
Oxygen Reduction Reactions of Catalysts with Asymmetric Atomic Structures: Mechanisms, Applications, and Challenges


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Pathway and Mechanism of Oxygen Reduction Reaction
2.1. Two-Electron Pathway and Mechanism of Oxygen Reduction Reaction
2.2. Four-Electron Pathway and Mechanism of Oxygen Reduction Reaction
2.3. Research on Reaction Pathways
2.3.1. Rotating Ring–Disk Electrode Technique
2.3.2. In Situ Raman Spectroscopy
2.3.3. In Situ ATR-SEIRAS
2.3.4. Density Functional Theory
2.3.5. X-Ray Absorption Near-Edge Structure
2.3.6. Fourier Transform Infrared Spectroscopy
3. Control of Oxygen Reduction Reaction Pathways
3.1. Control of Two-Electron Pathways
3.2. Control of Four-Electron Paths
3.2.1. Incorporation Forms of Nonmetallic Elements
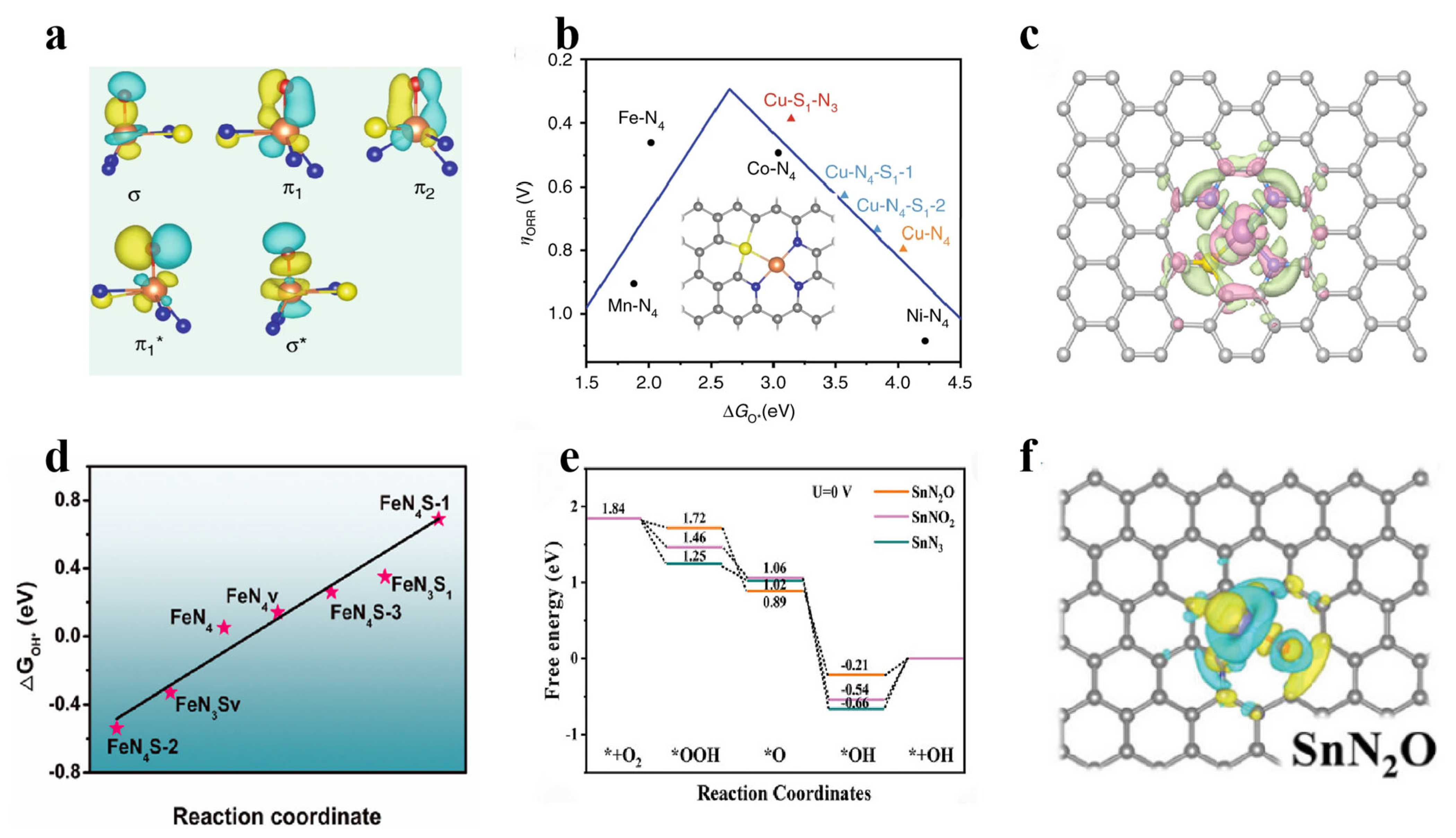
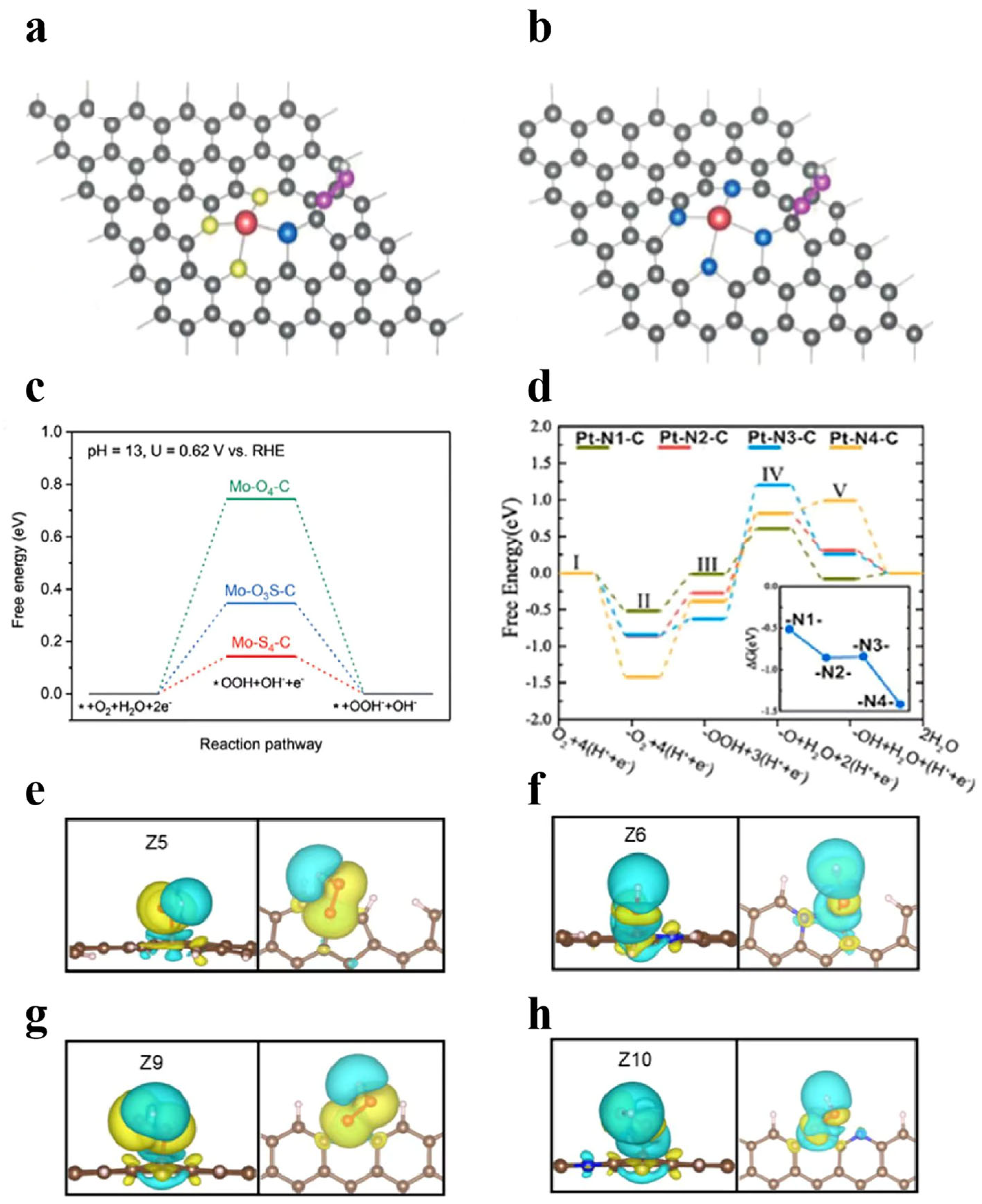
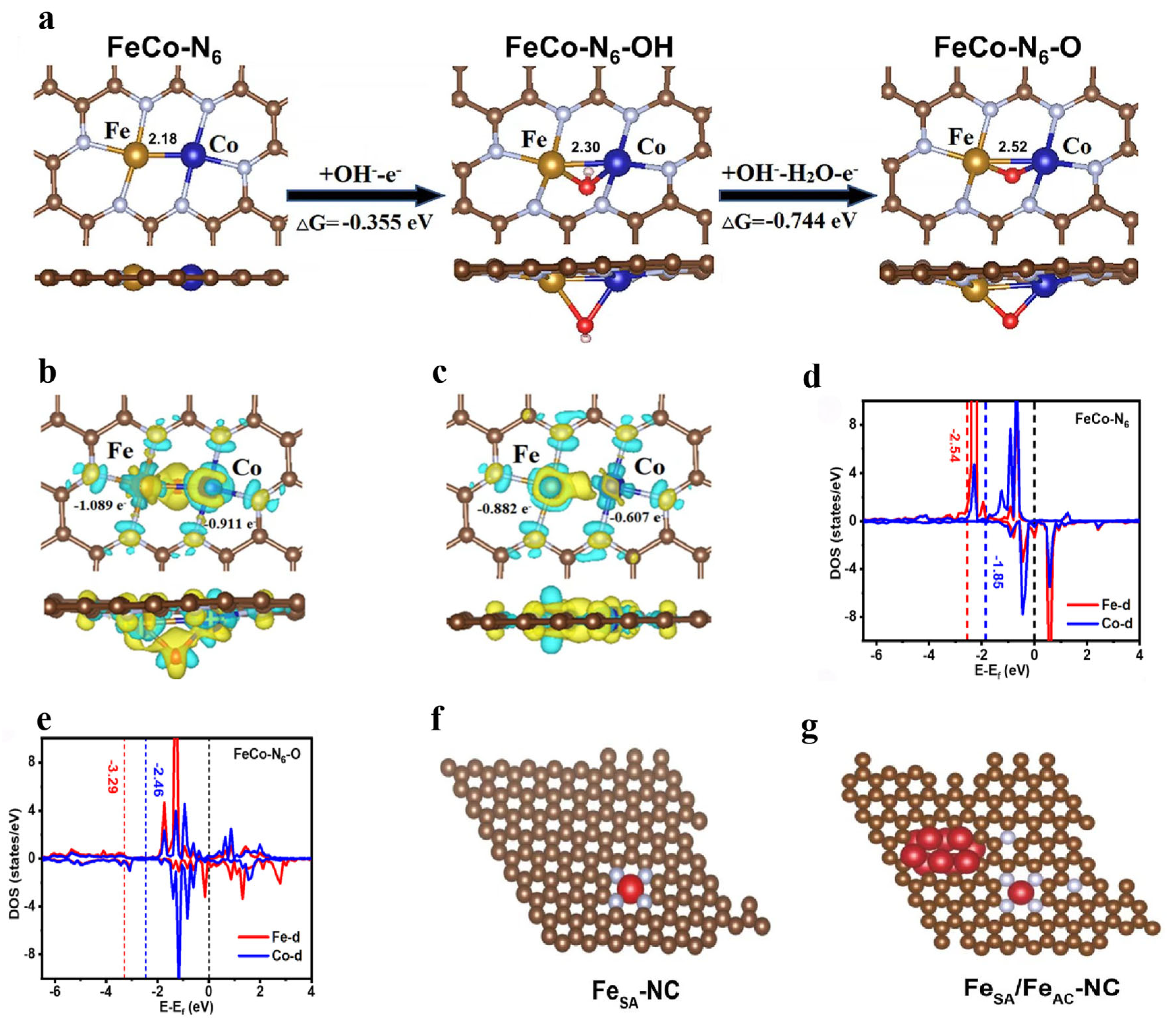
3.2.2. Co-Incorporation Forms of Nonmetallic and Metallic Elements
3.2.3. Other Incorporation Forms (Functional Groups, Defect Sites, Metallic Clusters, etc.)
4. Application of Oxygen Reduction Reaction
4.1. Device for Producing H2O2
4.2. Zinc–Air Battery
4.3. Hydrogen–Oxygen Fuel Cell
5. Conclusions and Prospects
- (i)
- Design Principles for Metal Element Doping: In ORR catalysts, transition metal elements are the predominant choice for constructing asymmetric atomic structures owing to their unique electronic configurations and chemical properties. The partially filled d-orbitals of transition metals can hybridize with the antibonding *π orbitals of O2, modulating the activation energy for O-O bond cleavage. The position of the d-band center critically governs the adsorption strength of oxygen intermediates (e.g., *OOH, *O, *OH). Furthermore, transition metals exhibit multiple oxidation states (e.g., Fe2+/Fe3+, Co2+/Co3+), empowering reversible electron transfer during ORR. This redox flexibility allows their oxidation states to dynamically adapt across reaction steps, thereby facilitating catalytic progression. Additionally, the atomic size and electronegativity of transition metals promote the formation of asymmetric coordination structures with other atoms or ligands. Such configurations induce asymmetric charge distribution and electronic effects, optimizing the adsorption and reactivity toward ORR intermediates. Finally, transition metals possess high chemical and thermal stability, enabling sustained catalytic activity under ORR operating conditions (e.g., acidic/alkaline environments, high potentials) while resisting structural degradation or corrosion.
- (ii)
- Design Principles for Nonmetal Element Doping: In the fabrication of asymmetric-atomic-structure catalysts, the incorporation of high-electronegativity nonmetal elements induces charge polarization through their strong electron-withdrawing capability, creating localized electron density differences. This effectively modulates the electronic structure of the catalyst, enhancing its adsorption capacity for reactants and catalytic activity. Additionally, synergistic co-doping of multiple high-electronegativity nonmetal elements can be considered to achieve more significant catalytic performance improvements. Interactions between different dopants may collectively modulate the electronic and geometric structures of the catalyst, generating synergistic effects. For low-electronegativity nonmetal elements, their weak electron-withdrawing or electron-donating capabilities can modulate the charge distribution, surface reactive sites, and intermediate adsorption behavior of the catalyst structure. Selecting elements with atomic radii matching the substrate can reduce the risk of lattice distortion and preserves structural stability.
- (iii)
- Design Principles for Other Dopants: In asymmetric-atomic-structure ORR catalysts, defect engineering amplifies catalytic activity and selectivity by introducing vacancies, edge sites, and other asymmetric configurations. These defects modulate electronic distribution, expose active sites, and optimize reaction pathways. Defects instigate localized states near the Fermi level, reducing the energy gap between O2’s d-orbitals and the catalyst’s electronic states (e.g., carbon vacancies shift the Fermi level downward by 0.2–0.5 eV), thereby enhancing charge transfer efficiency. Finally, defects in porous structures expose additional active sites (e.g., defects in mesoporous carbon walls enhance mass transport and active site density).
- (iv)
- In summary, for the 2e− pathway in the ORR, elements that weakly activate the O-O bond should be selected to reduce O-O bond cleavage capability, stabilize the *OOH intermediate, and retain the O-O bond, thereby enhancing H2O2 selectivity. The asymmetric configurations composed of N and O atoms in structures such as Co-N5, Zn-N3O and other structures promote the two-electron pathway of the oxygen reduction reaction. For the 4e− pathway, the O-O bond cleavage capability must be strengthened, and active sites should integrate dual functions of O2 adsorption and O-O bond scission to promote rapid adsorption/desorption equilibrium of oxygen-containing intermediates (*O, *OH). The presence of B and Fe elements in structures such as Co-N3B and Fe-B-Co/NC promotes the four-electron pathway of the oxygen reduction reaction. Additionally, precise regulation of defect type, location, and concentration can optimize the electronic structure and reaction kinetics of active sites, enabling directional selection of ORR pathways (2e−/4e−). Finally, the choice of carbon matrix must comprehensively consider the synergistic effects of multi-element doping, defect engineering, hierarchical structures, and coordination modulation to achieve electronic structure optimization, rapid mass transport, and efficient exposure of active sites.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Fu, J.; Cano, Z.P.; Park, M.G.; Yu, A.; Fowler, M.; Chen, Z. Electrically Rechargeable Zinc–Air Batteries: Progress, Challenges, and Perspectives. Adv. Mater. 2016, 29, 1604685. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Liang, R.; Liu, G.; Yu, A.; Bai, Z.; Yang, L.; Chen, Z. Recent Progress in Electrically Rechargeable Zinc–Air Batteries. Adv. Mater. 2018, 31, 1805230. [Google Scholar] [CrossRef] [PubMed]
- Haider, R.; Wen, Y.; Ma, Z.-F.; Wilkinson, D.P.; Zhang, L.; Yuan, X.; Song, S.; Zhang, J. High temperature proton exchange membrane fuel cells: Progress in advanced materials and key technologies. Chem. Soc. Rev. 2021, 50, 1138–1187. [Google Scholar] [CrossRef]
- Khan, M.A.; Al-Attas, T.; Roy, S.; Rahman, M.M.; Ghaffour, N.; Thangadurai, V.; Larter, S.; Hu, J.; Ajayan, P.M.; Kibria, M.G. Seawater electrolysis for hydrogen production: A solution looking for a problem? Energy Environ. Sci. 2021, 14, 4831–4839. [Google Scholar] [CrossRef]
- Shi, L.; Liu, D.; Lin, X.; Cheng, R.; Liu, F.; Kim, C.; Hu, C.; Qiu, J.; Amal, R.; Dai, L. Stable and High-performance Flow H2-O2 Fuel Cells with Coupled Acidic Oxygen Reduction and Alkaline Hydrogen Oxidation Reactions. Adv. Mater. 2024, 36, 2314077. [Google Scholar] [CrossRef]
- Wang, Q.; Kaushik, S.; Xiao, X.; Xu, Q. Sustainable zinc–air battery chemistry: Advances, challenges and prospects. Chem. Soc. Rev. 2023, 52, 6139–6190. [Google Scholar] [CrossRef]
- Xiao, F.; Wang, Y.C.; Wu, Z.P.; Chen, G.; Yang, F.; Zhu, S.; Siddharth, K.; Kong, Z.; Lu, A.; Li, J.C.; et al. Recent Advances in Electrocatalysts for Proton Exchange Membrane Fuel Cells and Alkaline Membrane Fuel Cells. Adv. Mater. 2021, 33, 2006292. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Liu, L. Recent Advances in Hybrid Seawater Electrolysis for Hydrogen Production. Adv. Mater. 2023, 36, 2308647. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, M.; Gu, X.; Shi, Y.; Deng, Z.; Cai, N. Water Electrolysis toward Elevated Temperature: Advances, Challenges and Frontiers. Chem. Rev. 2023, 123, 7119–7192. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, Y. Origin of Selective Production of Hydrogen Peroxide by Electrochemical Oxygen Reduction. J. Am. Chem. Soc. 2021, 143, 9423–9428. [Google Scholar] [CrossRef]
- Xu, S.; Lu, R.; Sun, K.; Tang, J.; Cen, Y.; Luo, L.; Wang, Z.; Tian, S.; Sun, X. Synergistic Effects in N,O-Comodified Carbon Nanotubes Boost Highly Selective Electrochemical Oxygen Reduction to H2O2. Adv. Sci. 2022, 9, 2201421. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Huang, S.; Wang, X.; Chen, S.; Wang, L.; Zhang, P.; Zhao, C. Activating the PdN4 single-atom sites for 4 electron oxygen reduction reaction via axial oxygen ligand modification. Chem. Eng. J. 2023, 472, 145129. [Google Scholar] [CrossRef]
- Tian, X.; Lu, X.F.; Xia, B.Y.; Lou, X.W. Advanced Electrocatalysts for the Oxygen Reduction Reaction in Energy Conversion Technologies. Joule 2020, 4, 45–68. [Google Scholar] [CrossRef]
- Zhai, Z.; Wang, Y.-J.; Pan, L.; Huang, F.; Liu, D.; Wang, B. Accelerating O-O bond dissociation in oxygen reduction reaction on the sp3-hybridized carbon. Appl. Surf. Sci. 2025, 691, 162668. [Google Scholar] [CrossRef]
- Yan, H.-M.; Wang, G.; Lv, X.-M.; Cao, H.; Qin, G.-Q.; Wang, Y.-G. Revealing the Potential-Dependent Rate-Determining Step of Oxygen Reduction Reaction on Single-Atom Catalysts. J. Am. Chem. Soc. 2025, 147, 3724–3730. [Google Scholar] [CrossRef]
- Cheng, J.; Lyu, C.; Li, H.; Wu, J.; Hu, Y.; Han, B.; Wu, K.; Hojamberdiev, M.; Geng, D. Steering the oxygen reduction reaction pathways of N-carbon hollow spheres by heteroatom doping. Appl. Catal. B Environ. 2023, 327, 122470. [Google Scholar] [CrossRef]
- Kumar, K.; Dubau, L.; Jaouen, F.; Maillard, F. Review on the Degradation Mechanisms of Metal-N-C Catalysts for the Oxygen Reduction Reaction in Acid Electrolyte: Current Understanding and Mitigation Approaches. Chem. Rev. 2023, 123, 9265–9326. [Google Scholar] [CrossRef]
- Wolker, T.; Brunnengräber, K.; Martinaiou, I.; Lorenz, N.; Zhang, G.-R.; Kramm, U.I.; Etzold, B.J.M. The effect of temperature on ionic liquid modified Fe-N-C catalysts for alkaline oxygen reduction reaction. J. Energy Chem. 2022, 68, 324–329. [Google Scholar] [CrossRef]
- Du, Y.X.; Yang, Q.; Lu, W.T.; Guan, Q.Y.; Cao, F.F.; Zhang, G. Carbon Black-Supported Single-Atom Co-N-C as an Efficient Oxygen Reduction Electrocatalyst for H2O2 Production in Acidic Media and Microbial Fuel Cell in Neutral Media. Adv. Funct. Mater. 2023, 33, 2300895. [Google Scholar] [CrossRef]
- Lim, C.; Fairhurst, A.R.; Ransom, B.J.; Haering, D.; Stamenkovic, V.R. Role of Transition Metals in Pt Alloy Catalysts for the Oxygen Reduction Reaction. ACS Catal. 2023, 13, 14874–14893. [Google Scholar] [CrossRef]
- Valério Neto, E.S.; Almeida, C.V.S.; Colmati, F.; Ciapina, E.G.; Salazar-Banda, G.R.; Eguiluz, K.I.B. Pt nanowires as electrocatalysts for proton-exchange membrane fuel cells applications: A review. J. Electroanal. Chem. 2022, 910, 116185. [Google Scholar] [CrossRef]
- Yin, S.; Song, Y.; Liu, H.; Cui, J.; Liu, Z.; Li, Y.; Xue, T.; Tang, W.; Zhang, D.; Li, H.; et al. Well-Defined PtCo@Pt Core-Shell Nanodendrite Electrocatalyst for Highly Durable Oxygen Reduction Reaction. Small 2025, 21, 2410080. [Google Scholar] [CrossRef] [PubMed]
- Chong, L.; Zhou, H.; Kubal, J.; Tang, Q.; Wen, J.; Yang, Z.; Bloom, I.D.; Abraham, D.; Zhu, H.; Zou, J.; et al. Highly durable fuel cell electrocatalyst with low-loading Pt-Co nanoparticles dispersed over single-atom Pt-Co-N-graphene nanofiber. Chem Catal. 2023, 3, 100541. [Google Scholar] [CrossRef]
- Bhuvanendran, N.; Ravichandran, S.; Lee, S.; Sanij, F.D.; Kandasamy, S.; Pandey, P.; Su, H.; Lee, S.Y. Recent progress in Pt-based electrocatalysts: A comprehensive review of supported and support-free systems for oxygen reduction. Coord. Chem. Rev. 2024, 521, 216191. [Google Scholar] [CrossRef]
- Liu, H.; Zhao, J.; Li, X. Controlled Synthesis of Carbon-Supported Pt-Based Electrocatalysts for Proton Exchange Membrane Fuel Cells. Electrochem. Energy Rev. 2022, 5, 13. [Google Scholar] [CrossRef]
- Sun, Q.; Li, X.-H.; Wang, K.-X.; Ye, T.-N.; Chen, J.-S. Inorganic non-carbon supported Pt catalysts and synergetic effects for oxygen reduction reaction. Energy Environ. Sci. 2023, 16, 1838–1869. [Google Scholar] [CrossRef]
- Peng, J.; Tao, P.; Song, C.; Shang, W.; Deng, T.; Wu, J. Structural evolution of Pt-based oxygen reduction reaction electrocatalysts. Chin. J. Catal. 2022, 43, 47–58. [Google Scholar] [CrossRef]
- Fu, Y.; Zhao, Q.; Wei, Q.; Bowen, C.R.; Wong, W.-Y.; Yang, W. Non-precious metal-based single-atom catalysts for oxygen reduction reaction: Fundamentals and applications. Mater. Sci. Eng. R Rep. 2024, 160, 100822. [Google Scholar] [CrossRef]
- Moghaddam, M.S.; Kafshgari, M.S.; Bahari, A.; Kafshgari, L.A.; Jafari, A. Types, properties, and applications of non-precious oxygen reduction reaction electrocatalyst: A review. J. Energy Chem. 2025, 107, 305–344. [Google Scholar] [CrossRef]
- Ao, X.; Wang, H.; Zhang, X.; Wang, C. Atomically Dispersed Metal–Nitrogen–Carbon Catalysts for Acidic Oxygen Reduction Reaction. ACS Appl. Mater. Interfaces 2025, 17, 2844–2862. [Google Scholar] [CrossRef]
- Yang, Z.; Chen, Y.; Zhang, S.; Zhang, J. Identification and Understanding of Active Sites of Non-Noble Iron-Nitrogen-Carbon Catalysts for Oxygen Reduction Electrocatalysis. Adv. Funct. Mater. 2023, 33, 2215185. [Google Scholar] [CrossRef]
- Xu, W.; Tang, H.; Gu, H.; Xi, H.; Wu, P.; Liang, B.; Liu, Q.; Chen, W. Research progress of asymmetrically coordinated single-atom catalysts for electrocatalytic reactions. J. Mater. Chem. A 2022, 10, 14732–14746. [Google Scholar] [CrossRef]
- Zhao, X.; Hao, Z.; Zhang, X.; Li, L.; Gao, Y.; Liu, L. Alkaline oxygen reduction/evolution reaction electrocatalysis: A critical review focus on orbital structure, non-noble metal catalysts, and descriptors. Chem. Eng. J. 2024, 497, 155005. [Google Scholar] [CrossRef]
- Yu, A.; Yang, Y. Atomically Dispersed Metal Catalysts for Oxygen Reduction Reaction: Two-Electron vs. Four-Electron Pathways. Angew. Chem. Int. Ed. 2025, 64, e202424161. [Google Scholar] [CrossRef]
- Zhu, P.; Xiong, X.; Wang, X.; Ye, C.; Li, J.; Sun, W.; Sun, X.; Jiang, J.; Zhuang, Z.; Wang, D.; et al. Regulating the FeN4 Moiety by Constructing Fe–Mo Dual-Metal Atom Sites for Efficient Electrochemical Oxygen Reduction. Nano Lett. 2022, 22, 9507–9515. [Google Scholar] [CrossRef]
- Buschermöhle, J.G.; Müller-Hülstede, J.; Schmies, H.; Schonvogel, D.; Zierdt, T.; Lucka, R.; Renz, F.; Wagner, P.; Wark, M. Fe–Sn–N–C Catalysts: Advancing Oxygen Reduction Reaction Performance. ACS Catal. 2025, 15, 4477–4488. [Google Scholar] [CrossRef]
- Smith, L.A.; Burrow, J.N.; Eichler, J.E.; Tang, F.; Lauro, S.N.; Zhan, X.; Warner, J.H.; Mullins, C.B. A Deep Dive Into the Study of Nitrogen-Doped Carbons as Electrocatalysts for the Oxygen Reduction Reaction via Design of Experiments. Small 2025, 21, 2410010. [Google Scholar] [CrossRef]
- Zong, L.; Fan, K.; Cui, L.; Lu, F.; Liu, P.; Li, B.; Feng, S.; Wang, L. Constructing Fe-N4 Sites through Anion Exchange-mediated Transformation of Fe Coordination Environments in Hierarchical Carbon Support for Efficient Oxygen Reduction. Angew. Chem. Int. Ed. 2023, 62, e202309784. [Google Scholar] [CrossRef]
- Wang, X.; Yang, C.; Wang, X.; Zhu, H.; Cao, L.; Chen, A.; Gu, L.; Zhang, Q.; Zheng, L.; Liang, H.-P. Green Synthesis of a Highly Efficient and Stable Single-Atom Iron Catalyst Anchored on Nitrogen-Doped Carbon Nanorods for the Oxygen Reduction Reaction. ACS Sustain. Chem. Eng. 2020, 9, 137–146. [Google Scholar] [CrossRef]
- Zhu, Y.; Deng, F.; Qiu, S.; Ma, F.; Zheng, Y.; Lian, R. Enhanced electro-Fenton degradation of sulfonamides using the N, S co-doped cathode: Mechanism for H2O2 formation and pollutants decay. J. Hazard. Mater. 2021, 403, 123950. [Google Scholar] [CrossRef]
- Wu, L.; Cao, J.; Fu, C.; Chen, Z.; Yin, C.; Lin, Q.; Pan, H.; Wang, K. Lead regulation of electronic properties and local structure of palladium (111) facet for enhanced direct hydrogen peroxide synthesis. J. Colloid Interface Sci. 2025, 682, 1205–1216. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Z.; Han, C.; Xu, Z.; Zhu, Y.; Attfield, J.P.; Zeng, J.; Yang, M. Ligand-modified nickel nitride for natural seawater H2O2 synthesis. Appl. Catal. B Environ. Energy 2025, 373, 125362. [Google Scholar] [CrossRef]
- Musaie, K.; Abbaszadeh, S.; Marais, K.; Nosrati-Siahmazgi, V.; Rezaei, S.; Xiao, B.; Dua, K.; Santos, H.A.; Shahbazi, M.A. H2O2-Generating Advanced Nanomaterials for Cancer Treatment. Adv. Funct. Mater. 2025, 2425866. [Google Scholar] [CrossRef]
- Yin, J.; Zhang, H.; Luo, M.; Zhao, J.; Zhou, X.; Wang, F.; Zhou, H.; Yuan, Y.; Liu, Y.; Shi, Y.; et al. Electro-Fenton process for wastewater treatment: From selective H2O2 generation to efficient *OH conversion. Chem. Eng. J. 2025, 507, 160709. [Google Scholar] [CrossRef]
- Chen, K.-Y.; Huang, Y.-X.; Jin, R.-C.; Huang, B.-C. Single atom catalysts for use in the selective production of hydrogen peroxide via two-electron oxygen reduction reaction: Mechanism, activity, and structure optimization. Appl. Catal. B Environ. 2023, 337, 122987. [Google Scholar] [CrossRef]
- Bu, Y.; Wang, Y.; Han, G.F.; Zhao, Y.; Ge, X.; Li, F.; Zhang, Z.; Zhong, Q.; Baek, J.B. Carbon-Based Electrocatalysts for Efficient Hydrogen Peroxide Production. Adv. Mater. 2021, 33, 171264. [Google Scholar] [CrossRef]
- Zhang, C.; Shen, W.; Guo, K.; Xiong, M.; Zhang, J.; Lu, X. A Pentagonal Defect-Rich Metal-Free Carbon Electrocatalyst for Boosting Acidic O2 Reduction to H2O2 Production. J. Am. Chem. Soc. 2023, 145, 11589–11598. [Google Scholar] [CrossRef]
- Kong, D.; Wang, Y.; Yan, Y.; Li, Z.; Lu, Y.; Chen, G. Breaking planar constraints: Curvature-tailored Co-N-C electrocatalysts for high-efficiency proton exchange membrane H2O2 production. Appl. Catal. B Environ. Energy 2025, 376, 125454. [Google Scholar] [CrossRef]
- Yan, L.; Wang, C.; Wang, Y.; Wang, Y.; Wang, Z.; Zheng, L.; Lu, Y.; Wang, R.; Chen, G. Optimizing the binding of the *OOH intermediate via axially coordinated Co-N5 motif for efficient electrocatalytic H2O2 production. Appl. Catal. B Environ. 2023, 338, 123078. [Google Scholar] [CrossRef]
- Yuan, Y.; Ma, J.; Kang, B.; Lee, J.Y. Unraveling the Common Nature of O and S Doping in Improving Electrochemical O2 Reduction Reaction Performance of FeN4C. ACS Catal. 2025, 15, 4039–4050. [Google Scholar] [CrossRef]
- Hu, H.; Wang, J.; Liao, K.; Chen, Z.; Zhang, S.; Sun, B.; Wang, X.; Ren, X.; Lin, J.; Han, X. Clarifying the Active Structure and Reaction Mechanism of Atomically Dispersed Metal and Nonmetal Sites with Enhanced Activity for Oxygen Reduction Reaction. Adv. Mater. 2024, 37, 2416126. [Google Scholar] [CrossRef]
- Tang, X.; Zhang, Y.; Tang, S.; Lützenkirchen-Hecht, D.; Yuan, K.; Chen, Y. Structural and Chemical Origin of Dual-Atom Sites for Enhanced Oxygen Electroreduction. ACS Catal. 2024, 14, 13065–13080. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, H.; Lin, L.; Sun, Z.; Sun, G. Single-atomic iron synergistic atom-cluster induce remote enhancement toward oxygen reduction reaction. J. Energy Chem. 2025, 102, 413–420. [Google Scholar] [CrossRef]
- Wu, L.; Wang, Y.; Shao, C.; Wang, L.; Li, B. Copper single atom-modulated functionalization of iron clusters on a porous carbon nanosheet for the oxygen reduction reaction. J. Mater. Chem. A 2025, 13, 5974–5986. [Google Scholar] [CrossRef]
- Lazaridis, T.; Stühmeier, B.M.; Gasteiger, H.A.; El-Sayed, H.A. Capabilities and limitations of rotating disk electrodes versus membrane electrode assemblies in the investigation of electrocatalysts. Nat. Catal. 2022, 5, 363–373. [Google Scholar] [CrossRef]
- Yu, Y.; Jiao, D.; Yi, L.; Zhao, D.; Zou, J.; Cui, X.; Hu, W. Unique electronic regulation in bimetallic MOF for efficient electrochemical synthesis of H2O2 at 500 mA cm−2. Nano Energy 2025, 141, 111096. [Google Scholar] [CrossRef]
- Xu, C.; Li, X.; Guo, P.-P.; Yang, K.-Z.; Zhao, Y.-M.; Chi, H.-M.; Xu, Y.; Wei, P.-J.; Wang, Z.-Q.; Xu, Q.; et al. Creating Asymmetric Fe–N3C–N Sites in Single-Atom Catalysts Boosts Catalytic Performance for Oxygen Reduction Reaction. ACS Appl. Mater. Interfaces 2024, 16, 37927–37937. [Google Scholar] [CrossRef]
- Yu, K.; Guan, S.; Zhang, W.; Zhang, W.; Meng, Y.; Lin, H.; Gao, Q. Engineering Asymmetric Electronic Structure of Co-N-C Single-Atomic Sites Toward Excellent Electrochemical H2O2 Production and Biomass Upgrading. Angew. Chem. Int. Ed. 2025, 64, e202502383. [Google Scholar] [CrossRef]
- Liu, C.; Yang, R.; Wang, J.; Liu, B.; Chang, X.; Feng, P.; Zhang, X.; Zhong, L.; Zhao, X.; Niu, L.; et al. Synergistic Catalysts with Fe Single Atoms and Fe3C Clusters for Accelerated Oxygen Adsorption Kinetics in Oxygen Reduction Reaction. Angew. Chem. Int. Ed. 2025, 64, e202501266. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, H.C.; Ma, X.; Li, J.; Yuan, Q.; Zhang, P.; Wang, M.; Li, J.; Li, M.; Wang, S.; et al. Vacancy Defects Inductive Effect of Asymmetrically Coordinated Single-Atom Fe-N3S1 Active Sites for Robust Electrocatalytic Oxygen Reduction with High Turnover Frequency and Mass Activity. Adv. Mater. 2023, 36, 2308243. [Google Scholar] [CrossRef]
- Zhao, Y.; Gao, Z.; Zhang, S.; Guan, X.; Xu, W.; Liang, Y.; Jiang, H.; Li, Z.; Wu, S.; Cui, Z.; et al. Asymmetric-Charge-Distributed Co-Mn Diatomic Catalyst Enables Efficient Oxygen Reduction Reaction. Adv. Funct. Mater. 2025, 518, 2504260. [Google Scholar] [CrossRef]
- Zhao, Y.; Adiyeri Saseendran, D.P.; Huang, C.; Triana, C.A.; Marks, W.R.; Chen, H.; Zhao, H.; Patzke, G.R. Oxygen Evolution/Reduction Reaction Catalysts: From In Situ Monitoring and Reaction Mechanisms to Rational Design. Chem. Rev. 2023, 123, 6257–6358. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Huang, M.; Kang, Y.; Fang, Y.; Zhao, T.; Wang, H.; Ou, J.; Liu, J.; Zhong, M.; Wang, T.; et al. Ligand effects enhancing low-temperature oxygen reduction kinetics in neutral conditions. Energy Environ. Sci. 2025, 18, 5298–5308. [Google Scholar] [CrossRef]
- Liao, X.; Lu, R.; Xia, L.; Liu, Q.; Wang, H.; Zhao, K.; Wang, Z.; Zhao, Y. Density Functional Theory for Electrocatalysis. Energy Environ. Mater. 2021, 5, 157–185. [Google Scholar] [CrossRef]
- Xie, L.; Zhou, W.; Huang, Y.; Qu, Z.; Li, L.; Yang, C.; Ding, Y.; Li, J.; Meng, X.; Sun, F.; et al. Elucidating the impact of oxygen functional groups on the catalytic activity of M–N4–C catalysts for the oxygen reduction reaction: A density functional theory and machine learning approach. Mater. Horiz. 2024, 11, 1719–1731. [Google Scholar] [CrossRef]
- Li, X.; Jiao, D.; Zhao, X. A Comprehensive Descriptor for Understanding High-Shell Heteroatom-Tuned Oxygen Reduction Reaction Activity on Diatomic FeCoN6 Sites. Renewables 2024, 2, 242–249. [Google Scholar] [CrossRef]
- Chen, K.; Huang, J.; Chen, J.; Gao, J.; Lu, Z.; Liu, X.; Lan, S.; Jia, G.; Ci, S.; Wen, Z. Neighboring Iron Single Atomic Sites Boost PtCo Intermetallic for High-Durability ORR Electrocatalysis. Energy Environ. Sci. 2025. [Google Scholar] [CrossRef]
- Yuan, M.; Liu, Y.; Du, Y.; Xiao, Z.; Li, H.; Liu, K.; Wang, L. Dual-Shelled Hollow Leafy Carbon Support with Atomically Dispersed (N,S)-Bridged Hydroxy-Coordinated Asymmetric Fe Sites for Oxygen Reduction. Adv. Funct. Mater. 2024, 34, 2401484. [Google Scholar] [CrossRef]
- Genz, N.S.; Kallio, A.J.; Oord, R.; Krumeich, F.; Pokle, A.; Prytz, Ø.; Olsbye, U.; Meirer, F.; Huotari, S.; Weckhuysen, B.M. Operando Laboratory-Based Multi-Edge X-Ray Absorption Near-Edge Spectroscopy of Solid Catalysts. Angew. Chem. Int. Ed. 2022, 61, e202209334. [Google Scholar] [CrossRef]
- Miao, C.; Xu, S.; An, Z.; Pan, X.; Li, Y.; Hu, N.; Li, L.; Zhou, Y.; Zhao, G. Self-Optimized Reconstruction of Metal–Organic Frameworks Introduces Cation Vacancies for Selective Electrosynthesis of Hydrogen Peroxide. Angew. Chem. Int. Ed. 2025, 64, e202501930. [Google Scholar] [CrossRef]
- Shang, H.; Zhou, X.; Dong, J.; Li, A.; Zhao, X.; Liu, Q.; Lin, Y.; Pei, J.; Li, Z.; Jiang, Z.; et al. Engineering unsymmetrically coordinated Cu-S1N3 single atom sites with enhanced oxygen reduction activity. Nat. Commun. 2020, 11, 3049. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Rodenbücher, C.; Wippermann, K.; Korte, C. Revealing Interfacial Reactions on Pt Electrodes in Ionic Liquids by In Situ Fourier-Transform Infrared Spectroscopy. Anal. Chem. 2023, 95, 16618–16624. [Google Scholar] [CrossRef]
- Li, Z.M.; Zhang, C.Q.; Liu, C.; Zhang, H.W.; Song, H.; Zhang, Z.Q.; Wei, G.F.; Bao, X.J.; Yu, C.Z.; Yuan, P. High-efficiency Electroreduction of O2 into H2O2 over ZnCo Bimetallic Triazole Frameworks Promoted by Ligand Activation. Angew. Chem. Int. Ed. 2023, 63, e202314266. [Google Scholar] [CrossRef]
- Liu, B.; Guo, P.; Dai, Y.; Liu, B.; Zhang, Z.; Cai, J.; Xia, Y.; Pan, Q.; Shen, L.; Zhang, Y.; et al. Tailoring asymmetric atomic strain of FeN4 sites for enhanced acidic oxygen reduction reaction. Chem. Eng. J. 2025, 507, 160174. [Google Scholar] [CrossRef]
- Du, H.; Li, C.; Liang, Y.; Wu, Z.-S. Key strategies and challenging perspectives of carbon-based electrocatalysts for sustainable H2O2 production. J. Energy Chem. 2025, 106, 864–879. [Google Scholar] [CrossRef]
- Liu, L.; Kang, L.; Feng, J.; Hopkinson, D.G.; Allen, C.S.; Tan, Y.; Gu, H.; Mikulska, I.; Celorrio, V.; Gianolio, D.; et al. Atomically dispersed asymmetric cobalt electrocatalyst for efficient hydrogen peroxide production in neutral media. Nat. Commun. 2024, 15, 4079. [Google Scholar] [CrossRef]
- Wei, G.; Liu, X.; Zhao, Z.; Men, C.; Ding, Y.; Gao, S. Constructing ultrahigh-loading unsymmetrically coordinated Zn-N3O single-atom sites with efficient oxygen reduction for H2O2 production. Chem. Eng. J. 2023, 455, 140721. [Google Scholar] [CrossRef]
- Zhang, W.; Choi, J.W.; Kim, S.; Le, T.T.; Nandy, S.; Hwang, C.-K.; Paek, S.Y.; Byeon, A.; Chae, K.H.; Lee, S.Y.; et al. Penta nitrogen coordinated cobalt single atom catalysts with oxygenated carbon black for electrochemical H2O2 production. Appl. Catal. B Environ. 2023, 331, 122712. [Google Scholar] [CrossRef]
- Li, J.; Zheng, W.; Meng, S.; Wang, S.; Zhan, S.; Sun, Y.; Hu, W.; Li, Y. Modulated Fe Central Spin State in FeCu4 Alloy Nanoparticles for Efficient and Selective Activation of H2O2. Adv. Funct. Mater. 2024, 35, 2417538. [Google Scholar] [CrossRef]
- Yang, X.; Priest, C.; Hou, Y.; Wu, G. Atomically dispersed dual-metal-site PGM-free electrocatalysts for oxygen reduction reaction: Opportunities and challenges. SusMat 2022, 2, 569–590. [Google Scholar] [CrossRef]
- Hu, H.; Zhang, C.; Liu, W.; Qi, H.; Wang, H.; Wang, X.; Zhang, L.; Liu, L.; Bao, L.; Alomar, M.; et al. Coaxial Metal-Nitrogen–Carbon Single-Atom Catalysts Boost Acid Hydrogen Peroxide Production. Adv. Funct. Mater. 2024, 35, 2419220. [Google Scholar] [CrossRef]
- Zhang, S.; Sun, B.; Liao, K.; Wang, X.; Chen, Z.; Wang, J.; Hu, W.; Han, X. Boosting Oxygen Reduction Reaction Performance of Fe Single-Atom Catalysts Via Precise Control of the Coordination Environment. Adv. Funct. Mater. 2025, 2425640. [Google Scholar] [CrossRef]
- Guan, G.; Liu, Y.; Li, F.; Shi, X.; Liu, L.; Wang, T.; Xu, X.; Zhao, M.; Ding, J.; Yang, H.B. Atomic Cobalt Metal Centers with Asymmetric N/B-Coordination for Promoting Oxygen Reduction Reaction. Adv. Funct. Mater. 2024, 34, 2408111. [Google Scholar] [CrossRef]
- Tang, C.; Jin, R.; Xia, Y.; Zhang, Q.; Wang, J.; Wu, M.; Cui, X.; Yang, Y. Asymmetric low-coordination tailoring of single-atom cobalt catalysts enabling efficient oxygen reduction reaction. Nano Energy 2025, 137, 110776. [Google Scholar] [CrossRef]
- Chen, L.; Yin, S.; Zeng, H.; Liu, J.; Xiao, X.; Cheng, X.; Huang, H.; Huang, R.; Yang, J.; Lin, W.-F.; et al. Engineering asymmetric electronic structure of cobalt coordination on CoN3S active sites for high performance oxygen reduction reaction. J. Energy Chem. 2024, 98, 494–502. [Google Scholar] [CrossRef]
- Liu, C.; Yang, X.; Jia, Z.; Pan, J.; Zhao, F.; Ban, Y.; Cui, X.; Bai, Z.; Yang, L. Boosted electron accumulation around Fe atom by asymmetry coordinated FeN3S1 for efficient oxygen reduction reaction. Int. J. Hydrog. Energy 2024, 90, 1003–1011. [Google Scholar] [CrossRef]
- Liu, Y.; Pang, H.; Chen, M.; Ji, X.; Meng, F. Hollow polyhedral Fe single-atom catalysts with S/N dual coordination boosting oxygen and iodide redox for high-efficiency zinc–air/iodide hybrid batteries. J. Mater. Chem. A 2025, 13, 16008–16017. [Google Scholar] [CrossRef]
- Lin, X.; Zhang, X.; Liu, D.; Shi, L.; Zhao, L.; Long, Y.; Dai, L. Asymmetric Atomic Tin Catalysts with Tailored p-Orbital Electron Structure for Ultra-Efficient Oxygen Reduction. Adv. Energy Mater. 2024, 14, 2303740. [Google Scholar] [CrossRef]
- Li, Y.; Sun, H.; Ren, L.; Sun, K.; Gao, L.; Jin, X.; Xu, Q.; Liu, W.; Sun, X. Asymmetric Coordination Regulating D-Orbital Spin-Electron Filling in Single-Atom Iron Catalyst for Efficient Oxygen Reduction. Angew. Chem. Int. Ed. 2024, 63, e202405334. [Google Scholar] [CrossRef]
- Brea, C.; Hu, G. Mechanistic Insight into Dual-Metal-Site Catalysts for the Oxygen Reduction Reaction. ACS Catal. 2023, 13, 4992–4999. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, N.; Shang, H.; Duan, H.; Sun, Z.; Zhang, L.; Lei, Y.; Luo, X.; Zhang, L.; Zhang, B.; et al. Precisely designing asymmetrical selenium-based dual-atom sites for efficient oxygen reduction. Nat. Commun. 2025, 16, 470. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, Z.; Liu, W.; Chang, C.; Tang, H.; Li, Z.; Chen, W.; Jia, C.; Yao, T.; Wei, S.; et al. Design of N-Coordinated Dual-Metal Sites: A Stable and Active Pt-Free Catalyst for Acidic Oxygen Reduction Reaction. J. Am. Chem. Soc. 2017, 139, 17281–17284. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.; Li, M.; Li, X.; Yan, X.; Hou, Z.; Huang, C. Introducing hydroxyl groups to tailor the d-band center of Ir atom through side anchoring for boosted ORR and HER. J. Energy Chem. 2024, 90, 144–151. [Google Scholar] [CrossRef]
- Cao, Y.; Zeng, J.; Zheng, X.; Liu, Y.; Lu, J.; Zhang, J.; Wang, Y.; Deng, Y.; Hu, W. Regulating d-orbital spin state of Fe in single-atom electrocatalyst for boosting oxygen reduction activity in neutral electrolyte. J. Mater. Sci. Technol. 2025, 227, 67–75. [Google Scholar] [CrossRef]
- An, Q.; Zhang, X.; Yang, C.; Su, H.; Zhou, W.; Liu, M.; Zhang, X.; Sun, X.; Bo, S.; Yu, F.; et al. Engineering Unsymmetrically Coordinated Fe Sites via Heteroatom Pairs Synergetic Contribution for Efficient Oxygen Reduction. Small 2023, 19, 2304303. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Qin, B.; Si, C.; Sun, X.; Li, B. Electrochemical reactions catalyzed by carbon dots from computational investigations: Functional groups, dopants, and defects. J. Mater. Chem. A 2024, 12, 2520–2560. [Google Scholar] [CrossRef]
- Li, M.; Hou, Z.; Li, X.; Gu, X.; Yan, X.; Lv, Q.; Huang, C. Asymmetrical anchor way of manganese atoms on carbon domain edge for enhanced oxygen reduction reaction. Appl. Catal. B Environ. Energy 2024, 356, 124249. [Google Scholar] [CrossRef]
- Bhardwaj, S.; Pathak, A.; Das, S.K.; Das, P.; Thapa, R.; Dey, R.S. Decoding Dual-Functionality in N-doped Defective Carbon: Unveiling Active Sites for Bifunctional Oxygen Electrocatalysis. Small 2025, 21, 2411035. [Google Scholar] [CrossRef]
- Yan, J.; Li, S.; Liu, M.; Zhou, H.; Su, H. Regulating electron effect by interface-induced dislocation in Fe2P/Fe to accelerate oxygen reduction reactions. Mater. Today Phys. 2025, 54, 101739. [Google Scholar] [CrossRef]
- Tu, H.; Zhang, H.; Song, Y.; Liu, P.; Hou, Y.; Xu, B.; Liao, T.; Guo, J.; Sun, Z. Electronic Asymmetry Engineering of Fe–N–C Electrocatalyst via Adjacent Carbon Vacancy for Boosting Oxygen Reduction Reaction. Adv. Sci. 2023, 10, 2305194. [Google Scholar] [CrossRef]
- Ji, S.; Wang, Y.; Liu, H.; Lu, X.; Guo, C.; Xin, S.; Horton, J.H.; Zhan, F.; Wang, Y.; Li, Z. Regulating the Electronic Synergy of Asymmetric Atomic Fe Sites with Adjacent Defects for Boosting Activity and Durability toward Oxygen Reduction. Adv. Funct. Mater. 2024, 34, 23146211. [Google Scholar] [CrossRef]
- Zhang, Y.; Feng, J.; Shang, Z.; Pang, B.; Zhang, S.; Dong, H.; Yu, L.; Dong, L. Synergistic effects of metal single atoms and clusters on graphene-supported electrocatalysts: Insights into the mechanism of oxygen reduction reaction. Carbon 2025, 233, 119830. [Google Scholar] [CrossRef]
- Tian, Q.; Jing, L.; Wang, W.; Ye, X.; Chai, X.; Zhang, X.; Hu, Q.; Yang, H.; He, C. Hydrogen Peroxide Electrosynthesis via Selective Oxygen Reduction Reactions Through Interfacial Reaction Microenvironment Engineering. Adv. Mater. 2024, 37, 2414490. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Han, G.F.; Li, F.; Bu, Y. Rational Design of Carbon-Based Electrocatalysts for H2O2 Production by Machine Learning and Structural Engineering. Adv. Energy Mater. 2025, 15, 2500953. [Google Scholar] [CrossRef]
- Wen, Y.; Zhang, T.; Wang, J.; Pan, Z.; Wang, T.; Yamashita, H.; Qian, X.; Zhao, Y. Electrochemical Reactors for Continuous Decentralized H2O2 Production. Angew. Chem. Int. Ed. 2022, 61, e202205972. [Google Scholar] [CrossRef]
- Oliveira Silva, T.; Granados-Fernández, R.; Lobato, J.; Lanza, M.R.V.; Rodrigo, M.A. Boosting New Electrochemical Reactor Designs to Improve the Performance in H2O2 Production Using Gas Diffusion Electrodes. ACS Sustain. Chem. Eng. 2025, 13, 3172–3182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sang, W.; Wang, T.; Wei, X.; Li, Z.; Chen, C.; Kou, Z.; Mu, S. Cu–N–C support confinement stabilizes active Co sites in oxygen reduction reaction. Nano Res. 2025, 18, 94907345. [Google Scholar] [CrossRef]
- Bao, Z.; Cao, Q.; Shao, Y.; Zhang, S.; Peng, X.; Li, Y.; Jiang, C.; Zhong, X.; Wang, J. Carbon and Oxygen-Doped Phosphorus Nitride (COPN) for Continuous Selective and Stable H2O2 Production. ACS Catal. 2023, 13, 14492–14502. [Google Scholar] [CrossRef]
- Mohd Shumiri, M.A.I.; Mohd Najib, A.S.; Fadil, N.A. Current status and advances in zinc anodes for rechargeable aqueous zinc-air batteries. Sci. Technol. Adv. Mater. 2025, 26, 2448418. [Google Scholar] [CrossRef]
- Tu, W.B.; Song, L.N.; Liang, S.; Wang, Y.; Sun, Y.; Wang, H.F.; Xu, J.J. Designing Hydrophobic Micro-Three-Phase Reaction Interfaces to Enhance the ORR Kinetics Toward Zinc-Air Battery. Adv. Energy Mater. 2025, 2404946. [Google Scholar] [CrossRef]
- Wang, D.; Li, X.; Ren, P.; Wei, Y.; Li, R.; Chen, Y.; Wang, W.; Yang, P.; Chen, Z. Asymmetric N, P-coordinated Co single-atom catalyst for efficient oxygen reduction reaction in Zn-air batteries. Appl. Catal. B Environ. Energy 2025, 375, 125436. [Google Scholar] [CrossRef]
- Zhang, Y.; Deng, Y.-P.; Wang, J.; Jiang, Y.; Cui, G.; Shui, L.; Yu, A.; Wang, X.; Chen, Z. Recent Progress on Flexible Zn-Air Batteries. Energy Storage Mater. 2021, 35, 538–549. [Google Scholar] [CrossRef]
- Wu, J.; Wu, W.-Y.; Wang, S.; Kai, D.; Ye, E.; Thitsartarn, W.; Tan, J.B.H.; Xu, J.; Yan, Q.; Zhu, Q.; et al. Polymer electrolytes for flexible zinc-air batteries: Recent progress and future directions. Nano Res. 2024, 17, 6058–6079. [Google Scholar] [CrossRef]
- Ahmed, S.; Beauger, C.; Zada, A.; Iqbal, W.; Ahmed, N.; Anwar, M.T.; Hassan, M. Recent advancements in designing high-performance proton exchange membrane fuel cells: A comprehensive review. Appl. Energy 2025, 390, 125753. [Google Scholar] [CrossRef]
- Zeng, W.; Guan, B.; Zhuang, Z.; Chen, J.; Zhu, L.; Ma, Z.; Hu, X.; Zhu, C.; Zhao, S.; Shu, K.; et al. Comprehensive review on the advances and comparisons of proton exchange membrane fuel cells (PEMFCs) and anion exchange membrane fuel cells (AFCs): From fundamental principles to key component technologies. Int. J. Hydrogen Energy 2025, 102, 222–246. [Google Scholar] [CrossRef]
- Sharma, R.; Morgen, P.; Chiriaev, S.; Lund, P.B.; Larsen, M.J.; Sieborg, B.; Grahl-Madsen, L.; Andersen, S.M. Insights into Degradation of the Membrane–Electrode Assembly Performance in Low-Temperature PEMFC: The Catalyst, the Ionomer, or the Interface? ACS Appl. Mater. Interfaces 2022, 14, 49658–49671. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, H.; Wen, S.; Fu, Q.; Zhao, X. Oxygen Reduction Reactions of Catalysts with Asymmetric Atomic Structures: Mechanisms, Applications, and Challenges. Catalysts 2025, 15, 615. https://doi.org/10.3390/catal15070615
Qiu H, Wen S, Fu Q, Zhao X. Oxygen Reduction Reactions of Catalysts with Asymmetric Atomic Structures: Mechanisms, Applications, and Challenges. Catalysts. 2025; 15(7):615. https://doi.org/10.3390/catal15070615
Chicago/Turabian StyleQiu, Hengxing, Shilong Wen, Qiuju Fu, and Xuebo Zhao. 2025. "Oxygen Reduction Reactions of Catalysts with Asymmetric Atomic Structures: Mechanisms, Applications, and Challenges" Catalysts 15, no. 7: 615. https://doi.org/10.3390/catal15070615
APA StyleQiu, H., Wen, S., Fu, Q., & Zhao, X. (2025). Oxygen Reduction Reactions of Catalysts with Asymmetric Atomic Structures: Mechanisms, Applications, and Challenges. Catalysts, 15(7), 615. https://doi.org/10.3390/catal15070615