
The Interaction Mechanism Between Modified Selective Catalytic Reduction Catalysts and Volatile Organic Compounds in Flue Gas: A Density Functional Theory Study

 and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Adsorption of Substances on Surfaces
2.1.1. Adsorption of Acetone
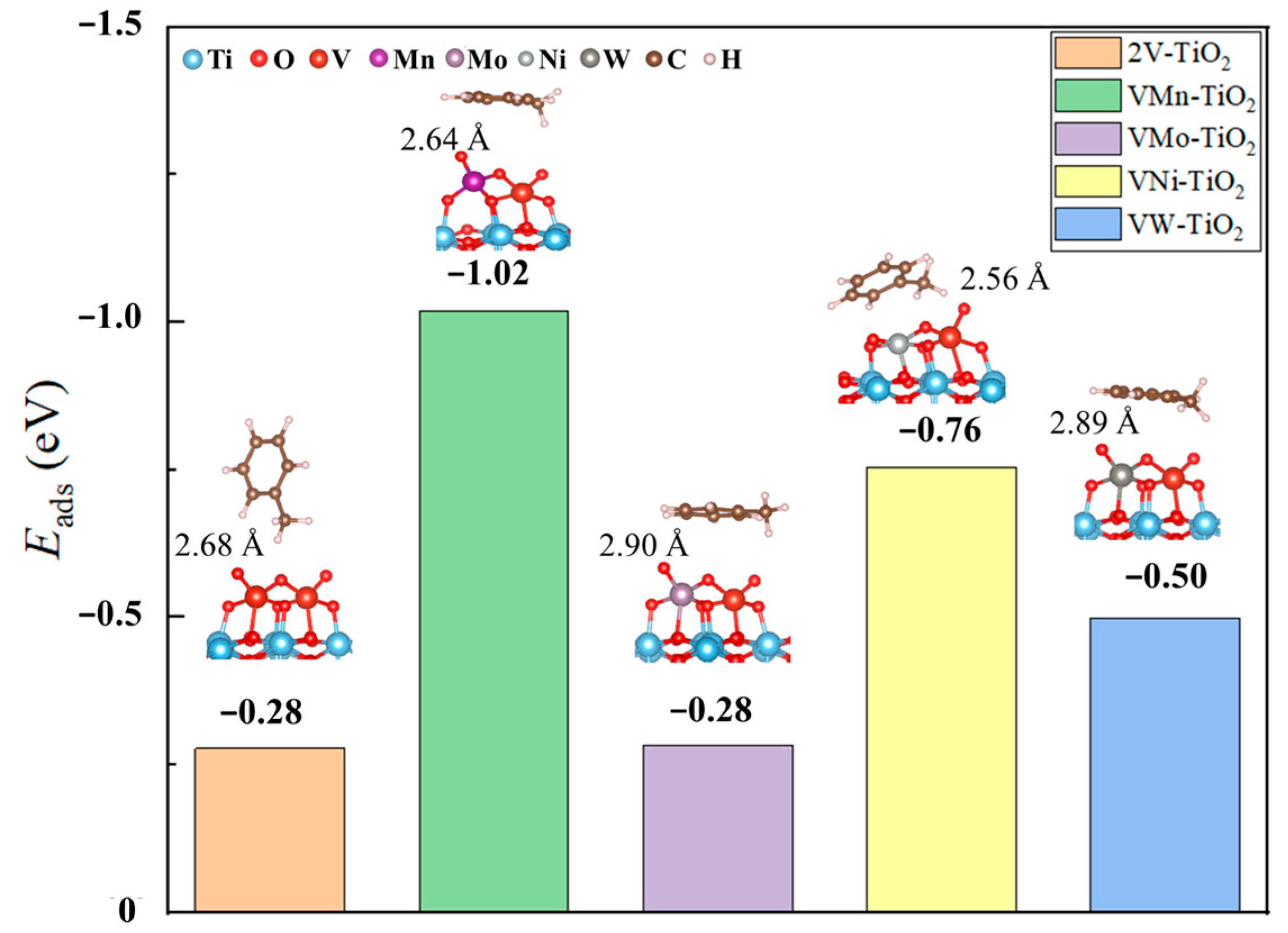
2.1.2. Adsorption of Toluene
2.2. Interaction of Acetone with Surfaces
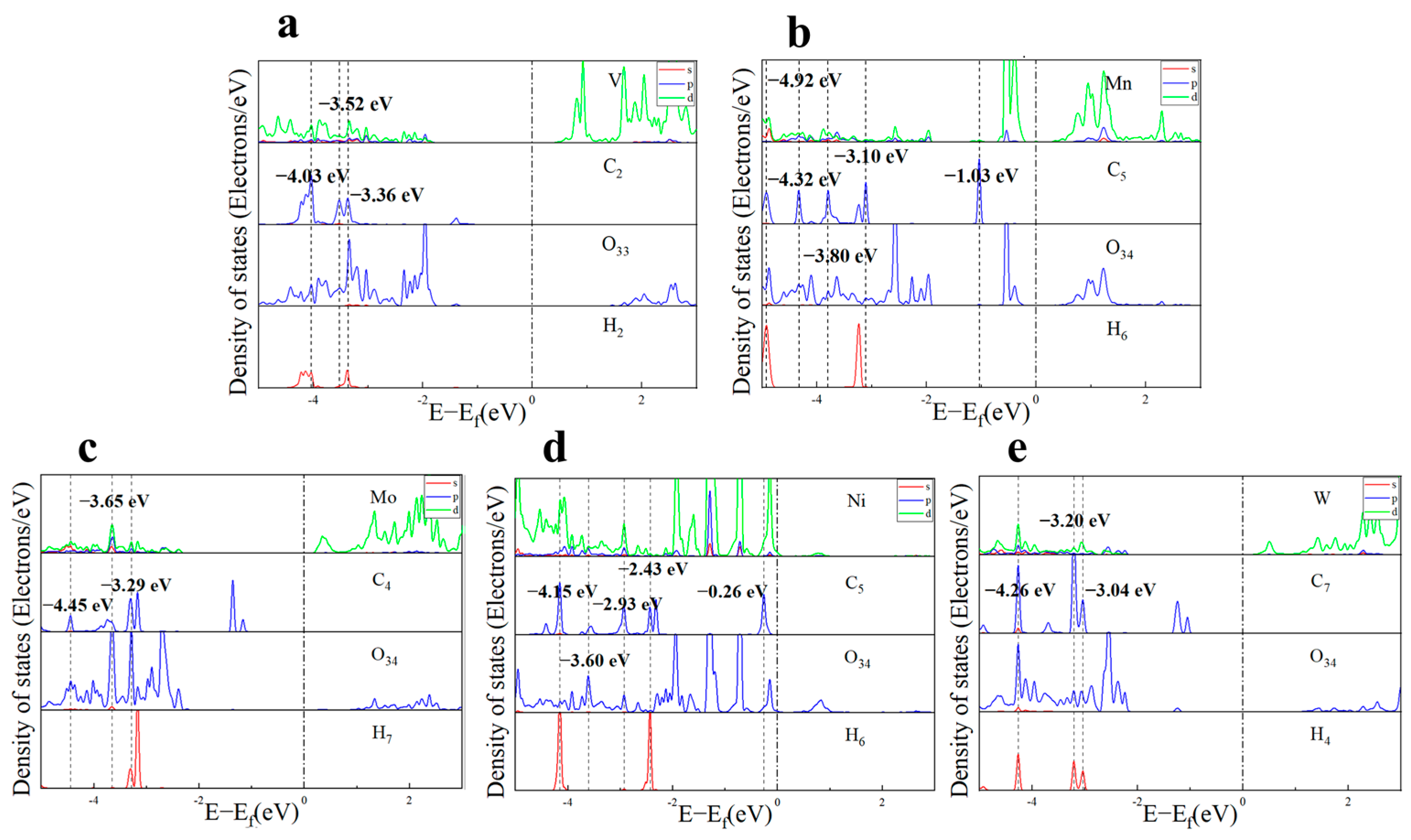
2.2.1. Electronic Structure Analysis of Acetone Adsorption
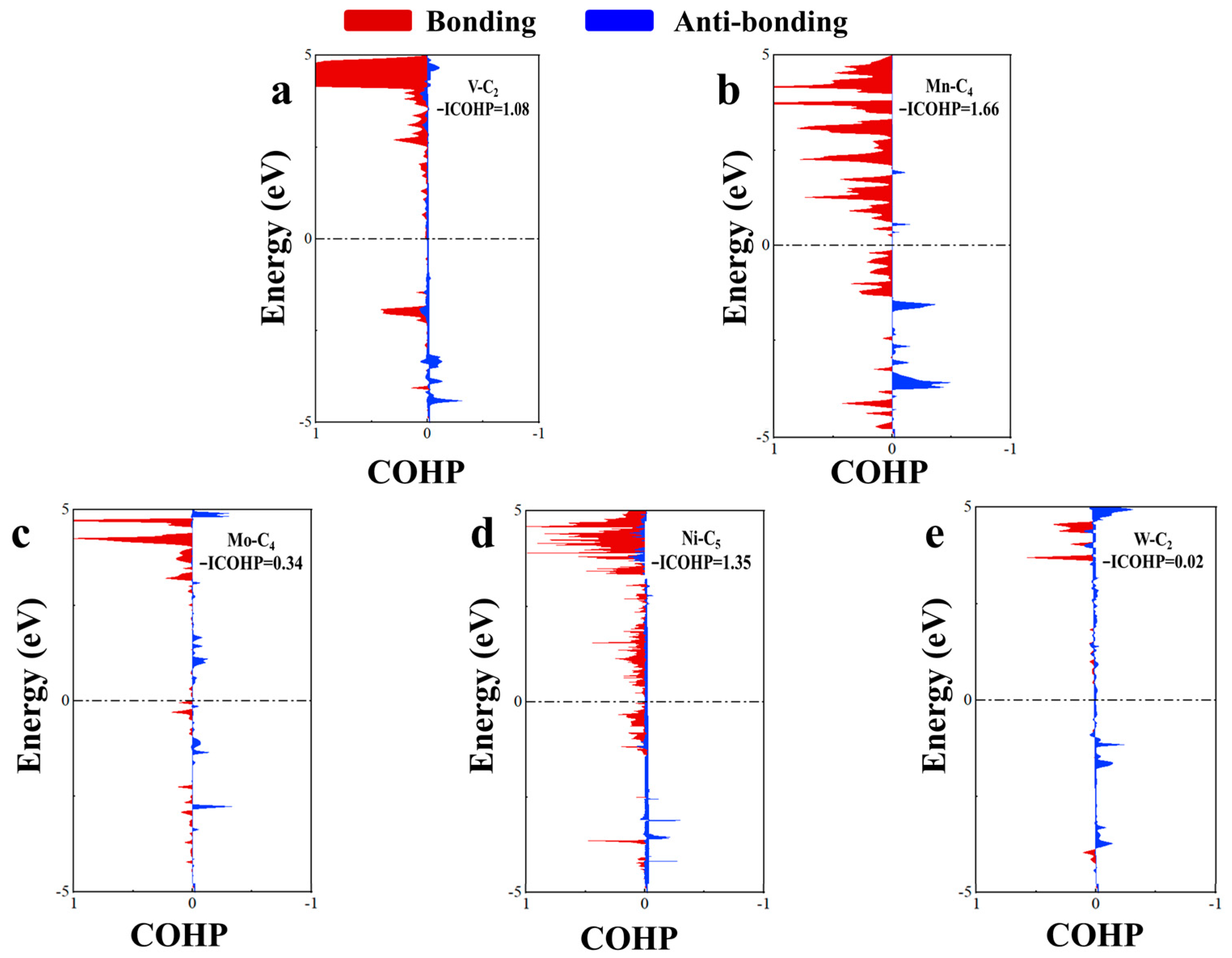
2.2.2. Bonding Analysis of Acetone Adsorption
2.3. Interaction of Toluene with Surfaces
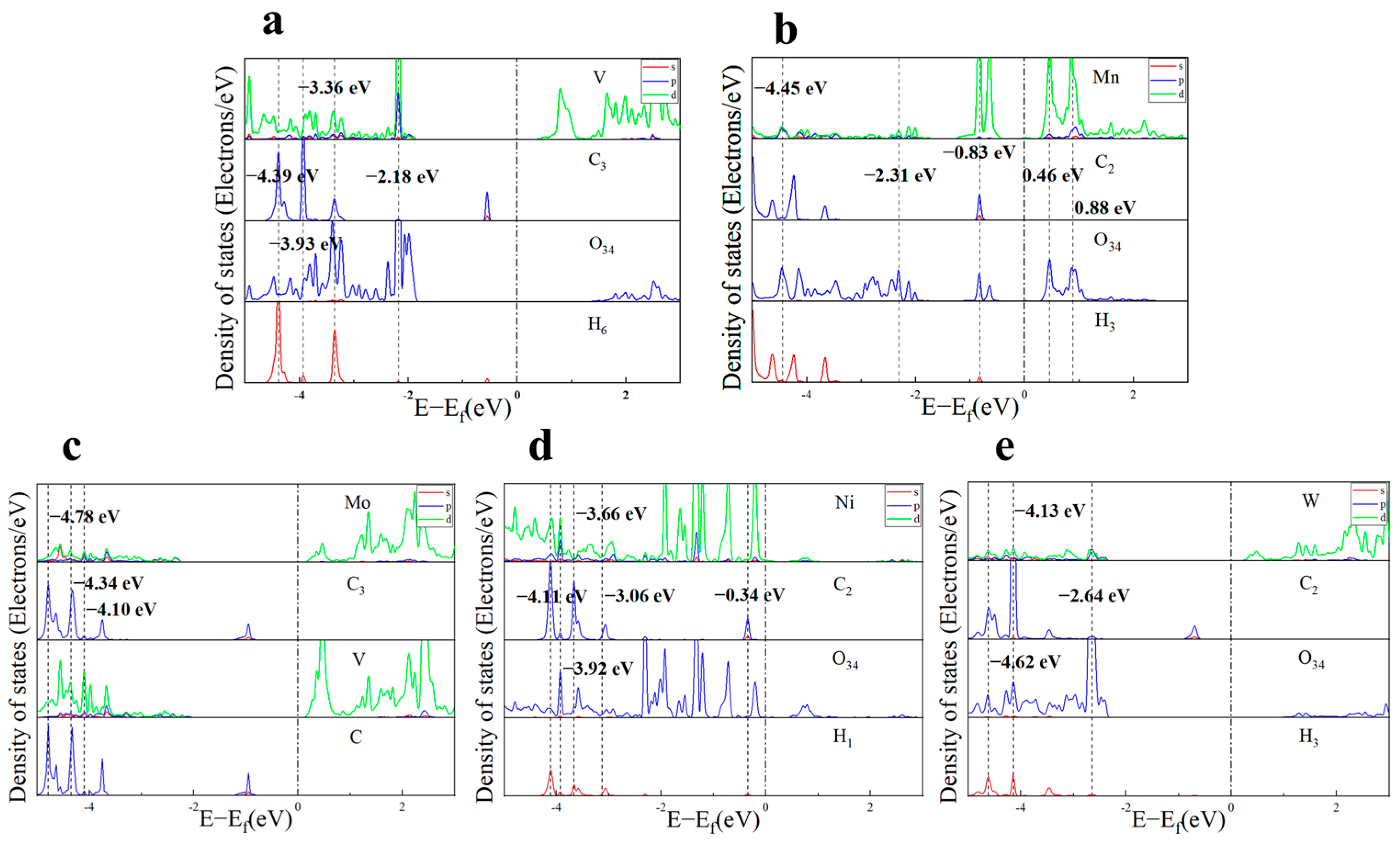
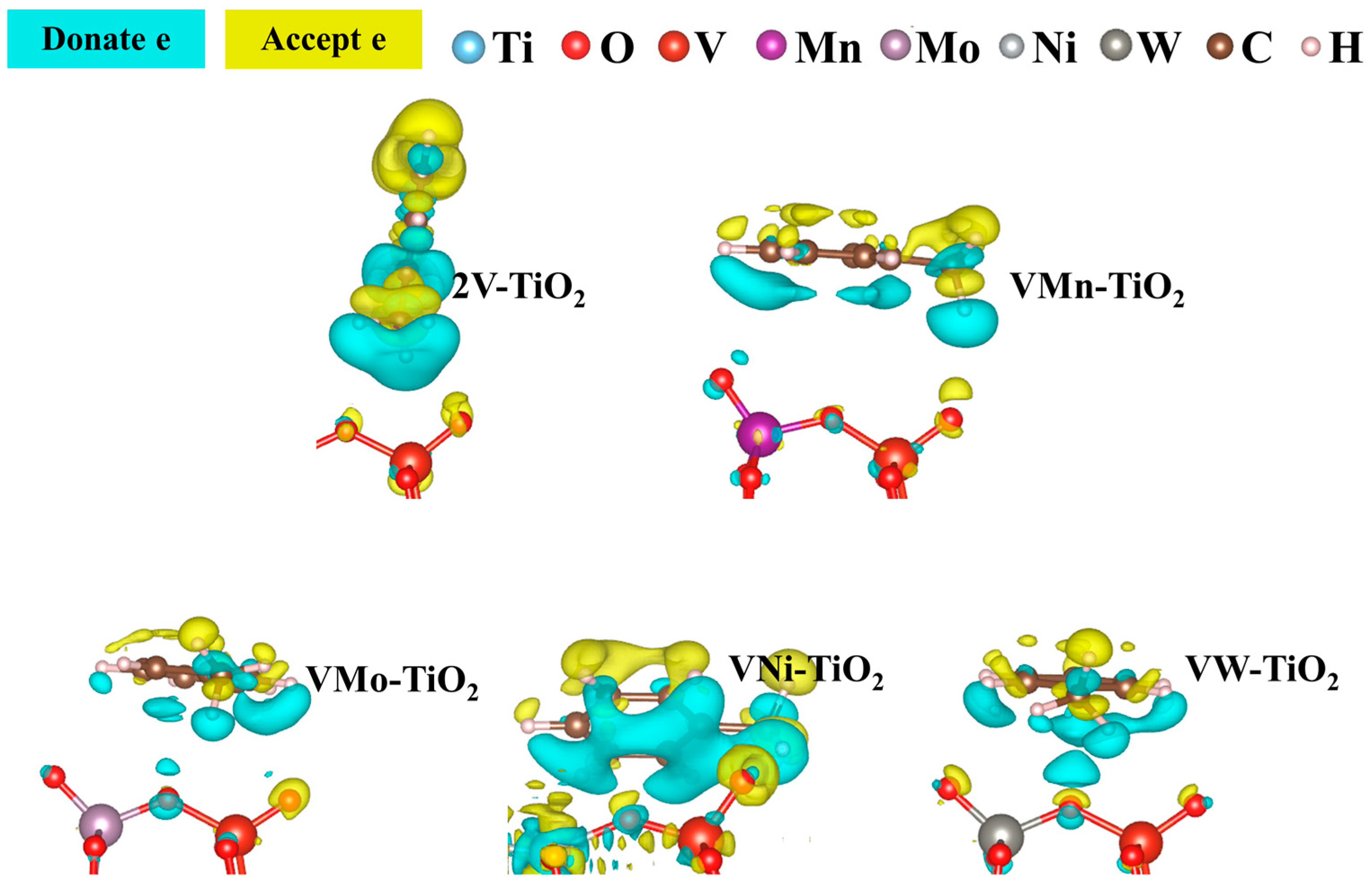
2.3.1. Electronic Structure Analysis of Toluene Adsorption
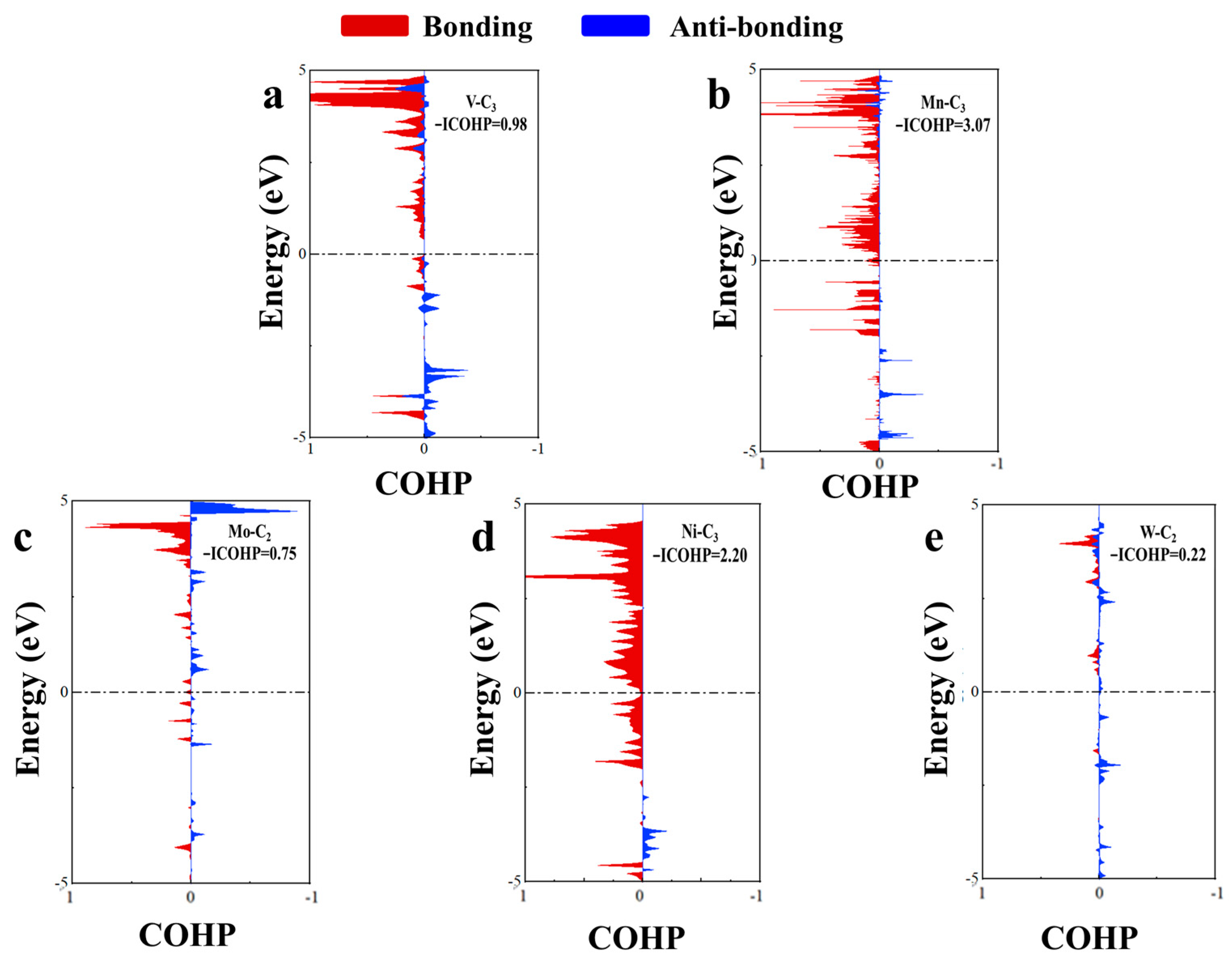
2.3.2. Bonding Analysis of Toluene Adsorption
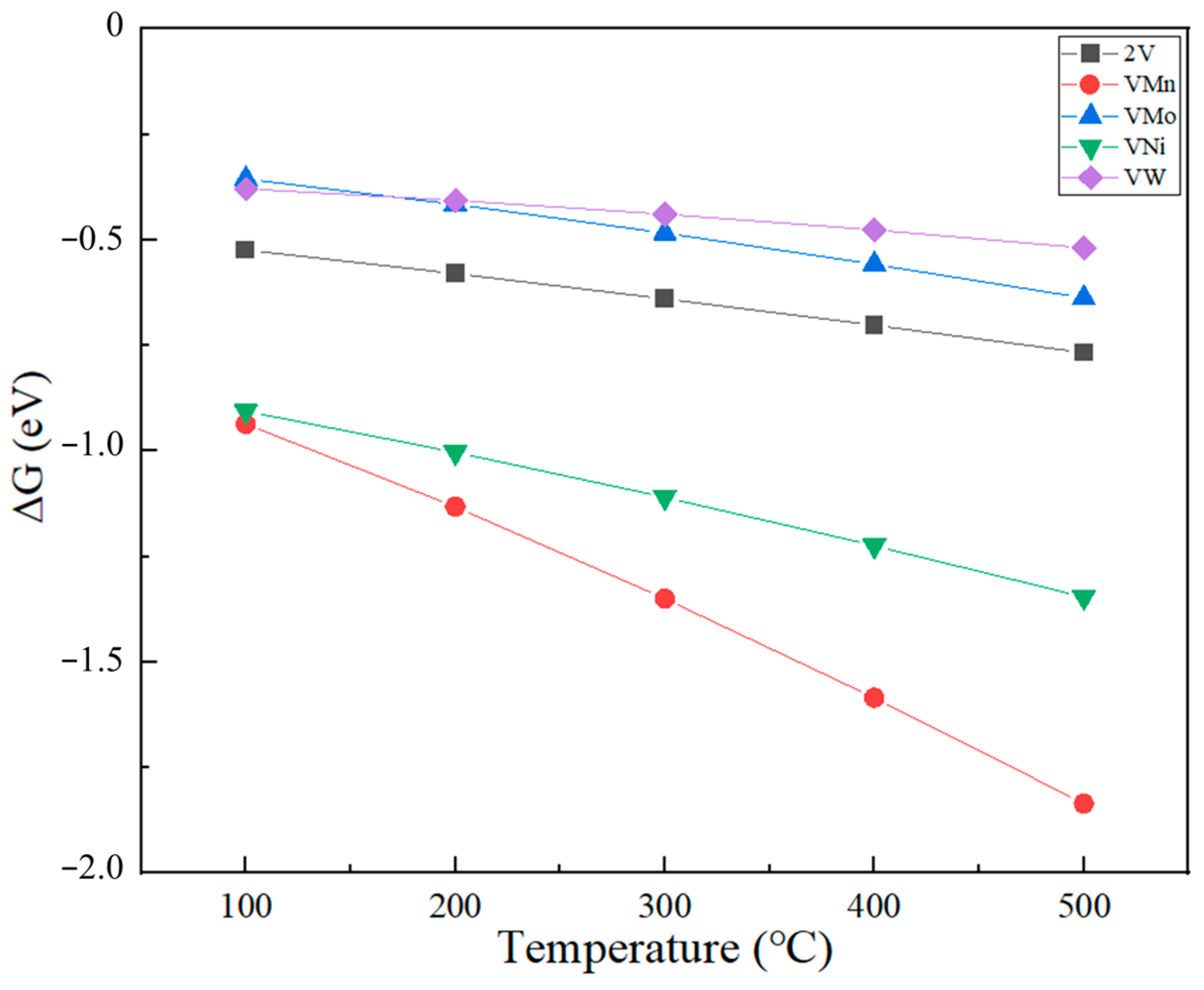
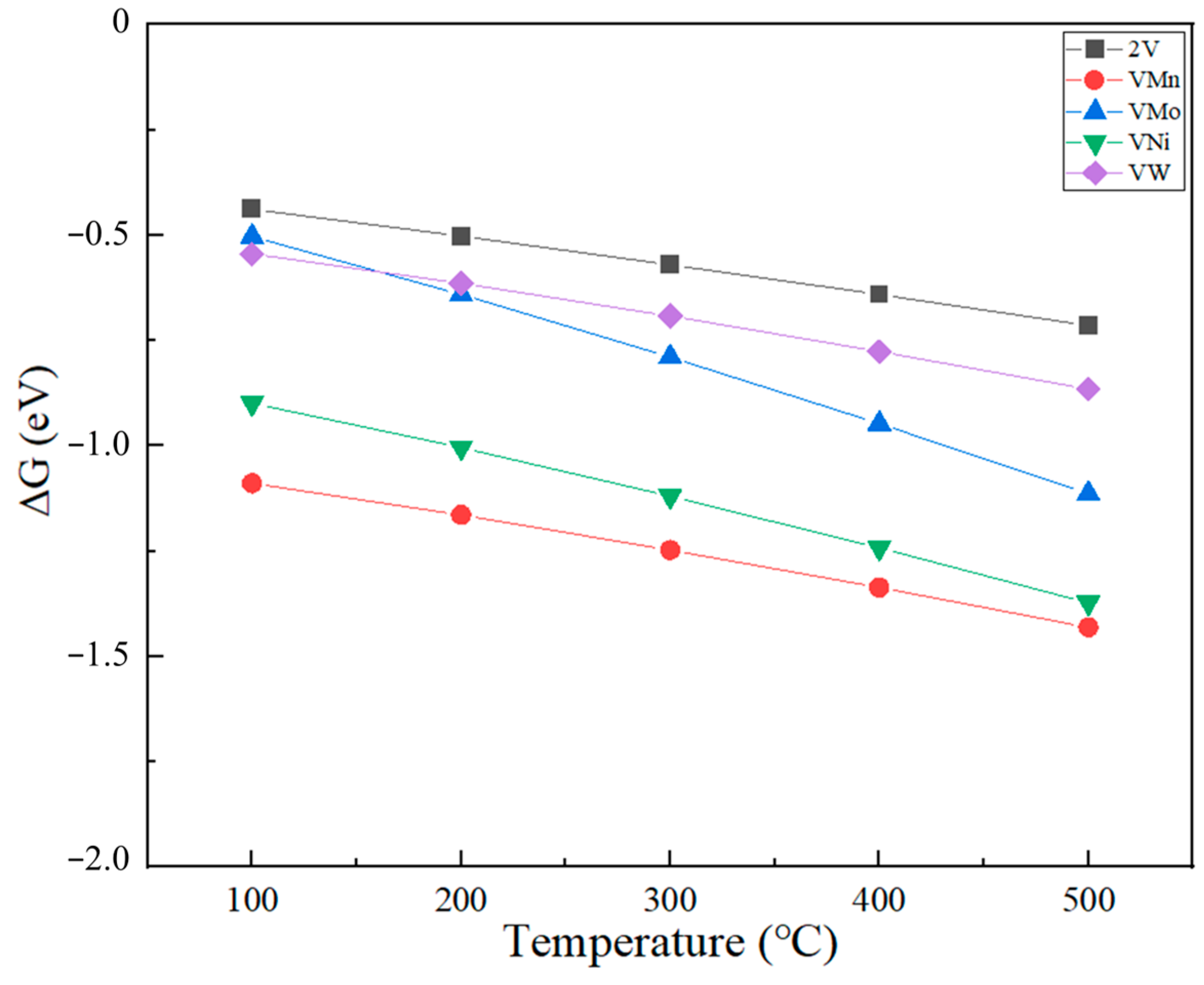
2.4. Effect of Temperature
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Li, J.; Chen, W.; Xu, C.; Hou, X.; Hu, X. Structural Effect of Cu-Mn/Al2O3 Catalysts on Enhancing Toluene Combustion Performance: Molecular Structure of Polyols and Hydrothermal Treatment. Catalysts 2024, 14, 443. [Google Scholar] [CrossRef]
- Paris, C.; Dib, H.; Bounoukta, C.E.; Genty, E.; Poupin, C.; Siffert, S.; Cousin, R. Benefit of LDH-Derived Mixed Oxides for the Co-Oxidation of Toluene and CO Exhausted from Biomass Combustion. Catalysts 2024, 14, 455. [Google Scholar] [CrossRef]
- Russell, C.K.; Rockey, J.L.; Hanna, R.N.; Miller, J.T. Impact of Co-Fed Hydrogen on High Conversion Propylene Aromatization on H-ZSM-5 and Ga/H-ZSM-5. Catalysts 2024, 14, 405. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Q.; Gao, W.; Ma, C.; Yang, M. Investigating the Role of Cs Species in the Toluene-Methanol Side Chain Alkylation Catalyzed by CsX Catalysts. Catalysts 2024, 14, 256. [Google Scholar] [CrossRef]
- Chen, W.-M.; Xi, B.-D.; Li, M.-X.; Ye, M.-Y.; Hou, J.-Q.; Wei, Y.-F.; Yu, C.-Z.; Meng, F.-H. Deciphering the Pivotal Reaction Conditions for Hydrogen Production from Tar Catalytic Cracking by Perovskite. Catalysts 2024, 14, 188. [Google Scholar] [CrossRef]
- Nomura, K.; Wang, X. Acyclic Diene Metathesis (ADMET) Polymerization for the Synthesis of Chemically Recyclable Bio-Based Aliphatic Polyesters. Catalysts 2024, 14, 97. [Google Scholar] [CrossRef]
- Lu, X.; Dang, Y.; Li, M.; Zhu, C.; Liu, F.; Tang, W.; Weng, J.; Ruan, M.; Steven, L.; Gao, P. Synergistic promotion of transition metal ion-exchange in TiO2 nanoarray-based monolithic catalysts for the selective catalytic reduction of NOx with NH3. Catal. Sci. Technol. 2022, 12, 5397–5407. [Google Scholar] [CrossRef]
- Manjunath, S.P.; Pooja, J.R.; Madhusoodana, C.D.; Thirupathi, G.; Binnal, P. Synthesis and Characterization of V2O5 Supported on TiO2 Selective Catalytic Reduction (SCR) Catalyst for De-NOx Application. J. Inst. Eng. Ser. E 2023, 104, 275–284. [Google Scholar] [CrossRef]
- Jana, G.; Pan, S.; Rodríguez-Kessler, P.L.; Merino, G.; Pratim, K. Chattaraj Adsorption of Molecular Hydrogen on Lithium–Phosphorus Double-Helices. J. Phys. Chem. C 2018, 122, 27941–27946. [Google Scholar] [CrossRef]
- Ortolan, A.O.; Madureira, L.; Rodríguez-Kessler, P.L. The nature of the central halide encapsulation in bambusuril hosts (BU [6]). Structural and interaction energy insights in BU [6]·X− (X = Cl, Br, I) from relativistic DFT calculations. Inorg. Chim. Acta 2023, 555, 121596. [Google Scholar] [CrossRef]
- Yu, L.; Zhong, Q.; Zhang, S.; Zhao, W.; Zhang, R. A CuO-V2O5/TiO2 catalyst for the selective catalytic reduction of NO with NH3. Combust. Sci. Technol. 2015, 187, 925–936. [Google Scholar]
- Grossale, A.; Nova, I.; Tronconi, E.; Chatterjee, D.; Weibel, M. The chemistry of the NO/NO2-NH3 “fast” SCR reaction over Fe-ZSM5 investigated by transient reaction analysis. J. Catal. 2008, 256, 312–322. [Google Scholar] [CrossRef]
- Huo, P.; Lu, Z.; Wang, H.; Pan, J.; Li, H.; Wu, X.; Yan, Y. Enhanced photodegradation of antibiotics solution under visible light with Fe2+/Fe3+ immobilized on TiO2/fly-ash cenospheres by using ions imprinting technology. Chem. Eng. J. 2011, 172, 615–622. [Google Scholar] [CrossRef]
- Yu, Y.; Miao, J.; Wang, J.; He, C.; Chen, J. Facile synthesis of CuSO4/TiO2 catalysts with superior activity and SO2 tolerance for NH3-SCR: Physicochemical properties and reaction mechanism. Catal. Sci. Technol. 2017, 7, 1590–1601. [Google Scholar] [CrossRef]
- Jiri, S.; Natale, F.; Pio, F.; Enrico, T.; Fiorenzo, B. Selective reduction of NOx by NH3 over honeycomb selective catalytic reduction catalysts. Ind. Eng. Chem. Res. 1993, 32, 1053–1060. [Google Scholar]
- Elsener, M.; Nuguid, R.J.G.; Kröcher, O.; Ferri, D. HCN production from formaldehyde during the selective catalytic reduction of NOx with NH3 over V2O5/WO3-TiO2. Appl. Catal. B Environ. 2021, 281, 119462. [Google Scholar] [CrossRef]
- Ciardelli, C.; Nova, I.; Tronconi, E.; Chatterjee, D.; Bandl-Konrad, B.; Weibel, M.; Krutzsch, B. Reactivity of NO/NO2-NH3 SCR system for diesel exhaust aftertreatment: Identification of the reaction network as a function of temperature and NO2 feed content. Appl. Catal. B Environ. 2007, 70, 80–90. [Google Scholar] [CrossRef]
- Fiore, A.M.; Naik, V.; Spracklen, D.V.; Steiner, A.; Unger, N.; Prather, M.; Zeng, G. Global air quality and climate. Chem. Soc. Rev. 2012, 41, 6663–6683. [Google Scholar] [CrossRef]
- Li, S.; Huang, W.; Xu, H.; Chen, T.; Ke, Y.; Qu, Z.; Yan, N. Alkali-induced deactivation mechanism of V2O5-WO3/TiO2 catalyst during selective catalytic reduction of NO by NH3 in aluminum hydrate calcining flue gas. Appl. Catal. B Environ. 2020, 270, 118872. [Google Scholar] [CrossRef]
- Stanciulescu, M.; Caravaggio, G.; Dobri, A.; Moir, J.; Burich, R.; Charland, J.P.; Bulsink, P. Low-temperature selective catalytic reduction of NOx with NH3 over Mn-containing catalysts. Appl. Catal. B Environ. 2012, 123, 229–240. [Google Scholar] [CrossRef]
- Xu, D.; Qu, W.; Liu, J.; Chen, J.; Fang, X.; Chen, L.; Chen, Y. Active sites of NO selective catalytic reduction over V2O5-WO3/TiO2. J. Mater. Chem. A. 2023, 11, 24644–24650. [Google Scholar] [CrossRef]
- Kong, M.; Liu, Q.; Jiang, L.; Tong, W.; Yang, J.; Ren, S.; Tian, Y. K+ deactivation of V2O5-WO3/TiO2 catalyst during selective catalytic reduction of NO with NH3, Effect of vanadium content. Chem. Eng. J. 2019, 370, 518–526. [Google Scholar] [CrossRef]
- Yu, B.; Liu, X.; Wu, S.; Yang, H.; Zhou, S.; Yang, L.; Liu, F. Study on Novel SCR Catalysts for Denitration of High Concentrated Nitrogen Oxides and Their Reaction Mechanisms. Catalysts 2024, 14, 406. [Google Scholar] [CrossRef]
- Baltakesmez, A.; Aykaç, C.; Güzeldir, B. Phase transition and changing properties of nanostructured V2O5 thin films deposited by spray pyrolysis technique, as a function of tungsten dopant. Appl. Phys. A 2019, 125, 1–18. [Google Scholar] [CrossRef]
- Sun, J.; Liu, Y.; Deng, J.; Bao, M.; Sun, Q.; Dai, H. PdPty/V2O5-TiO2, Highly Active Catalysts with Good Moisture-and Sulfur Dioxide-Resistant Performance in Toluene Oxidation. Catalysts 2022, 12, 1302. [Google Scholar] [CrossRef]
- Mandal, R.K.; Kundu, S.; Sain, S.; Pradhan, S.K. Enhanced photocatalytic performance of V2O5-TiO2 nanocomposites synthesized by mechanical alloying with morphological hierarchy. New J. Chem. 2019, 43, 2804–2816. [Google Scholar] [CrossRef]
- Tomlin, A.S. Air quality and climate impacts of biomass use as an energy source: A review. Energy Fuels 2021, 35, 14213–14240. [Google Scholar] [CrossRef]
- Chen, L.; Li, J.; Ge, M. The poisoning effect of alkali metals on nano V2O5-WO3/TiO2 catalysts on selective catalytic reduction of NOx by NH3. Chem. Eng. J. 2011, 170, 531–537. [Google Scholar] [CrossRef]
- Li, J.; Chang, H.; Ma, L.; Hao, J.; Yang, R.T. Low-temperature selective catalytic reduction of NOx with NH3 over metal oxide and zeolite catalysts: A review. Catal. Today 2011, 175, 147–156. [Google Scholar] [CrossRef]
- Li, L.; Chen, L.; Kong, M.; Liu, Q.; Ren, S. New insights into the deactivation mechanism of V2O5-WO3/TiO2 catalyst during selective catalytic reduction of NO with NH3, Synergies between arsenic and potassium species. RSC Adv. 2019, 9, 37724–37732. [Google Scholar] [CrossRef]
- Yi, X.; Wang, J.; Liu, Y.; Chen, Y.; Chen, J. Promotional effect of Fe and Ce co-doped on a V2O5-WO3/TiO2 catalyst for SCR of NOx with high K and Pb resistance. Catal. Sci. Technol. 2022, 12, 4169–4180. [Google Scholar] [CrossRef]
- Chen, L.; Li, J.; Ge, M. Promotional effect of Ce-doped V2O5-WO3/TiO2 with low vanadium loadings for selective catalytic reduction of NOx by NH3. J. Phys. Chem. C. 2009, 113, 21177–21184. [Google Scholar] [CrossRef]
- Dąbrowski, A. Adsorption: From theory to practice. Adv. Colloid Interface Sci. 2001, 93, 135–224. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Yang, L.; Jiang, W. Adsorption of acetone and toluene by N-functionalized porous carbon derived from ZIF-8. J. Ind. Eng. Chem. 2022, 111, 137–146. [Google Scholar] [CrossRef]
- Chelikowsky, J.R.; Cohen, M.L. Nonlocal pseudopotential calculations for the electronic structure of eleven diamond and zinc-blende semiconductors. Phys. Rev. B 1976, 14, 556. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–30. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Liu, Z.; Han, J.; Zhao, L.; Wu, Y.W.; Wang, H.X.; Pei, X.Q.; Yang, Y.P. Effects of Se and SeO2 on the denitrification performance of V2O5-WO3/TiO2 SCR catalyst. Appl. Catal A-Gen. 2019, 587, 117263. [Google Scholar] [CrossRef]
- Matijevic, E.; Good, R.J. Surface and Colloid Science; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Wang, V.; Xu, N.; Liu, J.C.; Tang, G.; Geng, W.T. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar] [CrossRef]
- Maintz, S.; Deringer, V.L.; Tchougréeff, A.L.; Dronskowski, R. LOBSTER: A tool to extract chemical bonding from plane-wave based, DFT. J. Comput. Chem. 2016, 37, 1030–1035. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhuang, K.; Wang, H.; Wu, Z.; Dong, Y.; Xu, Y.; Zhang, C.; Zhou, X.; Wu, Y.; Zhang, B. The Interaction Mechanism Between Modified Selective Catalytic Reduction Catalysts and Volatile Organic Compounds in Flue Gas: A Density Functional Theory Study. Catalysts 2025, 15, 728. https://doi.org/10.3390/catal15080728
Zhuang K, Wang H, Wu Z, Dong Y, Xu Y, Zhang C, Zhou X, Wu Y, Zhang B. The Interaction Mechanism Between Modified Selective Catalytic Reduction Catalysts and Volatile Organic Compounds in Flue Gas: A Density Functional Theory Study. Catalysts. 2025; 15(8):728. https://doi.org/10.3390/catal15080728
Chicago/Turabian StyleZhuang, Ke, Hanwen Wang, Zhenglong Wu, Yao Dong, Yun Xu, Chunlei Zhang, Xinyue Zhou, Yangwen Wu, and Bing Zhang. 2025. "The Interaction Mechanism Between Modified Selective Catalytic Reduction Catalysts and Volatile Organic Compounds in Flue Gas: A Density Functional Theory Study" Catalysts 15, no. 8: 728. https://doi.org/10.3390/catal15080728
APA StyleZhuang, K., Wang, H., Wu, Z., Dong, Y., Xu, Y., Zhang, C., Zhou, X., Wu, Y., & Zhang, B. (2025). The Interaction Mechanism Between Modified Selective Catalytic Reduction Catalysts and Volatile Organic Compounds in Flue Gas: A Density Functional Theory Study. Catalysts, 15(8), 728. https://doi.org/10.3390/catal15080728