
On the Reactivity Descriptors of Low-Coordinated Atoms on Foreign Solid Substrates as Models of Single-Atom Catalysts

 , ,
, , 
Abstract

1. Introduction
2. Results and Discussion
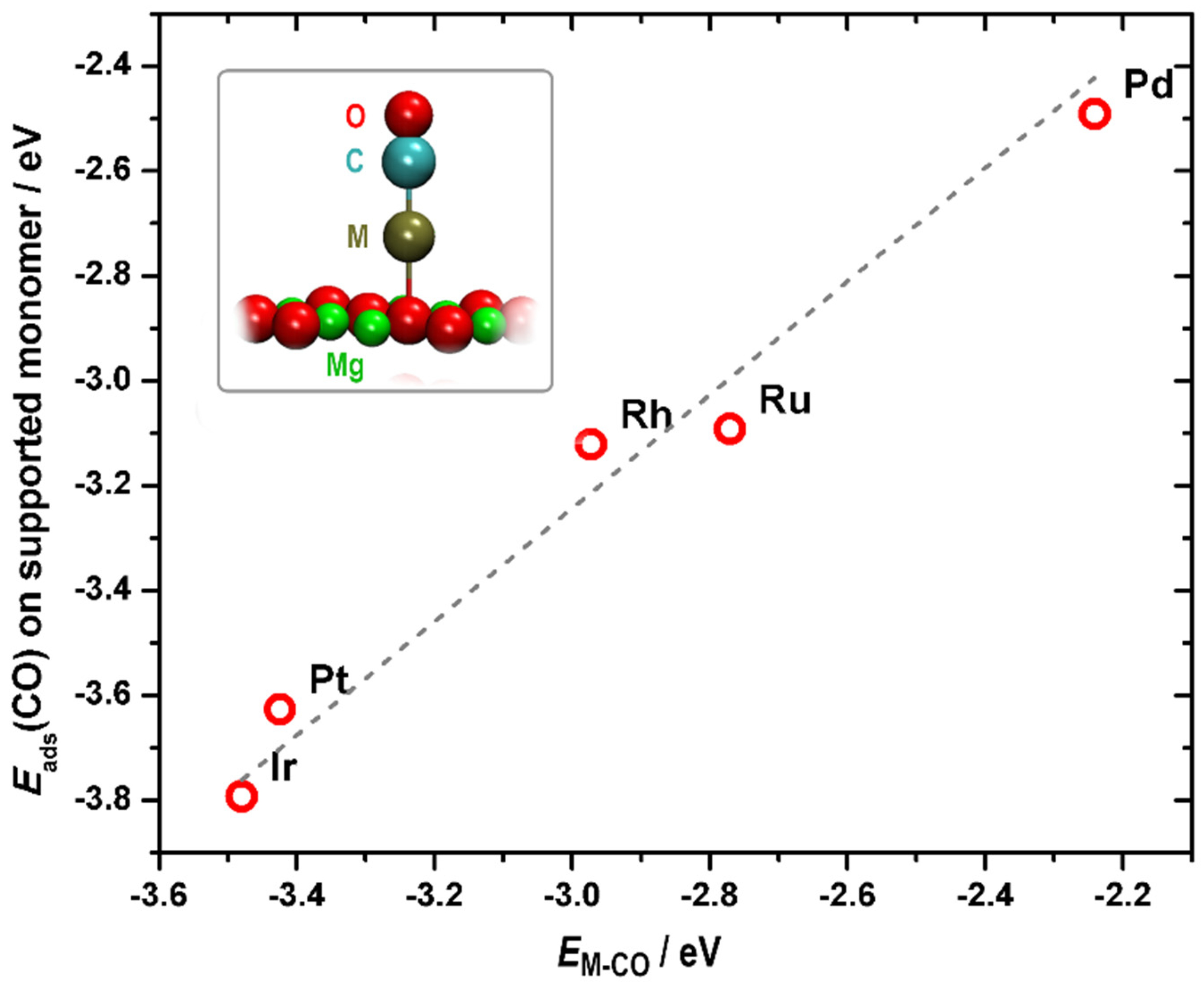
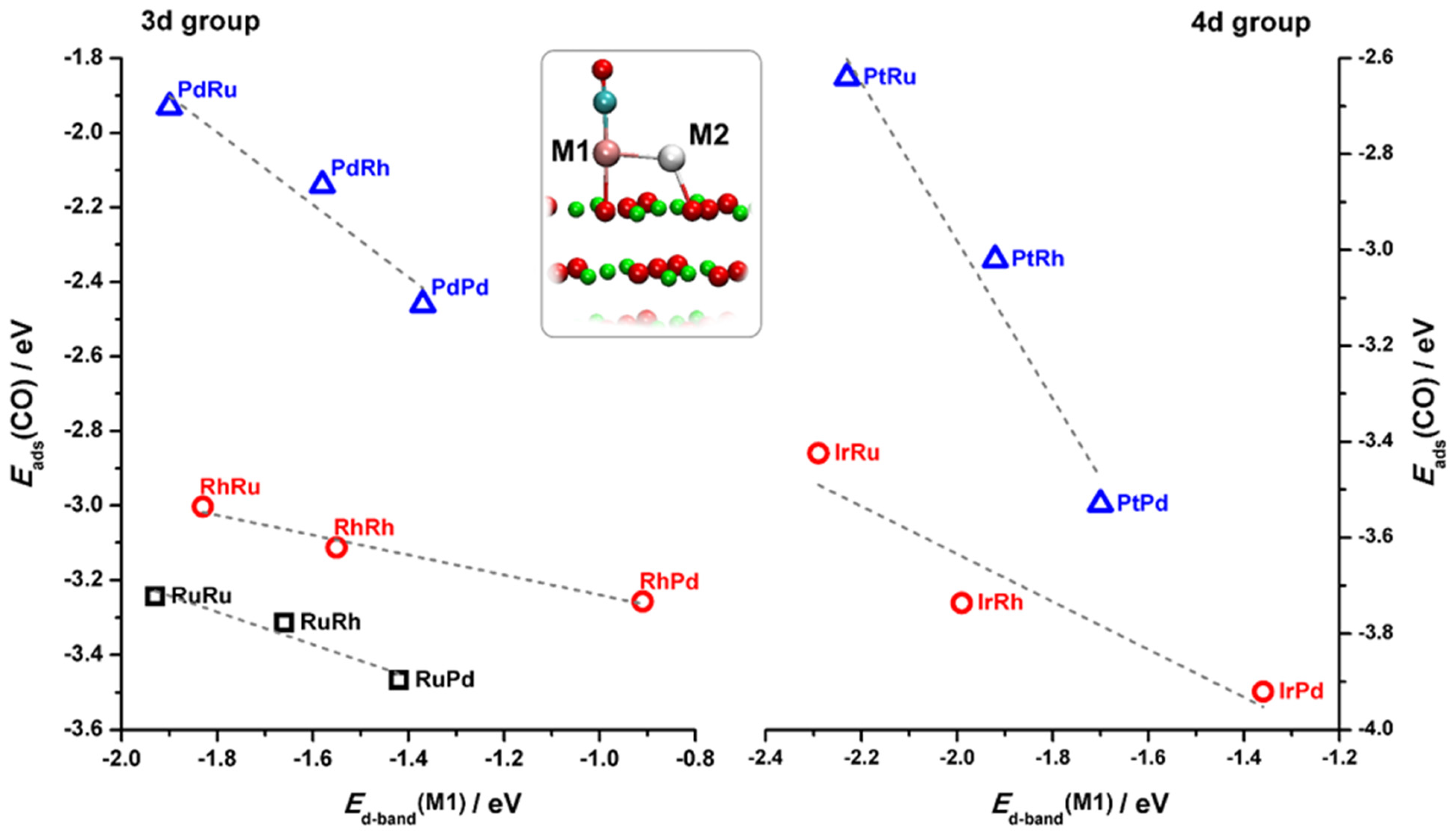
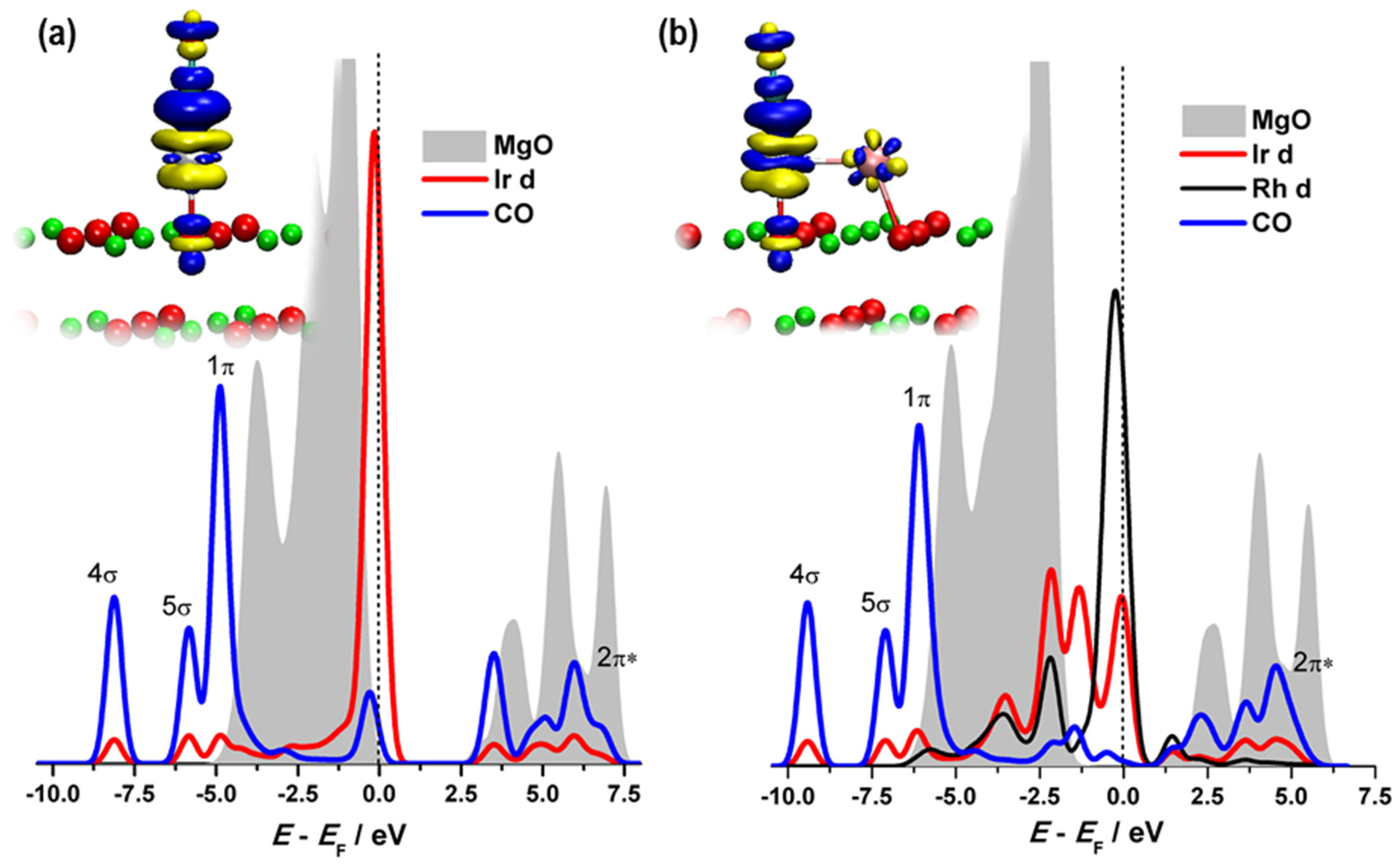
2.1. Reactivity of Metal Atoms and Dimers on MgO(001)
2.2. Tuning the Reactivity by Charging Metal Atoms—Cu Adatom on Mo-Supported NaF, MgO, and ScN Thin Films
2.3. CO Adsorption on Graphene and rGO-Supported Pt Monomers
3. Materials and Methods
3.1. MgO(001) Supported Metal Atoms and Bimetallic Dimers
3.2. Cu Adatom on Mo-Supported Thin Films
3.3. Single Pt Atoms on Oxygen-Functionalized Graphene
3.4. Interactions Quantified
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, J. Catalysis by Supported Single Metal Atoms. ACS Catal. 2017, 7, 34–59. [Google Scholar] [CrossRef]
- Roduner, E. Size Matters: Why Nanomaterials Are Different. Chem. Soc. Rev. 2006, 35, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Tauster, S.J.; Fung, S.C.; Baker, R.T.; Horsley, J.A. Strong Interactions in Supported-Metal Catalysts. Science 1981, 211, 1121–1125. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.T. Ultrathin Metal Films and Particles on Oxide Surfaces: Structural, Electronic and Chemisorptive Properties. Surf. Sci. Rep. 1997, 27, 1–111. [Google Scholar] [CrossRef]
- Gates, B.C.; Flytzani-Stephanopoulos, M.; Dixon, D.A.; Katz, A. Atomically Dispersed Supported Metal Catalysts: Perspectives and Suggestions for Future Research. Catal. Sci. Technol. 2017, 7, 4259–4275. [Google Scholar] [CrossRef]
- Copéret, C.; Allouche, F.; Chan, K.W.; Conley, M.P.; Delley, M.F.; Fedorov, A.; Moroz, I.B.; Mougel, V.; Pucino, M.; Searles, K.; et al. Bridging the Gap between Industrial and Well-Defined Supported Catalysts. Angew. Chem. Int. Ed. 2018, 57, 6398–6440. [Google Scholar] [CrossRef] [PubMed]
- Bordiga, S.; Damin, A.; Bonino, F.; Lamberti, C. Single Site Catalyst for Partial Oxidation Reaction: TS-1 Case Study. In Surface and Interfacial Organometallic Chemistry and Catalysis; Springer: Berlin/Heidelberg, Germany, 2005; pp. 37–68. [Google Scholar]
- Li, X.; Yang, X.; Huang, Y.; Zhang, T.; Liu, B. Supported Noble-Metal Single Atoms for Heterogeneous Catalysis. Adv. Mater. 2019, 31, 1902031. [Google Scholar] [CrossRef]
- Wang, S.; Wang, L.; Wang, D.; Li, Y. Recent Advances of Single-Atom Catalysts in CO2 Conversion. Energy Environ. Sci. 2023, 16, 2759–2803. [Google Scholar] [CrossRef]
- Liang, S.; Hao, C.; Shi, Y. The Power of Single-Atom Catalysis. ChemCatChem 2015, 7, 2559–2567. [Google Scholar] [CrossRef]
- Dobrota, A.S.; Pašti, I.A. Single-Atom Catalysts: Are You Really Single? J. Phys. Chem. Lett. 2024, 16, 77–86. [Google Scholar] [CrossRef]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-Atom Catalysis of CO Oxidation Using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef]
- Muratsugu, S.; Weng, Z.; Nakai, H.; Isobe, K.; Kushida, Y.; Sasaki, T.; Tada, M. Surface-Assisted Transfer Hydrogenation Catalysis on a γ-Al2O3-Supported Ir Dimer. Phys. Chem. Chem. Phys. 2012, 14, 16023. [Google Scholar] [CrossRef] [PubMed]
- Yardimci, D.; Serna, P.; Gates, B.C. Surface-Mediated Synthesis of Dimeric Rhodium Catalysts on MgO: Tracking Changes in the Nuclearity and Ligand Environment of the Catalytically Active Sites by X-Ray Absorption and Infrared Spectroscopies. Chem.—A Eur. J. 2013, 19, 1235–1245. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhang, G.; Gauquelin, N.; Chen, N.; Zhou, J.; Yang, S.; Chen, W.; Meng, X.; Geng, D.; Banis, M.N.; et al. Single-Atom Catalysis Using Pt/Graphene Achieved through Atomic Layer Deposition. Sci. Rep. 2013, 3, 1775. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Abild-Pedersen, F.; Studt, F.; Bligaard, T. Density Functional Theory in Surface Chemistry and Catalysis. Proc. Natl. Acad. Sci. USA 2011, 108, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Stamenkovic, V.R.; Fowler, B.; Mun, B.S.; Wang, G.; Ross, P.N.; Lucas, C.A.; Marković, N.M. Improved Oxygen Reduction Activity on Pt3Ni(111) via Increased Surface Site Availability. Science 2007, 315, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Kitchin, J.R.; Nørskov, J.K.; Barteau, M.A.; Chen, J.G. Role of Strain and Ligand Effects in the Modification of the Electronic and Chemical Properties of Bimetallic Surfaces. Phys. Rev. Lett. 2004, 93, 156801. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Waghmare, U.V.; Lee, S.-C. An Improved D-Band Model of the Catalytic Activity of Magnetic Transition Metal Surfaces. Sci. Rep. 2016, 6, 35916. [Google Scholar] [CrossRef]
- Peterson, A.A.; Nørskov, J.K. Activity Descriptors for CO 2 Electroreduction to Methane on Transition-Metal Catalysts. J. Phys. Chem. Lett. 2012, 3, 251–258. [Google Scholar] [CrossRef]
- Subbaraman, R.; Tripkovic, D.; Chang, K.-C.; Strmcnik, D.; Paulikas, A.P.; Hirunsit, P.; Chan, M.; Greeley, J.; Stamenkovic, V.; Markovic, N.M. Trends in Activity for the Water Electrolyser Reactions on 3d M(Ni,Co,Fe,Mn) Hydr(Oxy)Oxide Catalysts. Nat. Mater. 2012, 11, 550–557. [Google Scholar] [CrossRef]
- Calle-Vallejo, F.; Inoglu, N.G.; Su, H.-Y.; Martínez, J.I.; Man, I.C.; Koper, M.T.M.; Kitchin, J.R.; Rossmeisl, J. Number of Outer Electrons as Descriptor for Adsorption Processes on Transition Metals and Their Oxides. Chem. Sci. 2013, 4, 1245. [Google Scholar] [CrossRef]
- Fu, Q.; Saltsburg, H.; Flytzani-Stephanopoulos, M. Active Nonmetallic Au and Pt Species on Ceria-Based Water-Gas Shift Catalysts. Science 2003, 301, 935–938. [Google Scholar] [CrossRef]
- Sauer, J.; Döbler, J. Structure and Reactivity of V2O5: Bulk Solid, Nanosized Clusters, Species Supported on Silica and Alumina, Cluster Cations and Anions. Dalton Trans. 2004, 19, 3116–3121. [Google Scholar] [CrossRef]
- Yoon, B.; Häkkinen, H.; Landman, U.; Wörz, A.S.; Antonietti, J.M.; Abbet, S.; Judai, K.; Heiz, U. Charging Effects on Bonding and Catalyzed Oxidation of CO on Au8 Clusters on MgO. Science 2005, 307, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Zhai, H.J.; Döbler, J.; Sauer, J.; Wang, L.S. Probing the Electronic Structure of Early Transition-Metal Oxide Clusters: Polyhedral Cages of (V2O5)n− (n = 2–4) and (M2O5)2− (M = Nb, Ta). J. Am. Chem. Soc. 2007, 129, 13270–13276. [Google Scholar] [CrossRef]
- Molina, L.M.; Hammer, B. Some Recent Theoretical Advances in the Understanding of the Catalytic Activity of Au. Appl. Catal. A Gen. 2005, 291, 21–31. [Google Scholar] [CrossRef]
- Lin, X.; Yang, B.; Benia, H.-M.; Myrach, P.; Yulikov, M.; Aumer, A.; Brown, M.A.; Sterrer, M.; Bondarchuk, O.; Kieseritzky, E.; et al. Charge-Mediated Adsorption Behavior of CO on MgO-Supported Au Clusters. J. Am. Chem. Soc. 2010, 132, 7745–7749. [Google Scholar] [CrossRef]
- Pašti, I.A.; Baljozović, M.R.; Granda-Marulanda, L.P.; Skorodumova, N.V. Bimetallic Dimers Adsorbed on a Defect-Free MgO(001) Surface: Bonding, Structure and Reactivity. Phys. Chem. Chem. Phys. 2015, 17, 9666–9679. [Google Scholar] [CrossRef]
- Vorobyeva, E.; Chen, Z.; Mitchell, S.; Leary, R.K.; Midgley, P.; Thomas, J.M.; Hauert, R.; Fako, E.; López, N.; Pérez-Ramírez, J. Tailoring the Framework Composition of Carbon Nitride to Improve the Catalytic Efficiency of the Stabilised Palladium Atoms. J. Mater. Chem. A Mater. 2017, 5, 16393–16403. [Google Scholar] [CrossRef]
- Amft, M.; Skorodumova, N.V. Catalytic Activity of Small MgO-Supported Au Clusters towards CO Oxidation: A Density Functional Study. Phys. Rev. B Condens. Matter Mater. Phys. 2010, 81, 195443. [Google Scholar] [CrossRef]
- Frusteri, F.; Freni, S.; Spadaro, L.; Chiodo, V.; Bonura, G.; Donato, S.; Cavallaro, S. H2production for MC Fuel Cell by Steam Reforming of Ethanol over MgO Supported Pd, Rh, Ni and Co Catalysts. Catal. Commun. 2004, 5, 611–615. [Google Scholar] [CrossRef]
- Frusteri, F.; Freni, S.; Chiodo, V.; Spadaro, L.; Di Blasi, O.; Bonura, G.; Cavallaro, S. Steam Reforming of Bio-Ethanol on Alkali-Doped Ni/MgO Catalysts: Hydrogen Production for MC Fuel Cell. Appl. Catal. A Gen. 2004, 270, 1–7. [Google Scholar] [CrossRef]
- Ruban, A.; Hammer, B.; Stoltze, P.; Skriver, H.L.; Nørskov, J.K. Surface Electronic Structure and Reactivity of Transition and Noble Metals. J. Mol. Catal. A Chem. 1997, 115, 421–429. [Google Scholar] [CrossRef]
- Hammer, B.; Morikawa, Y.; Nørskov, J.K. CO Chemisorption at Metal Surfaces and Overlayers. Phys. Rev. Lett. 1996, 76, 2141–2144. [Google Scholar] [CrossRef]
- Molina, L.M.; Hammer, B. Active Role of Oxide Support during CO Oxidation at [Formula Presented]. Phys. Rev. Lett. 2003, 90, 4. [Google Scholar] [CrossRef]
- Molina, L.M.; Hammer, B. Theoretical Study of CO Oxidation on Au Nanoparticles Supported by MgO(100). Phys. Rev. B 2004, 69, 155424. [Google Scholar] [CrossRef]
- Pašti, I.A.; Skorodumova, N.V.; Mentus, S.V. Theoretical Studies in Catalysis and Electrocatalysis: From Fundamental Knowledge to Catalyst Design. React. Kinet. Mech. Catal. 2015, 115, 5–32. [Google Scholar] [CrossRef]
- Hammer, B.; Nørskov, J.K. Electronic Factors Determining the Reactivity of Metal Surfaces. Surf. Sci. 1995, 343, 211–220. [Google Scholar] [CrossRef]
- Mavrikakis, M.; Hammer, B.; Nørskov, J.K. Effect of Strain on the Reactivity of Metal Surfaces. Phys. Rev. Lett. 1998, 81, 2819–2822. [Google Scholar] [CrossRef]
- Pašti, I.; Mentus, S. DFT Study of Adsorption of Hydrogen and Carbon Monoxide on Pt XBi1-x/Pt(111) Bimetallic Overlayers: Correlation to Surface Electronic Properties. Phys. Chem. Chem. Phys. 2009, 11, 6225–6233. [Google Scholar] [CrossRef] [PubMed]
- Blyholder, G. Molecular Orbital View of Chemisorbed Carbon Monoxide. J. Phys. Chem. 1964, 68, 2772–2777. [Google Scholar] [CrossRef]
- Wu, X.; Senapati, L.; Nayak, S.K.; Selloni, A.; Hajaligol, M. A Density Functional Study of Carbon Monoxide Adsorption on Small Cationic, Neutral, and Anionic Gold Clusters. J. Chem. Phys. 2002, 117, 4010–4015. [Google Scholar] [CrossRef]
- Gan, L.Y.; Zhao, Y.J. Charge Effect in S Enhanced CO Adsorption: A Theoretical Study of CO on Au, Ag, Cu, and Pd (111) Surfaces Coadsorbed with S, O, Cl, and Na. J. Chem. Phys. 2010, 133, 094703. [Google Scholar] [CrossRef] [PubMed]
- Žguns, P.A.; Wessel, M.; Skorodumova, N.V. Cu Adatom Charging on Mo Supported ScN, MgO and NaF. RSC Adv. 2015, 5, 94436–94445. [Google Scholar] [CrossRef]
- Vasić, D.; Ristanović, Z.; Pašti, I.; Mentus, S. Systematic DFT-GGA Study of Hydrogen Adsorption on Transition Metals. Russ. J. Phys. Chem. A 2011, 85, 2373–2379. [Google Scholar] [CrossRef]
- Gajdoš, M.; Hafner, J. CO Adsorption on Cu(111) and Cu(001) Surfaces: Improving Site Preference in DFT Calculations. Surf. Sci. 2005, 590, 117–126. [Google Scholar] [CrossRef]
- Tang, Y.; Dai, X.; Yang, Z.; Pan, L.; Chen, W.; Ma, D.; Lu, Z. Formation and Catalytic Activity of Pt Supported on Oxidized Graphene for the CO Oxidation Reaction. Phys. Chem. Chem. Phys. 2014, 16, 7887–7895. [Google Scholar] [CrossRef]
- Dobrota, A.S.; Pašti, I.A.; Skorodumova, N.V. Oxidized Graphene as an Electrode Material for Rechargeable Metal-Ion Batteries—A DFT Point of View. Electrochim. Acta 2015, 176, 1092–1099. [Google Scholar] [CrossRef]
- Dobrota, A.S.; Pašti, I.A.; Mentus, S.V.; Skorodumova, N.V. A General View on the Reactivity of the Oxygen-Functionalized Graphene Basal Plane. Phys. Chem. Chem. Phys. 2016, 18, 6580–6586. [Google Scholar] [CrossRef]
- Pašti, I.A.; Gavrilov, N.M.; Mentus, S.V. Hydrogen Adsorption on Palladium and Platinum Overlayers: DFT Study. Adv. Phys. Chem. 2011, 2011, 305634. [Google Scholar] [CrossRef]
- Liu, S.; Huang, S. Theoretical Insights into the Activation of O2 by Pt Single Atom and Pt4 Nanocluster on Functionalized Graphene Support: Critical Role of Pt Positive Polarized Charges. Carbon 2017, 115, 11–17. [Google Scholar] [CrossRef]
- Ritopečki, M.S.; Dobrota, A.S.; Skorodumova, N.V.; Pašti, I.A. The Local Coordination Effects on the Reactivity and Speciation of Active Sites in Graphene-Embedded Single-Atom Catalysts over Wide PH and Potential Range. Nanomaterials 2022, 12, 4309. [Google Scholar] [CrossRef] [PubMed]
- Pašti, I.A.; Jovanović, A.; Dobrota, A.S.; Mentus, S.V.; Johansson, B.; Skorodumova, N.V. Atomic Adsorption on Graphene with a Single Vacancy: Systematic DFT Study through the Periodic Table of Elements. Phys. Chem. Chem. Phys. 2018, 20, 858–865. [Google Scholar] [CrossRef]
- Dobrota, A.S.; Skorodumova, N.V.; Mentus, S.V.; Pašti, I.A. Surface Pourbaix Plots of M@N4-Graphene Single-Atom Electrocatalysts from Density Functional Theory Thermodynamic Modeling. Electrochim. Acta 2022, 412, 140155. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A Modular and Open-Source Software Project for Quantum Simulations of Materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Buongiorno Nardelli, M.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced Capabilities for Materials Modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef]
- Wyckoff, R.W.G. Interscience Publishers, New York, New York Rocksalt Structure. In Crystal Structures; Interscience Publishers: New York, NY, USA, 1963; Volume 1, pp. 85–237. [Google Scholar]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Bengtsson, L. Dipole Correction for Surface Supercell Calculations. Phys. Rev. B Condens. Matter Mater. Phys. 1999, 59, 12301–12304. [Google Scholar] [CrossRef]
- Pašti, I.A.; Baljozović, M.; Skorodumova, N.V. Adsorption of Nonmetallic Elements on Defect-Free MgO(001) Surface—DFT Study. Surf. Sci. 2015, 632, 39–49. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab Initio Molecular-Dynamics Simulation of the Liquid-Metalamorphous- Semiconductor Transition in Germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B Condens. Matter Mater. Phys. 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules. In Encyclopedia of Computational Chemistry; John Wiley & Sons, Ltd.: Chichester, UK, 2002. [Google Scholar]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A Fast and Robust Algorithm for Bader Decomposition of Charge Density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Eb(M)/eV a | q(M)/e a | Ed-band(M)/eV a | Eads(CO)/eV a | EM–CO/eV | |
|---|---|---|---|---|---|
| Ru | −1.09 | −0.28 | −0.80 | −3.09 | −2.77 |
| Rh | −1.68 | −0.28 | −0.83 | −3.12 | −2.97 |
| Pd | −1.35 | −0.24 | −1.49 | −2.49 | −2.24 |
| Ir | −1.98 | −0.46 | −0.99 | −3.79 | −3.48 |
| Pt | −2.27 | −0.45 | −1.44 | −3.63 | −3.42 |
| M1 | M2 | Eb(M1) /eV | q(M1) /e b | Ed-band(M1) /eV b | Eads(CO) /eV |
|---|---|---|---|---|---|
| Ru | Ru | −4.12 | −0.23 a | −1.93 | −3.24 |
| Rh | −3.38 | −0.08 | −1.66 | −3.31 | |
| Pd | −2.11 | −0.12 | −1.42 | −3.47 | |
| Rh | Ru | −4.15 | −0.43 | −1.83 | −3.00 |
| Rh | −3.43 | −0.28 a | −1.55 | −3.11 | |
| Pd | −2.56 | −0.23 | −0.91 | −3.26 | |
| Pd | Ru | −2.71 | −0.37 | −1.90 | −1.93 b |
| Rh | −2.39 | −0.30 | −1.58 | −2.14 b | |
| Pd | −1.83 | −0.23 a | −1.37 | −2.46 b | |
| Ir | Ru | −5.01 | −0.59 | −2.29 | −3.42 |
| Rh | −4.11 | −0.53 | −1.99 | −3.74 | |
| Pd | −3.05 | −0.42 | −1.36 | −3.92 | |
| Pt | Ru | −4.15 | −0.66 | −2.23 | −2.64 b |
| Rh | −3.62 | −0.57 | −1.92 | −3.02 b | |
| Pd | −2.84 | −0.55 | −1.70 | −3.35 b |
| Substrate | Cu ads. Site a | Eb(Cu)/eV a | q(M)/|e| a | Eads(CO)/eV | Eads(H)/eV |
|---|---|---|---|---|---|
| NaF@Mo(001) | F on-top | −1.88 | −0.76 | −0.63 | −2.20 |
| MgO@Mo(001) | hollow | −1.47 | −0.67 | −0.31 | −2.30 |
| ScN@Mo(001) | hollow | −0.91 | −0.37 | −2.00 | −3.08 |
| ScN@Mo(001) | N on-top | −1.50 | −0.03 | −2.26 | −3.36 |
| isolated Cu | 0.00 | −0.78 | −2.88 |
| Surface Model | Eb(Pt)/eV | Pt ads. Site | q(Pt)/e | Ed-band(Pt)/eV | Eads(CO)/eV |
|---|---|---|---|---|---|
| pristine graphene | −1.56 | Bridge | −0.03 | −1.60 | −2.97 |
| epoxy-graphene-1 | −2.18 | Top | −0.10 | −1.57 | −2.59 |
| −1.85 | Bridge | −0.11 | −1.72 | −3.00 | |
| epoxy-graphene-2 | −2.82 | Top | −0.43 | −2.40 | −2.03 |
| −1.71 | Bridge | −0.14 | −1.44 | −2.82 | |
| −2.21 | Bridge | −0.11 | −1.60 | −2.56 | |
| hydroxyl-graphene | −2.40 | Bridge | −0.05 | −1.80 | −2.60 |
| −2.03 | Bridge | −0.06 | −1.68 | −2.77 | |
| −1.73 | Bridge | −0.17 | −1.73 | −3.25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Dobrota, A.S.; Jovanović, A.Z.; Johansson, B.; Skorodumova, N.V.; Pašti, I.A. On the Reactivity Descriptors of Low-Coordinated Atoms on Foreign Solid Substrates as Models of Single-Atom Catalysts. Catalysts 2026, 16, 278. https://doi.org/10.3390/catal16030278
Dobrota AS, Jovanović AZ, Johansson B, Skorodumova NV, Pašti IA. On the Reactivity Descriptors of Low-Coordinated Atoms on Foreign Solid Substrates as Models of Single-Atom Catalysts. Catalysts. 2026; 16(3):278. https://doi.org/10.3390/catal16030278
Chicago/Turabian StyleDobrota, Ana S., Aleksandar Z. Jovanović, Bӧrje Johansson, Natalia V. Skorodumova, and Igor A. Pašti. 2026. "On the Reactivity Descriptors of Low-Coordinated Atoms on Foreign Solid Substrates as Models of Single-Atom Catalysts" Catalysts 16, no. 3: 278. https://doi.org/10.3390/catal16030278
APA StyleDobrota, A. S., Jovanović, A. Z., Johansson, B., Skorodumova, N. V., & Pašti, I. A. (2026). On the Reactivity Descriptors of Low-Coordinated Atoms on Foreign Solid Substrates as Models of Single-Atom Catalysts. Catalysts, 16(3), 278. https://doi.org/10.3390/catal16030278