1. Introduction
The World Health Organization estimates that globally 663 million people do not have access to safe, clean water, which leads to a child dying every 90 seconds from water-related disease. Current wastewater treatment technologies such as adsorption concentrate or separate pollutants from water, but do not completely eliminate them.
Of particular concern is the widespread use of dyes, particularly in the textile, leather, paper and pharmaceutical sectors. The reoxygenation process and photosynthesis of living organisms in the aquatic ecosystem can be disrupted by the discharge of azo dye-containing effluent [
1]. In addition, due to their toxicity, carcinogenicity and mutagenicity, several synthetic dyes may pose human risks [
2]. For the treatment of wastewater containing azo dyes, it is still vital to continuously develop methods that are highly effective, ecologically safe and economically feasible.
A significant contribution to this problem has been made by utilizing alternative treatment methods that use highly reactive radical species—referred to as advanced oxidation processes (AOPs) [
3]. These include well-known methods such as the decomposition of H
2O
2 with a homogeneous catalyst in the Fenton reaction, electrochemical processes or using UV radiation with or without a photocatalyst, as well as less widespread methods such as plasma-initiated photolysis [
4] or the collapse of microbubbles [
5]. However, these AOPs have high running costs or are energy intensive, due to the use of expensive reagents (an excess of stoichiometric oxidants) or associated energy costs (for example, producing H
2 to generate hydrogen peroxide); a recent Special Issue, “Advanced Catalytic Material for Water Treatment”, published in Catalysts in 2023 [
6], describes recent research in this area.
One of the most widely studied AOPs is the use of photocatalysts that rely on the irradiation of UV or visible light to create electron–hole pairs in semiconducting catalysts. When the photocatalysts are irradiated by light with energy higher than the band gap, electrons are promoted from the valance band to the conductance band, leaving behind a positively charged hole in the valence band. The electrons and holes react with water and dissolved oxygen to form radical species that can degrade the pollutant molecules. This is potentially a low-cost process as it can use solar radiation; however, there are issues that hinder its effectiveness. The early studies used inexpensive TiO
2 photocatalysts, although these catalysts are limited by a large band gap that only allows light to be absorbed in the ultraviolet region, which makes up less than five percent of sunlight. Often, the TiO
2 is modified by dopants, either to hinder the fast recombination of electron–hole pairs or to decrease the band gap to utilize more of the visible spectrum [
7]. These disadvantages with TiO
2 and other single-component photocatalysts led to the development of second- and third-generation multicomponent photocatalysts to improve performance [
7]. However, even with these new catalysts, the reported performances that give >90% degradation take tens of minutes if not hours to achieve [
8]. There are also issues associated with treating water when it is dark or if the water is not clear, which limits the penetration depth of the light.
To address the limitations of photocatalysis, this study will investigate microwave-assisted catalysis as a low-cost, low-energy process. There have been several previous studies using microwave radiation to drive the removal of chemical pollutants. However, many of these utilise microwaves in conjunction with other methods (UV radiation or H
2O
2) which increase the cost and energy input of the processes [
8]. Microwave-assisted catalysis with semiconductors has the advantage of using the same mechanism as photocatalysis, the creation of electron–hole pairs, although these are created by heating the catalyst rather than using light, without the need for expensive additional oxidants. However, even though this field is maturing, many studies still require high microwave power (100–750 W) and long reaction times (5–60 min) [
8].
In this study, we report on the use of a simple NiO catalyst for the microwave-assisted degradation of two azo dyes, azorubine and methyl orange (MO), with conversions of >95% in only 10 s with 100 W microwave power.
2. Results
The NiO catalysts prepared using the microwave synthesis methods (NiO 1 and NiO 2) and the commercial material (NiO 3) were characterised using a combination of X-ray diffraction (XRD), Raman spectroscopy and scanning electron microscopy (SEM) to gain insights into their structure and properties.
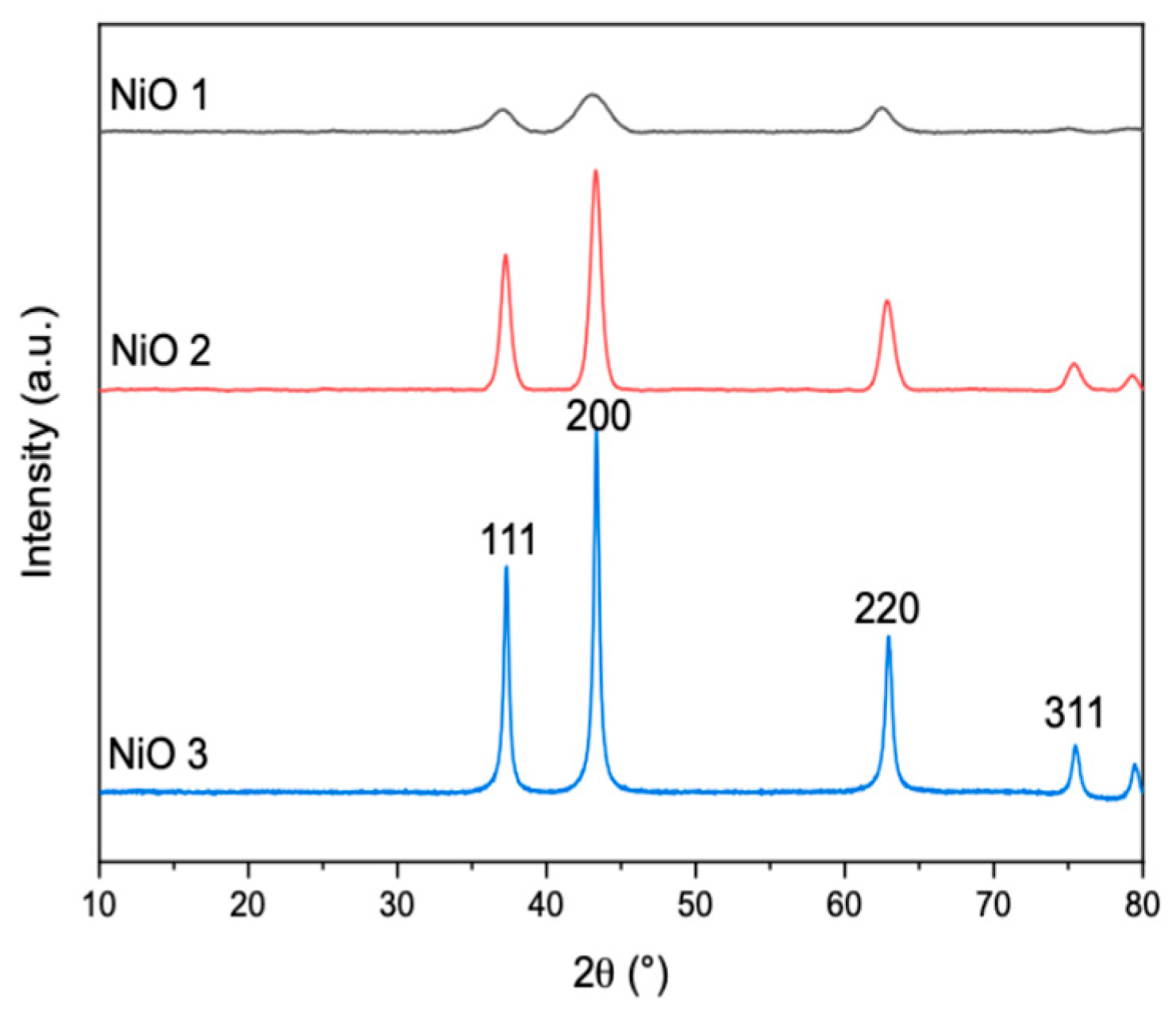
The reflections observed in the XRD patterns (
Figure 1) were consistent with previous studies [
9,
10,
11]. All the spectra had similar patterns, with reflections at around 37°, 44°, 63° and 76° 2θ, which can be indexed as the (111), (200), (220) and (311) crystal planes of the face-centred cubic NiO phase, respectively [
10], with the additional partial peak around 80° attributed to the (222) plane [
10].
From the signal-to-noise ratio, the commercial NiO appears to be more crystalline and the application of the Scherrer equation to the (200) reflection gave domain sizes for NiO 1, NiO 2 and NiO 3 of 67, 73 and 104 nm, respectively. It should be noted that the Scherrer equation has several limitations depending on the size and shape of the particles and the strain or disorder in the materials [
12]. The calculated domain sizes correlated with BET analysis, which gave higher surface areas for smaller primary particle sizes, as shown in
Table 1. In this study, NiO was found to be microporous and low-pressure measurements were used to obtain the surface area.
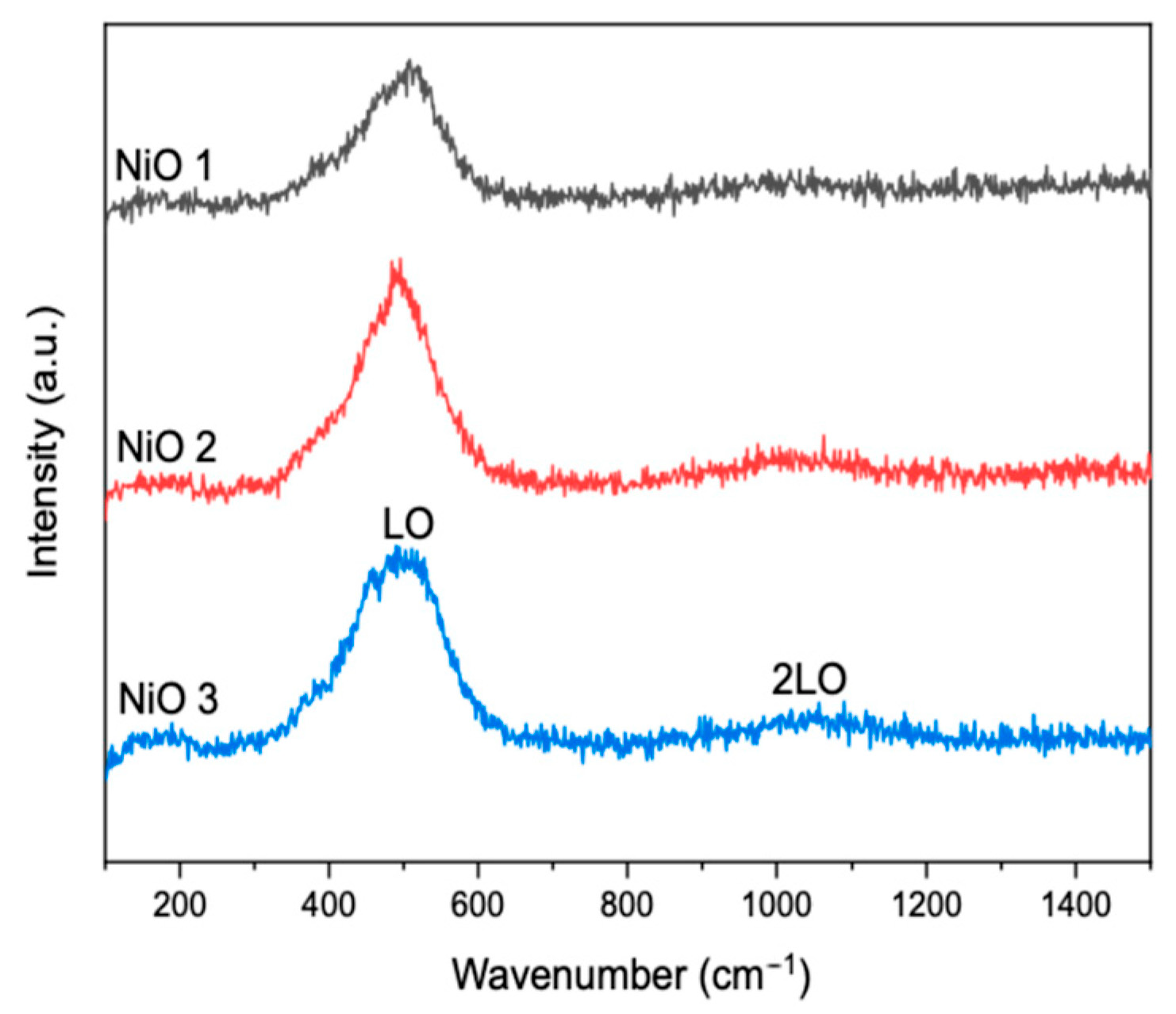
Raman spectroscopy was carried out to gain insight into the defect structure of the different NiO materials. The peaks observed in the Raman spectra (
Figure 2) were as previously reported, which further confirmed the existence of NiO [
13,
14]. The peaks at ~500 and ~1050 cm
−1 were attributed to first-order longitudinal optical (LO) and second-order longitudinal optical (2LO) phonon modes of NiO, respectively [
14,
15].
Liu et al. suggested that stoichiometric NiO should not give rise to first-order Raman scattering due to its rock salt structure [
16]. The first-order phonon LO scattering was proposed to be due to the presence of nickel (Ni
2+) vacancy defects, which break down the selection rules [
16,
17]. These defects are advantageous as they provide high energy sites for pollutant adsorption on the catalyst surface [
18]. In addition, Ghandi et al. reported that an increased intensity of the LO peak with a simultaneous decreased 2LO peak intensity, which was observed in the spectra of all the catalysts, was indicative of an increased percentage of Ni
2+ vacancies [
19].
The morphology of the catalysts was investigated using SEM and representative micrographs are shown in
Figure 3.
The three NiO materials studied were found to have very different morphologies. NiO 1 showed a flake-like morphology, with thin sheets of NiO arranged in layers. These thin layers gave rise to particles which appeared thinner than those for the NiO 2 and NiO 3 catalysts, which were three-dimensional. NiO 2 showed a more homogeneous particle size distribution of small, spherical particles. Although agglomeration was observed, since magnetic nanoparticles are likely to stick together, the primary particles were more uniform in size than NiO 1 and NiO 3. The commercial NiO 3 showed a powder morphology with random-shaped particles and a wide particle distribution.
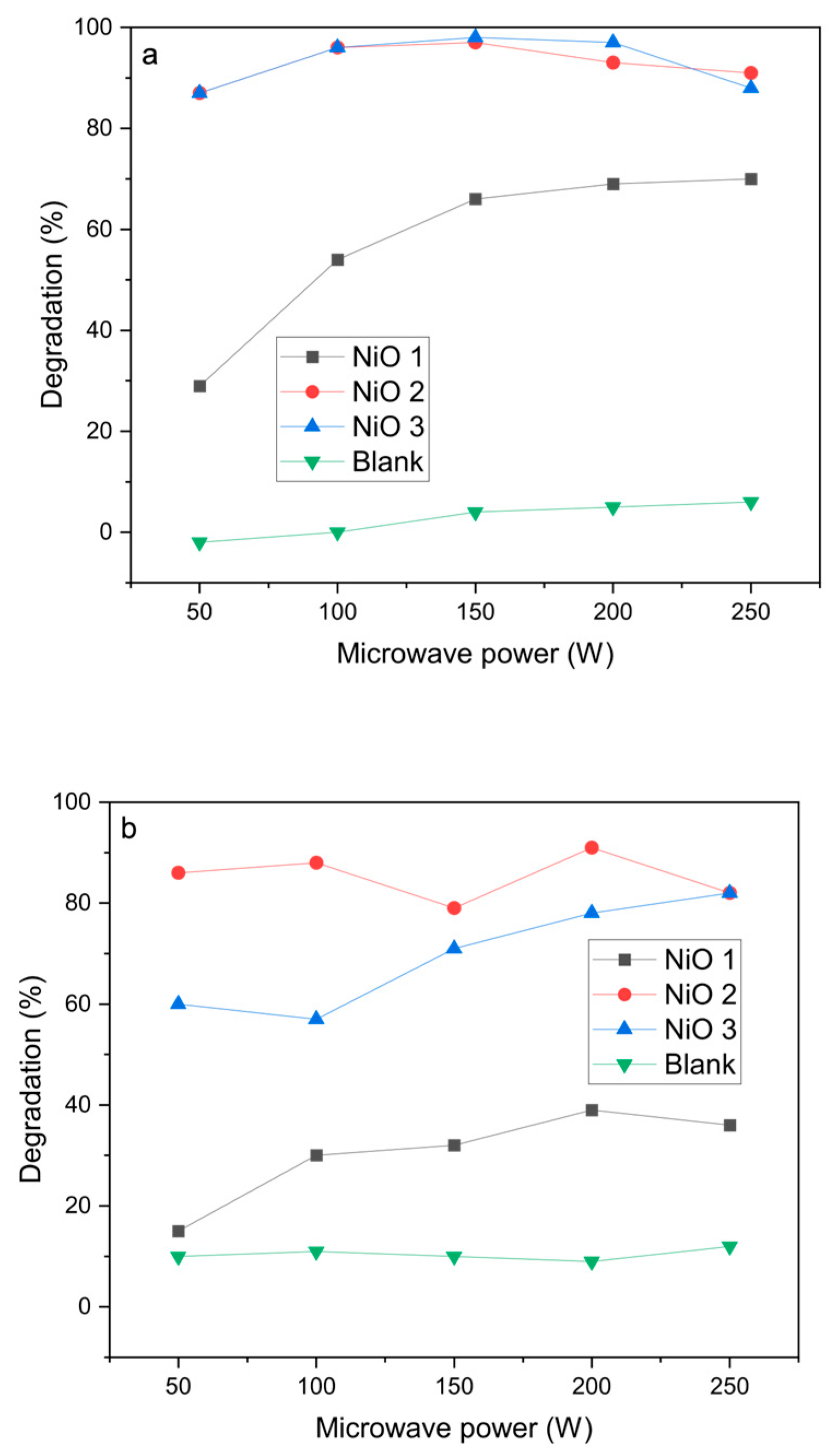
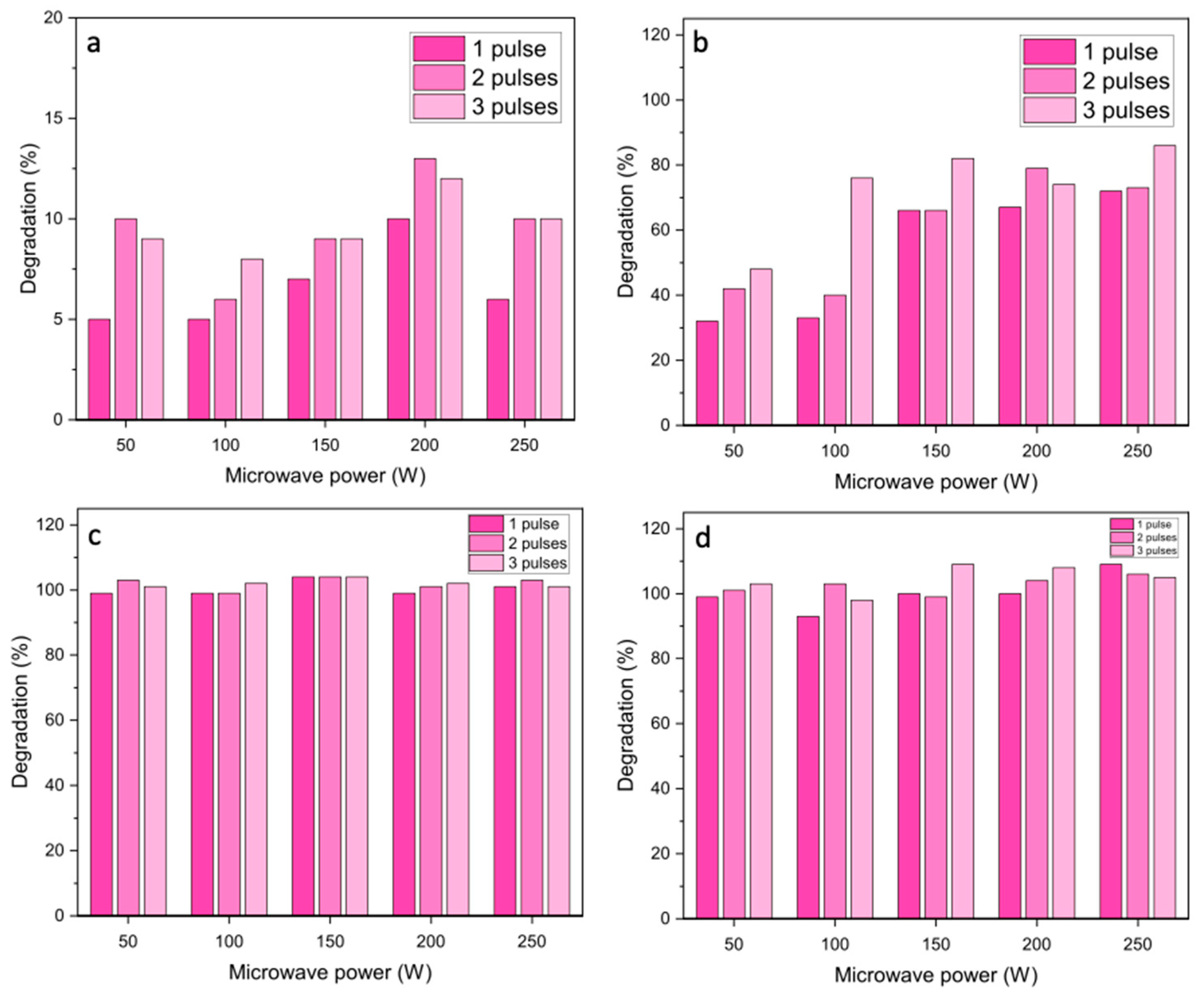
The NiO materials were then tested as catalysts for the microwave-assisted degradation of azo dyes. Initial experiments were carried out using a single 30 s pulse at different powers between 50 and 250 W (
Figure 4). Blank reactions without the catalysts showed very low degradation efficiencies under microwave heating, which confirms that microwave radiation alone is not able to decompose the azo dyes.
For azorubine, the degradation was very high, reaching 97% at 100 W power with both NiO 2 and NiO 3 catalysts. However, at higher power, the conversion decreased slightly, which was attributed to the pressure cut-off being reached in the sealed vessels due to autogenous pressure as the temperature increased. This led to the microwave radiation input not being constant over the 30 s pulse length, reducing the time the materials were irradiated and leading to a decrease in conversion. Methyl orange had a lower degradation rate under these conditions, but was still found to give >90% over the NiO 2 catalyst at 100 W microwave power. The conversion did not increase significantly at higher microwave powers, which was attributed to the temperature increase, which led to the microwave radiation not being constant over the 30 s pulse length.
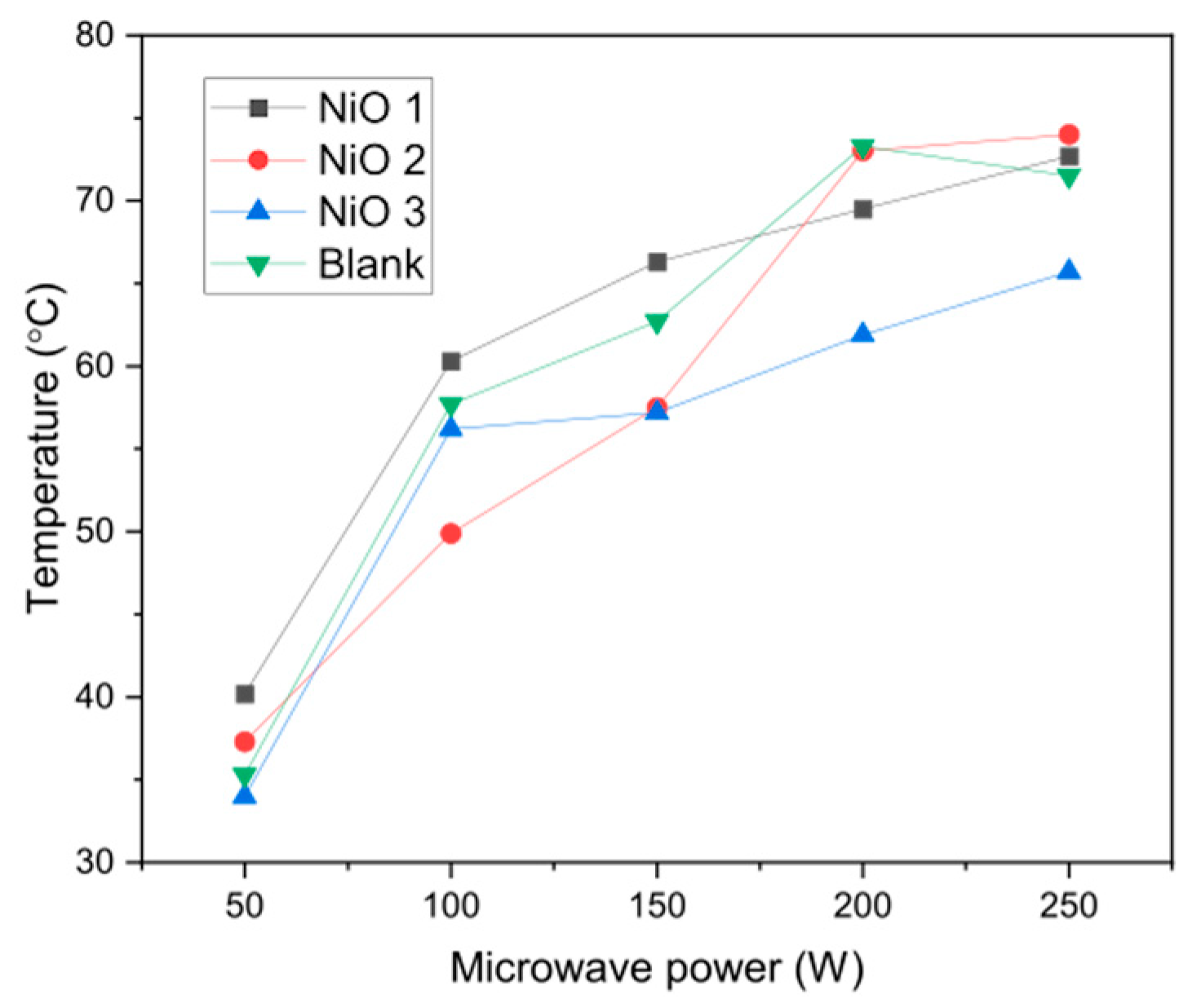
To confirm this hypothesis, the temperature of water after a 30 s pulse of microwave irradiation at different powers is shown in
Figure 5. The temperature reached around 75 °C at 200–250 W, although it is thought that selective heating of the NiO nanoparticles means that the active sites on the surface would be much higher than the bulk water temperature.
Following these initial observations, the experimental procedure was adapted to ensure that microwave power was constant for the whole of the reaction, allowing constant heating of the catalysts. Experiments were carried out using a duty cycle of three 10 s pulses with 1 min between pulses to limit the temperature of the water and avoid the pressure increases that stop the microwave power. Degradation was measured after each microwave pulse to study the effect of irradiation in 10 s increments.
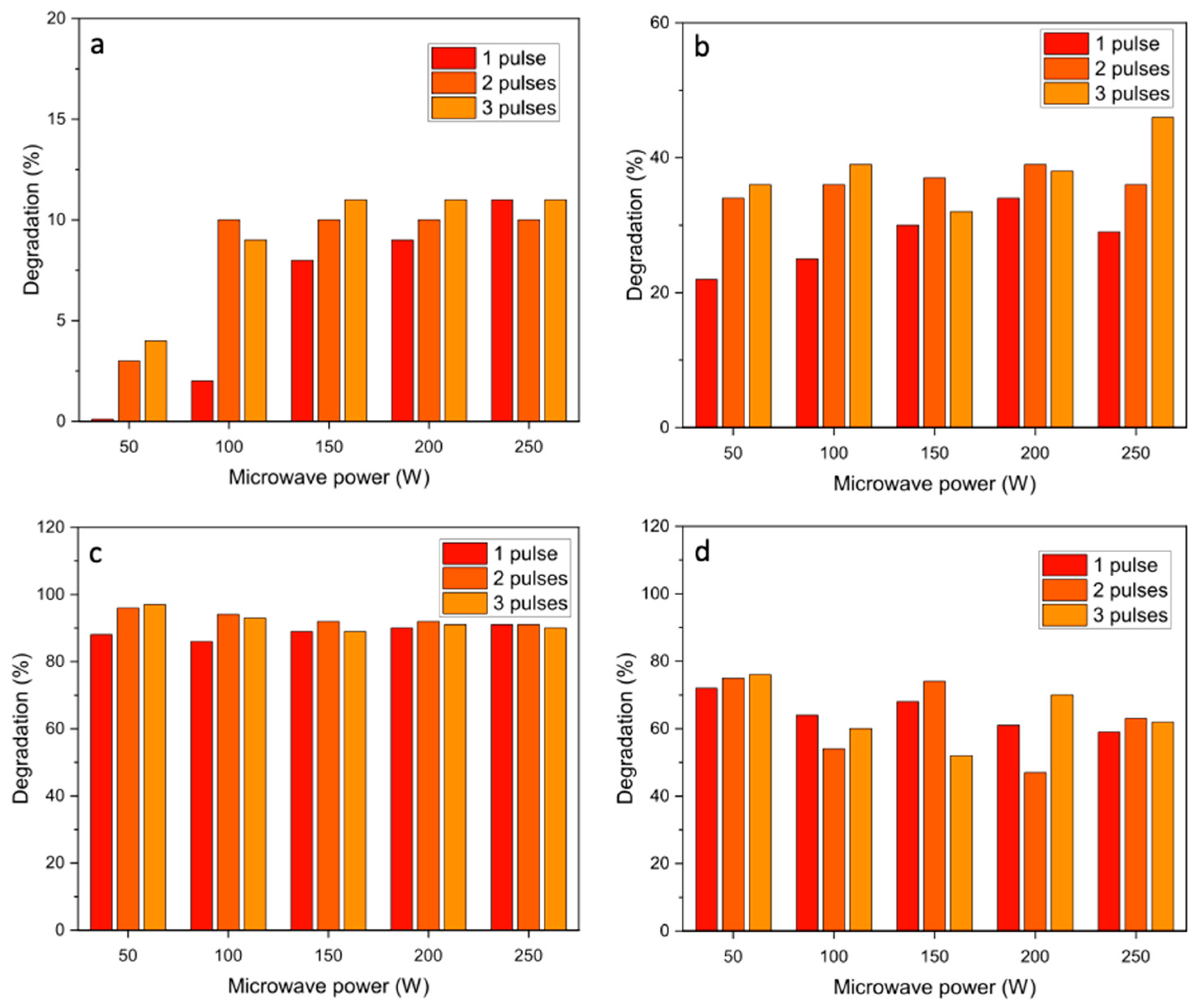
As expected from the 30 s pulse experiments, the absence of a catalyst resulted in low degradation of azorubine (
Figure 6). The presence of the NiO catalysts increased degradation efficiencies substantially, some more effectively than others. For the NiO 1 catalyst, the overall degradation typically showed a gradual increase over the three pulses to give a maximum of 80% conversion at microwave powers >80%. The final degradation after three 10 s pulses of microwave radiation was higher than for one 30 s pulse (
Figure 4), which is attributed to microwave irradiation being constant over the 30 s of pulsing, due to the lower water temperature during the reaction avoiding the pressure cut-off on the instrument. For the NiO 2 and NiO 3 catalysts, the degradation of azorubine reached ≥99% after the first 10 s pulse of irradiation at all microwave powers, demonstrating the efficiency of the microwave-assisted degradation performance. It is worth noting that since the degradation efficiencies with NiO 2 and NiO 3 were consistently reported at ≥99%, fluctuations observed in degradation are assumed to be within the experimental error, rather than an indication of catalyst deactivation.
Methyl orange degradation was not as efficient as azorubine degradation under these reaction conditions (
Figure 7), indicating that this azo dye is more difficult to break down. All the catalysts gave lower degradation rates for methyl orange than for azorubine, although NiO 2 showed a high conversion of both azo dyes, with >95% degradation observed after two 10 s pulses. Due to the very high conversions with short reactions, it was not possible to explore the kinetic behaviour using Arrhenius plots, which should have reaction rates determined at low conversions, and the methodology used did not allow reactions to be performed at different temperatures. However, the reaction rates for the microwave-assisted process have been calculated as the mass of azo dye converted per mass of catalyst per minute and compared with published studies of advanced oxidation processes that have investigated the degradation of methyl orange and azorubine in water (
Table 2 and
Table 3).
As can be seen, the rates of reaction for the microwave-assisted process are orders of magnitude better that for photocatalytic processes. Some studies using peroxymonosulfate (PMS) that is activated over catalysts to give highly reactive radical species show similar reaction rates, although this is a less sustainable route than the microwave-assisted process, which does not require additional reactants to carry out the degradation process. In our initial studies, we used high catalyst amounts, and there is scope for these to be considerably reduced as the process is studied further. Most degradation studies only report the conversion of the azo dye molecules and do not address the intermediate products formed, or the extent of mineralisation. However, some studies have used total organic carbon analysis or gas/liquid chromatography coupled with mass spectrometry to identify the reaction products [
25,
26,
27]. Although analysis of the products was not carried out in the current study, it is likely that a similar product profile of aromatic intermediates is formed that then further degrades to carbon dioxide and water.
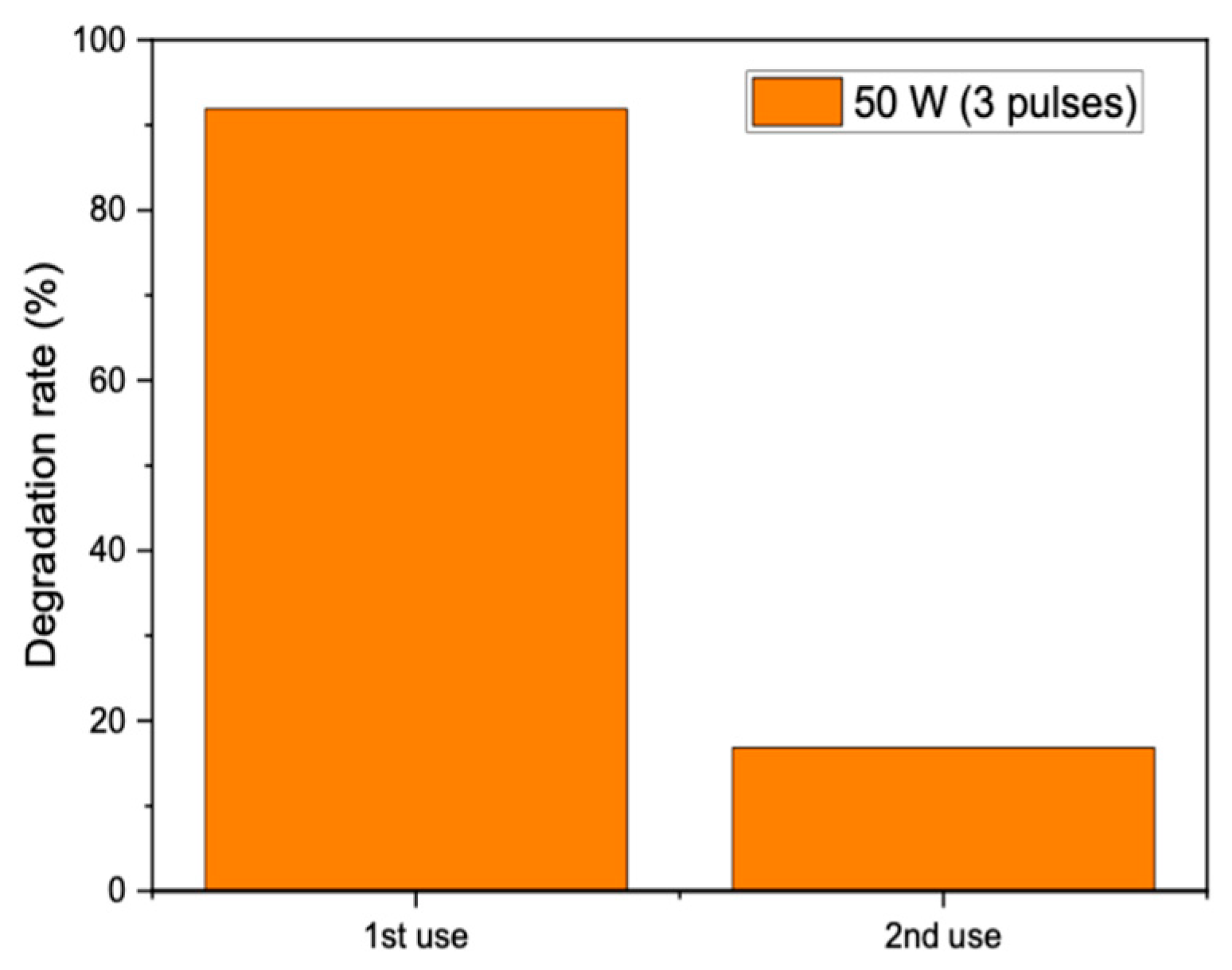
The reusability of the catalysts was probed by removing the catalyst from the solution after three 10 s pulses of microwave radiation and performing a second reaction with a fresh solution of methyl orange. Unfortunately, the catalyst was found to give poor performance on reuse, with the degradation dropping from 92% over the fresh catalyst to 17% when reused in a second reaction (
Figure 8). This could be due to poisoning by degradation products remaining adsorbed on the catalyst after the reaction or sintering of the catalyst on microwave heating, leading to a reduction in surface area. More experiments are needed to identify the deactivation mechanism and identify possible pretreatments to reduce the deactivation.
3. Discussion
For materials to be active for microwave-assisted catalysis, they need to be heated in microwave fields and contain active sites to perform the required reaction. The behaviour of materials under microwave heating is dependent on their complex permittivity and/or permeability, which indicate the ability to store energy and how well it converts the energy to heat in the microwave–electric and microwave–magnetic fields, respectively. This study used a standard laboratory microwave that utilises both the electric and magnetic components of the electromagnetic radiation and so materials could be heated by either of these mechanisms. NiO has suitable magnetic and dielectric properties and so can be heated in either the microwave–magnetic or microwave–electric fields. The microwave properties of NiO materials have been well studied, and it has been found that the magnetic loss tangent is an order of magnitude smaller than that of the electric loss tangent, which indicates that electric field heating will be more efficient for these materials [
28]. Microwaves typically have a fairly small penetration depth in water of only a few centimetres [
29]; however, the constant stirring and small diameter of the reaction vessel ensure that the NiO catalysts are constantly exposed to microwave radiation to allow them to be heated successfully.
In microwave-assisted degradation, several studies only consider heating as the mechanism for degrading molecules, and thermal cracking has been proposed as a mechanism [
30], as well as the production of hydroxyl radicals at hotspots on the catalyst surface through reaction with dissolved oxygen [
31]. However, there has also been the suggestion that metal-oxide-based semiconductor catalysts can interact with microwaves in a similar manner to photocatalysts (although wide-band semiconductors can be used) [
31,
32], with electron–hole pairs formed that can react with adsorbed water to form superoxide and hydroxyl radicals [
8]. However, the energy of microwave radiation is not high enough to directly promote electrons across the band gap, and this mechanism is again attributed to the permittivity of the metal oxides and their interaction with the microwave electric field to generate heat to promote electrons from the valence band to the conduction band. As NiO is a semiconductor with a bandgap of 3.5–3.8 eV [
33] that can act as a microwave susceptor, either mechanism is possible. Zhang et al. investigated the microwave degradation of methyl orange using TiO
2 supported on activated carbon (AC) [
34]. They proposed a mechanism where the AC gave rise to hotspots on the surface, which could mineralise the organic pollutants and form
•OH and
•H radicals from water, whilst at the same time heating the TiO
2 semiconductor to generate electron–hole pairs that would react with O
2 and H
2O to form
•OH and the superoxide radical anion (O
2•−). As TiO
2/AC is a composite material, the synergistic effect could be demonstrated over the individual components, which adds weight to this hypothesis.
For photocatalytic systems, the formation of electron–hole pairs can be probed in situ using photoluminescence (PL) spectroscopy [
35], which measures the light emitted when electrons fall back to the valence band from the conduction band, or electrochemical impedance spectroscopy (EIS) [
36], which can infer how quickly electrons and holes move in a material. Although it is relatively easy to undertake these spectroscopic methods while irradiating the sample with visible or UV radiation, it is not straightforward to carry out these in situ experiments with microwave-assisted catalysis.
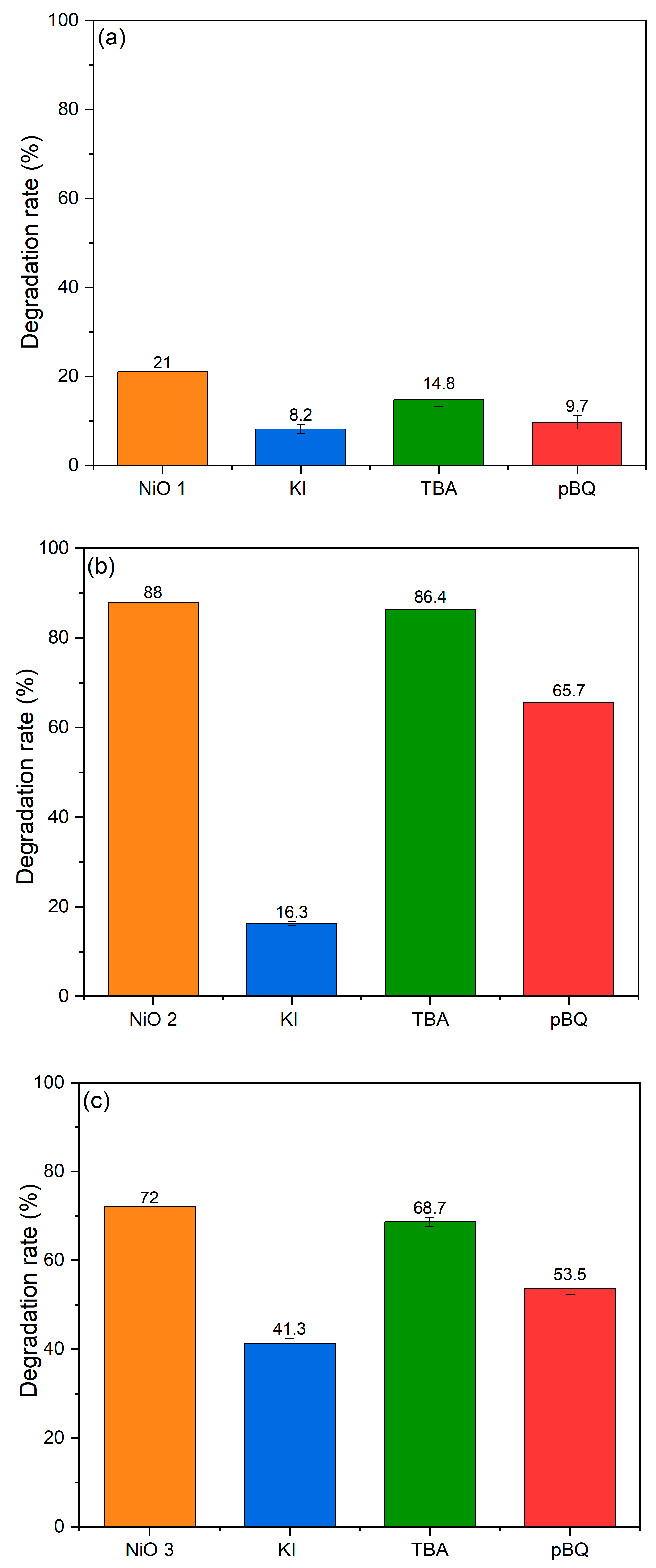
In this study, we have attempted to investigate the electron–hole pair formation using trapping agents as an indirect probe. The methyl orange degradation reaction was rerun, adding either potassium iodide (KI) to scavenge the positive holes (h
+), tertiary butanol (TBA) to scavenge hydroxyl radicals or benzoquinone (pBQ) to scavenge superoxide radicals (
Figure 9). Under these reaction conditions, observed reductions in degradation efficiency compared to the standard reaction could be attributed to the species that was removed during the trapping experiments. Over the NiO semiconductor, the different species can be produced from water and the dissolved oxygen in solution through reactions with the electron–hole pairs formed via the reactions below:
The azo dyes can then be degraded by reacting with either the holes or the radical species formed. The results of the trapping experiments in
Figure 9 clearly show that a combination of reactive species are playing a role in the degradation of methyl orange. When KI was added to scavenge the holes, there was a considerable drop of >80% in the degradation rate over NiO 2 compared with reductions of 60% over NiO 1 and 40% over NiO 3. When nBQ was used to scavenge the superoxide radicals, the reduction in degradation rates was smaller, with >50% reduction over NiO 1 and 25% reduction over NiO 2 and NiO 3. With TBA as a trapping agent, there was a very small reduction in the degradation efficiency over NiO 2 and NiO 3, but a 30% decrease in efficiency over the NiO 1 catalyst. It can be inferred from these results that electron–hole pairs are formed over all three catalysts, suggesting that the formation of electron–hole pairs is the underlying mechanism, with positively charged holes resulting in the bulk of the degradation for all catalysts.
Some general trends can be drawn from the structure–activity relationships of the three catalysts from the characterisation data. Maniammal et al. investigated the role of vacancies in NiO for the photodegradation of methylene blue [
37], which can be extrapolated to the present study that uses microwave radiation to heat to form the electron–hole pairs rather than UV light. The authors proposed that Ni
2+ vacancies can exist in either a 0, −1 or −2 charge state, which represents (i) uncompensated; (ii) partially compensated; and (iii) completely compensated conditions, respectively. Each uncompensated Ni
2+ vacancy generated two holes in the oxygen valence band (of NiO), even in the absence of irradiation [
37]. Irradiation and the subsequent absorption of photons results in the partially compensated Ni
2+ vacancies generating one hole, while completely compensated vacancies did not form holes. The holes generated on irradiation (or heating) are additional to the existing electron–hole pairs, which can enhance pollutant degradation by generating more
•OH. Furthermore, the presence of the vacancy defects can reduce the band gap energy for NiO, which facilitates the additional formation of electron–hole pairs [
9]. Therefore, due to the high peak intensity of LO and lower peak intensity of 2LO observed in the Raman spectra (
Figure 2), it can be concluded that all the NiO catalysts had a relatively high concentration of Ni
2+ vacancy defects on their surfaces. However, the LO peak for NiO 1 was less than those for NiO 2 and NiO 3, which suggested that it had a lower Ni
2+ vacancy concentration. This could explain why NiO 1 exhibited the lowest catalytic activity for azo dye degradation (
Figure 6 and
Figure 7). In a study of NiO properties under microwave heating by Yoshikawa and Sato, they found that at room temperature there was a small dielectric loss, but as the temperature increased, the conductance of NiO increased rapidly, indicating that the promotion of electrons across the band gap occurred rapidly [
38]. This suggests that the overriding mechanism is reliant on electron–hole pair generation, although hotspots generated on the catalysts could increase the rate of this process.
Microwave heating has also been shown to be influenced by the size and shape of the nanoparticles being heated. Previous studies on the temperature dependence and shape effect in the high-temperature microwave heating of nickel oxide powders found that spherical particles gave a uniform adsorption per volume, but shape effects can enhance the magnetic loss as the temperature increases [
39]. Hou et al. reviewed the morphological effects of metal oxides and concluded that elongated or irregular particles adsorb microwave radiation more efficiently than spherical particles [
40]. They suggest that thin 2D nanosheets are easier to heat in microwave fields due to their higher surface area and increased penetration depth. However, for nanocrystalline materials, the domain size within particles is also an important parameter for microwave heating [
41]. In the oscillating magnetic field used in this study with NiO materials, both electric-field and magnetic-field heating were important. The magnetic field affects the electron spin, domain wall and orientation of domains, with additional magnetic losses such as hysteresis, eddy current, domain wall resonance and electron spin resonance. For three-dimensional particles, there are more domain interactions, leading to larger heat loss interactions, generating uniform heating. For thicker materials, eddy current loss in particular is much more prevalent, leading to an increase in temperature [
42,
43]. This could explain the reason for the poorer performance of NiO 1, which has a platelet morphology.
Although NiO 2 and NiO 3 showed similar performance for azorubine degradation, there was a decrease in activity for the commercial material NiO 3 for methyl orange. For both the 30 s pulses with azorubine and the 10 s pulses with methyl orange, NiO 3 showed a general decrease in MO degradation with increased microwave radiation power. The MO degradation efficiencies obtained from this experiment suggested that NiO 3 was deactivating with increasing microwave radiation. One explanation could be the sintering of the catalyst nanoparticles with increasing microwave radiation power (and subsequent increasing temperature), which would result in an increase in domain size and a reduction in the catalytic surface area, which was previously reported by Akbari et al. [
44]. Sintering will also increase the crystallinity and reduce the number of defects in the structures, further reducing the ability of NiO 3 to be heated, decreasing the overall activity.
4. Materials and Methods
4.1. Catalyst Synthesis
Ni(II) acetate was dissolved in 100 mL distilled water to form a 0.2 M aqueous solution. Then, 25 mL of the aqueous Ni(II) acetate solution was mixed with 25 mL of a 0.4 M aqueous solution of sodium hydroxide under stirring. 25 mL of a 0.2 M aqueous solution of acetylacetone was then added to the reaction mixture, and the resulting blue–green solution was heated in a CEM Discover microwave synthesizer (CEM Microwave Technology Ltd., Buckingham, UK) at 300 W for 10 min. The resultant light green precipitate was recovered by filtration, washed with 500 mL distilled water and dried overnight in air at room temperature. The precipitate was then calcined in air at 400 °C for 4 h to obtain NiO. This material was denoted NiO 1.
Ni(NO3)2·6H2O was dissolved in 50 mL distilled water to form a 0.2 M aqueous solution. The solution was vigorously stirred and drops of 35 wt.% ammonia solution were added until the solution reached pH 9. The resulting solution was heated in a CEM Discover microwave synthesizer (CEM Microwave Technology Ltd., Buckingham, UK) at 300 W for 1 min. The resultant light green precipitate was recovered by centrifugation (VWR Mega Star 600, (Avantor, Lutterworth, UK) at 4200 rpm, washed several times with 20 mL distilled water and 20 mL ethanol and dried in an oven at 110 °C for 12 h. The precipitate was then calcined in air at 400 °C for 2 h to obtain NiO. This catalyst was denoted NiO 2.
For comparison, commercial nickel(II) oxide (Sigma Aldrich, Merck Life Science UK Ltd, Gillingham, UK) was used to benchmark the synthesised catalysts, and this material was denoted NiO 3.
4.2. Preparation of the Azo Dye Solutions
The carmoisine solution, which is commonly known as azorubine solution, was prepared by dissolving 25.0 mg of the dye in 1 L of distilled water, to produce a 49.76 μmol/L solution.
The methyl orange solution was prepared by dissolving 16.6 mg of the dye in 1 L of distilled water, to produce a 49.76 μmol/L solution.
4.3. Catalyst Testing for Azo Dye Degradation
The degradation experiments followed a standard procedure for both azo dye solutions and all catalysts.
The azo dye solution (3 mL, 49.76 μmol/L) and NiO catalyst (5 mg) were added to a 10 mL pressure vessel (CEM Microwave Technology Ltd., Buckingham, UK) designed for use in the CEM Discover microwave synthesizer (CEM Microwave Technology Ltd., Buckingham, UK). A magnetic stirrer was added to allow the uniform dispersion of the catalyst and the vessel placed in the microwave cavity and the solution was irradiated under a single 30 s pulse, at microwave powers of 50–250 W. Following reaction, the solution was centrifuged using a Camlab 1190945 D1008 Mini Centrifuge (Camlab Ltd., Cambridge, UK) to separate the NiO catalyst.
The same process was conducted with a cycle of three 10 s pulses with a 60 s dwell time between each pulse to avoid over-heating the solutions during microwave heating.
The degradation of the dyes was quantified using a UV–Visible AvaSpec-2048 Fiber Optic Spectrometer (Anglia Instruments Ltd., Soham, UK) with an AvaLight-DH-S-BAL deuterium halogen source, with the absorption of the azorubine and methyl orange dyes measured at 515 and 465 nm, respectively. The degradation percentage was calculated using the equation below:
where
A0 and
At are the initial absorbance of the dye and the absorbance at time
t, respectively.
4.4. Catalyst Characterisation
The XRD patterns of the catalysts were obtained using a PANalytical X’Pert Pro diffractometer (Malvern Panalytical, Malvern, UK) fitted with a CuKα radiation source (λ = 1.5406 A°) operating at 40 kV and 40 mA. The samples were placed in an Al sample holder and scans were taken from a 2θ angle of 5° to 80° at a scan rate of 2° min−1 with a step size of 0.0167°. PANalytical Highscore was used to match the XRD data to the crystal group and identify the sample phase using the International Centre for Diffraction Data (ICDD) database.
Scanning Electron Microscopy carried out using a Hitachi TM3030 Plus scanning electron microscope (Hitachi High-Tech, Daresbury, UK). Catalysts were mounted onto the sample holder using carbon tape for imaging.
Raman spectroscopy of the catalysts was acquired using a Renishaw InVia Raman microscope (Renishaw plc, Wotton-under-Edge, UK) using an argon ion laser (514 nm). Spectra were collected over the range 100–3200 cm−1, using 10% laser power, with 10 s exposure time and 5 accumulations.
Surface area determination was carried out by 5-point N2 adsorption at −196 °C using a Quantachrome ChemBET chemisorption analyser (Quantachrome UK Limited, Hook, UK). Approximately 50 mg of each sample was degassed at 150 °C for 3 h under vacuum prior to analysis.



 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}