
Experimental and Kinetic Modelling Study of the Heterogeneous Catalytic Conversion of Bioethanol into n-Butanol Using MgO–Al2O3 Mixed Oxide Catalyst
 , , , , , and
, , , , , and
Abstract

1. Introduction
2. Results
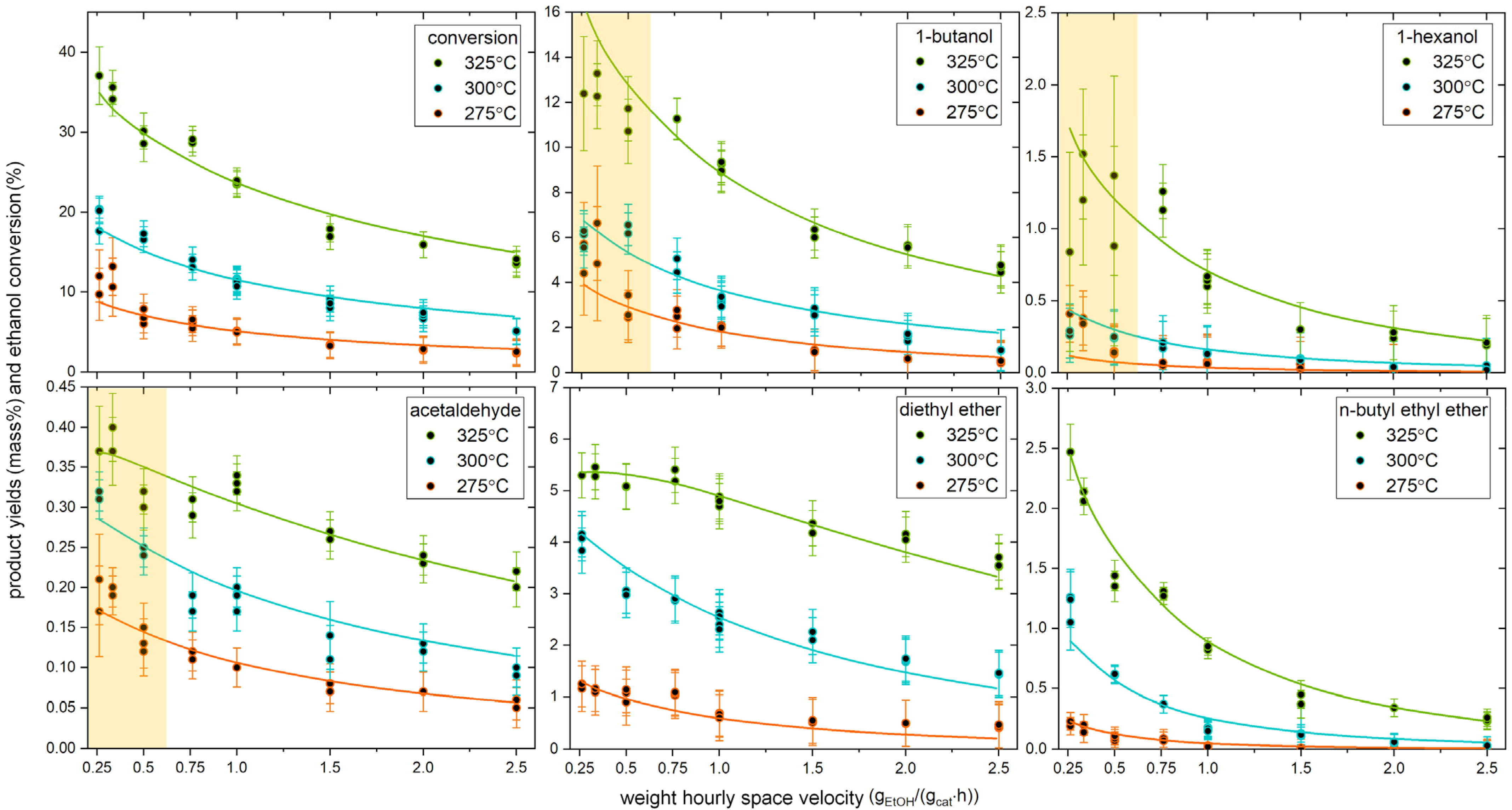
2.1. Experimental Results
2.2. Coupled Sorption–Reaction Kinetics Model
- No gas-phase chemistry is considered under the investigated relatively low-temperature conditions (T ≤ 325 °C), as justified by the simulations provided in the Supplementary Materials.
- Only one type of active site is assumed (denoted as *).
- All of the sorption and chemical reaction steps are reversible.
- The oxygenated species can adsorb and desorb (i.e., X + * X*, e.g., S1–10).
- Adsorbing species (Ai) react from and are produced in their adsorbed form (denoted with *). Consequently, reactions between adsorbing species follow the Langmuir–Hinshelwood mechanism (i.e., A1* + A2* …, e.g., R14–17, R21–27).
- Helium, hydrogen, and alkenes are considered non-adsorbing species.
- Non-adsorbing species (Ni) are produced in the gas phase (desorbed form) and can react only with adsorbed species via the Eley–Rideal mechanism (i.e., … Ni + Aj*, e.g., R11–13, R18–20).
- Alcohols can undergo dehydrogenation on the surface to produce aldehydes during a unimolecular step, which produces hydrogen gas (R11–13), or can react with acetaldehyde (R14–15rev). This reversible reaction was omitted for 1-hexanol, as hexanal was not identified among the products.
- Alcohols can dehydrate on the surface to form alkenes (R18–20). This reaction was also omitted for 1-hexanol, as hex-1-ene was not detected among the products.
- The aldol route in terms of Guerbet coupling: the aldol addition of acetaldehyde produces acetaldol (CH3-CH(OH)-CH2-CHO), which, however, was not detected among the products and was, therefore, assumed to undergo rapid dehydration. Consequently, these two steps were lumped into a single aldol condensation reaction, in which crotonaldehyde was formed directly from two acetaldehyde molecules (R21).
- The semi-direct Guerbet coupling mechanism I: the condensation of acetaldehyde with ethanol to form crotyl alcohol (R22).
- The semi-direct Guerbet coupling mechanism II: the condensation of ethanol with acetaldehyde to form butyraldehyde (R23).
- The direct Guerbet coupling of alcohols: the condensation of ethanol with ethanol and 1-butanol to form higher alcohols (R24–25).
- The etherification of ethanol with ethanol and 1-butanol (R26–27).
- Rate coefficients for the adsorption and desorption of alcohols (S2–5), aldehydes (S6–8), and ethers (S9–10).
- Rate coefficients for the dehydrogenation of alcohols to form aldehydes and H2 by the catalyst, and for the corresponding reverse reactions (R11–13).
- Rate coefficients for the hydrogenation of aldehydes by ethanol, and for the corresponding reverse reactions (R14–15).
- Rate coefficients for the hydrogenation of C=C bonds by ethanol and their reverse reactions (R16–17).
- Rate coefficients for the dehydration of alcohols and their reverse reactions (R18–20).
- These constraints reduced the number of independent Arrhenius parameters to 60.
2.3. Sorption–Reaction–Transport Model
2.4. Optimization of Model Parameters
2.5. Conversion and Liquid Product Yields
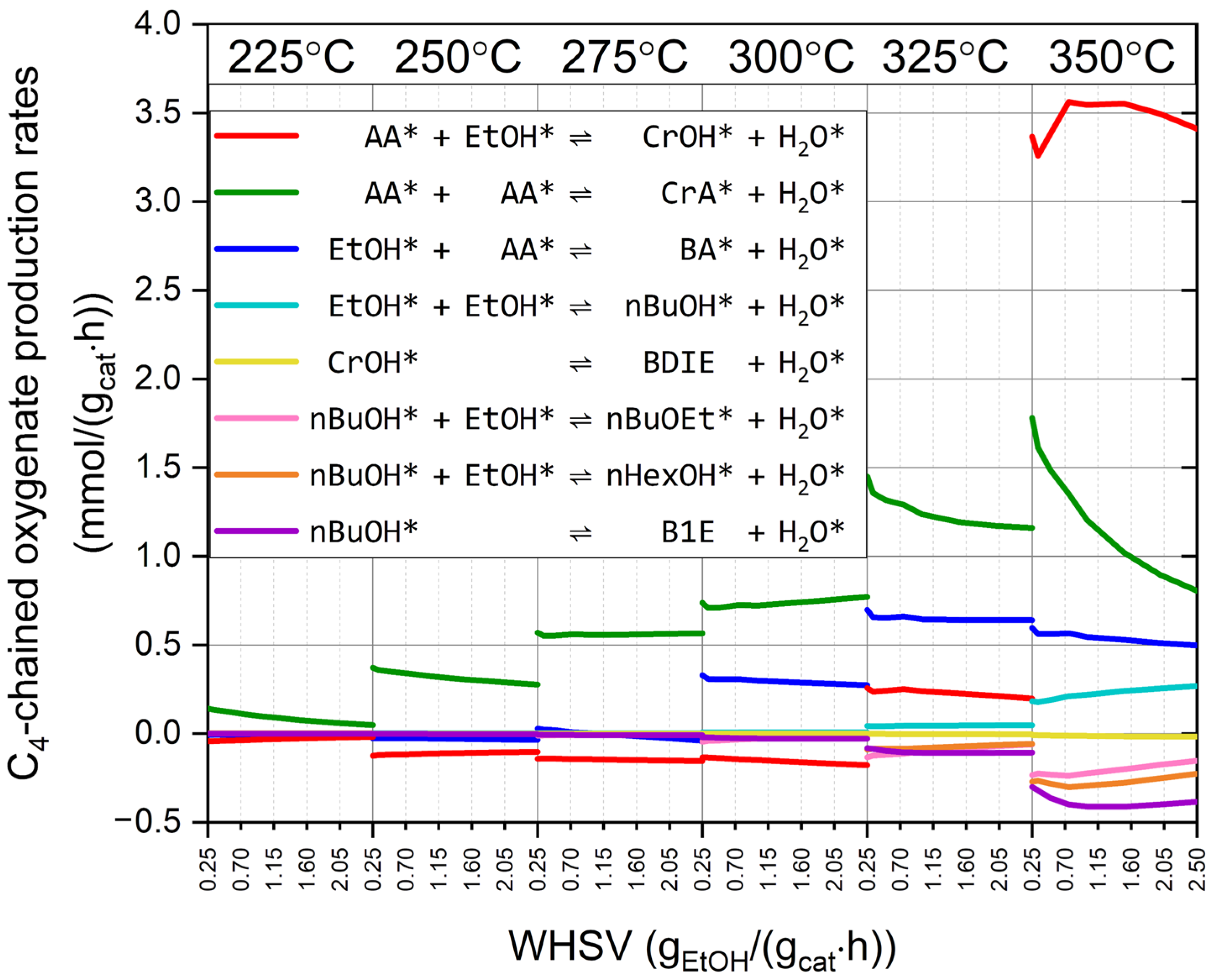
2.6. Gas-Phase Concentrations Along the Reactor
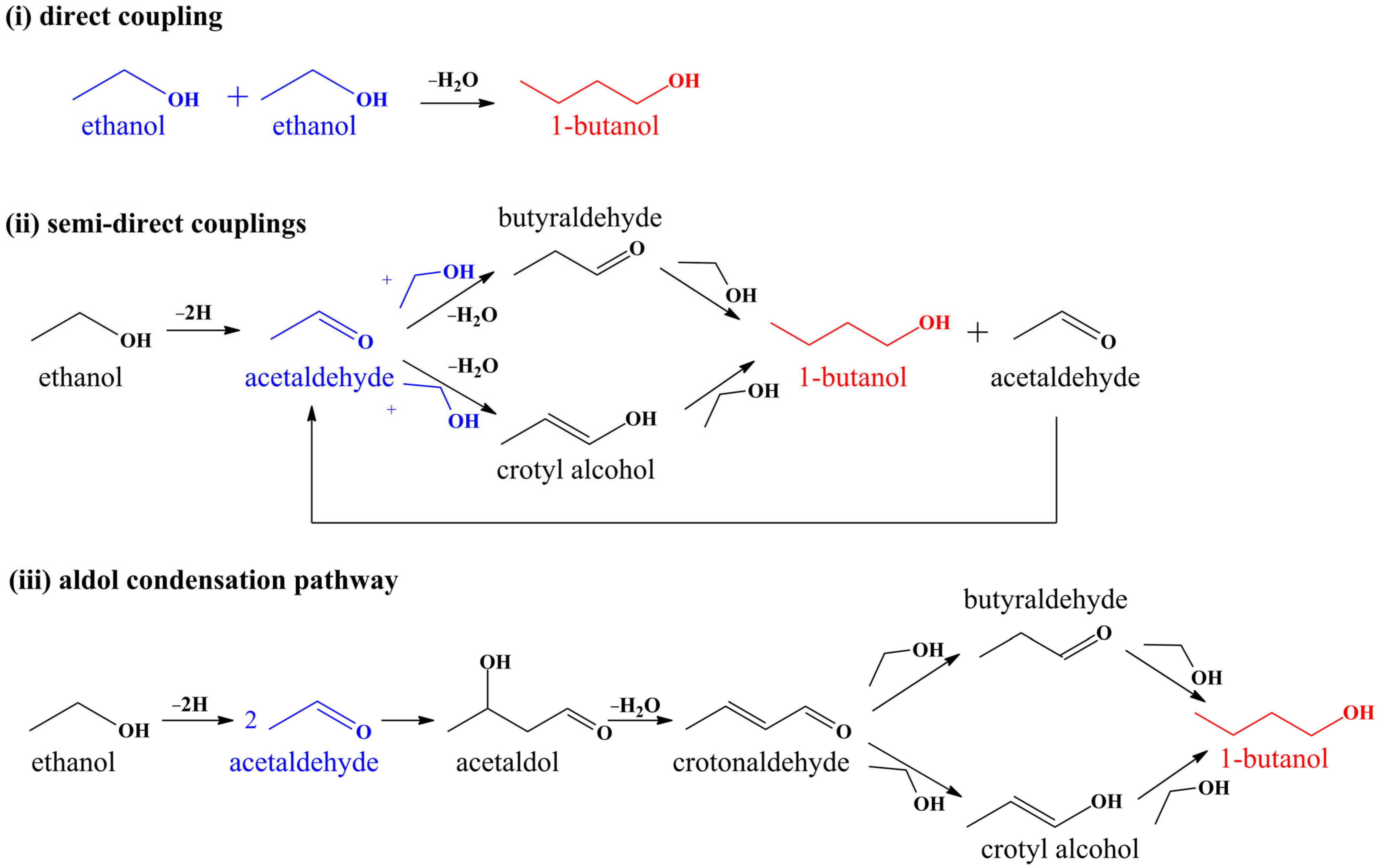
2.7. Mechanism of the Guerbet Reaction for 1-Butanol Formation
3. Materials and Methods
3.1. Materials and Experimental Methods
3.1.1. Synthesis of MgO–Al2O3 Mixed Oxide Catalyst
3.1.2. Catalyst Characterization
3.1.3. Catalytic Reaction
3.2. Reactive Sorption–Transport Model of the Flow-Through Packed-Bed Reactor
3.2.1. Energy and Momentum Balance
3.2.2. Material Balance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Schutyser, W.; Renders, T.; Van den Bosch, S.; Koelewijn, S.-F.; Beckham, G.; Sels, B.F. Chemicals from lignin: An interplay of lignocellulose fractionation, depolymerisation, and upgrading. Chem. Soc. Rev. 2018, 47, 852–908. [Google Scholar] [CrossRef]
- Wang, S.; Dai, G.; Yang, H.; Luo, Z. Lignocellulosic biomass pyrolysis mechanism: A state-of-the-art review. Prog. Energy Combust. Sci. 2017, 62, 33–86. [Google Scholar] [CrossRef]
- Jin, C.; Yao, M.; Liu, H.; Chia-fon, F.L.; Ji, J. Progress in the production and application of n-butanol as a biofuel. Renew. Sustain. Energy Rev. 2011, 15, 4080–4106. [Google Scholar] [CrossRef]
- Harvey, B.G.; Meylemans, H.A. The role of butanol in the development of sustainable fuel technologies. J. Chem. Technol. Biotechnol. 2011, 86, 2–9. [Google Scholar] [CrossRef]
- Hileman, J.I.; Stratton, R.W.; Donohoo, P.E. Energy content and alternative jet fuel viability. J. Propuls. Power 2010, 26, 1184–1196. [Google Scholar] [CrossRef]
- Siqueira, M.R.; Perrone, O.M.; Metzker, G.; de Oliveira Lisboa, D.C.; Thomeo, J.C.; Boscolo, M. Highly selective 1-butanol obtained from ethanol catalyzed by mixed metal oxides: Reaction optimization and catalyst structure behavior. Mol. Catal. 2019, 476, 110516. [Google Scholar] [CrossRef]
- Mück, J.; Kocík, J.; Hájek, M.; Tišler, Z.; Frolich, K.; Kašpárek, A. Transition metals promoting Mg-Al mixed oxides for conversion of ethanol to butanol and other valuable products: Reaction pathways. Appl. Catal. A Gen. 2021, 626, 118380. [Google Scholar] [CrossRef]
- Ramasamy, K.K.; Gray, M.; Job, H.; Smith, C.; Wang, Y. Tunable catalytic properties of bi-functional mixed oxides in ethanol conversion to high value compounds. Catal. Today 2016, 269, 82–87. [Google Scholar] [CrossRef]
- Lapuerta, M.; Rodríguez-Fernández, J.; Fernández-Rodríguez, D.; Patiño-Camino, R. Modeling viscosity of butanol and ethanol blends with diesel and biodiesel fuels. Fuel 2017, 199, 332–338. [Google Scholar] [CrossRef]
- Altun, S.; Oner, C.; Yasar, F.; Adin, H. Effect of n-butanol blending with a blend of diesel and biodiesel on performance and exhaust emissions of a diesel engine. Ind. Eng. Chem. Res. 2011, 50, 9425–9430. [Google Scholar] [CrossRef]
- Valihura, K.V.; Larina, O.V.; Kyriienko, P.I.; Yaremov, P.S.; Khalakhan, I.; Trachevskiy, V.V.; Soloviev, S.O.; Orlyk, S.M. Fast synthesis of MgO-Al2O3 systems: Effect on physicochemical characteristics and catalytic properties in Guerbet condensation of ethanol. Appl. Nanosci. 2023, 13, 6905–6918. [Google Scholar] [CrossRef]
- Bepari, S.; Basu, S.; Pradhan, N.C.; Dalai, A.K. Steam reforming of ethanol over cerium-promoted Ni-Mg-Al hydrotalcite catalysts. Catal. Today 2017, 291, 47–57. [Google Scholar] [CrossRef]
- Rubio-Rueda, J.A.; Quevedo-Hernandez, J.P.; López, M.B.; Galindo, J.F.; Hincapié-Triviño, G. Mg/Al and Cu-Mg/Al mixed oxides derived from hydrotalcites as catalysts to produce 1-butanol from ethanol. Mol. Catal. 2024, 569, 114528. [Google Scholar] [CrossRef]
- Palla, V.C.S.; Shee, D.; Maity, S.K. Conversion of n-butanol to gasoline range hydrocarbons, butylenes and aromatics. Appl. Catal. A Gen. 2016, 526, 28–36. [Google Scholar] [CrossRef]
- da Silva Trindade, W.R.; dos Santos, R.G. Review on the characteristics of butanol, its production and use as fuel in internal combustion engines. Renew. Sustain. Energy Rev. 2017, 69, 642–651. [Google Scholar] [CrossRef]
- Chakraborty, S.; Piszel, P.E.; Hayes, C.E.; Baker, R.T.; Jones, W.D. Highly selective formation of n-butanol from ethanol through the Guerbet process: A tandem catalytic approach. J. Am. Chem. Soc. 2015, 137, 14264–14267. [Google Scholar] [CrossRef]
- Nair, P.; Meenakshi, H. Review on the synthesis, performance and trends of butanol: A cleaner fuel additive for gasoline. Int. J. Ambient. Energy 2022, 43, 4207–4223. [Google Scholar] [CrossRef]
- García, V.; Päkkilä, J.; Ojamo, H.; Muurinen, E.; Keiski, R.L. Challenges in biobutanol production: How to improve the efficiency? Renew. Sustain. Energy Rev. 2011, 15, 964–980. [Google Scholar] [CrossRef]
- Veza, I.; Said, M.F.M.; Latiff, Z.A. Recent advances in butanol production by acetone-butanol-ethanol (ABE) fermentation. Biomass Bioenergy 2021, 144, 105919. [Google Scholar] [CrossRef]
- Xue, C.; Cheng, C. Butanol production by Clostridium. Adv. Bioenergy 2019, 4, 35–77. [Google Scholar] [CrossRef]
- Tian, W.; Herrera, J.E. Catalytic Relevance of Mg-Al-O Basic Centers in the Upgrade of Ethanol to n-Butanol. ChemCatChem 2024, 16, e202400225. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, Z.; Xin, X.; Bai, F.; Xue, C. Synergetic engineering of central carbon, energy, and redox metabolisms for high butanol production and productivity by Clostridium acetobutylicum. Ind. Eng. Chem. Res. 2020, 59, 17137–17146. [Google Scholar] [CrossRef]
- Chistyakov, A.; Nikolaev, S.; Zharova, P.; Tsodikov, M.; Manenti, F. Linear α-alcohols production from supercritical ethanol over Cu/Al2O3 catalyst. Energy 2019, 166, 569–576. [Google Scholar] [CrossRef]
- Zhang, Q.; Dong, J.; Liu, Y.; Wang, Y.; Cao, Y. Towards a green bulk-scale biobutanol from bioethanol upgrading. J. Energy Chem. 2016, 25, 907–910. [Google Scholar] [CrossRef]
- Kozlowski, J.T.; Davis, R.J. Heterogeneous catalysts for the Guerbet coupling of alcohols. ACS Catal. 2013, 3, 1588–1600. [Google Scholar] [CrossRef]
- Wu, X.; Fang, G.; Tong, Y.; Jiang, D.; Liang, Z.; Leng, W.; Liu, L.; Tu, P.; Wang, H.; Ni, J. Catalytic upgrading of ethanol to n-butanol: Progress in catalyst development. Chem. Sus. Chem 2018, 11, 71–85. [Google Scholar] [CrossRef]
- Lovón-Quintana, J.J.; Rodriguez-Guerrero, J.K.; Valença, P.G. Carbonate hydroxyapatite as a catalyst for ethanol conversion to hydrocarbon fuels. Appl. Catal. A Gen. 2017, 542, 136–145. [Google Scholar] [CrossRef]
- Galadima, A.; Muraza, O. Zeolite catalysts in upgrading of bioethanol to fuels range hydrocarbons: A review. J. Ind. Eng. Chem. 2015, 31, 1–14. [Google Scholar] [CrossRef]
- Birky, T.W.; Kozlowski, J.T.; Davis, R.J. Isotopic transient analysis of the ethanol coupling reaction over magnesia. J. Catal. 2013, 298, 130–137. [Google Scholar] [CrossRef]
- León, M.; Díaz, E.; Ordóñez, S. Ethanol catalytic condensation over Mg–Al mixed oxides derived from hydrotalcites. Catal. Today 2011, 164, 436–442. [Google Scholar] [CrossRef]
- Marcu, I.-C.; Tanchoux, N.; Fajula, F.; Tichit, D. Catalytic conversion of ethanol into butanol over M-Mg-Al mixed oxide catalysts (M = Pd, Ag, Mn, Fe, Cu, Sm, Yb) obtained from LDH precursors. Catal. Lett. 2013, 143, 23–30. [Google Scholar] [CrossRef]
- Pavel, O.D.; Tichit, D.; Marcu, I.-C. Acido-basic and catalytic properties of transition-metal containing Mg–Al hydrotalcites and their corresponding mixed oxides. Appl. Clay Sci. 2012, 61, 52–58. [Google Scholar] [CrossRef]
- Scheid, A.J.; Barbosa-Coutinho, E.; Schwaab, M.; Salau, N.P. Mechanism and kinetic modeling of ethanol conversion to 1-butanol over Mg and Al oxide derived from hydrotalcites. Ind. Eng. Chem. Res. 2019, 58, 12981–12995. [Google Scholar] [CrossRef]
- Vikár, A.; Lónyi, F.; Makoye, A.; Nagy, T.; Novodárszki, G.; Barthos, R.; Szabó, B.; Valyon, J.; Mihályi, M.R.; Deka, D. Ethanol Coupling Reactions over MgO–Al2O3 Mixed Oxide-Based Catalysts for Producing Biofuel Additives. Molecules 2023, 28, 3788. [Google Scholar] [CrossRef]
- Goitom, S.K.; Papp, M.; Kovács, M.; Nagy, T.; Zsély, I.G.; Turányi, T.; Pál, L. Efficient numerical methods for the optimisation of large kinetic reaction mechanisms. Combust. Theory Model. 2022, 26, 1071–1097. [Google Scholar] [CrossRef]
- Ergun, S. Fluid flow through packed columns. Chem. Eng. Prog. 1952, 48, 89. [Google Scholar]
- Taylor, G.I. Dispersion of soluble matter in solvent flowing slowly through a tube. Proc. R. Soc. London Ser. A. Math. Phys. Sci. 1953, 219, 186–203. [Google Scholar] [CrossRef]
- Ramasamy, K.K.; Gray, M.; Job, H.; Santosa, D.; Li, X.S.; Devaraj, A.; Karkamkar, A.; Wang, Y. Role of Calcination Temperature on the Hydrotalcite Derived MgO-Al2O3 in Converting Ethanol to Butanol. Top. Catal. 2016, 59, 46–54. [Google Scholar] [CrossRef]
- Quesada, J.; Faba, L.; Díaz, E.; Ordóñez, S. Role of the surface intermediates in the stability of basic mixed oxides as catalyst for ethanol condensation. Appl. Catal. A Gen. 2017, 542, 271–281. [Google Scholar] [CrossRef]
- Scalbert, J.; Thibault-Starzyk, F.; Jacquot, R.; Morvan, D.; Meunier, F. Ethanol condensation to butanol at high temperatures over a basic heterogeneous catalyst: How relevant is acetaldehyde self-aldolization? J. Catal. 2014, 311, 28–32. [Google Scholar] [CrossRef]
- Ndou, A.S.; Plint, N.; Coville, N.J. Dimerisation of ethanol to butanol over solid-base catalysts. Appl. Catal. A Gen. 2003, 251, 337–345. [Google Scholar] [CrossRef]
- Vlasenko, N.V.; Kyriienko, P.I.; Yanushevska, O.I.; Valihura, K.V.; Soloviev, S.O.; Strizhak, P.E. The Effect of Ceria Content on the Acid–Base and Catalytic Characteristics of ZrO2–CeO2 Oxide Compositions in the Process of Ethanol to n-Butanol Condensation. Catal. Lett. 2020, 150, 234–242. [Google Scholar] [CrossRef]
- Tsuchida, T.; Kubo, J.; Yoshioka, T.; Sakuma, S.; Takeguchi, T.; Ueda, W. Reaction of ethanol over hydroxyapatite affected by Ca/P ratio of catalyst. J. Catal. 2008, 259, 183–189. [Google Scholar] [CrossRef]
- Tsuchida, T.; Sakuma, S.; Takeguchi, T.; Ueda, W. Direct synthesis of n-butanol from ethanol over nonstoichiometric hydroxyapatite. Ind. Eng. Chem. Res. 2006, 45, 8634–8642. [Google Scholar] [CrossRef]
- Hucal, A.M.; Tian, W.; Ascherl, P.C.; Herrera, J.E. Tuning of Acid/Base Functionalities in the MgAlO System for Ethanol Upgrade to n-Butanol. Catalysts 2024, 14, 868. [Google Scholar] [CrossRef]
- Portillo Crespo, M.A.; Vidal-Barrero, F.; Azancot, L.; Reina, T.R.; Campoy, M. Insights on Guerbet Reaction: Production of Biobutanol From Bioethanol Over a Mg–Al Spinel Catalyst. Front. Chem. 2022, 10, 945596. [Google Scholar] [CrossRef] [PubMed]
- Fuller, E.N.; Schettler, P.D.; Giddings, J.C. A new method for prediction of binary gas-phase diffusion coefficients. Ind. Eng. Chem. 1966, 58, 18–27. [Google Scholar] [CrossRef]
- Cox, K.R.; Chapman, W.G. The Properties of Gases and Liquids, 5th Edition By Bruce E. Poling (University of Toledo), John M. Prausnitz (University of California at Berkeley), and John P. O’Connell (University of Virginia). McGraw-Hill: New York. 2001. 768 pp. $115.00. ISBN 0-07-011682-2. J. Am. Chem. Soc. 2001, 123, 6745. [Google Scholar] [CrossRef]
- Stiel, L.I.; Thodos, G. The viscosity of nonpolar gases at normal pressures. AIChE J. 1961, 7, 611–615. [Google Scholar] [CrossRef]
- Chung, T.-H.; Ajlan, M.; Lee, L.L.; Starling, K.E.; Ajlan, M. Generalized Multiparameter Correlation for Nonpolar and Polar Fluid Transport Properties. Ind. Eng. Chem. Res. 1988, 27, 671–679. [Google Scholar] [CrossRef]
- Neufeld, P.D.; Janzen, A.R.; Aziz, R.A. Empirical equations to calculate 16 of the transport collision integrals Ω(l,8)* for the lennard-jones (12-6) potential. J. Chem. Phys. 1972, 57, 1100–1102. [Google Scholar] [CrossRef]
- Pitzer, K.S. The Volumetric and Thermodynamic Properties of Fluids. I. Theoretical Basis and Virial Coefficients. J. Am. Chem. Soc. 1955, 77, 3427–3433. [Google Scholar] [CrossRef]
- Pitzer, K.S.; Lippmann, D.Z.; Curl, R.F.; Huggins, C.M.; Petersen, D.E. The Volumetric and Thermodynamic Properties of Fluids. II. Compressibility Factor, Vapor Pressure and Entropy of Vaporization. J. Am. Chem. Soc. 1955, 77, 3433–3440. [Google Scholar] [CrossRef]
- Joback, K.G.; Reid, R.C. Estimation of Pure-Component Properties from Group-Contributions. Chem. Eng. Commun. 1987, 57, 233–243. [Google Scholar] [CrossRef]
- Wilke, C.R.; Chang, P. Correlation of diffusion coefficients in dilute solutions. AIChE J. 1955, 1, 264–270. [Google Scholar] [CrossRef]
- Abdulla, T.J.; Cash, J.R.; Diamantakis, M.T. An MEBDF package for the numerical solution of large sparse systems of stiff initial value problems. Comput. Math. Appl. 2001, 42, 121–129. [Google Scholar] [CrossRef]
- Sarathy, S.M.; Oßwald, P.; Hansen, N.; Kohse-Höinghaus, K. Alcohol combustion chemistry. Prog. Energy Combust. Sci. 2014, 44, 40–102. [Google Scholar] [CrossRef]
- Sakai, Y.; Herzler, J.; Werler, M.; Schulz, C.; Fikri, M. A quantum chemical and kinetics modeling study on the autoignition mechanism of diethyl ether. Proc. Combust. Inst. 2017, 36, 195–202. [Google Scholar] [CrossRef]
- Goodwin, D.G.; Moffat, H.K.; Schoegl, I.; Speth, R.L.; Weber, B.W. Cantera: An Object-oriented Software Toolkit for Chemical Kinetics, Thermodynamics, and Transport Processes 2022. Available online: https://zenodo.org/records/6387882 (accessed on 20 July 2025).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | Value | Property | Value |
|---|---|---|---|
| Reactor length (l) | 9.1 cm | Specific pore volume (Vspec) | 0.25 cm3/g |
| Catalyst bed length (lbed) | 4.74 cm | Specific surface area (Aspec) | 218 m2/g |
| Reactor diameter (d) | 9.00 mm | Crystal density (ρcryst) | 3.3 g/cm3 |
| Thermocouple diameter (dTC) | 2.40 mm | Volume density (ρvol) | 0.69 g/cm3 |
| Reduced eff. diameter (dred) | 8.67 mm | Grain density (ρgrain) | 1.81 g/cm3 |
| Catalyst bed cross-section (A) | 0.590 cm2 | Crystal volume (Vcryst) | 0.606 cm3 |
| Catalyst volume (V) | 2.91 cm3 | Pore volume (Vpore) | 0.506 cm3 |
| Catalyst mass (mcat) | 2.00 g | Void volume (Vvoid) | 1.798 cm3 |
| Grain diameter (dgrain) | 0.5–0.8 mm | Void fraction ) | 0.618 |
| # | Short Notation | Product Phase 1 | Name | Group Formula | Class Formula |
|---|---|---|---|---|---|
| 1 | He | G | helium | He | – |
| 2 | H2 | G | hydrogen | H2 | |
| 3 | H2O | L | water | H2O | |
| 4 | EtOH | L | ethanol | C2H5OH | ROH |
| 5 | nBuOH | L | 1-butanol | nC4H9OH | |
| 6 | nHexOH | L | 1-hexanol | nC6H13OH | |
| 7 | CrOH | L | crotyl alcohol | CH3CH=CHCH2OH | |
| 8 | AA | L | acetaldehyde | CH3CHO | RCHO |
| 9 | CrA | L | crotonaldehyde | CH3CH=CHCHO | |
| 10 | BA | L | butyraldehyde | CH3CH2CH2CHO | |
| 11 | Et2O | L | diethyl ether | C2H5OC2H5 | R1OR2 |
| 12 | nBuOEt | L | n-butyl ethyl ether | nC4H9OC2H5 | |
| 13 | C2H4 | G | ethylene | CH2=CH2 | RC2H3 |
| 14 | B1E | G | but-1-ene | CH2=CHCH2CH3 | |
| 15 | BDIE | G | but-1,3-diene | CH2=CHCH=CH2 |
| # | Reactions | log10 kfor (cm,s,mol) | nfor4 (z) | Efor/R (K) | log10 krev (cm,s,mol) | Erev/R (K) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S1 | H2O | + | * | H2O * | −0.183 | 0.5 | 10,687 | 0.384 | 21,483 | |||
| S2–5 | ROH | + | * | ROH * | 5.007 | 9927 | 2.657 | 10,984 | ||||
| S6–8 | RCHO | + | * | RCHO * | 3.847 | 26,980 | 2.246 | 8511 | ||||
| S9–10 | R1OR2 | + | * | R1OR2 * | 4.157 | 17,646 | 2.531 | 17,426 | ||||
| 1 R11–13 | RCH2OH * | RCHO * | + | H2 | 0.044 | 0 | 2 | 9.685 | 6 | |||
| 2 R14–15 | RCHO * | + | C2H5OH * | RCH2OH * | + | CH3CHO * | 10.316 | 29,995 | 6.826 | 1070 | ||
| 3 R16–17 | nC3H5X * | + | C2H5OH * | nC3H7X * | + | CH3CHO * | 9.675 | 9857 | 5.752 | 24,926 | ||
| 1 R18–20 | RC2H4OH * | RC2H3 | + | H2O * | 2.375 | 1842 | 7.433 | 6359 | ||||
| R21 | AA * | + | AA * | CrA * | + | H2O * | 12.453 | 25 | 7.434 | 2398 | ||
| R22 | AA * | + | EtOH * | CrOH * | + | H2O * | 9.944 | 30,000 | 9.410 | 23,775 | ||
| R23 | EtOH * | + | AA * | BA * | + | H2O * | 9.384 | 4 | 8.357 | 73 | ||
| R24 | EtOH * | + | EtOH * | nBuOH * | + | H2O * | 5.380 | 29,822 | 4.023 | 20,025 | ||
| R25 | nBuOH * | + | EtOH * | nHexOH * | + | H2O * | 7.380 | 12,964 | 8.062 | 16,396 | ||
| R26 | EtOH * | + | EtOH | Et2O * | + | H2O * | 8.189 | 2546 | 9.778 | 10,303 | ||
| R27 | nBuOH * | + | EtOH * | nBuOEt * | + | H2O * | 7.599 | 1 | 8.766 | 11,444 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makoye, A.; Vikár, A.; Nacsa, A.B.; Barthos, R.; Valyon, J.; Lónyi, F.; Nagy, T. Experimental and Kinetic Modelling Study of the Heterogeneous Catalytic Conversion of Bioethanol into n-Butanol Using MgO–Al2O3 Mixed Oxide Catalyst. Catalysts 2025, 15, 709. https://doi.org/10.3390/catal15080709
Makoye A, Vikár A, Nacsa AB, Barthos R, Valyon J, Lónyi F, Nagy T. Experimental and Kinetic Modelling Study of the Heterogeneous Catalytic Conversion of Bioethanol into n-Butanol Using MgO–Al2O3 Mixed Oxide Catalyst. Catalysts. 2025; 15(8):709. https://doi.org/10.3390/catal15080709
Chicago/Turabian StyleMakoye, Amosi, Anna Vikár, András Bence Nacsa, Róbert Barthos, József Valyon, Ferenc Lónyi, and Tibor Nagy. 2025. "Experimental and Kinetic Modelling Study of the Heterogeneous Catalytic Conversion of Bioethanol into n-Butanol Using MgO–Al2O3 Mixed Oxide Catalyst" Catalysts 15, no. 8: 709. https://doi.org/10.3390/catal15080709
APA StyleMakoye, A., Vikár, A., Nacsa, A. B., Barthos, R., Valyon, J., Lónyi, F., & Nagy, T. (2025). Experimental and Kinetic Modelling Study of the Heterogeneous Catalytic Conversion of Bioethanol into n-Butanol Using MgO–Al2O3 Mixed Oxide Catalyst. Catalysts, 15(8), 709. https://doi.org/10.3390/catal15080709