Abstract
Sorption-enhanced reverse water–gas shift (SE-RWGS), designated as COMAX, was studied using a Pt4A bifunctional catalyst (reactive adsorbent). The bifunctional Pt4A catalyst integrates CO2 activation and reaction with water adsorption functionality, where the active phase is loaded onto a carrier that provides a surface area for Pt dispersion as well as H2O adsorption capacity. The 0.3 wt% Pt-4A molecular sieve reactive sorbent was tested at a kg scale in a pressure swing (reactive) adsorption–regeneration process. More than 400 cycles over 50 days of operation were successfully demonstrated without significant decay. Cyclic stability was achieved, provided that the regeneration temperature was sufficiently high to ensure near-complete dehydration. The single-bead structure withstood the pressure swing operation effectively, with only a maximum of 2% of the total recovered reactive sorbent turning to fines (<500 μm). The successful integration of catalytic activity and water adsorption capacity into a single particle presents opportunities for the further intensification of sorption-enhanced reactions for CO2 conversion.
1. Introduction
The conversion of carbon dioxide (CO2) to transport fuels using hydrogen has the potential to integrate CO2 capture with energy storage and conversion [1,2,3]. In particular, synthetic aviation fuel (SAF) production using renewable H2 and CO2 is a promising pathway. The aviation industry faces significant challenges in reducing its climate impact. While direct electrification is feasible for other transportation sectors, hydrocarbons will remain the predominant fuel source for aircraft. SAF, produced via renewable hydrogen and carbon dioxide, is one of the few viable alternatives. This drop-in fuel can be synthesized via the well-established Fischer–Tropsch process [4] or through the emerging methanol-to-jet route [5]. Both pathways rely on syngas containing CO and H2, which is produced via the selective CO2 reduction process to CO. Traditional thermochemical conversion is achieved through the endothermic reverse water–gas shift (RWGS) reaction, which is limited by thermodynamic equilibrium, operating above 850 °C with limited single-pass CO2 conversion. This high-temperature process demands significant energy input, complicates electrification and increases the risk of catalyst deactivation. A key challenge is optimizing a low-temperature process that maintains high conversion and selectivity. Conversion at lower temperatures (300–400 °C) can be significantly enhanced if the reaction product, water, is selectively removed [6]. The principle of separation enhancement, based on Le Chatelier’s principle, shifts an equilibrium-limited reaction toward higher conversion by selectively removing reaction products [7,8]. Hydrophilic zeolites, such as Linde Type A (LTA) and Faujasite (FAU), exhibit strong water adsorption under RWGS conditions [9,10]. Recently, proof-of-concept studies have demonstrated the feasibility of a bifunctional catalyst (reactive sorbent) for sorption-enhanced RWGS, designated as COMAX, which integrates CO2 activation and water adsorption in a single bead [11,12]. Platinum and copper are both recognized as effective catalysts for the (reverse) water–gas shift reaction [13,14]. Achieving sorption-enhanced RWGS activity requires custom-tailored zeolite-supported Pt and Cu catalysts that function as bifunctional reactive sorbents. A key consideration in designing these sorbents for CO2 hydrogenation is preserving the water sorption capacity while introducing active components for CO2 activation and reaction. Platinum is preferred due to its high intrinsic activity, allowing for lower loadings that minimize the disruption of water sorption sites. Additionally, Pt is less prone to deactivation at the preferred reactive adsorption temperatures (350–400 °C). This study presents the first cyclic demonstration of the longevity of the COMAX process with a bifunctional reactive sorbent under pressure swing mode in a single-column adsorption–blowdown–purge–repressurization cycle.
2. Results and Discussion
Figure 1 gives an example of the reactive adsorption result with a 0.3 wt%Pt-4A and a H2:CO2 of 3, at 25 bar(a) and 390 °C. Approximately a maximum of 93% conversion of CO2 to CO is achieved by the simultaneous RWGS reaction and adsorption of the product water in the initial pre-breakthrough period. During the course of the reaction, the water product displaces a part of the CO2 from the reaction sites, and the CO2 and the CO first slowly proceed from the initially very high concentration toward near steady state concentration levels by the end of the 120 min. In the last part of this adsorption sequence, this process is accelerated by water breakthrough, i.e., the saturation of the sorbent. The excess of hydrogen in the reaction with CO2 is aligned with the anticipated H2:CO product gas required for downstream methanol synthesis and/or the Fischer–Tropsch reaction as part of a process integration towards SAF.
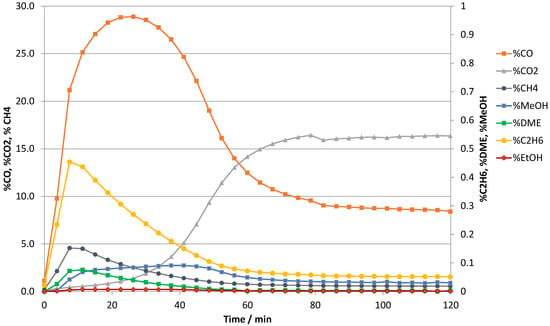
Figure 1.
CO2 and products in the reactive adsorption step on 0.3 wt%Pt_4A molecular sieve using 71.2% H2, 23.8% CO2 and 5.0% inert tracer at 390 °C, 25 bara, 250 h−1 and 80 sccm.
Process and cycle design focuses on the initial pre-breakthrough period in the reactive adsorption step with very high or maximum CO productivity, e.g., the initial 40 min that is shown in Figure 1. The overall product distribution is listed in Table 1. The amount of carbon that ends up in the methane becomes significant at the highest temperature of 390 °C.

Table 1.
Product distribution obtained with 0.3wt%Pt_4A based on 40 min. (outlet flow integrated over time) of production using 69.3% H2, 25.7% CO2 and 5.0% inert tracer at 25 bara, 250 h−1 and 80 sccm.
Subsequently, 0.3 wt%Pt_4A on the kg scale (2 kg; 200 cm length, 3.8 cm internal diameter) was tested in pressure swing cyclic mode following the sequence of (reactive) adsorption (60 min), blowdown (10 min), purge (36 min) and repressurization (3 min), with the adsorption step feed flow from top to bottom of the column and the regeneration steps in reversed flow direction, and with a higher space velocity of 600 h−1 in the reactive adsorption step. From the preliminary screening of temperature, it was decided that the focus would be on the reactive sorption temperatures of 360 and 380 °C.
Figure 2 shows the products (GC analysis) with the H2:CO2 of 3 in the reactive adsorption step but only at the condition 600 h−1 with a 100% H2 purge flow and a 100% H2 repressed flow condition. Figure 2 shows the performance of the cyclic COMAX operation over time and at this fixed set of conditions only. The many periods without data in the graph represent the periods where the reactive adsorbent was exposed to other test conditions (feed and purge flow variations) that, although used at TNO for model validation, are out of the scope of this paper. The setpoint temperature is 360 °C on the left hand side of the dashed line and 380 °C on the right side of the dashed line. The concentration at various moments in time during the course of the experimental program is shown. The graph represents multiple measurements points per cycle, and throughout the cycle, the CO production decreases while the adsorbent becomes more saturated. This results in a range of concentrations illustrated by the ascending (CO2) and descending (CO) patterns. From the maximum CO yield, it follows that the trend is initially somewhat downwards and the decay of the CO maximum concentration in the (approximately) 1100 h on the stream period seems to level off and eventually stabilizes at approximately 25% CO at 360 °C.
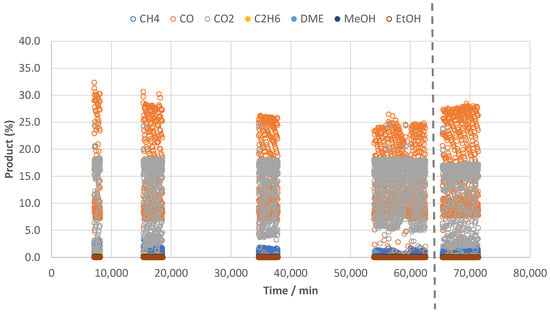
Figure 2.
Products monitored by µGC during the reactive adsorption at 600 h−1 with H2 purge and H2 repress only; setpoint heating at 360 °C. The last part to the right of the dashed line was obtained with setpoint heating at 380 °C.
The post-test unloading of the reactor revealed that roughly 2% of the total recovered reactive sorbent were fines (sizes less than 500 μm). A test with 100 mbar steam in inert gas at 370 °C with the pre-test sorbent and post-test unloaded reactive sorbent after 50 days of operation did not indicate a loss of water sorption capacity. Tests with up to four times higher GHSV showed that the steady state concentrations of CO and CO2 are the same: The catalytic functionality in the present study is largely over dimensioned and the decay of the CO yield in Figure 2 is largely determined by sorption characteristics. As much as the largest part of the initial decay shown in Figure 2 relates to incomplete regeneration. Figure 3 gives an example of the CO evolution (NDIR analysis) during reactive adsorption at the setpoint of 360 °C and the regeneration together with the temperature profiles monitored at 70 cm and 100 cm in the reactor during these periods. During the reactive adsorption process, the temperature increases first and then it levels off to the setpoint, which was 360 °C during these cycles. During the regeneration, the temperature drops to 330 °C and then it reaches 360 °C only at the very end of the purge period. This means that for the majority of the purge time, the temperature was far below the setpoint and, as is known to the experts in the field, typically on the low side to fully regenerate molecular sieves in a time scale of minutes [15].
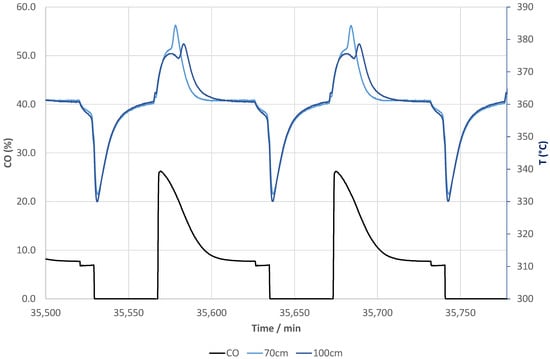
Figure 3.
CO product (NDIR) and temperatures across the reactor column during cyclic operation at a setpoint of 360 °C.
In Figure 4, the main trends obtained for the categories of side products during cyclic operation in the same period are shown. While most of these instantaneously disappear from the gas phase during regeneration (blowdown and purge), a very small fraction of the methanol appears to remain in the sorbent at the start of the next reactive adsorption step.
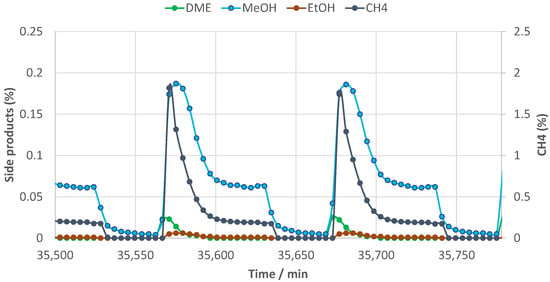
Figure 4.
Typical methanol (and other trace component) trends during the 600 h−1 H2 purge H2 repress cycling; 360 °C.
When we now make the same analysis for the sequence monitored at setpoint 380 °C, the positive impact of the higher temperature on the cyclic stability becomes obvious. First when we look at the right hand side of the dashed line in Figure 2, the CO production monitored by µGC does not indicate any decay during the cycling, i.e., it appears very constant during the 60 cycles included in this sequence. This is shown in much more detail with the higher data resolution analysis as available from the NDIR gas analysis running in parallel and compiled in Figure 5, which indeed shows a perfectly stable CO production.
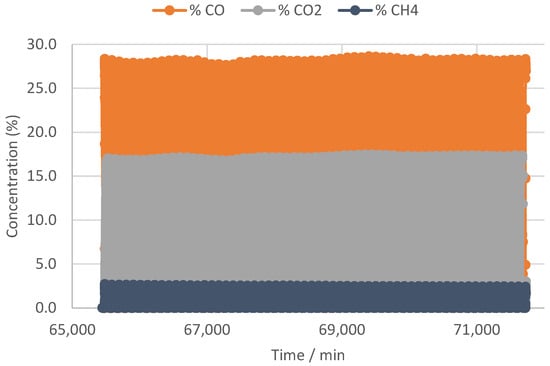
Figure 5.
Products monitored by NDIR during reactive adsorption at 600 h−1 with H2 purge and H2 repress and the setpoint heating at 380 °C.
Figure 6 shows an excerpt of the CO evolution (NDIR analysis) during reactive adsorption at 380 °C and the regeneration together with the temperature profiles monitored at 70 cm and 100 cm in the reactor during these periods. During the reactive adsorption process, the temperature increases first and then it levels off to the setpoint, which was 380 °C during these cycles. During the regeneration, the temperature drops to 350 °C and then it reaches 380 °C only at the very end of the purge. This means that for the majority of the purge time, the temperature was far below the setpoint temperature but the temperature never decreased below 350 °C, which is important to ensure the (near) complete regeneration of molecular sieves in a time scale of minutes.
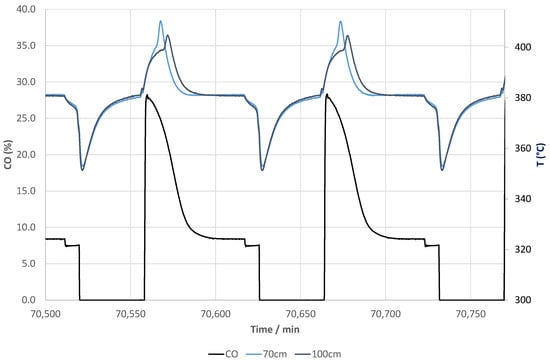
Figure 6.
CO product (NDIR) and temperatures across the reactor column during cyclic operation at a setpoint of 380 °C.
In Figure 7, the main trends obtained for the categories of side products during cyclic operation at 380 °C in the same period are shown. Similar to the situation at 360 °C, a very small fraction of the methanol appears to remain in the sorbent at the start of the next reactive adsorption step.
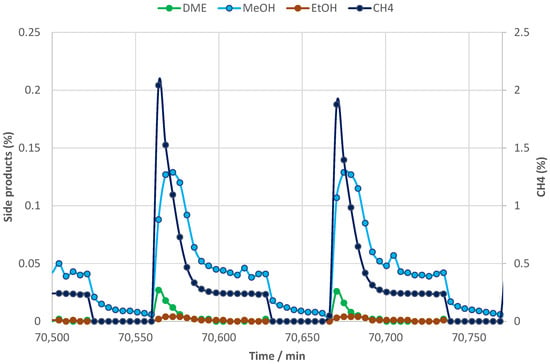
Figure 7.
Typical methanol (and other trace component) trends during the 600 h−1 H2 purge H2 repress cycling; 380 °C.
The formation of methane is slightly higher at the higher temperature of reactive sorption with approximately a 2.1% maximum, but this is only for a very short time in the very initial part of the sorption enhancement period that levels off to an almost 10-fold lower concentration within minutes on stream. A process design with COMAX integrated with a downstream FT reaction or methanol synthesis reaction would disfavor the formation of methane since, at best, it would be inert and would begin accumulating in (recycle) loops. Moreover, any loss of precious green hydrogen through formation of hydrocarbon should be avoided. Overall, it appears that good cyclic stability can be obtained in pressure swing cyclic operation, provided that the temperature during the regeneration steps is high enough to ensure near-complete dehydration (>350 °C).
3. Materials and Methods
Molecular sieve beads 4A zeolite (2–3 mm) were used for incipient wetness impregnation with H2PtCl6.6H2O to obtain the reactive sorbent for COMAX with 0.3 wt% Pt loading. Prior to impregnation, the molecular sieve was first dried at 105 °C overnight. After the impregnation with the metal solution the obtained reactive sorbents were dried at 120 °C. Subsequently, the reactive sorbent was calcined in a furnace oven (Heraeus D-6450) at 400 °C (5 °C/min, 6 h dwell). The reactive sorbent was activated in situ with 10 °C/min in N2 to 325 °C and subsequently in 10% N2, 84% H2 and 6% CO2 with a dwell time of 4 h. Experimental runs with reactive adsorption steps at 25 bar(a) pressure were conducted on two high-pressure test rigs (see Supplementary Materials). The first test rig, at a gram scale of reactive sorbent operated at low GHSV (250 h−1), was used to analyze the type of products and side products in the reactive adsorption step and used to judge overall performance as a function of temperature of the reactive sorbent, prior to scaling up the synthesis. Here, adsorbent regeneration was performed in a co-current mode by a combination of both temperature and pressure swing (TPSA) by periodic switching to dry nitrogen, decreasing the pressure to 3 bar(a) and increasing the temperature to 400 °C. The second test rig, at a kilogram scale and 200 cm column length, enabled the use of up to 20 L/min total flow during the reactive adsorption step and the ability to adjust gas direction for counter-current regeneration with up to 50 L/min purge (hydrogen) gas following the pressure swing to atmospheric pressure. Heating was provided with electrical trace heating. During adsorption, this reactor was primarily operated at a higher space velocity of typically 600 h−1 at temperatures up to 390 °C. Gas analysis was performed by a gas chromatograph (µGC, equipped with TCD and FID), non-dispersive IR analyzer (NDIR) and a mass spectrometer (MS).
4. Conclusions
The cyclic operation of the Pt4A bifunctional catalyst in the sorption-enhanced reverse water–gas shift (COMAX) process was successfully demonstrated under pressure swing conditions. More than 400 cycles over 50 days of operation were completed without a significant decay in stability. Cyclic stability was maintained, provided that the regeneration temperature was high enough to ensure near-complete dehydration. Methane formation and minor by-products were observed but remained within controllable limits for process integration. The single-bead bifunctional sorbent design eliminates challenges associated with mixing separate sorbent and catalyst particles, such as settling, channeling and mass transfer limitations, and the differences in hardness which lead to abrasion. This design simplifies the scale-up for industrial applications. The COMAX process thus presents a promising pathway for enhancing CO2-to-syngas conversion efficiency at mild operating conditions, supporting sustainable aviation fuel (SAF) production. The further optimization of adsorption–regeneration cycles and integration with downstream catalytic processes will enhance the industrial viability of this technology.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/catal15050480/s1. Figures S1 and S2: High pressure test rig gr. scale; Figures S3 and S4: High pressure test rig kgr. scale.
Author Contributions
Conceptualization, J.A.Z.P.; methodology, J.A.Z.P. and V.D.; formal analysis, J.A.Z.P., S.B. and G.D.E. investigation, J.A.Z.P., S.B. and G.D.E.; writing—original draft preparation, J.A.Z.P.; writing—review and editing, J.A.Z.P., A.G. and J.B.; project administration, J.A.Z.P.; funding acquisition, G.S., J.B. and J.A.Z.P. All authors have read and agreed to the published version of the manuscript.
Funding
The work was carried out with subsidies from the Ministry of Economic Affairs and Climate Policy and the Ministry of Agriculture, Nature and Food Quality, National regulations EZK and LNV subsidies, Topsector Energy carried out by the Netherlands Enterprise Agency.
Data Availability Statement
The data presented in this study are available upon request from the corresponding author due to restrictions imposed by the publishing entities.
Acknowledgments
The authors thank Jan-Jaap Riegman (T.EN) and Mehdi Hamdi (T.EN) for the fruitful discussions. Martien Koppes (TNO) is thanked for assistance with the high pressure experiments.
Conflicts of Interest
Author Andreas Geisbauer was employed by Clariant Produkte (Deutschland) GmbH. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| SE-RWGS | Sorption-Enhanced Reverse Water–Gas Shift; |
| CO2 | Carbon Dioxide; |
| H2 | Hydrogen; |
| RWGS | Reverse Water–Gas Shift; |
| SAF | Synthetic Aviation Fuel; |
| LTA | Linde Type A (zeolite); |
| FAU | Faujasite (zeolite); |
| Pt4A | Platinum on 4A molecular sieve; |
| GHSV | Gas Hourly Space Velocity; |
| TPSA | Temperature and Pressure Swing Adsorption; |
| NDIR | Non-Dispersive Infrared Analyzer; |
| µGC | Micro Gas Chromatograph; |
| TCD | Thermal Conductivity Detector; |
| FID | Flame Ionization Detector |
References
- Yu, K.M.K.; Curcic, I.; Gabriel, J.; Tsang, S.C.E. Recent advances in CO2 capture and utilization. ChemSusChem 2008, 1, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Detz, R.J.; Reek, J.N.H.; van der Zwaan, B.C.C. The future of solar fuels: When could they become competitive? Energy Environ. Sci. 2018, 11, 1653–1669. [Google Scholar] [CrossRef]
- Van Os, P. Accelerating low carbon industrial growth through carbon capture, utilization and storage (CCUS). Greenh. Gases Sci. Technol. 2018, 8, 994–997. [Google Scholar] [CrossRef]
- Pearson, R.; Coe, A.; Paterson, J. Innovation in Fischer–Tropsch: A sustainable approach to fuels production a cost-effective method of converting any carbon source into high-quality liquid hydrocarbon fuels. Johns. Matthey Technol. Rev. 2021, 65, 395–403. [Google Scholar] [CrossRef]
- Schmidt, P.; Batteiger, V.; Roth, A.; Weindorf, W.; Raksha, T. Power-to-liquids as renewable fuel option for aviation: A review. Chem. Ing. Tech. 2018, 90, 127–140. [Google Scholar] [CrossRef]
- Carvill, B.T.; Hufton, J.R.; Anand, M.; Sircar, S. Sorption-enhanced reaction process. AIChE J. 1996, 42, 2765. [Google Scholar] [CrossRef]
- Campbell, J.A. Le Chatelier’s Principle, Temperature Effects, and Entropy. J. Chem. Ed. 1985, 62, 231–232. [Google Scholar] [CrossRef]
- Van Kampen, J.; Boon, J.; van Berkel, F.; Vente, J.; Van Sint Annaland, M. Steam separation enhanced reactions: Review and outlook. Chem. Eng. J. 2019, 374, 1286–1303. [Google Scholar] [CrossRef]
- Ghodhbene, M.; Bougie, F.; Fongarland, P.; Iliuta, M.C. Hydrophilic zeolite sorbents for In-situ water removal in high temperature processes. Can. J. Chem. Eng. 2017, 95, 1842–1849. [Google Scholar] [CrossRef]
- Van Kampen, J.; Boon, J.; Van Sint Annaland, M. Steam adsorption on molecular sieve 3A for sorption enhanced reaction processes. Adsorption 2020, 27, 577–589. [Google Scholar] [CrossRef]
- Pieterse, J.A.Z.; Elzinga, G.D.; Booneveld, S.; Van Kampen, J.; Boon, J. Reactive Water Sorbents for the Sorption-Enhanced Reverse Water–Gas Shift. Catal. Lett. 2021, 152, 460–466. [Google Scholar] [CrossRef]
- Pieterse, J.A.Z.; Elzinga, G.D. Multifunctional Catalyst for the Conversion of Carbon Dioxide. WO2021240017A1, 2 December 2021. [Google Scholar]
- Arunajatesan, V.; Subramaniam, B.; Hutchenson, K.W.; Herkes, F.E. In situ FTIR investigations of reverse water gas shift reaction activity at supercritical conditions. Chem. Eng. Sci. 2007, 62, 5062–5069. [Google Scholar] [CrossRef]
- Dietz, L.; Piccinin, S.; Maestri, M. Mechanistic Insights into CO2 activation via reverse water–gas shift on metal surfaces. J. Phys. Chem. C 2015, 119, 4959–4966. [Google Scholar] [CrossRef]
- Guffanti, S.; Visconti, C.G.; van Kampen, J.; Boon, J.; Groppi, G. Reactor modelling and design for sorption enhanced dimethyl ether synthesis. Chem. Eng. J. 2021, 404, 126573. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).