
Stability of an Ultra-Low-Temperature Water–Gas Shift Reaction SILP Catalyst


Abstract
1. Introduction
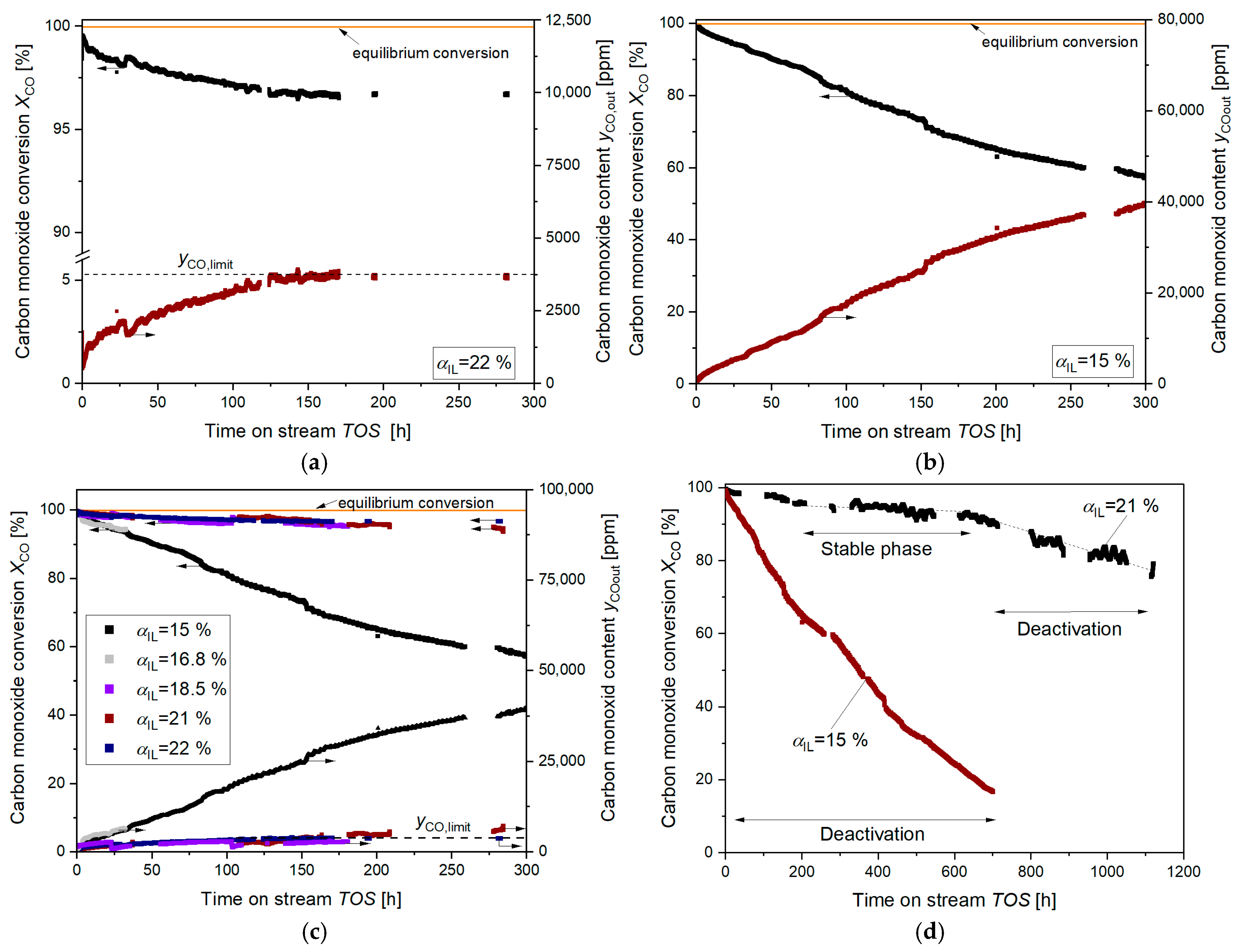
2. Results
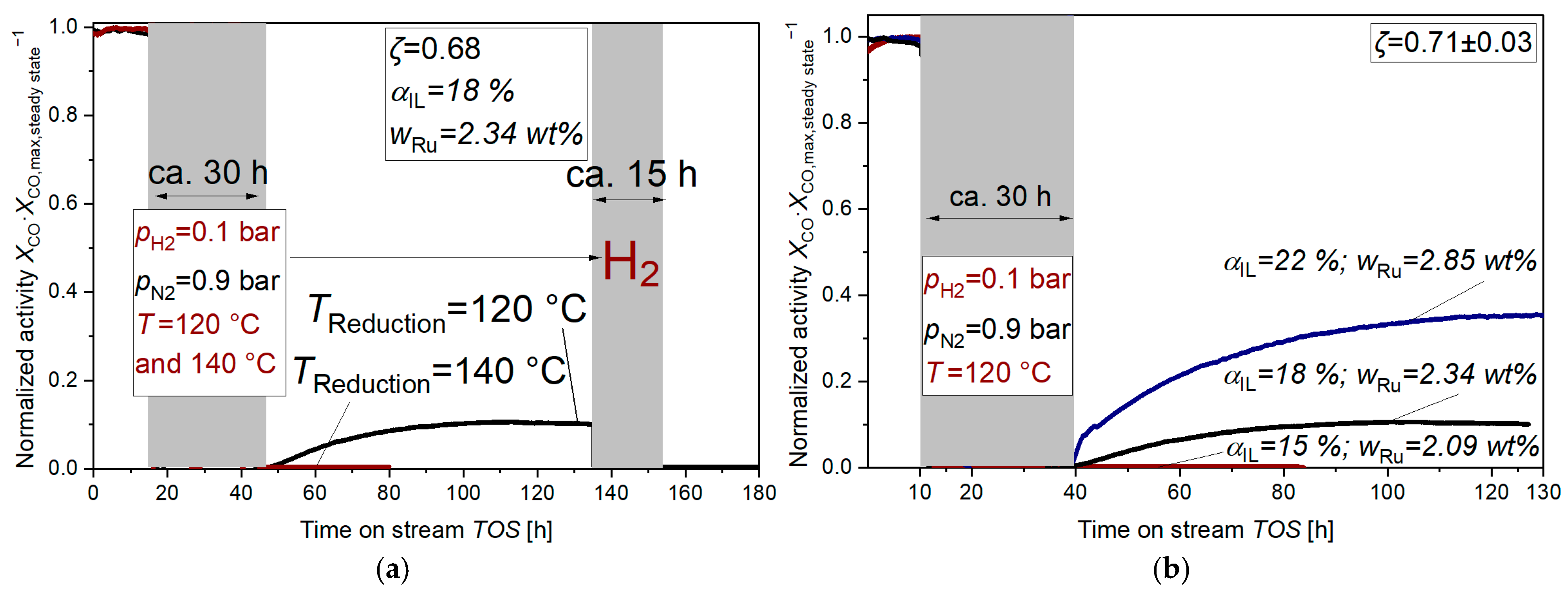
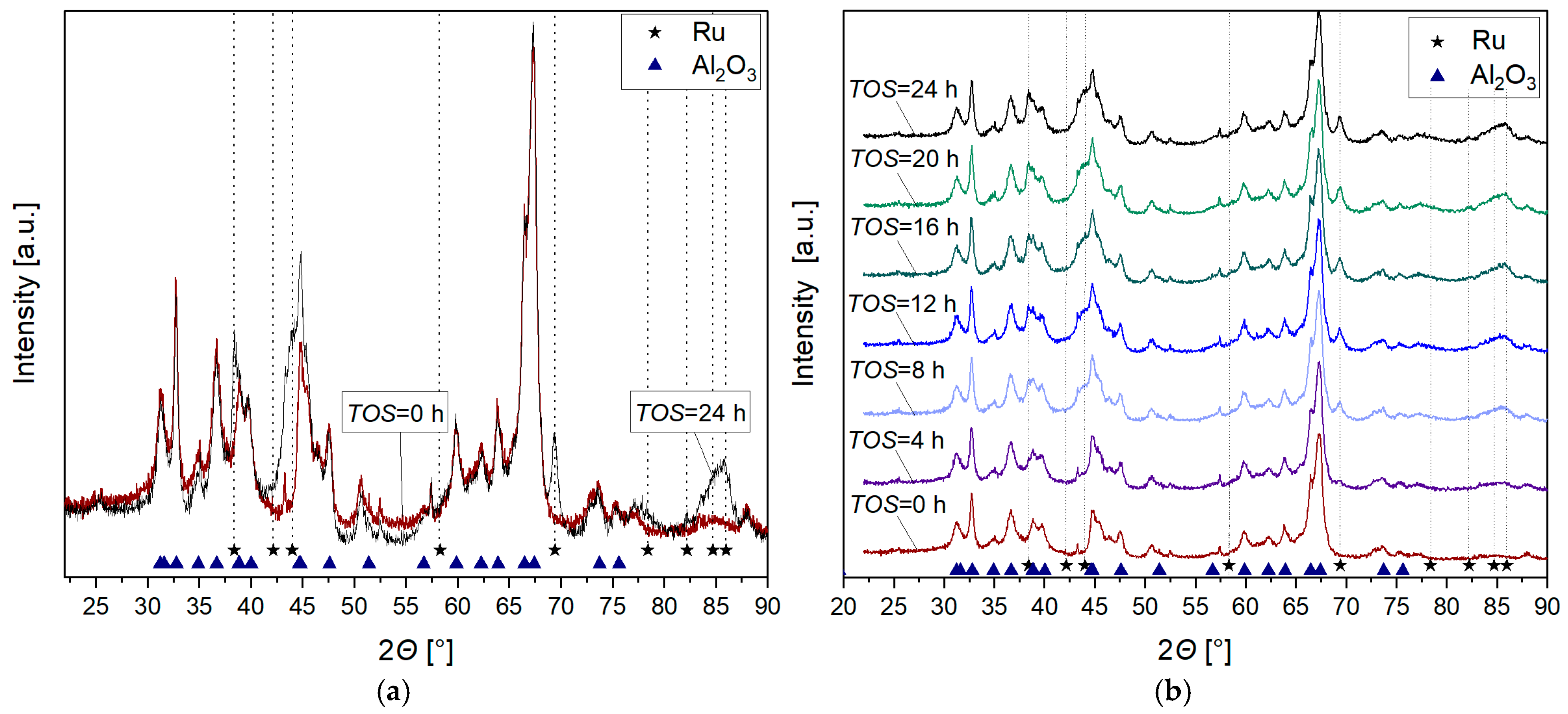
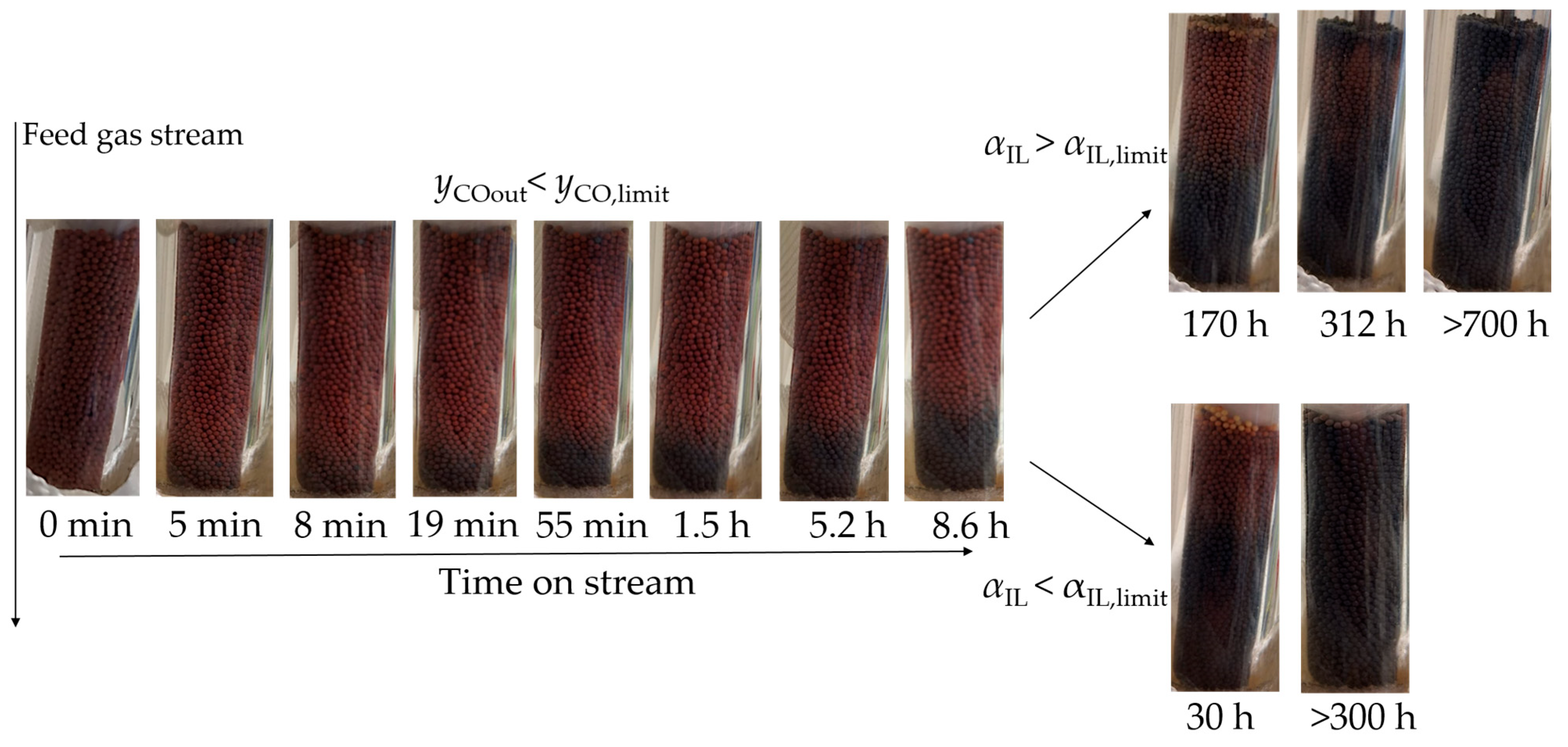
2.1. Nanoparticle Formation Under Reducing Conditions
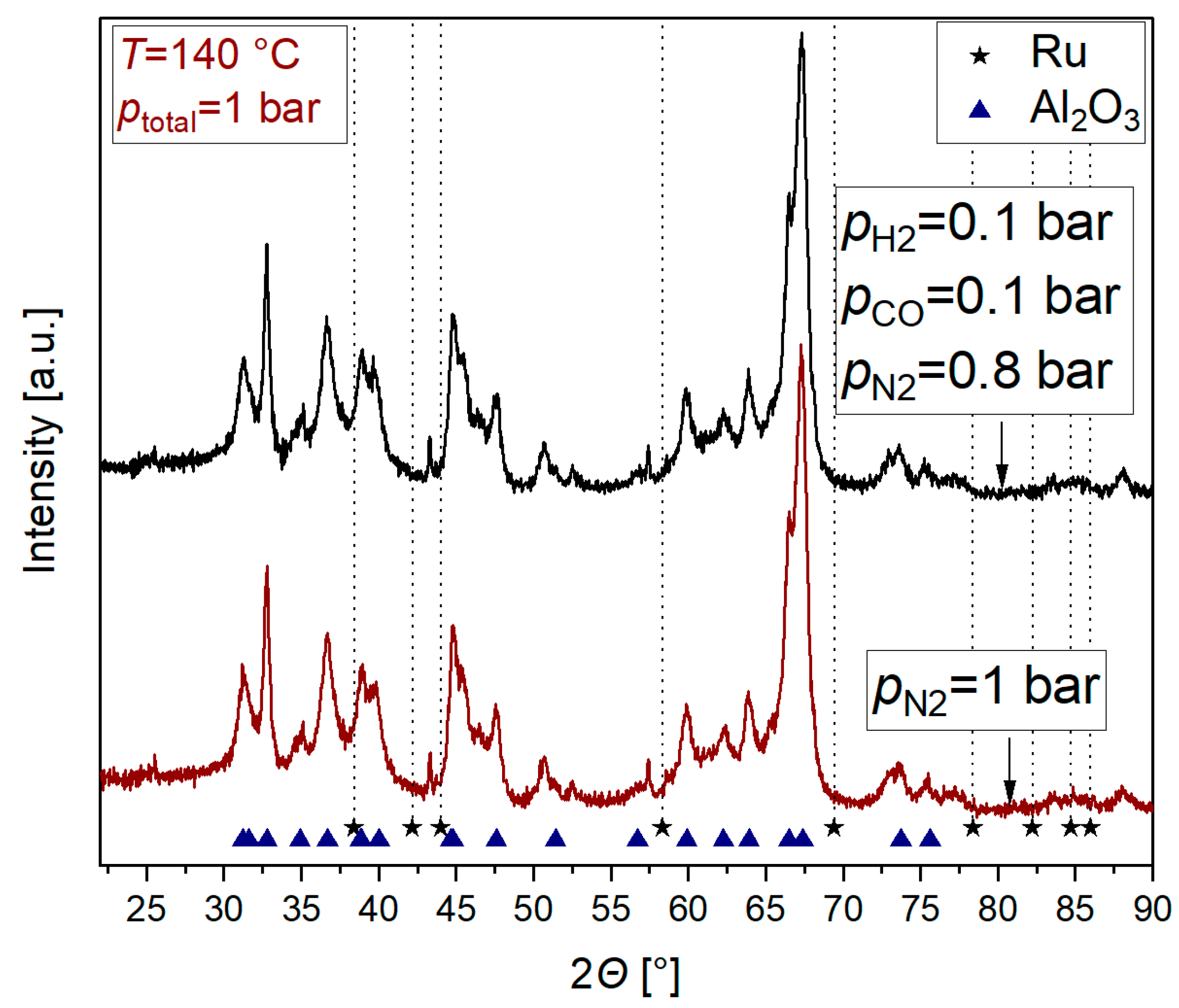
2.2. Influence of CO on Ru Nanoparticle Formation
3. Discussion
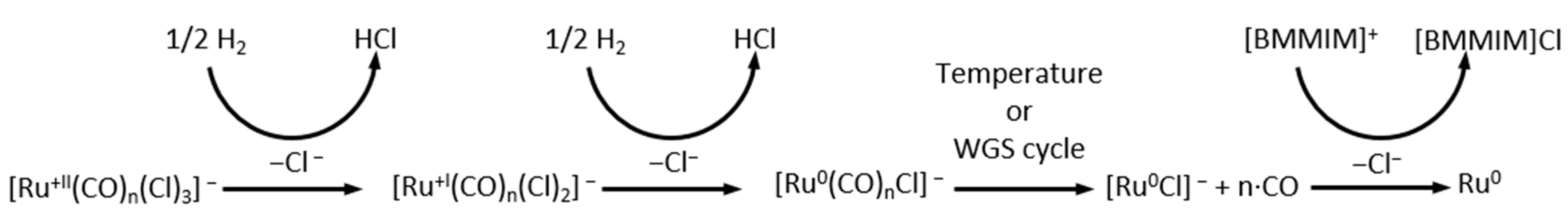
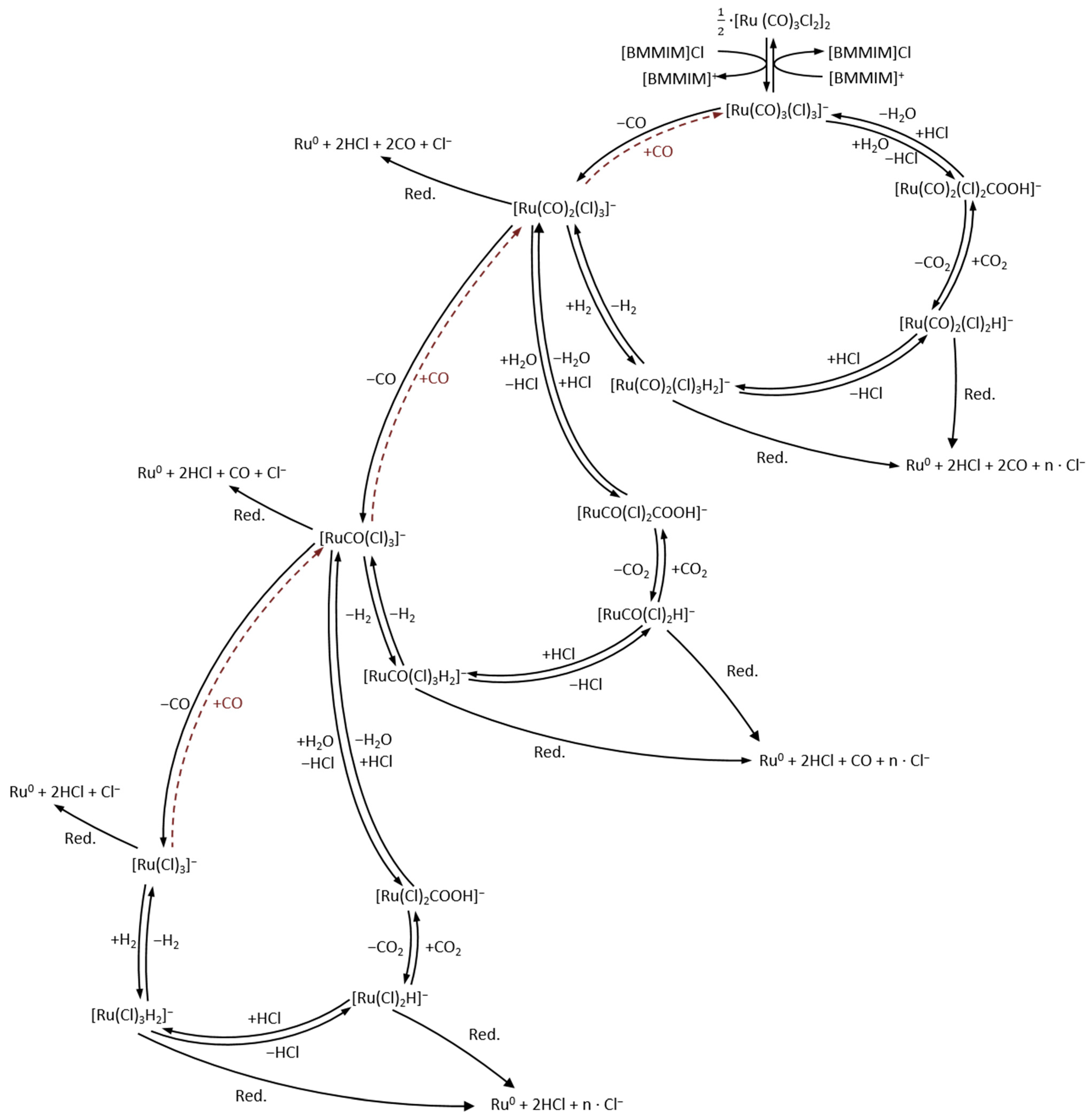
3.1. Nanoparticle Formation
3.2. Nanoparticle Formation Mechanism in the Absence of CO
4. Materials and Methods
4.1. Catalyst Preparation
4.2. Experimental Setup
4.3. Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DCM | Dichlormethane |
| IL | Ionic liquid |
| LT | Low temperature |
| No | By the number |
| NP | Nanoparticle |
| PEM | Proton exchange membrane |
| SILP | Supported ionic liquid phase |
| TEM | Transmission electron microscopy |
| TOS | Time on stream |
| WGSR | Water–gas shift reaction |
| XRD | X-ray diffraction |
References
- Ratnasamy, C.; Wagner, J. Water Gas Shift Catalysis. Catal. Rev. Sci. Eng. 2009, 51, 325–440. [Google Scholar] [CrossRef]
- Divisek, J.; Oetjen, H.-F.; Peinecke, V.; Schmidt, V.M.; Stimming, U. Components for PEM Fuel Cell Systems Using Hydrogen and CO Containing Fuels. Electrochim. Acta 1998, 43, 3811–3815. [Google Scholar] [CrossRef]
- Werner, S.; Szesni, N.; Kaiser, M.; Fischer, R.W.; Haumann, M.; Wasserscheid, P. Ultra-Low-Temperature Water-Gas Shift Catalysis Using Supported Ionic Liquid Phase (SILP) Materials. ChemCatChem 2010, 2, 1399–1402. [Google Scholar] [CrossRef]
- Yun, S.; Ted Oyama, S. Correlations in Palladium Membranes for Hydrogen Separation: A Review. J. Memb. Sci. 2011, 375, 28–45. [Google Scholar] [CrossRef]
- Mishra, A.P.R. A Review on Preferential Oxidation of Carbon Monoxide in Hydrogen Rich Gases. Chem. React. Eng. Catal. 2011, 6, 1–14. [Google Scholar] [CrossRef]
- Steinrück, H.P.; Wasserscheid, P. Ionic Liquids in Catalysis. Catal. Lett. 2015, 145, 380–397. [Google Scholar] [CrossRef]
- Riisager, A.; Fehrmann, R.; Haumann, M.; Wasserscheid, P. Supported Ionic Liquid Phase (SILP) Catalysis: An Innovative Concept for Homogeneous Catalysis in Continuous Fixed-Bed Reactors. Eur. J. Inorg. Chem. 2006, 2006, 695–706. [Google Scholar] [CrossRef]
- Werner, S.; Szesni, N.; Bittermann, A.; Schneider, M.J.; Härter, P.; Haumann, M.; Wasserscheid, P. Screening of Supported Ionic Liquid Phase (SILP) Catalysts for the Very Low Temperature Water-Gas-Shift Reaction. Appl. Catal. A Gen. 2010, 377, 70–75. [Google Scholar] [CrossRef]
- Werner, S. Ultra-Low Temperature Water-Gas Shift Reaction with Supported Ionic Liquid Phase (SILP) Catalysts Wassergas-Shift-Katalyse Mit Geträgerten Ionischen Flüssigkeiten Unter Extrem Milden Reaktionsbedingungen. Ph.D. Thesis, Universität Erlangen, Erlangen, Germany, 2011. [Google Scholar]
- Werner, S.; Szesni, N.; Fischer, R.W.; Haumann, M.; Wasserscheid, P. Homogeneous Ruthenium-Based Water-Gas Shift Catalysts via Supported Ionic Liquid Phase (SILP) Technology at Low Temperature and Ambient Pressure. Phys. Chem. Chem. Phys. 2009, 11, 10817–10819. [Google Scholar] [CrossRef]
- Fischer, F.; Jess, A. Kinetic Study of the Water-Gas Shift Reaction at Ultralow Temperature over a Ru-Based Supported Ionic Liquid Phase Catalyst. Chem. Ing. Tech. 2022, 94, 1695–1703. [Google Scholar] [CrossRef]
- Scholten, J.D.; Leal, B.C.; Dupont, J. Transition Metal Nanoparticle Catalysis in Ionic Liquids. ACS Catal. 2012, 2, 184–200. [Google Scholar] [CrossRef]
- He, Z.; Alexandridis, P. Nanoparticles in Ionic Liquids: Interactions and Organization. Phys. Chem. Chem. Phys. 2015, 17, 18238–18261. [Google Scholar] [CrossRef] [PubMed]
- Seitkalieva, M.M.; Samoylenko, D.E.; Lotsman, K.A.; Rodygin, K.S.; Ananikov, V.P. Metal Nanoparticles in Ionic Liquids: Synthesis and Catalytic Applications. Coord. Chem. Rev. 2021, 445, 213982. [Google Scholar] [CrossRef]
- Dupont, J.; Scholten, J.D. On the Structural and Surface Properties of Transition-Metal Nanoparticles in Ionic Liquids. Chem. Soc. Rev. 2010, 39, 1780–1804. [Google Scholar] [CrossRef]
- Wegner, S.; Janiak, C. Metal Nanoparticles in Ionic Liquids. Top. Curr. Chem. 2017, 375, 65. [Google Scholar] [CrossRef]
- Janiak, C. Ionic Liquids for the Synthesis and Stabilization of Metal Nanoparticles. Z. Naturforschung—Sect. B J. Chem. Sci. 2013, 68, 1059–1089. [Google Scholar] [CrossRef]
- Kratzer, E.; Schötz, S.; Maisel, S.; Blaumeiser, D.; Khan Antara, S.; Ewald, L.; Dotzel, D.; Haumann, M.; Görling, A.; Korth, W.; et al. Wilkinson-Type Catalysts in Ionic Liquids for Hydrogenation of Small Alkenes: Understanding and Improving Catalyst Stability. Catal. Sci. Technol. 2023, 13, 2053–2069. [Google Scholar] [CrossRef]
- Marinkovic, J.M.; Riisager, A.; Franke, R.; Wasserscheid, P.; Haumann, M. Fifteen Years of Supported Ionic Liquid Phase-Catalyzed Hydroformylation: Material and Process Developments. Ind. Eng. Chem. Res. 2019, 58, 2409–2420. [Google Scholar] [CrossRef]
- Bauer, T.; Stepic, R.; Wolf, P.; Kollhoff, F.; Karawacka, W.; Wick, C.R.; Haumann, M.; Wasserscheid, P.; Smith, D.M.; Smith, A.S.; et al. Dynamic Equilibria in Supported Ionic Liquid Phase (SILP) Catalysis: In Situ IR Spectroscopy Identifies [Ru(CO)XCly]n Species in Water Gas Shift Catalysis. Catal. Sci. Technol. 2018, 8, 344–357. [Google Scholar] [CrossRef]
- Stepić, R.; Wick, C.R.; Strobel, V.; Berger, D.; Vučemilović-Alagić, N.; Haumann, M.; Wasserscheid, P.; Smith, A.; Smith, D.M. Mechanism of the Water–Gas Shift Reaction Catalyzed by Efficient Ruthenium Based Catalysts: A Computational and Experimental Study. Angew. Chem. 2018, 58, 741–745. [Google Scholar] [CrossRef]
- Upadhyay, P.R.; Srivastava, V. Ionic Liquid Mediated In Situ Synthesis of Ru Nanoparticles for CO2 Hydrogenation Reaction. Catal. Lett. 2017, 147, 1051–1060. [Google Scholar] [CrossRef]
- Nandanwar, S.U.; Chakraborty, M.; Mukhopadhyay, S.; Shenoy, K.T. Stability of Ruthenium Nanoparticles Synthesized by Solvothermal Method. Cryst. Res. Technol. 2011, 46, 393–399. [Google Scholar] [CrossRef]
- Garbis, P.; Kern, C.; Jess, A. Kinetics and Reactor Design Aspects of Selective Methanation of CO over a Ru/γ-Al2O3 Catalyst in CO2/H2 Rich Gases. Energies 2019, 12, 469. [Google Scholar] [CrossRef]
- Sobota, M.; Schernich, S.; Schulz, H.; Hieringer, W.; Paape, N.; Wasserscheid, P.; Görling, A.; Laurin, M.; Libuda, J. Preparation and Characterization of Ultrathin [Ru(CO)3Cl2]2 and [BMIM][Tf2N] Films on Al2O3/NiAl(110) under UHV Conditions. Phys. Chem. Chem. Phys. 2012, 14, 10603–10612. [Google Scholar] [CrossRef]
- Heym, F. Dampfdruck Und Thermische Zersetzung Reiner Und Geträgerter Ionischer Flüssigkeiten. Ph.D. Thesis, Universität Bayreuth, Bayreuth, Germany, 2013. [Google Scholar]
- Gerritsen, L.A.; Herman, J.M.; Scholten, J.J.F. Hydroformylation with Supported Liquid Phase Rhodium Catalysts Part III Influence of the Type of Support, the Degree of Pore Filling and Organic Additives on the Catalytic Performance. J. Mol. Catal. 1980, 9, 241–256. [Google Scholar] [CrossRef]
- Ohlin, C.A.; Dyson, P.J.; Laurenczy, G. Carbon Monoxide Solubility in Ionic Liquids: Determination, Prediction and Relevance to Hydroformylation. Chem. Commun. 2004, 4, 1070–1071. [Google Scholar] [CrossRef]
- Haumann, M.; Riisager, A. Hydroformylation in Room Temperature Ionic Liquids (RTILs): Catalyst and Process Developments. Chem. Rev. 2008, 108, 1474–1497. [Google Scholar] [CrossRef]
- Strobel, V. Towards Rational Design of SILP Catalysts: A Water-Gas Shift Reaction Case Study Reaktionstechnische Modellierung von SILP Katalysatoren Am Beispiel Der Wasser-Gas Shift Reaktion. Ph.D. Thesis, Universität Erlangen, Erlangen, Germany, 2020. [Google Scholar]
- Rietzler, F.; May, B.; Steinrück, H.P.; Maier, F. Switching Adsorption and Growth Behavior of Ultrathin [C2C1Im][OTf] Films on Au(111) by Pd Deposition. Phys. Chem. Chem. Phys. 2016, 18, 25143–25150. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | αIL | Feed Gas Composition (Rest N2), Period Duration and Temperature | XCO, P1 in % | XCO, P3 in % | Rel. Activity (XCO, P3/XCO, P1) in % | ||
|---|---|---|---|---|---|---|---|
| Period 1 | Period 2 | Period 3 | |||||
| Ref. | 0.18 | WGSR: 10% CO, 20% H2O steady state b 120 °C | WGSR: 10% CO, 20% H2O, 60 h, 120 °C | WGSR: 10% CO, 20% H2O 10–90 h c 120 °C | 13.6 | 13.6 | 100 |
| 1 | 0.18 | 10% H2, 30 h, 120 °C | 13.3 | 1.38 | 10 | ||
| 0.18 | 10% H2, 45 h a, 120 °C | 13.3 | 0 | 0 | |||
| 0.18 | 10% H2, 30 h, 140 °C | 14 | 0 | 0 | |||
| 0.15 | 10% H2, 30 h, 120 °C | 12.2 | 0 | 0 | |||
| 0.22 | 10% H2, 30 h, 120 °C | 12.5 | 4.4 | 35 | |||
| 0.22 | 10% H2, 45 h a, 120 °C | 12.5 | 2.4 | 19 | |||
| 2 | 0.18 | 10% H2, 10% CO, 30 h, 120 °C | 14 | 13.7 | 98 | ||
| 0.18 | 90% H2, 10% CO, 30 h, 120 °C | 13.2 | 12 | 91 | |||
| 3 | 0.18 | 10% CO, 40 h, 120 °C | 13.9 | 13.6 | 98 | ||
| 4 | 0.18 | 10% H2O, 30 h, 120 °C | 12 | 7 | 58 | ||
| 0.11 | 10% H2, 10% H2O; 30 h, 120 °C | 10 | 0 | 0 | |||
| 0.18 | 10% H2, 10% H2O; 30 h, 120 °C | 13.5 | 0 | 0 | |||
| 5 | 0.18 | 10% H2, 10% CO2, 30 h, 120 °C | 12.2 | 0 | 0 | ||
| 6 | 0.11 | 56% H2, 20% H2O, 10% CO, 9% CO2, 5 h, 140 °C + 25 h, 120 °C | 8.7 | 7.7 | 89 | ||
| 0.18 | 70% H2, 20% H2O, 10% CO 30 h, 120 °C | 14.5 | 13.9 | 93 | |||
| 7 | 0.18 | 100% N2, 60 h, 120 °C | 8.5 | 8.2 | 96 | ||
| 0.18 | 100% N2, 75 h, 40 °C | 13.4 | 13.1 | 98 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fischer, F.; Thiessen, J.; Korth, W.; Jess, A. Stability of an Ultra-Low-Temperature Water–Gas Shift Reaction SILP Catalyst. Catalysts 2025, 15, 602. https://doi.org/10.3390/catal15060602
Fischer F, Thiessen J, Korth W, Jess A. Stability of an Ultra-Low-Temperature Water–Gas Shift Reaction SILP Catalyst. Catalysts. 2025; 15(6):602. https://doi.org/10.3390/catal15060602
Chicago/Turabian StyleFischer, Ferdinand, Johannes Thiessen, Wolfgang Korth, and Andreas Jess. 2025. "Stability of an Ultra-Low-Temperature Water–Gas Shift Reaction SILP Catalyst" Catalysts 15, no. 6: 602. https://doi.org/10.3390/catal15060602
APA StyleFischer, F., Thiessen, J., Korth, W., & Jess, A. (2025). Stability of an Ultra-Low-Temperature Water–Gas Shift Reaction SILP Catalyst. Catalysts, 15(6), 602. https://doi.org/10.3390/catal15060602