammonia from the Catalyst to the System. Catalysts 2022, 12, 1015. [Google Scholar] [CrossRef]
- Wang, M.; Liu, S.; Qian, T.; Liu, J.; Zhou, J.; Ji, H.; Xiong, J.; Zhong, J.; Yan, C. Over 56.55% Faradaic efficiency of ambient ammonia synthesis enabled by positively shifting the reaction potential. Nat. Commun. 2019, 10, 341. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Chang, Q.; Dodelet, J.-P.; Chenitz, R. Recent Advances in Electrocatalysts for Oxygen Reduction Reaction. Chem. Rev. 2016, 116, 3594–3657. [Google Scholar] [CrossRef]
- Yang, D.; Chen, T.; Wang, Z. Electrochemical reduction of aqueous nitrogen (N2) at a low overpotential on (110)-oriented Mo nanofilm. J. Mater. Chem. A 2017, 5, 18967–18971. [Google Scholar] [CrossRef]
- Yao, Y.; Wang, H.; Yuan, X.-z.; Li, H.; Shao, M. Electrochemical Nitrogen Reduction Reaction on Ruthenium. ACS Energy Lett. 2019, 4, 1336–1341. [Google Scholar] [CrossRef]
- Wang, J.; Yu, L.; Hu, L.; Chen, G.; Xin, H.; Feng, X. Ambient ammonia synthesis via palladium-catalyzed electrohydrogenation of dinitrogen at low overpotential. Nat. Commun. 2018, 9, 1795. [Google Scholar] [CrossRef]
- Shi, M.-M.; Bao, D.; Wulan, B.-R.; Li, Y.-H.; Zhang, Y.-F.; Yan, J.-M.; Jiang, Q. Au Sub-Nanoclusters on TiO2 toward Highly Efficient and Selective Electrocatalyst for N2 Conversion to NH3 at Ambient Conditions. Adv. Mater. 2017, 29, 1606550. [Google Scholar] [CrossRef]
- Rutledge, H.L.; Tezcan, F.A. Electron Transfer in Nitrogenase. Chem. Rev. 2020, 120, 5158–5193. [Google Scholar] [CrossRef]
- Shi, Z.; Yang, W.; Gu, Y.; Liao, T.; Sun, Z. Metal–Nitrogen–Doped Carbon Materials as Highly Efficient Catalysts: Progress and Rational Design. Adv. Sci. 2020, 7, 2001069. [Google Scholar] [CrossRef]
- Wu, D.; He, B.; Wang, Y.; Lv, P.; Ma, D.; Jia, Y. Double-atom catalysts for energy-related electrocatalysis applications: A theoretical perspective. J. Phys. D Appl. Phys. 2022, 55, 203001. [Google Scholar] [CrossRef]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef]
- Chen, Y.; He, T.; Liu, Y.; Liu, Y.; Liu, Y.-N.; Liu, C.; Zhang, Y. Electronic engineering to enhance bifunctional activity of carbon-based single-atom catalysts in rechargeable zinc-air batteries. Curr. Opin. Electrochem. 2023, 37, 101206. [Google Scholar] [CrossRef]
- Chen, Y.; Ji, S.; Chen, C.; Peng, Q.; Wang, D.; Li, Y. Single-Atom Catalysts: Synthetic Strategies and Electrochemical Applications. Joule 2018, 2, 1242–1264. [Google Scholar] [CrossRef]
- Yang, X.-F.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T. Single-Atom Catalysts: A New Frontier in Heterogeneous Catalysis. Acc. Chem. Res. 2013, 46, 1740–1748. [Google Scholar] [CrossRef]
- Long, X.; Huang, F.; Yao, Z.; Li, P.; Zhong, T.; Zhao, H.; Tian, S.; Shu, D.; He, C. Advancements in Electrocatalytic Nitrogen Reduction: A Comprehensive Review of Single-Atom Catalysts for Sustainable Ammonia Synthesis. Small 2024, 20, 2400551. [Google Scholar] [CrossRef]
- Zhao, Y.; Yan, L.; Zhao, X. Development of Carbon-Based Electrocatalysts for Ambient Nitrogen Reduction Reaction: Challenges and Perspectives. ChemElectroChem 2022, 9, e202101126. [Google Scholar] [CrossRef]
- Guan, W.; Shao, H.; Zhang, C.; Qiu, X.; Zhao, J.; Wang, Y.; Zhang, L.; Shao, M.; Hu, J. Strategies for the regulation of specific active sites in metal–nitrogen–carbon. Nano Energy 2024, 120, 109149. [Google Scholar] [CrossRef]
- Yang, X.J.; Wen, Z.; Wang, Z.L.; Jiang, Q. Os1B11N12/C2N as an Efficient Electrocatalyst for Nitrogen Reduction Reaction. ChemSusChem 2022, 15, e202102648. [Google Scholar] [CrossRef]
- Mushtaq, M.A.; Arif, M.; Fang, X.; Yasin, G.; Ye, W.; Basharat, M.; Zhou, B.; Yang, S.; Ji, S.; Yan, D. Photoelectrochemical reduction of N2 to NH3 under ambient conditions through hierarchical MoSe2@g-C3N4 heterojunctions. J. Mater. Chem. A 2021, 9, 2742–2753. [Google Scholar] [CrossRef]
- Kumar, P.; Singh, G.; Guan, X.; Lee, J.; Bahadur, R.; Ramadass, K.; Kumar, P.; Kibria, M.G.; Vidyasagar, D.; Yi, J.; et al. Multifunctional carbon nitride nanoarchitectures for catalysis. Chem. Soc. Rev. 2023, 52, 7602–7664. [Google Scholar] [CrossRef]
- Askins, E.J.; Zoric, M.R.; Li, M.; Luo, Z.; Amine, K.; Glusac, K.D. Toward a mechanistic understanding of electrocatalytic nanocarbon. Nat. Commun. 2021, 12, 3288. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Wang, Z.; Guan, J. Optimizing the Electrocatalytic Selectivity of Carbon Dioxide Reduction Reaction by Regulating the Electronic Structure of Single-Atom M–N–C Materials. Adv. Funct. Mater. 2022, 32, 2111504. [Google Scholar] [CrossRef]
- Zhu, C.; Fu, S.; Shi, Q.; Du, D.; Lin, Y. Single-Atom Electrocatalysts. Angew. Chem. Int. Ed. Engl. 2017, 56, 13944–13960. [Google Scholar] [CrossRef]
- Wu, G.; Zelenay, P. Activity versus stability of atomically dispersed transition-metal electrocatalysts. Nat. Rev. Mater. 2024, 9, 643–656. [Google Scholar] [CrossRef]
- Faraji, S.; Wang, Z.; Lopez-Rivera, P.; Liu, M. Advancements in computational approaches for rapid metal site discovery in carbon-based materials for electrocatalysis. Energy Adv. 2023, 2, 1781–1799. [Google Scholar] [CrossRef]
- Chhetri, A.; Biswas, A.; Podder, S.; Dey, R.S.; Mitra, J. Strategic design of VO2 encased in N-doped carbon as an efficient electrocatalyst for the nitrogen reduction reaction in neutral and acidic media. Nanoscale 2024, 16, 9426–9435. [Google Scholar] [CrossRef]
- Choi, C.; Gu, G.H.; Noh, J.; Park, H.S.; Jung, Y. Understanding potential-dependent competition between electrocatalytic dinitrogen and proton reduction reactions. Nat. Commun. 2021, 12, 4353. [Google Scholar] [CrossRef] [PubMed]
- Geng, Z.; Liu, Y.; Kong, X.; Li, P.; Li, K.; Liu, Z.; Du, J.; Shu, M.; Si, R.; Zeng, J. Achieving a Record-High Yield Rate of 120.9 for N2 Electrochemical Reduction over Ru Single-Atom Catalysts. Adv. Mater. 2018, 30, 1803498. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Xiao, C.; Ding, J.; Huang, Z.; Yang, X.; Zhang, T.; Mitlin, D.; Hu, W. Review of Carbon Support Coordination Environments for Single Metal Atom Electrocatalysts (SACS). Adv. Mater. 2024, 36, 2301477. [Google Scholar] [CrossRef]
- Majumder, M.; Saini, H.; Dědek, I.; Schneemann, A.; Chodankar, N.R.; Ramarao, V.; Santosh, M.S.; Nanjundan, A.K.; Kment, Š.; Dubal, D.; et al. Rational Design of Graphene Derivatives for Electrochemical Reduction of Nitrogen to Ammonia. ACS Nano 2021, 15, 17275–17298. [Google Scholar] [CrossRef]
- Han, X.; Zhang, T.; Wang, X.; Zhang, Z.; Li, Y.; Qin, Y.; Wang, B.; Han, A.; Liu, J. Hollow mesoporous atomically dispersed metal–nitrogen–carbon catalysts with enhanced diffusion for catalysis involving larger molecules. Nat. Commun. 2022, 13, 2900. [Google Scholar] [CrossRef]
- Tang, T.; Wang, Z.; Guan, J. Structural optimization of carbon-based diatomic catalysts towards advanced electrocatalysis. Coord. Chem. Rev. 2023, 492, 215288. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, Z.; She, F.; Wei, L.; Zeng, Z.; Li, H. Design of molecular MNC dual-atom catalysts for nitrogen reduction starting from surface state analysis. J. Colloid Interface Sci. 2023, 640, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Shi, L.; Bai, X.; Li, Q.; Ling, C.; Wang, J. Highly Efficient Photo-/Electrocatalytic Reduction of Nitrogen into Ammonia by Dual-Metal Sites. ACS Cent. Sci. 2020, 6, 1762–1771. [Google Scholar] [CrossRef]
- Zhao, X.; Levell, Z.H.; Yu, S.; Liu, Y. Atomistic Understanding of Two-dimensional Electrocatalysts from First Principles. Chem. Rev. 2022, 122, 10675–10709. [Google Scholar] [CrossRef]
- Marzari, N.; Ferretti, A.; Wolverton, C. Electronic-structure methods for materials design. Nat. Mater. 2021, 20, 736–749. [Google Scholar] [CrossRef]
- Jain, A.; Shin, Y.; Persson, K.A. Computational predictions of energy materials using density functional theory. Nat. Rev. Mater. 2016, 1, 15004. [Google Scholar] [CrossRef]
- Chen, Z.; Zhao, J.; Cabrera, C.R.; Chen, Z. Computational Screening of Efficient Single-Atom Catalysts Based on Graphitic Carbon Nitride (g-C3N4) for Nitrogen Electroreduction. Small Methods 2019, 3, 1800368. [Google Scholar] [CrossRef]
- Wang, S.; Qian, C.; Zhou, S. Accelerating the development of electrocatalysts for electrochemical nitrogen fixation through theoretical and computational approaches. Mater. Chem. Front. 2023, 7, 4259–4280. [Google Scholar] [CrossRef]
- Lv, C.; Zhong, L.; Yao, Y.; Liu, D.; Kong, Y.; Jin, X.; Fang, Z.; Xu, W.; Yan, C.; Dinh, K.N.; et al. Boosting Electrocatalytic Ammonia Production through Mimicking “π Back-Donation”. Chem 2020, 6, 2690–2702. [Google Scholar] [CrossRef]
- Légaré, M.-A.; Bélanger-Chabot, G.; Dewhurst, R.D.; Welz, E.; Krummenacher, I.; Engels, B.; Braunschweig, H. Nitrogen fixation and reduction at boron. Science 2018, 359, 896–900. [Google Scholar] [CrossRef]
- Tong, W.; Huang, B.; Wang, P.; Li, L.; Shao, Q.; Huang, X. Crystal-Phase-Engineered PdCu Electrocatalyst for Enhanced Ammonia Synthesis. Angew. Chem. Int. Ed. Engl. 2020, 59, 2649–2653. [Google Scholar] [CrossRef]
- Li, J.; Chen, S.; Quan, F.; Zhan, G.; Jia, F.; Ai, Z.; Zhang, L. Accelerated Dinitrogen Electroreduction to Ammonia via Interfacial Polarization Triggered by Single-Atom Protrusions. Chem 2020, 6, 885–901. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, C.; Sun, L.; Wang, T. Progress of Experimental and Computational Catalyst Design for Electrochemical Nitrogen Fixation. ACS Catal. 2022, 12, 8936–8975. [Google Scholar] [CrossRef]
- Ellingsson, V.; Iqbal, A.; Skúlason, E.; Abghoui, Y. Nitrogen Reduction Reaction to Ammonia on Transition Metal Carbide Catalysts. ChemSusChem 2023, 16, e202300947. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Guo, K.; Xie, Y.; Liu, S.; Chen, L.; Xu, J. High efficiency carbon nanotubes-based single-atom catalysts for nitrogen reduction. Sci. Rep. 2023, 13, 9926. [Google Scholar] [CrossRef]
- Zang, W.; Yang, T.; Zou, H.; Xi, S.; Zhang, H.; Liu, X.; Kou, Z.; Du, Y.; Feng, Y.P.; Shen, L.; et al. Copper Single Atoms Anchored in Porous Nitrogen-Doped Carbon as Efficient pH-Universal Catalysts for the Nitrogen Reduction Reaction. ACS Catal. 2019, 9, 10166–10173. [Google Scholar] [CrossRef]
- Dong, H.; Sun, H.; Xing, G.; Liu, S.; Duan, X.; Liu, J. First-Principles Study of Bimetallic Pairs Embedded on Graphene Co-Doped with N and O for N2 Electroreduction. Molecules 2024, 29, 779. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, T. Toward High-Performance Electrochemical Ammonia Synthesis by Circumventing the Surface H-Mediated N2 Reduction. JACS Au 2024, 4, 4023–4031. [Google Scholar] [CrossRef]
- Barman, N.; Kapse, S.; Thapa, R. Electronic Descriptor to Identify the Activity of SACs for E-NRR and Effect of BF3 as Electrolyte Ion. ChemSusChem 2025, 18, e202400902. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, B.; Sun, B.; Wang, N.; Hu, W.; Komarneni, S. N-Doped Porous Carbon Self-Generated on Nickel Oxide Nanosheets for Electrocatalytic N2 Fixation with a Faradaic Efficiency beyond 30%. ACS Sustain. Chem. Eng. 2019, 7, 18874–18883. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, Y.; Zhang, Z.; Hou, D.; Bai, F.; Han, Y.; Zhang, C.; Zhang, Y.; Hu, J. Metal–support interactions for heterogeneous catalysis: Mechanisms characterization techniques and applications. J. Mater. Chem. A 2023, 11, 8540–8572. [Google Scholar] [CrossRef]
- Shen, H.; Gracia-Espino, E.; Ma, J.; Zang, K.; Luo, J.; Wang, L.; Gao, S.; Mamat, X.; Hu, G.; Wagberg, T.; et al. Synergistic Effects between Atomically Dispersed Fe–N–C and C–S–C for the Oxygen Reduction Reaction in Acidic Media. Angew. Chem. Int. Ed. Engl. 2017, 56, 13800–13804. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.B.; Yang, L.; Yu, L.B.; Song, E.H.; Jiang, Q. The VN3 embedded graphane with the improved selectivity for nitrogen fixation. Appl. Surf. Sci. 2020, 513, 145855. [Google Scholar] [CrossRef]
- Guo, X.; Gu, J.; Lin, S.; Zhang, S.; Chen, Z.; Huang, S. Tackling the Activity and Selectivity Challenges of Electrocatalysts toward the Nitrogen Reduction Reaction via Atomically Dispersed Biatom Catalysts. J. Am. Chem. Soc. 2020, 142, 5709–5721. [Google Scholar] [CrossRef]
- Seh, Z.W.; Kibsgaard, J.; Dickens, C.F.; Chorkendorff, I.; Nørskov, J.K.; Jaramillo, T.F. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 2017, 355, eaad4998. [Google Scholar] [CrossRef]
- van Deelen, T.W.; Hernández Mejía, C.; de Jong, K.P. Control of metal–support interactions in heterogeneous catalysts to enhance activity and selectivity. Nat. Catal. 2019, 2, 955–970. [Google Scholar] [CrossRef]
- Ren, Y.; Yu, C.; Tan, X.; Huang, H.; Wei, Q.; Qiu, J. Strategies to suppress hydrogen evolution for highly selective electrocatalytic nitrogen reduction: Challenges and perspectives. Energy Environ. Sci. 2021, 14, 1176–1193. [Google Scholar] [CrossRef]
- Yang, J.; Li, W.; Wang, D.; Li, Y. Electronic Metal–Support Interaction of Single-Atom Catalysts and Applications in Electrocatalysis. Adv. Mater. 2020, 32, 2003300. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Z.; Hwang, S.; Li, X.; Wang, C.; Samarakoon, W.; Karakalos, S.; Li, D.; Chen, M.; He, Y.; Wang, M.; et al. 3D porous graphitic nanocarbon for enhancing the performance and durability of Pt catalysts: A balance between graphitization and hierarchical porosity. Energy Environ. Sci. 2019, 12, 2830–2841. [Google Scholar] [CrossRef]
- Jiang, L.; Bai, X.; Zhi, X.; Jiao, Y. New Mechanistic Insights into Electrokinetic Competition Between Nitrogen Reduction and Hydrogen Evolution Reactions. Adv. Energy Mater. 2024, 14, 2303809. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- He, Y.; Liu, S.; Wang, M.; Ji, H.; Zhang, L.; Cheng, Q.; Qian, T.; Yan, C. Advancing the Electrochemistry of Gas-Involved Reactions through Theoretical Calculations and Simulations from Microscopic to Macroscopic. Adv. Funct. Mater. 2022, 32, 2208474. [Google Scholar] [CrossRef]
- Han, Y.; Xu, H.; Li, Q.; Du, A.; Yan, X. DFT-assisted low-dimensional carbon-based electrocatalysts design and mechanism study: A review. Front. Chem. 2023, 11, 1286257. [Google Scholar] [CrossRef]
- Jinnouchi, R.; Anderson, A.B. Electronic structure calculations of liquid–solid interfaces: Combination of density functional theory and modified Poisson–Boltzmann theory. Phys. Rev. B 2008, 77, 245417. [Google Scholar] [CrossRef]
- Ojha, K.; Arulmozhi, N.; Aranzales, D.; Koper, M.T.M. Double Layer at the Pt(111)–Aqueous Electrolyte Interface: Potential of Zero Charge and Anomalous Gouy-Chapman Screening. Angew. Chem. Int. Ed. Engl. 2020, 59, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Sundararaman, R.; Goddard, W.A., III; Arias, T.A. Grand canonical electronic density-functional theory: Algorithms and applications to electrochemistry. J. Chem. Phys. 2017, 146, 114104. [Google Scholar] [CrossRef] [PubMed]
- Melander, M.M.; Kuisma, M.J.; Christensen, T.E.K.; Honkala, K. Grand-canonical approach to density functional theory of electrocatalytic systems: Thermodynamics of solid–liquid interfaces at constant ion and electrode potentials. J. Chem. Phys. 2019, 150, 041706. [Google Scholar] [CrossRef]
- Zeng, L.; Wu, T.; Ye, T.; Mo, T.; Qiao, R.; Feng, G. Modeling galvanostatic charge-discharge of nanoporous supercapacitors. Nat. Comput. Sci. 2021, 1, 725–731. [Google Scholar] [CrossRef]
- Chen, J.W.; Zhang, Z.; Yan, H.M.; Xia, G.J.; Cao, H.; Wang, Y.G. Pseudo-adsorption and long-range redox coupling during oxygen reduction reaction on single atom electrocatalyst. Nat. Commun. 2022, 13, 1734. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Li, Y.; Dong, H.; Ding, L.; Li, Y. Ruthenium single-atom catalysis for electrocatalytic nitrogen reduction unveiled by grand canonical density functional theory. J. Mater. Chem. A 2020, 8, 20402–20407. [Google Scholar] [CrossRef]
- Wu, T.; Melander, M.M.; Honkala, K. Coadsorption of NRR and HER Intermediates Determines the Performance of Ru-N4 toward Electrocatalytic N2 Reduction. ACS Catal. 2022, 12, 2505–2512. [Google Scholar] [CrossRef]
- Re Fiorentin, M.; Bianchi, M.G.; Christiansen, M.A.H.; Ciotti, A.; Risplendi, F.; Wang, W.; Jónsson, E.Ö.; Jónsson, H.; Cicero, G. Methodological Frameworks for Computational Electrocatalysis: From Theory to Practice. Small Methods 2026, 10, e01542. [Google Scholar] [CrossRef] [PubMed]
- Garcia Carcamo, R.A.; Shi, J.; Estejab, A.; Xie, T.; Bhattacharjee, S.; Biswas, S.; Bodenschatz, C.J.; Chen, X.; Maurya, M.; Zhang, X.; et al. A Perspective on Multiscale Modeling of Explicit Solvation-Enabled Simulations of Catalysis at Liquid–Solid Interfaces. ACS Catal. 2025, 15, 7448–7457. [Google Scholar] [CrossRef]
- Qian, S.-J.; Cao, H.; Chen, J.-W.; Chen, J.-C.; Wang, Y.-G.; Li, J. Critical Role of Explicit Inclusion of Solvent and Electrode Potential in the Electrochemical Description of Nitrogen Reduction. ACS Catal. 2022, 12, 11530–11540. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Z.; Li, Y.; Qu, Y.; Li, Y.; Li, W.; Zhao, M. Screening of Transition-Metal Single-Atom Catalysts Anchored on Covalent–Organic Frameworks for Efficient Nitrogen Fixation. ACS Appl. Mater. Interfaces 2022, 14, 1024–1033. [Google Scholar] [CrossRef]
- Xue, Z.; Zhang, X.; Qin, J.; Liu, R. High-throughput identification of high activity and selectivity transition metal single-atom catalysts for nitrogen reduction. Nano Energy 2021, 80, 105527. [Google Scholar] [CrossRef]
- Kuai, S.; Dong, H.; Duan, X.; Wang, M.; Liu, J. Single TM–N4 Anchored on Topological Defective Graphene for Electrocatalytic Nitrogen Reduction: A DFT Study. Catalysts 2025, 15, 1135. [Google Scholar] [CrossRef]
- Chen, Z.W.; Lu, Z.; Chen, L.X.; Jiang, M.; Chen, D.; Singh, C.V. Machine-learning-accelerated discovery of single-atom catalysts based on bidirectional activation mechanism. Chem Catal. 2021, 1, 183–195. [Google Scholar] [CrossRef]
- Han, L.; Ren, Z.; Ou, P.; Cheng, H.; Rui, N.; Lin, L.; Liu, X.; Zhuo, L.; Song, J.; Sun, J.; et al. Modulating Single-Atom Palladium Sites with Copper for Enhanced Ambient Ammonia Electrosynthesis. Angew. Chem. Int. Ed. Engl. 2021, 60, 345–350. [Google Scholar] [CrossRef]
- Xu, N.; He, Y.; Wang, M.; Cheng, C.; Cheng, Q.; Liu, S.; Ji, H.; Yan, C.; Rosei, F. Breaking scaling relations in nitrogen reduction with asymmetrical heterobimetallic FeCo sites to boost ammonia synthesis. Mater. Chem. Front. 2024, 8, 851–858. [Google Scholar] [CrossRef]
- Zhang, Z.; Xu, X. g-C3N4-Supported Metal-Pair Catalysts toward Efficient Electrocatalytic Nitrogen Reduction: A Computational Evaluation. Adv. Theory Simul. 2022, 5, 2100579. [Google Scholar] [CrossRef]
- Chen, Z.W.; Yan, J.M.; Jiang, Q. Single or Double: Which Is the Altar of Atomic Catalysts for Nitrogen Reduction Reaction? Small Methods 2019, 3, 1800291. [Google Scholar] [CrossRef]
- Sun, C.N.; Wang, Z.L.; Lang, X.Y.; Wen, Z.; Jiang, Q. Synergistic Effect of Active Sites of Double-Atom Catalysts for Nitrogen Reduction Reaction. ChemSusChem 2021, 14, 4593–4600. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, J.; Li, H.; Li, Y.; Li, J.; Wei, K.; Peng, F.; Gao, F. Nitrogen Reduction Reaction: Heteronuclear Double-Atom Electrocatalysts. Small Struct. 2023, 4, 2200306. [Google Scholar] [CrossRef]
- Han, B.; Meng, H.; Li, F.; Zhao, J. Fe3 Cluster Anchored on the C2N Monolayer for Efficient Electrochemical Nitrogen Fixation. Catalysts 2020, 10, 974. [Google Scholar] [CrossRef]
- Pei, W.; Hou, L.; Yu, X.; Xia, W.; Wang, Z.; Liu, Y.; Zhou, S.; Tu, Y.; Zhao, J. Graphitic carbon nitride supported trimeric metal clusters as electrocatalysts for N2 reduction reaction. J. Catal. 2024, 429, 115232. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, M.; Wang, Z.; Jiang, Q. Mo Cluster Support on C2N as a Highly-efficient Catalyst for Electrocatalytic Nitrogen Reduction Reaction. ChemPhysChem 2023, 24, e202300012. [Google Scholar] [CrossRef]
- Wang, X.; Qiu, S.; Feng, J.; Tong, Y.; Zhou, F.; Li, Q.; Song, L.; Chen, S.; Wu, K.H.; Su, P.; et al. Confined Fe–Cu Clusters as Sub-Nanometer Reactors for Efficiently Regulating the Electrochemical Nitrogen Reduction Reaction. Adv. Mater. 2020, 32, 2004382. [Google Scholar] [CrossRef]
- Nong, W.; Qin, S.; Huang, F.; Liang, H.; Yang, Z.; Qi, C.; Li, Y.; Wang, C. Designing C3N-supported single atom catalysts for efficient nitrogen reduction based on descriptor of catalytic activity. Carbon 2021, 182, 297–306. [Google Scholar] [CrossRef]
- Yang, H.; Liu, Y.; Luo, Y.; Lu, S.; Su, B.; Ma, J. Achieving High Activity and Selectivity of Nitrogen Reduction via Fe–N3 Coordination on Iron Single-Atom Electrocatalysts at Ambient Conditions. ACS Sustain. Chem. Eng. 2020, 8, 12809–12816. [Google Scholar] [CrossRef]
- Kong, Y.; Li, Y.; Sang, X.; Yang, B.; Li, Z.; Zheng, S.; Zhang, Q.; Yao, S.; Yang, X.; Lei, L.; et al. Atomically Dispersed Zinc(I) Active Sites to Accelerate Nitrogen Reduction Kinetics for Ammonia Electrosynthesis. Adv. Mater. 2022, 34, e2103548. [Google Scholar] [CrossRef]
- Li, L.; Yu, W.; Gong, W.; Wang, H.; Chiang, C.-L.; Lin, Y.; Zhao, J.; Zhang, L.; Lee, J.-M.; Zou, G. Sulfur-induced electron redistribution of single molybdenum atoms promotes nitrogen electroreduction to ammonia. Appl. Catal. B 2023, 321, 122038. [Google Scholar] [CrossRef]
- Li, Y.; Ji, Y.; Zhao, Y.; Chen, J.; Zheng, S.; Sang, X.; Yang, B.; Li, Z.; Lei, L.; Wen, Z.; et al. Local Spin-State Tuning of Iron Single-Atom Electrocatalyst by S-Coordinated Doping for Kinetics-Boosted Ammonia Synthesis. Adv. Mater. 2022, 34, e2202240. [Google Scholar] [CrossRef]
- Li, Y.; Li, J.; Huang, J.; Chen, J.; Kong, Y.; Yang, B.; Li, Z.; Lei, L.; Chai, G.; Wen, Z.; et al. Boosting Electroreduction Kinetics of Nitrogen to Ammonia via Tuning Electron Distribution of Single-Atomic Iron Sites. Angew. Chem. Int. Ed. Engl. 2021, 60, 9078–9085. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.; Ma, N.; Li, Y.; Liang, B.; Luo, S.; Fan, J. Establishing an orbital-level understanding of active origins of heteroatom-coordinated single-atom catalysts: The case of N2 reduction. J. Colloid Interface Sci. 2023, 650, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, S.; Han, C.; Lu, Q.; Fu, Q.; Jiang, H.; Yang, L.; Xing, Y.; Zheng, Q.; Shen, J.; et al. Effect of boron nitrogen modulation in metal atoms anchoring on flower-like carbon superstructure for efficient ammonia electrosynthesis. Chem. Eng. J. 2023, 468, 143517. [Google Scholar] [CrossRef]
- Gu, Y.; Xi, B.; Tian, W.; Zhang, H.; Fu, Q.; Xiong, S. Boosting Selective Nitrogen Reduction via Geometric Coordination Engineering on Single-Tungsten-Atom Catalysts. Adv. Mater. 2021, 33, e2100429. [Google Scholar] [CrossRef]
- Han, L.; Hou, M.; Ou, P.; Cheng, H.; Ren, Z.; Liang, Z.; Boscoboinik, J.A.; Hunt, A.; Waluyo, I.; Zhang, S.; et al. Local Modulation of Single-Atomic Mn Sites for Enhanced Ambient Ammonia Electrosynthesis. ACS Catal. 2021, 11, 509–516. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, W.; Sun, C. Synergetic effect between non-metals and dual metal catalysts for nitrogen reduction reaction. Inorg. Chem. Front. 2023, 10, 4746–4753. [Google Scholar] [CrossRef]
- Shi, L.; Bi, S.; Qi, Y.; He, R.; Ren, K.; Zheng, L.; Wang, J.; Ning, G.; Ye, J. Anchoring Mo Single-Atom Sites on B/N Codoped Porous Carbon Nanotubes for Electrochemical Reduction of N2 to NH3. ACS Catal. 2022, 12, 7655–7663. [Google Scholar] [CrossRef]
- Wang, X.; Yang, L.-M. Efficient modulation of the catalytic performance of electrocatalytic nitrogen reduction with transition metals anchored on N/O-codoped graphene by coordination engineering. J. Mater. Chem. A 2022, 10, 1481–1496. [Google Scholar] [CrossRef]
- Ma, Z.; Lv, P.; Wu, D.; Li, X.; Chu, K.; Ma, D.; Jia, Y. V (Nb) Single Atoms Anchored by the Edge of a Graphene Armchair Nanoribbon for Efficient Electrocatalytic Nitrogen Reduction: A Theoretical Study. Inorg. Chem. 2022, 61, 17864–17872. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, H.; Chen, X. Inspired by nitrogenase: Who is the better electron donor for FeMo active site in nitrogen reduction reaction? Mater. Today Energy 2025, 49, 101839. [Google Scholar] [CrossRef]
- Li, X.; Zhou, Q.; Wang, S.; Li, Y.; Liu, Y.; Gao, Q.; Wu, Q. Tuning the Coordination Environment to Effect the Electrocatalytic Behavior of a Single-Atom Catalyst toward the Nitrogen Reduction Reaction. J. Phys. Chem. C 2021, 125, 11963–11974. [Google Scholar] [CrossRef]
- Liu, X.; Jiao, Y.; Zheng, Y.; Jaroniec, M.; Qiao, S.Z. Building Up a Picture of the Electrocatalytic Nitrogen Reduction Activity of Transition Metal Single-Atom Catalysts. J. Am. Chem. Soc. 2019, 141, 9664–9672. [Google Scholar] [CrossRef]
- Yuan, S.; Meng, G.; Liu, D.; Zhao, W.; Zhu, H.; Chi, Y.; Ren, H.; Guo, W. Synergy of Substrate Chemical Environments and Single-Atom Catalysts Promotes Catalytic Performance: Nitrogen Reduction on Chiral and Defected Carbon Nanotubes. ACS Appl. Mater. Interfaces 2022, 14, 52544–52552. [Google Scholar] [CrossRef]
- Kong, Y.; Wu, L.; Yang, X.; Li, Y.; Zheng, S.; Yang, B.; Li, Z.; Zhang, Q.; Zhou, S.; Lei, L.; et al. Accelerating Protonation Kinetics for Ammonia Electrosynthesis on Single Iron Sites Embedded in Carbon with Intrinsic Defects. Adv. Funct. Mater. 2022, 32, 2205409. [Google Scholar] [CrossRef]
- Chen, Z.W.; Chen, L.X.; Wen, Z.; Jiang, Q. Understanding electro-catalysis by using density functional theory. Phys. Chem. Chem. Phys. 2019, 21, 23782–23802. [Google Scholar] [CrossRef]
- Chen, H.; Yang, Y.; Jiao, C.; Zhuo, Z.; Mao, J.; Liu, Y. Theoretical screening of cooperative N-bridged dual-atom sites for efficient electrocatalytic nitrogen reduction with remolding insight. Nano Res. 2024, 17, 3413–3422. [Google Scholar] [CrossRef]
- Li, S.; Zhang, H.; Holby, E.F.; Zelenay, P.; Kort-Kamp, W.J.M. Machine learning-guided design, synthesis, and characterization of atomically dispersed electrocatalysts. Curr. Opin. Electrochem. 2024, 48, 101578. [Google Scholar] [CrossRef]
- Ling, C.; Ouyang, Y.; Li, Q.; Bai, X.; Mao, X.; Du, A.; Wang, J. A General Two-Step Strategy–Based High-Throughput Screening of Single Atom Catalysts for Nitrogen Fixation. Small Methods 2018, 3, 1800376. [Google Scholar] [CrossRef]
- Mukherjee, M.; Dutta, S.; Ghosh, M.; Basuchowdhuri, P.; Datta, A. Performance of the nitrogen reduction reaction on metal bound g-C6N6: A combined approach of machine learning and DFT. Phys. Chem. Chem. Phys. 2022, 24, 17050–17058. [Google Scholar] [CrossRef]
- Wang, S.; Qian, C.; Zhou, S. Machine Learning Design of Single-Atom Catalysts for Nitrogen Fixation. ACS Appl. Mater. Interfaces 2023, 15, 40656–40664. [Google Scholar] [CrossRef]
- Xu, L.; Huang, Y.; Lin, H.; Du, R.; Wang, M.; Ma, F.; Wei, X.; Zhu, G.; Zhang, J. High-throughput screening of carbon nitride single-atom catalysts for nitrogen fixation based on machine learning. J. Mater. Chem. A 2024, 12, 33053–33065. [Google Scholar] [CrossRef]
- Liu, H.; Li, J.; Arbiol, J.; Yang, B.; Tang, P. Catalytic reactivity descriptors of metal–nitrogen–doped carbon catalysts for electrocatalysis. EcoEnergy 2023, 1, 154–185. [Google Scholar] [CrossRef]
- Choi, J.; Suryanto, B.H.R.; Wang, D.; Du, H.-L.; Hodgetts, R.Y.; Ferrero Vallana, F.M.; MacFarlane, D.R.; Simonov, A.N. Identification and elimination of false positives in electrochemical nitrogen reduction studies. Nat. Commun. 2020, 11, 5546. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Qiao, S.-Z. How to explore ambient electrocatalytic nitrogen reduction reliably and insightfully. Chem. Soc. Rev. 2019, 48, 3166–3180. [Google Scholar] [CrossRef]
- Andersen, S.Z.; Čolić, V.; Yang, S.; Schwalbe, J.A.; Nielander, A.C.; McEnaney, J.M.; Enemark-Rasmussen, K.; Baker, J.G.; Singh, A.R.; Rohr, B.A.; et al. A rigorous electrochemical ammonia synthesis protocol with quantitative isotope measurements. Nature 2019, 570, 504–508. [Google Scholar] [CrossRef]
- Kolen, M.; Ripepi, D.; Smith, W.A.; Burdyny, T.; Mulder, F.M. Overcoming Nitrogen Reduction to Ammonia Detection Challenges: The Case for Leapfrogging to Gas Diffusion Electrode Platforms. ACS Catal. 2022, 12, 5726–5735. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Lu, S.; Zhang, P.; Tian, J.; Shi, L.; Ling, C.; Zhou, Q.; Wang, J. Accelerated Discovery of Single-Atom Catalysts for Nitrogen Fixation via Machine Learning. Energy Environ. Mater. 2023, 6, e12304. [Google Scholar] [CrossRef]
- Lv, X.; Wei, W.; Huang, B.; Dai, Y.; Frauenheim, T. High-Throughput Screening of Synergistic Transition Metal Dual-Atom Catalysts for Efficient Nitrogen Fixation. Nano Lett. 2021, 21, 1871–1878. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Ma, N.; Fan, J. Directly predicting N2 electroreduction reaction free energy using interpretable machine learning with non-DFT calculated features. J. Energy Chem. 2024, 97, 139–148. [Google Scholar] [CrossRef]
- He, C.; Chen, D.; Zhang, W.X. Machine learning-driven shortening the screening process towards high-performance nitrogen reduction reaction electrocatalysts with four-step screening strategy. J. Colloid Interface Sci. 2024, 676, 22–32. [Google Scholar] [CrossRef]
- Singh, A.N.; Anand, R.; Zafari, M.; Ha, M.; Kim, K.S. Progress in Single/Multi Atoms and 2D-Nanomaterials for Electro/Photocatalytic Nitrogen Reduction: Experimental, Computational and Machine Learning Developments. Adv. Energy Mater. 2024, 14, 2304106. [Google Scholar] [CrossRef]
- Ouyang, R.; Curtarolo, S.; Ahmetcik, E.; Scheffler, M.; Ghiringhelli, L.M. SISSO: A compressed-sensing method for identifying the best low-dimensional descriptor in an immensity of offered candidates. Phys. Rev. Mater. 2018, 2, 083802. [Google Scholar] [CrossRef]
- Purcell, T.A.R.; Scheffler, M.; Ghiringhelli, L.M. Recent advances in the SISSO method and their implementation in the SISSO++ code. J. Chem. Phys. 2023, 159, 114110. [Google Scholar] [CrossRef]
- Zhao, W.; Chen, L.; Zhang, W.; Yang, J. Single Mo1(W1, Re1) atoms anchored in pyrrolic-N3 doped graphene as efficient electrocatalysts for the nitrogen reduction reaction. J. Mater. Chem. A 2021, 9, 6547–6554. [Google Scholar] [CrossRef]
- Lin, X.; Wang, Y.; Chang, X.; Zhen, S.; Zhao, Z.J.; Gong, J. High-Throughput Screening of Electrocatalysts for Nitrogen Reduction Reactions Accelerated by Interpretable Intrinsic Descriptor. Angew. Chem. Int. Ed. Engl. 2023, 62, e202300122. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, J.A.; Ringe, S.; Dickens, C.F.; Garza, A.J.; Bell, A.T.; Head-Gordon, M.; Nørskov, J.K.; Chan, K. Challenges in Modeling Electrochemical Reaction Energetics with Polarizable Continuum Models. ACS Catal. 2019, 9, 920–931. [Google Scholar] [CrossRef]
- Unke, O.T.; Chmiela, S.; Sauceda, H.E.; Gastegger, M.; Poltavsky, I.; Schütt, K.T.; Tkatchenko, A.; Müller, K.-R. Machine Learning Force Fields. Chem. Rev. 2021, 121, 10142–10186. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.; Pérez-Ramírez, J. Atomically precise control in the design of low-nuclearity supported metal catalysts. Nat. Rev. Mater. 2021, 6, 969–985. [Google Scholar] [CrossRef]
- Lin, X.; Du, X.; Wu, S.; Zhen, S.; Liu, W.; Pei, C.; Zhang, P.; Zhao, Z.-J.; Gong, J. Machine learning-assisted dual-atom sites design with interpretable descriptors unifying electrocatalytic reactions. Nat. Commun. 2024, 15, 8169. [Google Scholar] [CrossRef]
- Xu, H.; Cheng, D.; Cao, D.; Zeng, X.C. Revisiting the universal principle for the rational design of single-atom electrocatalysts. Nat. Catal. 2024, 7, 207–218. [Google Scholar] [CrossRef]
- Liu, X.; Qi, L.; Song, E.; Gao, W. Effective Descriptor for Nitrogen Reduction on Atomic Catalysts. Catal. Lett. 2022, 153, 300–310. [Google Scholar] [CrossRef]
- Kim, M.; Yeo, B.C.; Park, Y.; Lee, H.M.; Han, S.S.; Kim, D. Artificial Intelligence to Accelerate the Discovery of N2 Electroreduction Catalysts. Chem. Mater. 2020, 32, 709–720. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Yi, J.; Wen, Z.; Jiang, Q. Theoretical Insights and Design Strategies of Metal–Nitrogen–Carbon Catalysts for Electrochemical Nitrogen Reduction Reaction. Catalysts 2026, 16, 456. https://doi.org/10.3390/catal16050456
Yi J, Wen Z, Jiang Q. Theoretical Insights and Design Strategies of Metal–Nitrogen–Carbon Catalysts for Electrochemical Nitrogen Reduction Reaction. Catalysts. 2026; 16(5):456. https://doi.org/10.3390/catal16050456
Chicago/Turabian StyleYi, Jianhui, Zi Wen, and Qing Jiang. 2026. "Theoretical Insights and Design Strategies of Metal–Nitrogen–Carbon Catalysts for Electrochemical Nitrogen Reduction Reaction" Catalysts 16, no. 5: 456. https://doi.org/10.3390/catal16050456
APA StyleYi, J., Wen, Z., & Jiang, Q. (2026). Theoretical Insights and Design Strategies of Metal–Nitrogen–Carbon Catalysts for Electrochemical Nitrogen Reduction Reaction. Catalysts, 16(5), 456. https://doi.org/10.3390/catal16050456