


Supramolecular Perylene Diimides for Photocatalytic Hydrogen Production

Abstract

1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Photocatalyst | Preparation Method | Structure and Form | Photocatalytic Hydrogen Production Efficiency | Light Source | Wavelength | Electron Source for H2 Evolution | Reference |
|---|---|---|---|---|---|---|---|
| PTA and Cl-PTA | Hydrolysis-acidification reassembly strategy | Nanosheets | PTA: 15.2 mmol·g−1·h−1 Cl-PTA: 27.1 mmol·g−1·h−1 | CEL-HXF300 300W xenon lamp 1 | Full-spectrum irradiation (UV + visible light) | Ascorbic acid | [48] |
| PyBpDBSO-5 | Suzuki coupling reaction | Flaky amorphous structures | 48.5 mmol·g−1·h−1 | Visible light | >420 nm | TEOA | [49] |
| PTCDIs/Pt/g-C3N4 | Self-assembly method | Amorphous structure | 0.015 mmol·g−1·h−1 | 300 W xenon lamp (CEL-S500, Beijing AULTT) with 420 nm cutoff optical filter attached 1 | >420 nm | TEOA | [56] |
| PDI-phthalic | Self-assembly method | Nanosheets | 1.1 mmol·g−1·h−1 | 300/500 W xenon lamp with a 420 nm cut-off filter 2 | >420 nm | Ascorbic acid | [66] |
| PDI/Zn0.8Cd0.2S | Self-assembly method | Nanorods/nanospheres | 71.98 μmol·g−1·h−1 | 300W xenon lamp (1000 mW·cm−2) | >420 nm | Water | [67] |
| PDI/Zn0.7Cd0.3S | Co-precipitation-hydrothermal method | Nanoparticles | 5.166 mmol·g−1·h−1 | Visible-light irradiation | Monochromatic light at 420 nm | Na2S and Na2SO3 aqueous solution | [68] |
| N-APDI | Self-assembly method | Nanosheets | 61.49 mmol·g−1·h−1 | 300 W xenon lamp | >400 nm | Ascorbic acid | [69] |
| P-PMPDI | Self-assembly method | Multilayer nanobelts | 11.7 mmol·g−1·h−1 | Visible light | 400 nm–780 nm | Ascorbic acid | [50] |
| P-PMPDI-Zr | Self-assembly method | Multilayer nanobelts | 50.46 mmol·g−1·h−1 | CEL-HXF300 300 W xenon lamp 1 | 400 nm–780 nm | Ascorbic acid | [54] |
| CBZ-PDCA-PT2 | Heating + evaporating + filtration, Suzuki coupling | Nanoparticles | 30 mmol·g−1·h−1 | Sun simulator SCIENCETECH SF-300-A equipped with airmass filter AM1.5 G 3 | Visible light | Ascorbic acid | [70] |
| TPPS/PDI | Self-assembly method | Nanowires | 30.36 mmol·g−1·h−1 | Full-spectrum light | Full-spectrum irradiation | Ascorbic acid | [71] |
| g-C3N4/rGO/PDIP | Wet-chemistry reduction + solvent evaporation + heat treatment | Nanosheets/nanorods (core/shell) | 15.8 μmol·g−1·h−1 | Visible light | ≥420 nm | Water | [72] |
| PDI/g-C3N4 | Self-assembly method | Nanorods/nanosheets | 1649.93 μmol·g−1·h−1 | Full-spectrum light | Full-spectrum irradiation | TEOA | [73] |
| COF (TATF-COF)/PUP | Imidazole solvent method + via in situ coupling | Nanosheets/nanorods | 94.5 mmol·g−1·h−1 | 350 W xenon lamp | >420 nm | Ascorbic acid | [74] |
| PDI-TiO2 | Solvent compounding method | Nanosheets/nanoparticle | 238 mmol·g−1·h−1 | 300 W xenon lamp | Visible light | Water | [75] |
| ZIS/PDIIM | Hydrothermal method | Nanosheets/nanorods | 13.04 mmol·g−1·h−1 | PLS-SXE300 300 W xenon lamp (0.6 W/cm2)1 | 320 nm–780 nm | Na2S and Na2SO3 aqueous solution | [76] |
| PDINH/TiO2 | Hydrothermal method + mixing | Nanosheets/nanorods | 1.2 mmol·g−1·h−1 | PLS-SXE 300 300 W Xe arc lamp 4 | UV–VIS light irradiation | Methanol | [77] |
| GQDs/PDI-14% | Self-assembly method + mixing | Zero-dimensional quantum dots/nanofibers | 1.6 mmol·g−1·h−1 | Visible light | >420 nm | Ascorbic acid | [78] |
| PTA | Facile hydrolysis reassembly of PTCDA | Nanosheets | 118.9 mmol·g−1·h−1 | 300 W Xe lamp (~530 mW cm−2) | ≥300 nm | Ascorbic acid | [79] |
| CN-P-0.2% | Thermal condensing of cyanamide | Porous structure | 17.7 mmol·g−1·h−1 | LED | 450 nm | TEOA | [80] |
2. Fundamentals
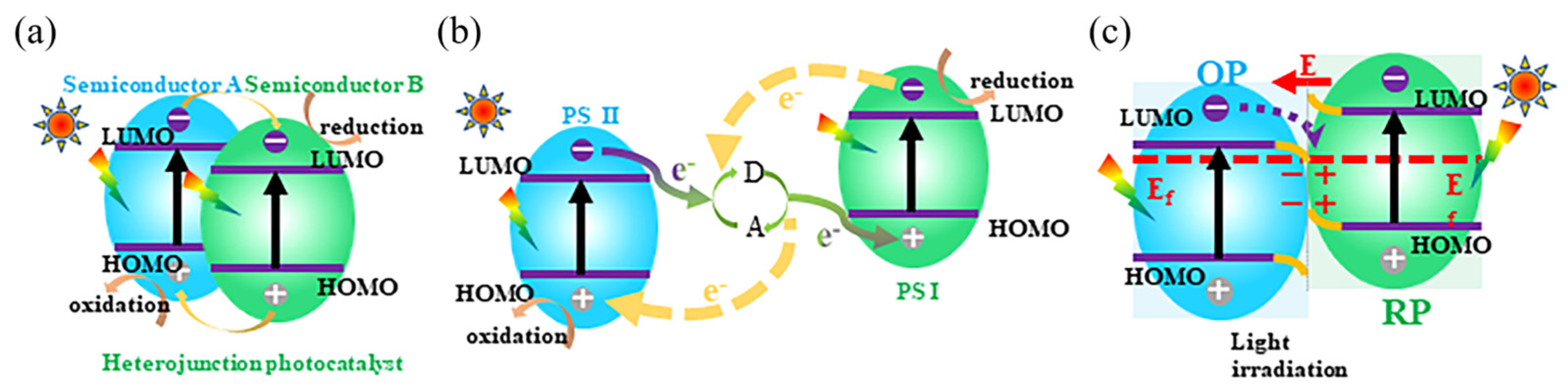
2.1. The Process of Photocatalytic Water Splitting
2.2. The Structure/Property of PDI
3. Modification Strategies
3.1. Molecular-Level Modifications
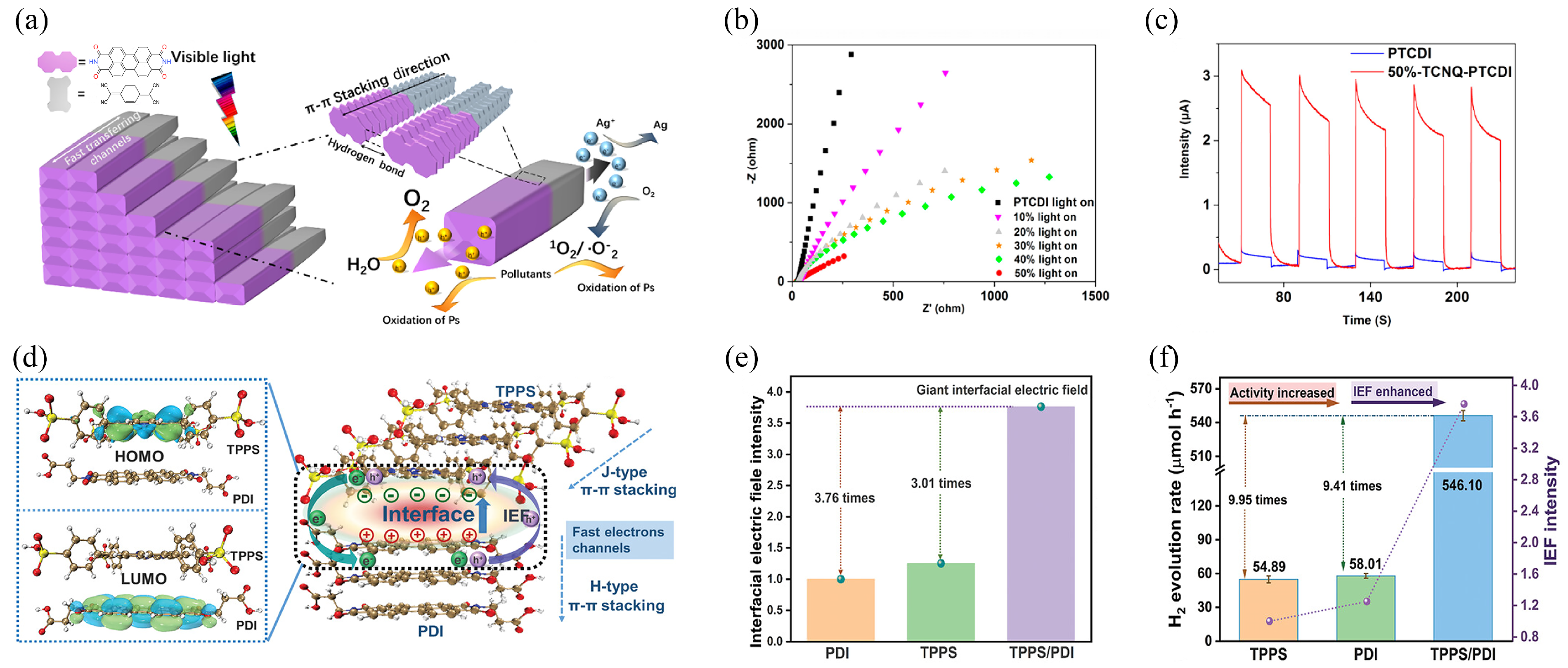
3.2. Nanostructuring
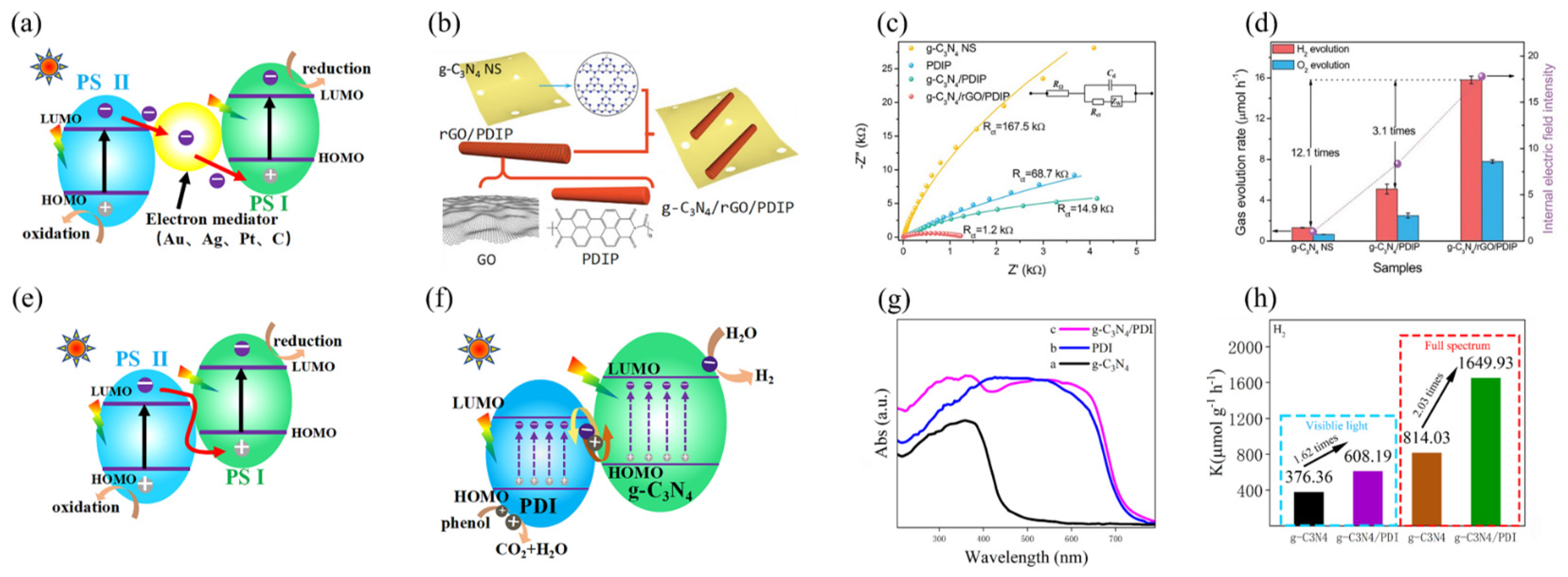
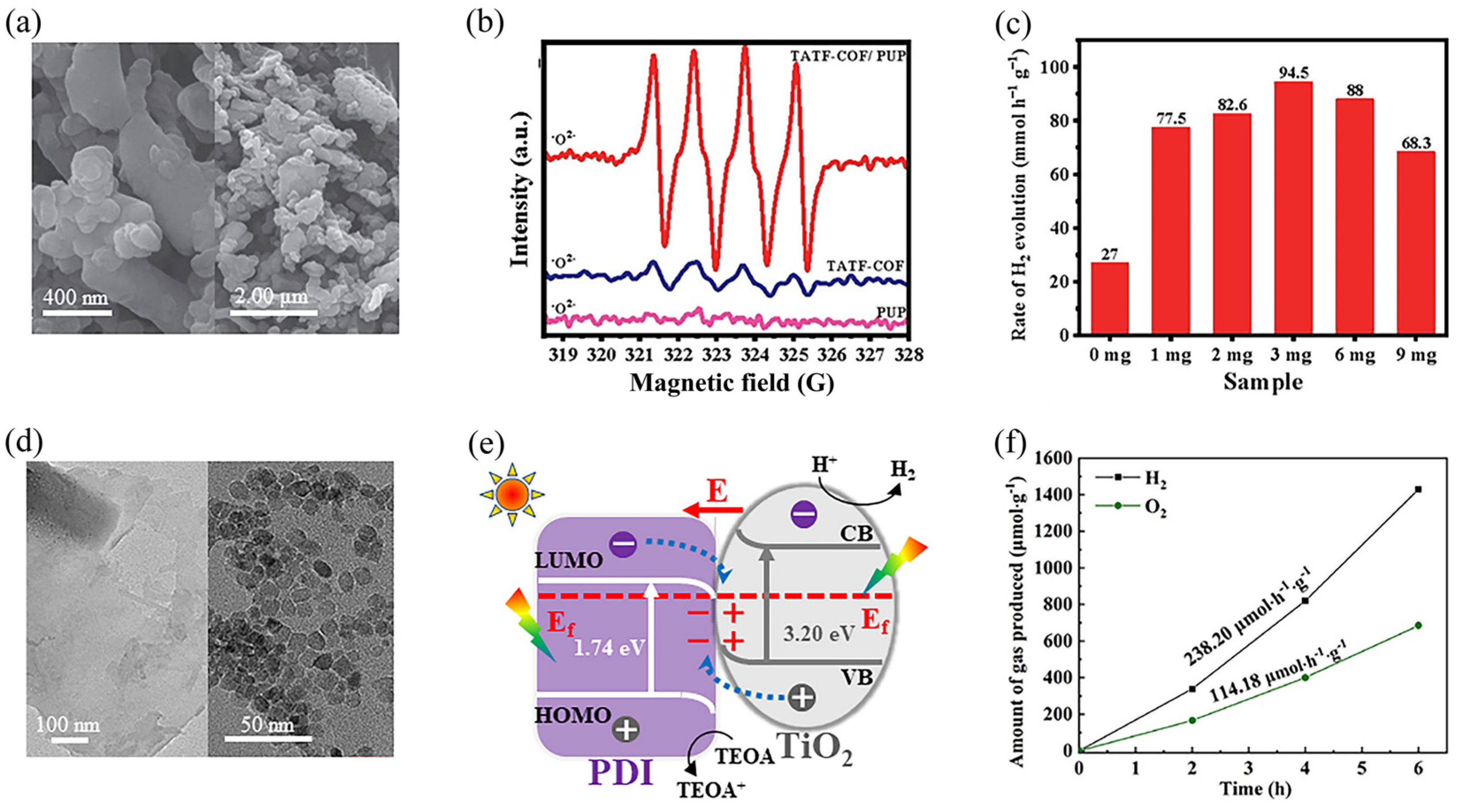
3.3. Heterostructuring
4. Conclusions and Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| PTA | 3,4,9,10-perylenetetracarboxylic acid |
| Cl-PTA | 1,6,7,12-tetrachloro-3,4,9,10-perylenetetracarboxylic acid |
| PyBpDBSO-X | Polymer from reaction of perylene diimide, biphenyl, and dibenzothiophene-S,S-dioxide; X represents content of perylene diimide (PDI) |
| PTCDI(s)/PDINH/PDI | Perylene-3, 4, 9, 10-tetracarboxylic diimide |
| N-APDI | Pyrrole-phosphoric acid-PDI |
| P-PMPDI | Pyrrole substituted N,N′-bis(phosphonomethyl)-3,4,9,10-perylenediimide |
| CBZ-PDCA-PT2 | Complex of 1,7-bis [4-(carbazol-9-yl)phenyl]-N,N-bis(2-ethylhexyl)perylene-3,4:9,10-tetracarboxylic acid diimide (Cbz-PDI) and Pt |
| TPPS | Tetra(4-sulfonatophenyl)porphyrin |
| g-C3N4/rGO/PDIP | Graphitic carbon nitride/rGO/perylene diimide polymer |
| COF | Covalent organic framework |
| TATF-COF | Triazine-based imine-linked COF |
| PUP | Perylene diimide urea polymer |
| ZIS | ZnIn2S4 |
| PDIIM | Perylene-3,4,9,10-tetracarboxylic dianhydride (PTCDA) reacting with 3-(1H-imidazol-1-yl)propan-1-amine |
| GQDs | Graphene quantum dots |
| CP-N | Carbon nitride (mpg-C3N4)/PTCDA composite photocatalyst |
| TEOA | Triethanolamine |
References
- Singh, R.; Dutta, S. A review on H2 production through photocatalytic reactions using TiO2/TiO2-assisted catalysts. Fuel 2018, 220, 607–620. [Google Scholar] [CrossRef]
- Ismael, M. Latest progress on the key operating parameters affecting the photocatalytic activity of TiO2-based photocatalysts for hydrogen fuel production: A comprehensive review. Fuel 2021, 303, 121207. [Google Scholar] [CrossRef]
- Kim, D.; Sakimoto, K.K.; Hong, D.; Yang, P. Artificial Photosynthesis for Sustainable Fuel and Chemical Production. Angew. Chem. Int. Ed. 2015, 54, 3259–3266. [Google Scholar] [CrossRef]
- Lewis, N.S.; Nocera, D.G. Powering the planet: Chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. USA 2006, 103, 15729–15735. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Zhang, Q.; Wang, Y. Semiconductor-based nanocomposites for photocatalytic H2 production and CO2 conversion. Phys. Chem. Chem. Phys. 2013, 15, 2632–2649. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xu, H.; Shang, H.; Jin, L.; Chen, C.; Wang, Y.; Yuan, M.; Du, Y. Ir-Doped Pd Nanosheet Assemblies as Bifunctional Electrocatalysts for Advanced Hydrogen Evolution Reaction and Liquid Fuel Electrocatalysis. Inorg. Chem. 2020, 59, 3321–3329. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhu, C.; Wu, Z.; Stavitski, E.; Lui, Y.H.; Kim, T.-H.; Liu, H.; Huang, L.; Luan, X.; Zhou, L.; et al. Integrating Rh Species with NiFe-Layered Double Hydroxide for Overall Water Splitting. Nano Lett. 2020, 20, 136–144. [Google Scholar] [CrossRef]
- Bae, S.-Y.; Mahmood, J.; Jeon, I.-Y.; Baek, J.-B. Recent advances in ruthenium-based electrocatalysts for the hydrogen evolution reaction. Nanoscale Horiz. 2020, 5, 43–56. [Google Scholar] [CrossRef]
- Wang, Q.; Pornrungroj, C.; Linley, S.; Reisner, E. Strategies to improve light utilization in solar fuel synthesis. Nat. Energy 2022, 7, 13–24. [Google Scholar] [CrossRef]
- Chen, C.; Wu, A.; Yan, H.; Xiao, Y.; Tian, C.; Fu, H. Trapping [PMo12O40]3− clusters into pre-synthesized ZIF-67 toward MoxCoxC particles confined in uniform carbon polyhedrons for efficient overall water splitting. Chem. Sci. 2018, 9, 4746–4755. [Google Scholar] [CrossRef]
- Navidpour, A.H.; Hao, D.; Li, X.; Li, D.; Huang, Z.; Zhou, J.L. Key factors in improving the synthesis and properties of visible-light activated g-C3N4 for photocatalytic hydrogen production and organic pollutant decomposition. Catal. Rev. 2024, 66, 1665–1736. [Google Scholar] [CrossRef]
- Sahu, A.K.; Song, Z.X.; Upadhyayula, S. Ceria-based photocatalysts in water-splitting for hydrogen production and carbon dioxide reduction. Catal. Rev. 2024, 66, 1400–1477. [Google Scholar] [CrossRef]
- Davis, S.J.; Caldeira, K.; Matthews, H.D. Future CO2 Emissions and Climate Change from Existing Energy Infrastructure. Science 2010, 329, 1330–1333. [Google Scholar] [CrossRef] [PubMed]
- McGlade, C.; Ekins, P. The geographical distribution of fossil fuels unused when limiting global warming to 2 °C. Nature 2015, 517, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Takata, T.; Domen, K. Particulate photocatalysts for overall water splitting. Nat. Rev. Mater. 2017, 2, 17050. [Google Scholar] [CrossRef]
- Zhu, Y.; Lv, C.; Yin, Z.; Ren, J.; Yang, X.; Dong, C.-L.; Liu, H.; Cai, R.; Huang, Y.-C.; Theis, W.; et al. A [001]-Oriented Hittorf’s Phosphorus Nanorods/Polymeric Carbon Nitride Heterostructure for Boosting Wide-Spectrum-Responsive Photocatalytic Hydrogen Evolution from Pure Water. Angew. Chem. Int. Ed. 2020, 59, 868–873. [Google Scholar] [CrossRef]
- Kosco, J.; Bidwell, M.; Cha, H.; Martin, T.; Howells, C.T.; Sachs, M.; Anjum, D.H.; Gonzalez Lopez, S.; Zou, L.; Wadsworth, A.; et al. Enhanced photocatalytic hydrogen evolution from organic semiconductor heterojunction nanoparticles. Nat. Mater. 2020, 19, 559–565. [Google Scholar] [CrossRef]
- Yang, W.; Ma, G.; Fu, Y.; Peng, K.; Yang, H.; Zhan, X.; Yang, W.; Wang, L.; Hou, H. Rationally designed Ti3C2 MXene@TiO2/CuInS2 Schottky/S-scheme integrated heterojunction for enhanced photocatalytic hydrogen evolution. Chem. Eng. J. 2022, 429, 132381. [Google Scholar] [CrossRef]
- Lee, W.H.; Lee, C.W.; Cha, G.D.; Lee, B.-H.; Jeong, J.H.; Park, H.; Heo, J.; Bootharaju, M.S.; Sunwoo, S.-H.; Kim, J.H.; et al. Floatable photocatalytic hydrogel nanocomposites for large-scale solar hydrogen production. Nat. Nanotechnol. 2023, 18, 754–762. [Google Scholar] [CrossRef]
- Swathi, S.; Babu, E.S.; Yuvakkumar, R.; Ravi, G.; Chinnathambi, A.; Alharbi, S.A.; Velauthapillai, D. Branched and unbranched ZnO nanorods grown via chemical vapor deposition for photoelectrochemical water-splitting applications. Ceram. Int. 2021, 47 Pt A, 9785–9790. [Google Scholar] [CrossRef]
- Goktas, S.; Goktas, A. A comparative study on recent progress in efficient ZnO based nanocomposite and heterojunction photocatalysts: A review. J. Alloys Compd. 2021, 863, 158734. [Google Scholar] [CrossRef]
- Du, Z.; Pan, J.; Ma, C.; Guan, Y.; Sun, M.; Gao, Z.; Tang, H.; Yan, X. CdS nanorods embedded in ZnIn2S4 nanosheets to construct n-n heterojunction for ultrahigh photocatalytic H2 and H2O2 production. Colloids Surf. A Physicochem. Eng. Asp. 2025, 706, 135774. [Google Scholar] [CrossRef]
- Wang, K.; Zhao, N.; Wang, J.; Xie, H.; Wang, M.; Jin, Z. “Electron trap” modified type-I heterojunction CdS/CoSe/graphdiyne photocatalyst synergistically promotes photocatalytic hydrogen production. J. Environ. Chem. Eng. 2025, 13, 114967. [Google Scholar] [CrossRef]
- Ghosh, S.; Das, P.S.; Bera, S.; Sarkar, D.; Roy, K.; Nath, S.; Ghosh, P.; Ghosh, C.K.; Allu, A.R. Conjugated Polymer-Supported Doped Bi2WO6 S-Scheme Heterojunction for Proficient Water Splitting via Dual Regulation of Band Gap Engineering and Improved Charge Separation. ACS Appl. Energy Mater. 2024, 7, 10906–10920. [Google Scholar] [CrossRef]
- Huang, Y.J.; He, B.; Li, J.X.; Song, S.Y.; Cao, J.; Zheng, Y.Y.; Wang, J.J.; Zhu, M.; Pan, J.Q.; Li, C.R. The Bi2WO6/CdS hollow core-shell S-scheme nano-heterojunction towards enhanced visible light photocatalytic H2 evolution and degradation via template induction. Int. J. Hydrogen Energy 2024, 82, 1331–1340. [Google Scholar] [CrossRef]
- Chiu, M.-H.; Kuo, C.-C.; Huang, C.-W.; Yang, W.-D. Preparation of CuS/PbS/ZnO Heterojunction Photocatalyst for Application in Hydrogen Production. Catalysts 2022, 12, 1677. [Google Scholar] [CrossRef]
- Gahlawat, A.; Kumar, D.; Lokhande, P.E.; Sharma, R.; Verma, B.; Rednam, U.; Ghotekar, S.; Ghfar, A.A.; Kumar, Y.A.; Praveenkumar, S. Fabrication of Graphene Oxide on CdS- and PbS-Doped Bismuth Titanates for Photocatalytic Hydrogen Production. J. Electron. Mater. 2024, 53, 7753–7761. [Google Scholar] [CrossRef]
- Chen, Y.; Yan, C.; Dong, J.; Zhou, W.; Rosei, F.; Feng, Y.; Wang, L.-N. Structure/Property Control in Photocatalytic Organic Semiconductor Nanocrystals. Adv. Funct. Mater. 2021, 31, 2104099. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, X.; Liu, D. Recent developments of perylene diimide (PDI) supramolecular photocatalysts: A review. J. Photochem. Photobiol. C Photochem. Rev. 2021, 48, 100436. [Google Scholar] [CrossRef]
- Chen, Y.; Jiang, D.; Gong, Z.; Li, Q.; Shi, R.; Yang, Z.; Lei, Z.; Li, J.; Wang, L.-N. Visible-light responsive organic nano-heterostructured photocatalysts for environmental remediation and H2 generation. J. Mater. Sci. Technol. 2020, 38, 93–106. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; De Sarkar, S.; Das, A. Supramolecular Engineering and Self-Assembly Strategies in Photoredox Catalysis. ACS Catal. 2021, 11, 710–733. [Google Scholar] [CrossRef]
- Yang, H.; Sun, S.; Lyu, J.; Yang, Q.; Cui, J. Mechanism insight into triple S-Scheme intermolecular carbon nitride homojunction with robust built-in electric field for highly enhanced photocatalytic hydrogen evolution. Chem. Eng. J. 2024, 481, 148297. [Google Scholar] [CrossRef]
- Lu, X.-J.; Xu, L.; Ullah, I.; Li, H.-B.; Xu, A.-W. Sulfur-doped g-C3N4 photocatalyst for significantly steered visible light photocatalytic H2 evolution from water splitting. Catal. Sci. Technol. 2024, 14, 606–614. [Google Scholar] [CrossRef]
- Zhang, M.F.; Liang, X.F.; Gao, Y.; Liu, Y. C60- and CdS-Co-Modified Nano-Titanium Dioxide for Highly Efficient Photocatalysis and Hydrogen Production. Materials 2024, 17, 1206. [Google Scholar] [CrossRef]
- Shi, L.J.; Guo, P.; Zheng, J.M.; Zhao, P.J.; Jiang, Z.Y.; Shen, L. C60 surface-supported TM@Si16 (TM = Ti, Zr, Hf) superatoms as self-assembled photocatalysts. Appl. Surf. Sci. 2023, 616, 156465. [Google Scholar] [CrossRef]
- Nikoloudakis, E.; Coutsolelos, A.G.; Stratakis, E. Mini-Review on Catalytic Hydrogen Evolution from Porphyrin-Graphene Structures. Energy Fuels 2024, 38, 19222–19235. [Google Scholar] [CrossRef]
- Liu, Y.S.; Lv, X.P.; Zhong, Y.; Wang, G.Y.; Liu, S.H.; Chen, S.D.; Qi, C.; He, M.; Ping, S.G.; Luo, Z.Q.; et al. Self-Assembly Regulated Photocatalysis of Porphyrin-TiO2 Nanocomposites. Molecules 2024, 29, 3872. [Google Scholar] [CrossRef]
- Miao, X.; Wang, B.B.; Fan, H.T.; Zhang, P.; Bai, S.Q.; Liu, W.S. Tremella-like Boron-doped hierarchical CN and dispersion Co phthalocyanine assembling heterojunction for photocatalytic hydrogen evolution. Chem. Eng. J. 2023, 465, 142775. [Google Scholar] [CrossRef]
- Kharissova, O.V.; Méndez, Y.P.; Kharisov, B.I.; Nikolaev, A.L.; Luévano-Hipólito, E.; González, L.T. Porphyrins, phthalocyanines, and related covalent-organic frameworks in the photochemical and electrochemical water splitting: A review. Particuology 2024, 90, 236–265. [Google Scholar] [CrossRef]
- Tang, K.; Shao, J.Y.; Li, J.K.; Li, S.M.; Tang, J.H.; Duan, R.; Yao, J.N.; Zhong, Y.W. Pyridine-terminated small molecular photocatalyst for water reduction. Chem 2024, 10, 1925–1939. [Google Scholar] [CrossRef]
- Tateishi, I.; Kuwahara, S.; Furukawa, M.; Katsumata, H.; Kaneco, S. O-doped g-C3N4 prepared in pyridine for efficiently photocatalytic hydrogen production. Environ. Technol. 2024, 45, 5063–5073. [Google Scholar] [CrossRef] [PubMed]
- Camara, F.; Aguirre-Araque, J.S.; Fortage, J.; Collomb, M.N. Enhancing the stability of photocatalytic systems for hydrogen evolution in water by using a tris-phenyl-phenanthroline sulfonate ruthenium photosensitizer. Sustain. Energy Fuels 2024, 8, 1457–1472. [Google Scholar] [CrossRef]
- Tong, L.; Duan, L.; Zhou, A.; Thummel, R.P. First-row transition metal polypyridine complexes that catalyze proton to hydrogen reduction. Coord. Chem. Rev. 2020, 402, 213079. [Google Scholar] [CrossRef]
- Kommula, B.; Durairaj, P.; Mishra, S.; Kar, S.; Sury, A.; Kumar, A.; De, A.K.; Sarkar, S.; Bhattacharyya, S. Self-Assembled Oligothiophenes for Photocatalytic Hydrogen Production and Simultaneous Organic Transformation. ACS Appl. Nano Mater. 2022, 5, 14746–14758. [Google Scholar] [CrossRef]
- Ye, D.N.; Liu, L.; Peng, Q.M.; Qiu, J.B.; Gong, H.; Zhong, A.G.; Liu, S.Y. Effect of Controlling Thiophene Rings on D-A Polymer Photocatalysts Accessed via Direct Arylation for Hydrogen Production. Molecules 2023, 28, 4507. [Google Scholar] [CrossRef] [PubMed]
- Piao, H.W.; Zhao, J.; Zhang, S.J.; Quan, Q.; Hu, J.E.; Huang, Q.L.; Zhu, R.Y.; Fan, L.P.; Xiao, C.F. Polypyrrole/cadmium sulfide/nickel hollow fiber as an enhanced and recyclable intrinsic photocatalyst for pollutant removal and high-effective hydrogen evolution. Int. J. Hydrogen Energy 2024, 52, 173–189. [Google Scholar] [CrossRef]
- Pal, S.; Das, P.S.; Naskar, M.K.; Ghosh, S. Metal oxide nanocrystals embedded polypyrrole nanohybrid: Exploring role of interface in photocatalytic hydrogen generation. Mater. Today Sustain. 2024, 25, 100610. [Google Scholar] [CrossRef]
- Lu, J.; Yu, H.; Liu, L.; Li, G.; Wang, H.; Cui, W. Molecular structure regulation through bay-substitution with chlorine to enhance the photocatalytic H2 production of perylene tetracarboxylic acid-based supramolecular photocatalyst. Int. J. Hydrogen Energy 2024, 66, 612–624. [Google Scholar] [CrossRef]
- Fan, Y.; Kong, C.; Zhang, L.; Wu, H.; Li, J.; Guo, J.; Yi, Q. Enhancing photocatalytic hydrogen evolution performance for D-π-A conjugated polymers based on the perylene diimide. Sep. Purif. Technol. 2025, 355, 129721. [Google Scholar] [CrossRef]
- Kong, K.; Zhang, S.; Chu, Y.; Hu, Y.; Yu, F.; Ye, H.; Ding, H.; Hua, J. A self-assembled perylene diimide nanobelt for efficient visible-light-driven photocatalytic H2 evolution. Chem. Commun. 2019, 55, 8090–8093. [Google Scholar] [CrossRef]
- Zhang, M.-X.; Zhao, G.-J. Modification of n-Type Organic Semiconductor Performance of Perylene Diimides by Substitution in Different Positions: Two-Dimensional π-Stacking and Hydrogen Bonding. ChemSusChem 2012, 5, 879–887. [Google Scholar] [CrossRef]
- Wang, J.; Liu, D.; Zhu, Y.; Zhou, S.; Guan, S. Supramolecular packing dominant photocatalytic oxidation and anticancer performance of PDI. Appl. Catal. B Environ. 2018, 231, 251–261. [Google Scholar] [CrossRef]
- Jones, B.A.; Ahrens, M.J.; Yoon, M.H.; Facchetti, A.; Marks, T.J.; Wasielewski, M.R. High-mobility air-stable n-type semiconductors with processing versatility: Dicyanoperylene-3,4:9,10-bis(dicarboximides). Angew. Chem. - Int. Ed. 2004, 43, 6363–6366. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Wang, Z.; Kong, K.; Feng, S.; Xu, L.; Ye, H.; Wu, W.; Gong, X.; Hua, J. Efficient and stable photocatalytic H2 evolution by self-assembly of zirconium(iv) coordination with perylene diimide supramolecules under visible light irradiation. J. Mater. Chem. A 2021, 9, 7675–7683. [Google Scholar] [CrossRef]
- Li, R.; Luan, J.; Zhang, Y.; Jiang, L.; Yan, H.; Chi, Q.; Yan, Z. A review of efficient photocatalytic water splitting for hydrogen production. Renew. Sustain. Energy Rev. 2024, 206, 114863. [Google Scholar] [CrossRef]
- Chen, S.; Wang, C.; Bunes, B.R.; Li, Y.; Wang, C.; Zang, L. Enhancement of visible-light-driven photocatalytic H2 evolution from water over g-C3N4 through combination with perylene diimide aggregates. Appl. Catal. A Gen. 2015, 498, 63–68. [Google Scholar] [CrossRef]
- Chen, S.; Li, Y.; Wang, C. Visible-light-driven photocatalytic H2 evolution from aqueous suspensions of perylene diimide dye-sensitized Pt/TiO2 catalysts. RSC Adv. 2015, 5, 15880–15885. [Google Scholar] [CrossRef]
- Liu, D.; Wang, J.; Bai, X.J.; Zong, R.L.; Zhu, Y.F. Self-Assembled PDINH Supramolecular System for Photocatalysis under Visible Light. Adv. Mater. 2016, 28, 7284–7290. [Google Scholar] [CrossRef]
- Chen, S.; Slattum, P.; Wang, C.; Zang, L. Self-Assembly of Perylene Imide Molecules into 1D Nanostructures: Methods, Morphologies, and Applications. Chem. Rev. 2015, 115, 11967–11998. [Google Scholar] [CrossRef]
- Bahadur, V.; Yadav, N.; Chavali, M.; Kumar, P.; Singh, B.K. Perylene bisimides—Advanced synthesis and photoelectric applications. Dye. Pigment. 2024, 231, 112389. [Google Scholar] [CrossRef]
- Wei, W.; Ouyang, S.; Zhang, T. Perylene diimide self-assembly: From electronic structural modulation to photocatalytic applications. J. Semicond. 2020, 41, 091708. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, X.; Wang, Z.; Yang, W.; Chen, K.; Zhao, J.; Gurzadyan, G.G. Precise Control of the Electronic Coupling Magnitude between the Electron Donor and Acceptor in Perylenebisimide Derivatives via Conformation Restriction and Its Effect on Photophysical Properties. J. Phys. Chem. C 2018, 122, 3756–3772. [Google Scholar] [CrossRef]
- Zhang, F.; Li, W.; Jiang, T.; Li, X.; Shao, Y.; Ma, Y.; Wu, J. Real roles of perylene diimides for improving photocatalytic activity. RSC Adv. 2020, 10, 23024–23037. [Google Scholar] [CrossRef]
- Yang, B.; Lu, L.; Liu, S.; Cheng, W.; Liu, H.; Huang, C.; Meng, X.; Rodriguez, R.D.; Jia, X. Recent progress in perylene diimide supermolecule-based photocatalysts. J. Mater. Chem. A 2024, 12, 3807–3843. [Google Scholar] [CrossRef]
- Li, Z.; Liu, F.; Lu, Y.; Hu, J.; Feng, J.; Shang, H.; Sun, B.; Jiang, W. Molecular Design of Perylene Diimide Derivatives for Photocatalysis. ACS Catal. 2025, 15, 1829–1840. [Google Scholar] [CrossRef]
- Sheng, Y.; Li, W.; Zhu, Y.; Zhang, L. Ultrathin perylene imide nanosheet with fast charge transfer enhances photocatalytic performance. Appl. Catal. B Environ. 2021, 298, 120585. [Google Scholar] [CrossRef]
- Liu, Z.; Lin, X.; Chen, Y.; Song, N.; Hao, Y.; Jia, S.; Sun, H.; Sun, Y.; Yan, Y.; Li, Y.; et al. Synthesis of PDI/Zn0.8Cd0.2S composites for efficient visible light-driven photocatalytic overall water splitting. J. Taiwan Inst. Chem. Eng. 2023, 143, 104693. [Google Scholar] [CrossRef]
- Song, J.; Chen, Y.; Sun, D.; Li, X. Perylenetetracarboxylic diimide modified Zn0.7Cd0.3S hybrid photocatalyst for efficient hydrogen production from water under visible light irradiation. Inorg. Chem. Commun. 2018, 92, 27–34. [Google Scholar] [CrossRef]
- Xu, H.; Wang, Z.; Feng, S.; Liu, X.; Gong, X.; Hua, J. High photocatalytic hydrogen evolution via strong built-in electric field induced by high molecular dipoles of heteroatom-annulated perylene imide supramolecule. Int. J. Hydrogen Energy 2023, 48, 8071–8081. [Google Scholar] [CrossRef]
- Hoffman, E.; Kozakiewicz, K.; Rybczyńska, M.; Mońka, M.; Grzywacz, D.; Liberek, B.; Bojarski, P.; Serdiuk, I.E. Photochemical transformation of a perylene diimide derivative beneficial for the in situ formation of a molecular photocatalyst of the hydrogen evolution reaction. J. Mater. Chem. A 2024, 12, 5233–5243. [Google Scholar] [CrossRef]
- Yang, J.; Jing, J.; Li, W.; Zhu, Y. Electron Donor-Acceptor Interface of TPPS/PDI Boosting Charge Transfer for Efficient Photocatalytic Hydrogen Evolution. Adv. Sci. 2022, 9, 2201134. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, J.; Chai, Y.; Zhang, Z.; Zhu, Y. Efficient Photocatalytic Overall Water Splitting Induced by the Giant Internal Electric Field of a g-C3N4/rGO/PDIP Z-Scheme Heterojunction. Adv. Mater. 2021, 33, 2007479. [Google Scholar] [CrossRef]
- Miao, H.; Yang, J.; Sheng, Y.; Li, W.; Zhu, Y. Controlled Synthesis of Higher Interfacial Electron Transfer Graphite-Like Carbon Nitride/Perylenetetracarboxylic Diimide Heterogeneous for Enhanced Photocatalytic Activity. Sol. RRL 2021, 5, 2000453. [Google Scholar] [CrossRef]
- Liang, Z.; Shen, R.; Zhang, P.; Li, Y.; Li, N.; Li, X. All-organic covalent organic frameworks/perylene diimide urea polymer S-scheme photocatalyst for boosted H2 generation. Chin. J. Catal. 2022, 43, 2581–2591. [Google Scholar] [CrossRef]
- Liu, L.; Liu, J.; Zong, S.; Huang, Z.; Feng, X.; Zheng, J.; Fang, Y. Step-scheme perylenediimide supramolecular nanosheet and TiO2 nanoparticle composites for boosted water splitting performance. Int. J. Hydrogen Energy 2022, 47, 39486–39498. [Google Scholar] [CrossRef]
- Liu, L.; Wu, Y.; Song, R.; Zhang, Y.; Ma, Y.; Wan, J.; Zhang, M.; Cui, H.; Yang, H.; Chen, X.; et al. Morphology engineering and photothermal effect derived from perylene diimide based derivative for boosting photocatalytic hydrogen evolution of ZnIn2S4. J. Colloid Interface Sci. 2022, 628, 701–711. [Google Scholar] [CrossRef]
- Li, X.; Lv, X.; Zhang, Q.; Huang, B.; Wang, P.; Qin, X.; Zhang, X.; Dai, Y. Self-assembled supramolecular system PDINH on TiO2 surface enhances hydrogen production. J. Colloid Interface Sci. 2018, 525, 136–142. [Google Scholar] [CrossRef]
- Yang, J.; Miao, H.; Jing, J.; Zhu, Y.; Choi, W. Photocatalytic activity enhancement of PDI supermolecular via π-π action and energy level adjusting with graphene quantum dots. Appl. Catal. B Environ. 2021, 281, 119547. [Google Scholar] [CrossRef]
- Guo, Y.; Zhou, Q.; Nan, J.; Shi, W.; Cui, F.; Zhu, Y. Perylenetetracarboxylic acid nanosheets with internal electric fields and anisotropic charge migration for photocatalytic hydrogen evolution. Nat. Commun. 2022, 13, 2067. [Google Scholar] [CrossRef]
- Ye, C.; Li, J.-X.; Wu, H.-L.; Li, X.-B.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Enhanced Charge Separation Efficiency Accelerates Hydrogen Evolution from Water of Carbon Nitride and 3,4,9,10-Perylene-tetracarboxylic Dianhydride Composite Photocatalyst. ACS Appl. Mater. Interfaces 2018, 10, 3515–3521. [Google Scholar] [CrossRef]
- Bastard, G.; Mendez, E.E.; Chang, L.L.; Esaki, L.; Mikhnenko, O.V.; Blom, P.W.M.; Nguyen, T.-Q. Exciton binding energy in quantum wells. Phys. Rev. B 1982, 26, 1974. [Google Scholar] [CrossRef]
- Le Bahers, T.; Rérat, M.; Sautet, P. Semiconductors Used in Photovoltaic and Photocatalytic Devices: Assessing Fundamental Properties from DFT. J. Phys. Chem. C 2014, 118, 5997–6008. [Google Scholar] [CrossRef]
- Takanabe, K. Photocatalytic Water Splitting: Quantitative Approaches toward Photocatalyst by Design. ACS Catal. 2017, 7, 8006–8022. [Google Scholar] [CrossRef]
- Clarke, T.M.; Durrant, J.R. Charge Photogeneration in Organic Solar Cells. Chem. Rev. 2010, 110, 6736–6767. [Google Scholar] [CrossRef] [PubMed]
- Puschnig, P.; Ambrosch-Draxl, C. Excitons in organic semiconductors. Comptes Rendus Phys. 2009, 10, 504–513. [Google Scholar] [CrossRef]
- Molaei, M.J. Recent advances in hydrogen production through photocatalytic water splitting: A review. Fuel 2024, 365, 131159. [Google Scholar] [CrossRef]
- Zhao, C.X.; Chen, Z.P.; Shi, R.; Yang, X.F.; Zhang, T.R. Recent Advances in Conjugated Polymers for Visible-Light-Driven Water Splitting. Adv. Mater. 2020, 32, 1907296. [Google Scholar] [CrossRef]
- Walter, M.G.; Warren, E.L.; McKone, J.R.; Boettcher, S.W.; Mi, Q.X.; Santori, E.A.; Lewis, N.S. Solar Water Splitting Cells. Chem. Rev. 2010, 110, 6446–6473. [Google Scholar] [CrossRef]
- Kudo, A.; Miseki, Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 2009, 38, 253–278. [Google Scholar] [CrossRef]
- Zhou, W.; Jing, Q.; Li, J.; Chen, Y.; Hao, G.; Wang, L. Organic Photocatalysts for Solar Water Splitting: Molecular- and Aggregate-Level Modifications. Acta Phys. -Chim. Sin. 2023, 39, 2211010. [Google Scholar] [CrossRef]
- Martell, M.; Ocheje, M.U.; Gelfand, B.S.; Rondeau-Gagné, S.; Welch, G.C. Sidechain engineering of N-annulated perylene diimide molecules. New J. Chem. 2021, 45, 21001–21005. [Google Scholar] [CrossRef]
- Balakrishnan, K.; Datar, A.; Naddo, T.; Huang, J.L.; Oitker, R.; Yen, M.; Zhao, J.C.; Zang, L. Effect of side-chain substituents on self-assembly of perylene diimide molecules: Morphology control. J. Am. Chem. Soc. 2006, 128, 7390–7398. [Google Scholar] [CrossRef] [PubMed]
- Würthner, F.; Saha-Möller, C.R.; Fimmel, B.; Ogi, S.; Leowanawat, P.; Schmidt, D. Perylene Bisimide Dye Assemblies as Archetype Functional Supramolecular Materials. Chem. Rev. 2016, 116, 962–1052. [Google Scholar] [CrossRef]
- Huang, C.; Barlow, S.; Marder, S.R. Perylene-3,4,9,10-tetracarboxylic Acid Diimides: Synthesis, Physical Properties, and Use in Organic Electronics. J. Org. Chem. 2011, 76, 2386–2407. [Google Scholar] [CrossRef] [PubMed]
- Vajiravelu, S.; Ramunas, L.; Juozas Vidas, G.; Valentas, G.; Vygintas, J.; Valiyaveettil, S. Effect of substituents on the electron transport properties of bay substituted perylene diimide derivatives. J. Mater. Chem. 2009, 19, 4268–4275. [Google Scholar] [CrossRef]
- Guo, Y.; Han, G.; Duan, R.; Geng, H.; Yi, Y. Boosting the electron mobilities of dimeric perylenediimides by simultaneously enhancing intermolecular and intramolecular electronic interactions. J. Mater. Chem. A 2018, 6, 14224–14230. [Google Scholar] [CrossRef]
- Wu, H.; Xue, L.; Shi, Y.; Chen, Y.; Li, X. Organogels Based on J- and H-Type Aggregates of Amphiphilic Perylenetetracarboxylic Diimides. Langmuir 2011, 27, 3074–3082. [Google Scholar] [CrossRef]
- Kaiser, T.E.; Wang, H.; Stepanenko, V.; Würthner, F. Supramolecular Construction of Fluorescent J-Aggregates Based on Hydrogen-Bonded Perylene Dyes. Angew. Chem. Int. Ed. 2007, 46, 5541–5544. [Google Scholar] [CrossRef]
- Chen, Z.; Stepanenko, V.; Dehm, V.; Prins, P.; Siebbeles, L.D.A.; Seibt, J.; Marquetand, P.; Engel, V.; Würthner, F. Photoluminescence and Conductivity of Self-Assembled π–π Stacks of Perylene Bisimide Dyes. Chem. Eur. J. 2007, 13, 436–449. [Google Scholar] [CrossRef]
- Sautter, A.; Thalacker, C.; Würthner, F. Control of liquid crystallinity of diazadibenzoperylene dyes by covalent and hydrogen-bonded attachment of mesogens. Angew. Chem. Int. Ed. 2001, 40, 4425–4428. [Google Scholar] [CrossRef]
- Lin, H.; Camacho, R.; Tian, Y.; Kaiser, T.E.; Würthner, F.; Scheblykin, I.G. Collective Fluorescence Blinking in Linear J-Aggregates Assisted by Long-Distance Exciton Migration. Nano Lett. 2010, 10, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Wang, J.; Zhao, J.; Zhuang, Y.; Liu, B.; Zhu, Y.; Jia, H.; Wu, K.; Shen, J.; Fu, X.; et al. Molecular Dipole-Induced Photoredox Catalysis for Hydrogen Evolution over Self-Assembled Naphthalimide Nanoribbons. Angew. Chem. Int. Ed. 2022, 61, e202117645. [Google Scholar] [CrossRef]
- Pu, Y.; Bao, F.; Wang, D.; Zhang, X.; Guo, Z.; Chen, X.; Wei, Y.; Wang, J.; Zhang, Q. The asymmetrical-structure of supramolecular precursor to improve internal electric field for simultaneously enhancing contaminant degradation and H2O2 production performance. J. Environ. Chem. Eng. 2022, 10, 107123. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhang, S.; Liu, Y.; Mei, J.; Chen, S.; Lu, P.; Qin, A.; Ma, Y.; Sun, J.Z.; Tang, B.Z. Tetraphenylethenyl-modified perylene bisimide: Aggregation-induced red emission, electrochemical properties and ordered microstructures. J. Mater. Chem. 2012, 22, 7387–7394. [Google Scholar] [CrossRef]
- Ghosh, S.; Li, X.-Q.; Stepanenko, V.; Wuerthner, F. Control of H- and J-Type π Stacking by Peripheral Alkyl Chains and Self-Sorting Phenomena in Perylene Bisimide Homo- and Heteroaggregates. Chem. Eur. J. 2008, 14, 11343–11357. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shi, W.; Liu, D.; Zhang, Z.; Zhu, Y.; Wang, D. Supramolecular organic nanofibers with highly efficient and stable visible light photooxidation performance. Appl. Catal. B Environ. 2017, 202, 289–297. [Google Scholar] [CrossRef]
- Mikhnenko, O.V.; Blom, P.W.M.; Nguyen, T.-Q. Exciton diffusion in organic semiconductors. Energy Environ. Sci. 2015, 8, 1867–1888. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, D.; Liu, W.; Lou, Y.; Zhang, Y.; Dong, Y.; Xu, J.; Pan, C.; Zhu, Y. Create a strong internal electric-field on PDI photocatalysts for boosting phenols degradation via preferentially exposing π-conjugated planes up to 100%. Appl. Catal. B Environ. 2022, 300, 120762. [Google Scholar] [CrossRef]
- Meng, S.; Hu, Y.; Zhao, H.; Yao, H.; Wu, Y.; Xue, J.; Shen, Q. Double hydrogen bonding-induced compact H-type 7C-7C stacking enhancing rapid carrier transfer in perylene diimide supramolecules achieving high oxygen evolution performance. J. Colloid Interface Sci. 2024, 666, 201–209. [Google Scholar] [CrossRef]
- Marschall, R. Semiconductor Composites: Strategies for Enhancing Charge Carrier Separation to Improve Photocatalytic Activity. Adv. Funct. Mater. 2014, 24, 2421–2440. [Google Scholar] [CrossRef]
- Low, J.; Yu, J.; Jaroniec, M.; Wageh, S.; Al-Ghamdi, A.A. Heterojunction Photocatalysts. Adv. Mater. 2017, 29, 1601694. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, J.; Liu, D.; Luo, W.; Zhang, M.; Jiang, W.; Zhu, Y. Highly Efficient Organic Photocatalyst with Full Visible Light Spectrum through π-π Stacking of TCNQ-PTCDI. ACS Appl. Mater. Interfaces 2016, 8, 30225–30231. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Zhang, L.; Cheng, B.; Fan, J.; Yu, J. S-Scheme Heterojunction Photocatalyst. Chem 2020, 6, 1543–1559. [Google Scholar] [CrossRef]
- Bard, A.J. Photoelectrochemistry and heterogeneous photo-catalysis at semiconductors. J. Photochem. 1979, 10, 59–75. [Google Scholar] [CrossRef]
- Tada, H.; Mitsui, T.; Kiyonaga, T.; Akita, T.; Tanaka, K. All-solid-state Z-scheme in CdS-Au-TiO2 three-component nanojunction system. Nat. Mater. 2006, 5, 782–786. [Google Scholar] [CrossRef]
- Fu, J.; Xu, Q.; Low, J.; Jiang, C.; Yu, J. Ultrathin 2D/2D WO3/g-C3N4 step-scheme H2-production photocatalyst. Appl. Catal. B Environ. 2019, 243, 556–565. [Google Scholar] [CrossRef]
- Wakerley, D.W.; Kuehnel, M.F.; Orchard, K.L.; Ly, K.H.; Rosser, T.E.; Reisner, E. Solar-driven reforming of lignocellulose to H2 with a CdS/CdOx photocatalyst. Nat. Energy 2017, 2, 17021. [Google Scholar] [CrossRef]
- Kobayashi, A. Photoredox Cascade Catalyst for Efficient Hydrogen Production with Biomass Photoreforming. Angew. Chem. Int. Ed. 2023, 62, e202313014. [Google Scholar] [CrossRef]
- Hong, Y.H.; Lee, Y.-M.; Nam, W.; Fukuzumi, S. Molecular Photocatalytic Water Splitting by Mimicking Photosystems I and II. J. Am. Chem. Soc. 2022, 144, 695–700. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, L.; Meng, Q.; Zhou, W.; Hu, B.; Jiang, Z.; Cai, Y.; Liu, X.; Chen, Y. Supramolecular Perylene Diimides for Photocatalytic Hydrogen Production. Catalysts 2025, 15, 463. https://doi.org/10.3390/catal15050463
Tian L, Meng Q, Zhou W, Hu B, Jiang Z, Cai Y, Liu X, Chen Y. Supramolecular Perylene Diimides for Photocatalytic Hydrogen Production. Catalysts. 2025; 15(5):463. https://doi.org/10.3390/catal15050463
Chicago/Turabian StyleTian, Long, Qing Meng, Wenjie Zhou, Bang Hu, Zichun Jiang, Yulong Cai, Xiaoguang Liu, and Yingzhi Chen. 2025. "Supramolecular Perylene Diimides for Photocatalytic Hydrogen Production" Catalysts 15, no. 5: 463. https://doi.org/10.3390/catal15050463
APA StyleTian, L., Meng, Q., Zhou, W., Hu, B., Jiang, Z., Cai, Y., Liu, X., & Chen, Y. (2025). Supramolecular Perylene Diimides for Photocatalytic Hydrogen Production. Catalysts, 15(5), 463. https://doi.org/10.3390/catal15050463