1. Introduction
Proton exchange membrane fuel cells (PEMFCs) are among the most promising fuel cell technologies for applications in unmanned vehicles (UVs), unmanned aerial vehicles (UAVs), unmanned underwater vehicles (UUVs), advanced combat man systems (ACMSs), backup power systems, and disaster relief power systems. PEMFCs operate on high-purity hydrogen (H
2), an ideal fuel that electrochemically reacts with oxygen (O
2) from the air to generate electrical energy, with water (H
2O) as the sole by-product [
1,
2,
3,
4]. However, challenges persist in hydrogen production, storage, and transportation. Currently, hydrogen is primarily stored in high-pressure vessels. While effective, this method is suboptimal for portable applications such as UVs due to the low hydrogen density (HD) and the considerable weight of storage vessels.
Extensive research has been conducted to identify effective materials for hydrogen storage. Among the various candidates, complex hydrides such as NaBH
4 [
5,
6,
7,
8,
9,
10], LiBH
4 [
11], NaAlH
4 [
12], MgH
2 [
13,
14,
15], and NH
3BH
3 [
16,
17] have been widely investigated as promising hydrogen storage materials. Sodium borohydride (NaBH
4) has attracted significant attention due to its high hydrogen content (up to 10.8 wt.%), excellent stability in alkali solutions, non-toxicity, and fire safety. Moreover, NaBH
4 is the most cost-effective and commercially available hydride among these materials.
Furthermore, in the presence of a suitable catalyst, the hydrolysis of NaBH
4 proceeds exothermically under ambient pressure and temperature, with half of the generated hydrogen originating from water. Various acids and metal-based catalysts have been investigated for this hydrolysis process [
5]. Among these, noble metal catalysts, including Pt, Rh, and Ru (with or without supports), have demonstrated the highest hydrogen generation rates (HGR) for NaBH
4 hydrolysis [
18,
19]. However, the high cost of these noble metal catalysts has driven research efforts toward the development of non-noble metal alternatives. Our analysis of advancements in NaBH
4 hydrolysis suggests that cobalt-based catalysts emerge as promising alternatives due to their high catalytic activity and cost-effectiveness.
The design and fabrication of nanomaterials play a crucial role in heterogeneous catalysis to achieve improved performance. Traditionally, efforts have focused on reducing particle size to increase the available reaction surface area, a concept known as size-dependent catalytic chemistry [
20]. While cobalt (Co) has been explored as a promising alternative to precious metal catalysts due to its favorable properties, it presents certain challenges. These challenges include a lengthy synthesis process and the pyrophoric nature of nano-sized elemental cobalt, which rapidly oxidizes upon exposure to air [
9].
To address these limitations, various forms of cobalt-based catalysts have been investigated, including metallic Co [
21], cobalt boride (B) [
22,
23], cobalt boride alloys [
24,
25,
26,
27,
28,
29], and Co supported on carbon [
30,
31,
32,
33], resin [
34], metal oxides [
19,
35], or a copper sheet [
36]. Among these, carbon-based supports are particularly attractive due to their lower densities. The impregnation chemical reduction method is usually employed for synthesizing carbon-supported Co-based catalyst [
30,
31,
37]. This process typically involves four main steps: (1) mixing cobalt salts with carbon substrates, (2) reducing cobalt using an NaBH
4 solution, (3) separating, washing, and drying the as-synthesized catalyst, and (4) annealing in an inert atmosphere to enhance catalytic performance. However, this method is costly and challenging to scale up for mass production.
Recent studies have highlighted cobalt oxide (Co
3O
4) as a promising catalyst for the hydrolysis reaction of NaBH
4 [
9,
38,
39,
40,
41,
42,
43]. The superior catalytic behavior of cobalt oxide in this reaction primarily results from the formation of cobalt boride (Co
2B), which occurs upon the reduction of oxides by NaBH
4 [
41]. Krishnan et al. reported that Co
3O
4 formed by the thermal decomposition of Co(NO
3)
2 under flowing air in a tubular furnace at 600 °C showed the highest catalytic capability [
40]. The enhanced performance of Co
3O
4 synthesized at 600 °C is attributed to its improved crystallinity, which facilitates a quicker transformation into Co
2B during the NaBH
4 hydrolysis reaction compared to Co
3O
4 synthesized at a lower temperature (prepared at 200 °C and 400 °C) [
40]. Groven et al. reported that a cobalt oxide catalyst precursor for the NaBH
4 hydrolysis reaction could be synthesized using the solution combustion method and found that the hydrogen generation rate of this Co
3O
4 is as high as 1.93 L min
−1 g
cat−1 under experimental conditions of 20 °C, with a- NaBH
4 aqueous solution (0.6 wt.%) and a catalyst precursor loading of 5 wt.%. Remarkably, this activity exceeded that exhibited by commercial cobalt oxide and cobalt metal catalysts [
42].
Various synthetic strategies have been developed for producing nanoscale oxide powders, including the Pechini method [
44], hydrothermal synthesis [
45], microwave-assisted synthesis [
46], spray pyrolysis [
47], co-precipitation [
48], sol-gel processing [
49], gel-casting techniques [
50,
51,
52,
53,
54,
55], and thermal plasma [
56]. Gel-casting has emerged as a particularly attractive route due to its ability to rapidly yield highly homogeneous nanopowders. For instance, the gel-casting technique has been successfully used to synthesize perovskite materials such as La
0.72Sr
0.18MnO
3−δ (LSM) [
50] and(La
0.75Sr
0.25)
1−x(Cr
0.5Mn
0.5)O
3−δ (LSCM) [
57]. Importantly, LSM and LSCM powders synthesized via gel-casting exhibit superior electrochemical catalytic performance compared with counterparts synthesized via conventional solid-state reaction routes. Additionally, the particle size of the synthesized nanopowders can be tailored by controlling the ratio of organic precursors to metal salts during synthesis [
51].
In this paper, nanoscale cobalt oxide powders were synthesized using an aqueous gel-casting technique. The effects of the molar ratio of monomer to metal nitrate, initiator concentration relative to the monomer, calcination temperature, and dwelling time on particle size and catalytic performance were investigated in detail. The synthesized cobalt oxide nanopowders were thoroughly characterized via Brunauer–Emmett–Teller (BET) surface area analysis, X-ray diffraction (XRD), thermogravimetric analysis (TGA), and transmission electron microscopy (TEM). Furthermore, the catalytic effectiveness of these nanopowders was evaluated by measuring the hydrogen generation rate (HGR) during the hydrolysis reaction of an alkaline-stabilized NaBH4 solution.
2. Results and Discussion
2.1. Thermal Analysis
Thermal gravimetric analysis (TGA) provides critical insights into the thermal stability and decomposition behavior of polymer-based precursors, such as poly(acrylamide) (PAM), which are used in synthesizing catalyst nanoparticles [
58]. As shown in
Figure 1, the TGA curve of the PAM sample displays four distinct weight-loss stages, each corresponding to specific decomposition processes.
In the initial stage, occurring below 190 °C, a weight loss of approximately 6.23 wt.% was observed. This is primarily due to the removal of physically adsorbed moisture and chemically bound H
2O within the polymer matrix. This observation is consistent with existing literature on hydrophilic polymers, which tend to retain significant amounts of moisture through hydrogen bonding and physical adsorption mechanisms [
59,
60].
The second transition region, corresponding to a temperature range of 190 to 350 °C, results in an additional weight loss of about 2.9 wt.%. This phase is attributed to the thermally induced cleavage and release of hydroxyl and amino functionalities attached to the branched chains of PAM. Functional groups like hydroxyl and amino are sensitive to thermal activation, facilitating their early detachment from the polymer backbone as decomposition initiates.
At temperatures above 350 °C, thermal degradation accelerates significantly, as clearly delineated by the third and fourth peaks in the TGA curve. These peaks correspond primarily to the breakdown of the polymer backbone itself, as covalent bonds within the acrylamide structure undergo cleavage, releasing gaseous decomposition products [
50]. Ultimately, at 700 °C, the polymer completely decomposes, as indicated by the total weight loss (100 wt.%). This comprehensive decomposition underscores the suitability of PAM as a polymeric precursor that leaves minimal residue, which is crucial for generating highly pure inorganic nanopowders upon calcination [
50].
Although complete decomposition occurs at 700 °C, the actual calcination step was strategically conducted at 600 °C. This slightly lower temperature was chosen to balance the need for complete removal of organic residues with the necessity of preserving the nanostructure and high specific surface area of cobalt oxide nanoparticles. At 600 °C, the TGA curve indicates that substantial decomposition of the polymer backbone has already taken place, significantly reducing the organic content to negligible levels. Additionally, maintaining calcination at this temperature helps prevent excessive sintering and aggregation of cobalt oxide particles, which would otherwise diminish catalytic activity by reducing the active surface area. Consequently, a 2 h calcination at 600 °C provides sufficient thermal energy for thorough organic decomposition while concurrently allowing controlled crystal growth, yielding cobalt oxide nanoparticles with optimal catalytic properties.
Further validation of the efficiency of the synthesis protocol and calcination conditions is provided by subsequent TGA measurements of the synthesized cobalt oxide nanopowders up to 700 °C. The measured cobalt oxide content across all samples ranged from 92 to 98.6 wt.% (
Table 1), demonstrating a high degree of purity and consistency in the nanoparticle synthesis process. This significant cobalt oxide yield highlights the effectiveness of the gel-casting synthesis route combined with the carefully optimized thermal treatment.
These findings collectively underscore the critical interplay between precursor composition, thermal decomposition pathways, and calcination conditions in achieving high-purity and homogeneous cobalt oxide nanopowders suitable for catalytic applications. The precise control of these parameters ensures reproducible nanoparticle properties, which are essential for catalytic performance optimization and practical implementation in hydrogen generation reactions.
2.2. Phase Formation
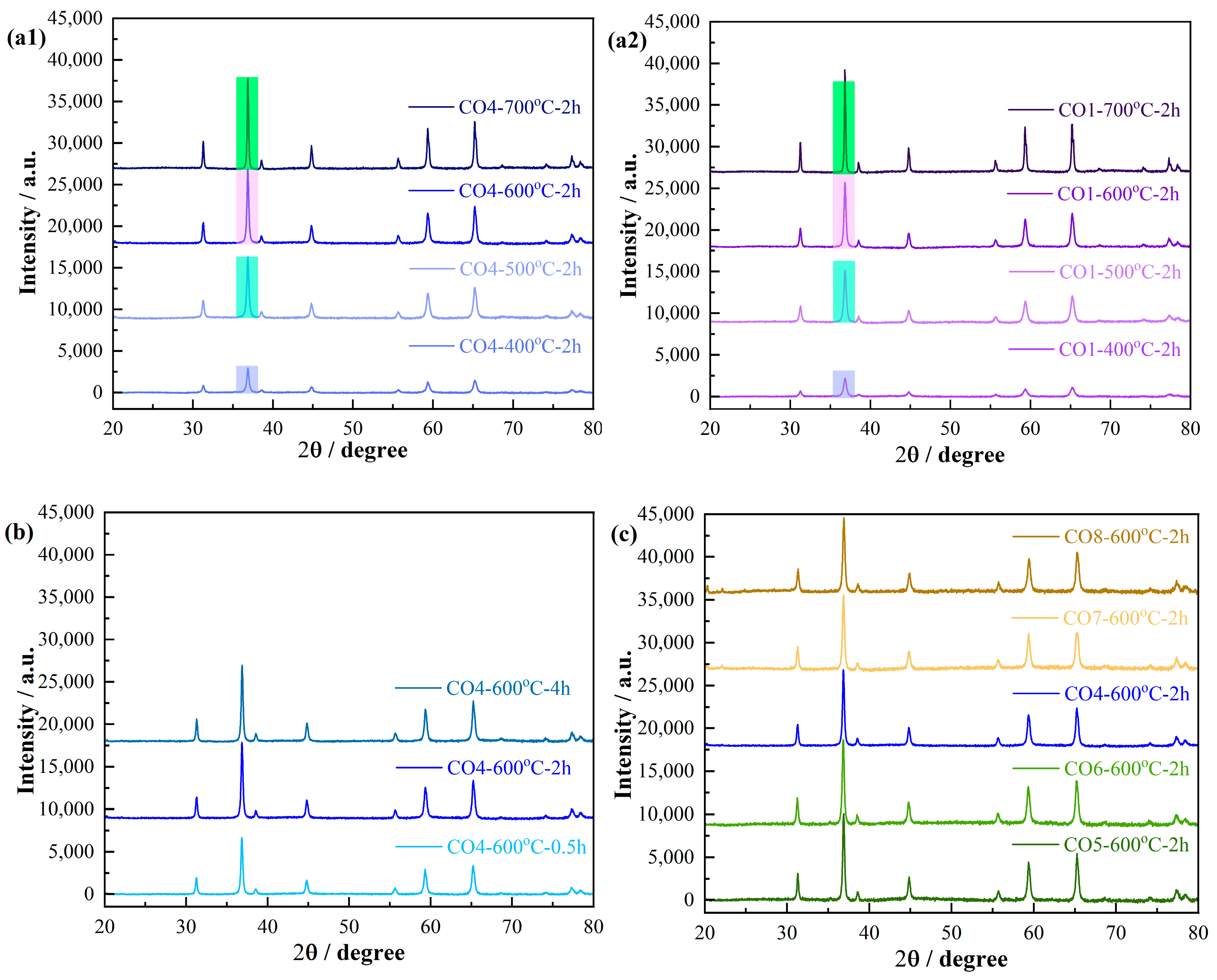
To investigate the phase evolution of the powders synthesized via aqueous gel-casting, XRD measurements were performed on samples heat-treated at various temperatures.
Figure 2 presents the XRD patterns of COx powders calcined at different temperatures for 2 h in air. As shown in
Figure 2a1, the CO4 powder calcined at 400 °C exhibits the characteristic diffraction peaks corresponding to the Co
3O
4 phase. Notably, this phase remains stable and persists as the sole detectable phase even when the calcination temperature is increased to 700 °C, indicating the remarkable thermal stability of the Co
3O
4 phase. The observed diffraction peaks align well with the standard Co
3O
4 data (JCPDS Card No. 80-1541), and no secondary phases were detected, confirming the phase purity of the synthesized material. The XRD patterns exhibit characteristic diffraction peaks at 2θ values of approximately 31.3°, 36.8°, 38.5°, 44.8°, 55.6°, 59.3°, and 65.2°, corresponding to the (220), (311), (222), (400), (422), (511), and (440) planes, respectively. The absence of additional phases confirms the high purity and effectiveness of the aqueous gel-casting approach in synthesizing phase-pure Co
3O
4 nanopowders.
For the CO1 powders synthesized using different molar ratios of organic monomer to metal nitrates relative to CO4 and calcined at various temperatures for 2 h in air, the XRD patterns consistently exhibit the characteristic Co
3O
4 phase peaks (
Figure 2a2). This observation indicates that the synthesis process consistently yields the Co
3O
4 phase under the specified calcination conditions, highlighting the robustness and reproducibility of the synthesis method in producing high-purity Co
3O
4 nanopowders.
According to the Scherrer equation, the relative intensity and broadening of diffraction peaks are influenced by crystallinity and crystal size. The Scherrer equation is given as follows:
where
D is the crystallite size,
K is the shape factor (usually taken as 0.89),
λ is the X-ray wavelength,
β is the full width at half maximum (FWHM) of the diffraction peak (in radians), and
θ is the diffraction angle. The Scherrer equation reveals that crystallite size is inversely related to peak broadening, meaning that sharper and more intense diffraction peaks are indicative of larger crystallite sizes. This relationship arises from the fundamental principle that well-crystallized materials exhibit reduced peak broadening due to the coherent scattering of X-rays over longer distances within the crystal lattice. Consequently, narrower and higher peaks signify improved crystallinity and increased grain size, reflecting the optimized synthesis conditions that promote crystal growth.
As shown in
Figure 2a, noticeable differences in the relative intensity and peak width of the diffraction patterns are observed between CO4 and CO1 powders calcined at the same temperature. Specifically, the diffraction peak intensity of CO4 at 36.848° is notably higher than that of CO1 when both powders are calcined at 400 °C, 500 °C, and 600 °C. This discrepancy can be attributed to the differences in the monomer-to-metal (Co
2+) ratio, where the denser and more interconnected organic network structure in CO4 promotes a reduction in the phase formation temperature. Consequently, CO4 exhibits superior crystallinity and larger crystal size compared to CO1, indicating that the synthesis parameters significantly influence the crystalline quality and structural characteristics of the final product.
Upon increasing the calcination temperature to 700 °C, as highlighted by the green regions in
Figure 2a1,a2, the diffraction peak intensity of CO1 surpasses that of CO4. This phenomenon can be explained by the incomplete decomposition of the organic network within CO4 during preheating on the hot plate, where the decomposition temperature remained below 400 °C, while complete network breakdown required temperatures up to 700 °C (see
Figure 1). The high-density polymeric network in CO4 effectively isolates the Co
3O
4 particles, preventing their interaction and aggregation, thereby limiting grain growth. In contrast, the CO1 sample, with significantly lower organic content, provides fewer barriers to diffusion and aggregation, facilitating the formation of larger crystalline domains. As a result, CO1 exhibits higher diffraction peak intensity at elevated temperatures, reflecting more significant particle coarsening.
Additionally, the extended calcination time promotes crystal growth, as evidenced by the increased peak intensity of CO4 calcined at 600 °C for 4 h compared to those calcined for a shorter duration (
Figure 2b). This effect can be attributed to prolonged thermal exposure, which enhances particle mobility and diffusion, allowing for grain coalescence and the formation of larger crystalline domains. Furthermore, the precursor composition also plays a crucial role. As shown in
Figure 2c, the relative intensity of the diffraction peaks decreases as the weight percentage of APS increases from 0.5 wt.% to 4 wt.%, which is attributed to the formation of smaller wet gel units during polymerization due to increased cross-linking density. These smaller units lead to finer crystalline structures after calcination, thereby reducing peak intensity. However, a further increase in APS concentration from 4 wt.% to 6 wt.% does not reduce the peak intensity, suggesting a saturation effect in the polymerization process, where excessive APS does not significantly alter the gel unit size, likely due to a limitation in available monomer molecules.
These insights illustrate the complex interplay between calcination parameters and precursor composition, emphasizing the ability to precisely control crystallinity and particle size to optimize Co3O4 nanopowders. An optimal combination, identified as a monomer-to-metal nitrate molar ratio of 4:1 and an APS concentration of 2 wt.%, with calcination conditions of 600 °C for 2 h, yields nanopowders exhibiting superior crystalline quality and desirable microstructural properties, which are essential for practical catalytic applications.
2.3. Morphology of As-Synthesized Powders
The controlled design and synthesis of nanomaterials are pivotal in advancing heterogeneous catalysis, as they significantly influence catalytic performance. Conventionally, minimizing particle size has been considered an effective strategy to maximize the active surface area, a fundamental concept in size-dependent catalytic chemistry. The increased exposure of surface atoms resulting from smaller particles generally leads to enhanced catalytic activity. However, optimizing particle size necessitates a delicate balance to mitigate challenges such as agglomeration and instability, which can negate the advantages of a larger surface area.
In this study, the particle size was systematically optimized by examining the influence of two critical parameters: (1) the ratio of acrylamide (AM) to metal nitrate and (2) the initiator weight percentage within the organic monomer system. By carefully adjusting these factors, we aimed to achieve a stable and highly active catalytic material with a well-controlled particle size distribution.
Figure 3a–d shows the TEM micrographs of COx powders synthesized via aqueous gel-casting and calcined in air at 600 °C for 2 h. The COx powders were synthesized with various molar ratios of monomer to metal nitrate. All samples displayed spherical particle morphology. The average particle sizes were measured as 40 ± 15 nm, 22 ± 12 nm, 20 ± 10 nm, and 16 ± 5 nm for the COx powders synthesized with monomer-to-metal nitrate molar ratios of 1:1, 2:1, 3:1, and 4:1, respectively, designated as CO1, CO2, CO3, and CO4. A clear decreasing trend in particle size was observed with increasing monomer-to-metal nitrate ratio. This phenomenon can be attributed to the formation of a more densely crosslinked polymeric network at higher monomer content, which restricts particle growth during calcination, thereby leading to smaller particle sizes.
Figure 3e–h displays the TEM micrographs of COx powders synthesized with different weight percentages of APS relative to AM and calcined in air at 600 °C for 2 h. The average particle sizes were 83 ± 27 nm, 65 ± 21 nm, 16 ± 5 nm, 17 ± 5 nm, and 17 ± 7 nm for APS-to-AM weight percentages of 0.5 wt.%, 1 wt.%, 2 wt.%, 4 wt.%, and 6 wt.%, respectively, designated as CO5, CO6, CO4, CO7, and CO8. These results clearly demonstrate that the particle size of the COx powders is strongly influenced by the APS content. A significant reduction in particle size is observed as the APS concentration increases from 0.5 wt.% to 2 wt.%. However, further increasing the APS concentration beyond 4 wt.% does not result in any noticeable decrease in particle size, suggesting that a saturation effect occurs at higher APS levels.
The relationship between particle size and APS concentration can be understood through the role of APS in the gelation process. Cheng et al. previously reported that wet gels formed via chain propagation and crosslinking reactions typically exhibit homogeneous and fine circular units within the polymeric network [
51]. They concluded that the particle size remained unaffected by crosslinking agents when the concentration had already reached a saturation threshold, where additional crosslinking no longer alters the network structure due to the limitation of reactive sites.
In contrast, our results clearly demonstrate that particle size is significantly influenced by the use of APS as an initiator (
Figure 3e–h). This divergence can be attributed to the distinct function of APS compared to conventional crosslinking agents. Unlike crosslinkers, APS primarily regulates chain initiation, propagation, and termination reactions, which govern the size and density of circular units formed during gelation. At elevated APS concentrations, more initiation sites are generated, resulting in the formation of a greater number of smaller gel units. Consequently, finer particles are obtained after calcination.
However, when the APS concentration exceeds 4 wt.%, the chain propagation and termination reactions likely reach a dynamic equilibrium, where the availability of monomer molecules becomes the limiting factor. As a result, further increases in APS concentration do not significantly affect the gel unit size, leading to a plateau in particle size at higher APS levels. This phenomenon underscores the existence of a saturation point beyond which additional APS fails to impact gelation or particle size, analogous to the threshold effect observed for crosslinking agents.
2.4. Preliminary Results of the Catalytic Property of COx Nanopowders
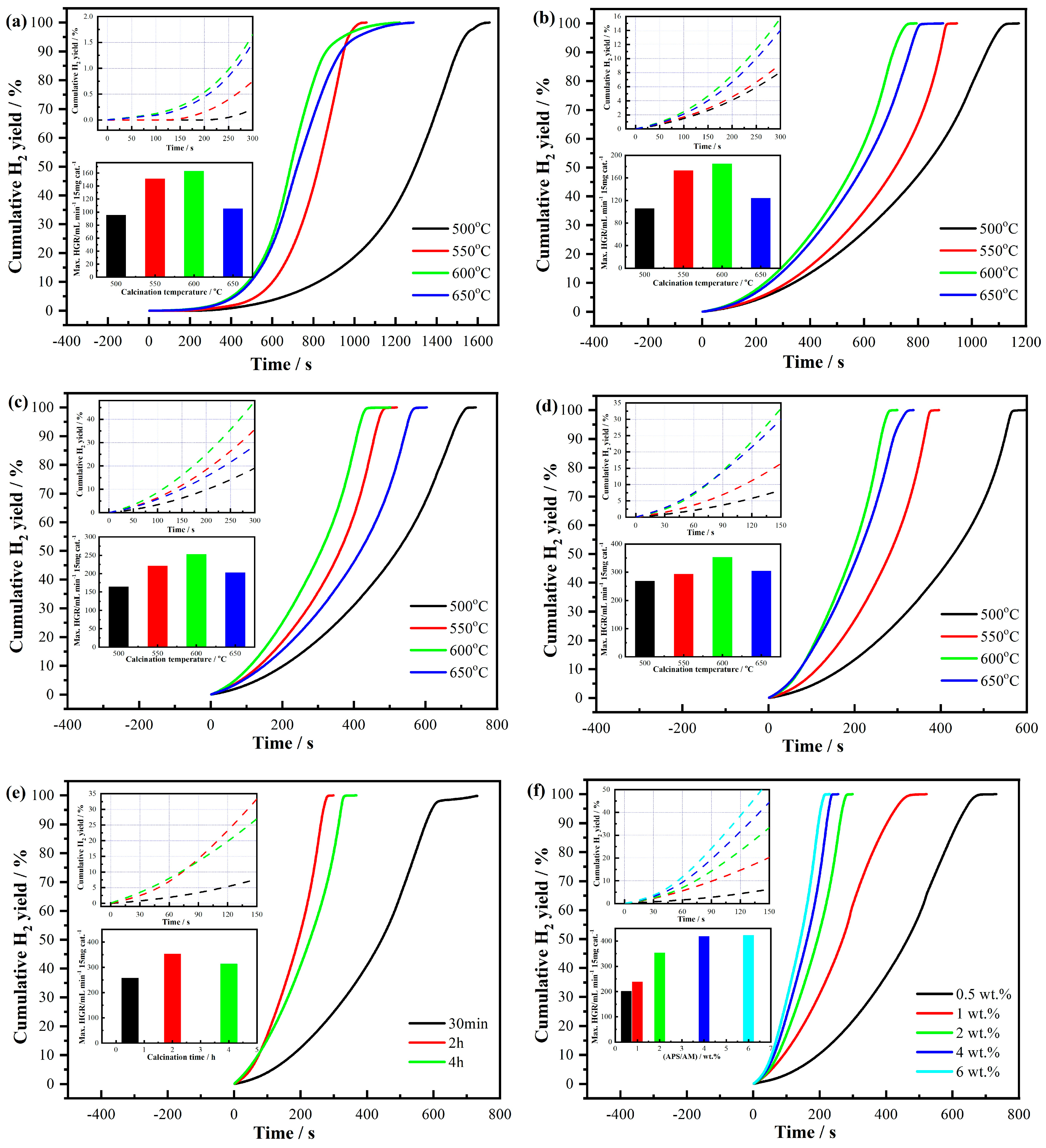
The cumulative hydrogen yield from the hydrolysis of NaBH
4, catalyzed by cobalt boride formed in situ from COx powders, exhibits significant variation depending on several synthesis parameters. These parameters include the monomer-to-metal molar ratio, APS/AM weight percentage, calcination temperature, and dwelling time of the COx nanopowders.
Figure 4a–d presents the cumulative hydrogen yield as a function of time during sodium borohydride hydrolysis over CO1, CO2, CO3, and CO4 powders, calcined at 500 °C, 550 °C, 600 °C, and 650 °C, respectively. The results clearly demonstrate a pronounced dependence of hydrogen production efficiency on both the synthesis and calcination conditions, highlighting the critical role of material processing in optimizing catalytic performance.
As shown in
Figure 4a, the induction period (or activation period), during which the cumulative hydrogen yield remains negligible, is approximately 250 s when CO1 calcined at 500 °C is employed to form cobalt boride in situ. Notably, the induction period decreases significantly as the calcination temperature increases, reaching its minimum at 600 °C. However, a slight increase in the induction period is observed when the calcination temperature exceeds 600 °C. This trend indicates that while increasing the calcination temperature initially enhances catalyst activation by enhancing crystallinity and surface properties, excessive calcination may lead to grain coarsening or a reduction in active surface area, ultimately diminishing catalyst efficiency.
The maximum HGR of NaBH4 hydrolysis over CO1 calcined at 500 °C, 550 °C, 600 °C, and 650 °C reaches 95.6 mL min−1 (15 mg cat.)−1 (i.e., 6.37 L min−1 g cat.−1), 151 mL min−1 (15 mg cat.)−1 (i.e., 10.07 L min−1 g cat.−1), 163 mL min−1 (15 mg cat.)−1 (i.e., 10.87 L min−1 g cat.−1), and 105.6 mL min−1 (15 mg cat.)−1 (i.e., 7.04 L min−1 g cat.−1), respectively. These results clearly demonstrate that the optimal calcination temperature for CO1 is 600 °C, as it yields the highest HGR, attributed to the optimal balance between crystallinity and active surface area.
As illustrated in
Figure 4b–d, the maximum HGR from NaBH
4 hydrolysis catalyzed by cobalt boride formed in situ from CO2, CO3, and CO4 powders reaches 185 mL min
−1 (15 mg cat.)
−1 (i.e., 12.33 L min
−1 g
cat.
−1), 252.9 mL min
−1 (15 mg cat.)
−1 (i.e., 16.86 L min
−1 g
cat.
−1) and 353.5 mL min
−1 (15 mg cat.)
−1 (i.e., 23.57 L min
−1 g
cat.
−1), respectively, when calcined at 600 °C. These observations clearly indicate that the monomer-to-metal nitrate molar ratio significantly influences the catalytic performance, with CO4 (molar ratio 4:1) achieving the highest HGR. This enhancement is attributed to the smaller particle size and optimized network structure of CO4, which provide a large active surface area and greater accessibility to active sites.
Moreover, as depicted in
Figure 4e,f, the HGR and cumulative hydrogen yield from NaBH
4 catalyzed by cobalt boride formed in situ from COx calcined at 600 °C were further examined as functions of dwelling time and initiator weight percentage to monomer. The maximum HGR from the hydrolysis of NaBH
4 by cobalt boride formed in situ from CO9, CO4, and CO10 powders reaches the highest values of 257.9 mL min
−1 (15 mg cat.)
−1 (i.e., 17.19 L min
−1 g
cat.
−1), 353.3 mL min
−1 (15 mg cat.)
−1 (i.e., 23.55 L min
−1 g
cat.
−1), and 313.5 mL min
−1 (15 mg cat.)
−1 (i.e., 20.90 L min
−1 g
cat.
−1), respectively. These findings suggest that the optimal dwelling time for calcination is 2 h within the investigation range. Prolonged calcination beyond this duration likely induces grain growth and reduces the active surface area, thereby counteracting the benefits of enhanced crystallinity.
Additionally, the maximum HGR increases with an increasing APS/AM weight percentage when the APS concentrations range from 0.5 wt.% to 4 wt.%. However, further raising the APS concentration to 6 wt.% does not result in additional improvements in HGR, indicating the presence of a saturation effect. This phenomenon is consistent with previous observations where (i) greater grain maturity was shown to enhance catalytic performance (see
Figure 2) and (ii) smaller particle sizes were demonstrated to improve HGR due to the increased active surface area (see
Figure 3).
The results clearly demonstrate that optimal synthesis parameters, including the monomer-to-metal ratio, calcination temperature, calcination duration, and APS concentration, play a critical role in determining catalytic performance. The CO4 sample (4:1 ratio), calcined at 600 °C for 2 h with 2 wt.% APS, exhibits the highest hydrogen generation rate and the shortest induction period, reflecting the synergistic effect of enhanced crystallinity and optimal surface properties. These findings provide valuable insights into the design of high-performance cobalt-based catalysts for hydrogen production.
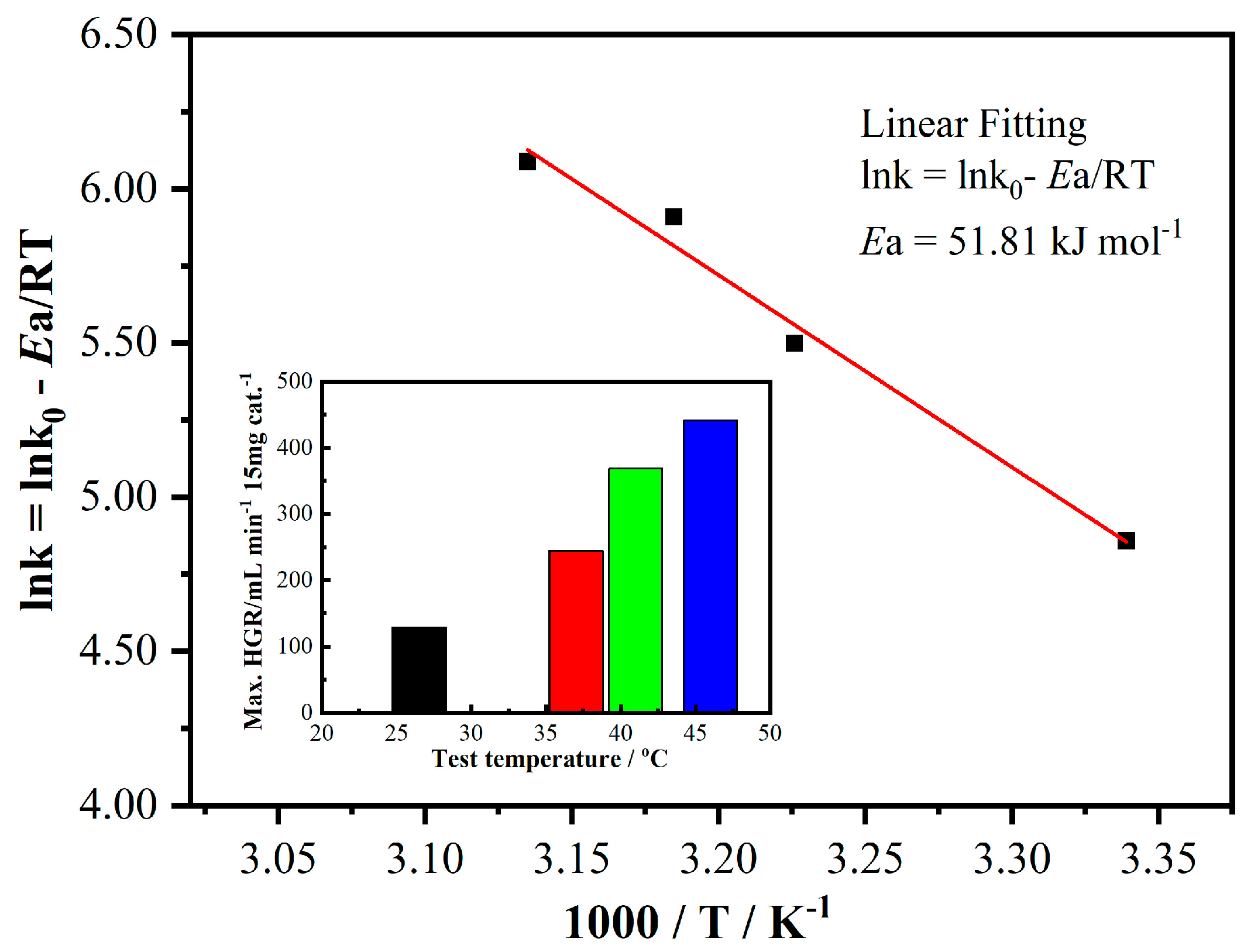
The effect of reaction temperature on the hydrogen generation performance of CO4 calcined at 600 °C for 2 h in air is presented in
Figure 5. According to the Arrhenius equation, the reaction rate constant
k can be expressed as follows:
where
k0 is the pre-exponential parameter (frequency factor),
Ea is the activation energy of the reaction (kJ/mol),
R is the universal gas constant (8.314 J/mol·K), and
T is the reaction temperature (in Kelvin).
The activation energy (
Ea) for the hydrolysis of NaBH
4 using CO4 as a catalyst was determined from the slope of the Arrhenius plot to be 51.81 kJ mol
−1 (see
Figure 5). This value is comparable to the reported activation energy of 47 kJ mol
−1 for Ru/resin, a conventional catalyst commonly used to enhance the hydrolysis of NaBH
4. These results indicate that CO4 exhibits competitive catalytic performance compared to traditional catalysts, demonstrating its potential as an efficient alternative for hydrogen generation.
3. Practical Applications and Technological Implications
The experimental results demonstrate that cobalt boride catalysts possess considerable potential for practical hydrogen generation, offering a cost-effective and scalable solution with high efficiency. Below, the key technological implications are discussed in detail, supported by data from the study.
3.1. Sustainability and Cost Efficiency
Cobalt boride catalysts represent a promising alternative to Ru-based catalysts, which are often employed in hydrogen generation but face limitations due to high costs and limited availability. As cobalt is abundant and economically viable, replacing noble metal catalysts with cobalt boride significantly lowers material expenses. The activation energy (Ea) for hydrogen generation using cobalt boride catalysts was calculated to be 51.81 kJ mol−1, which closely matches the Ea of 47 kJ mol−1 reported for Ru-based catalysts. This comparable efficiency, combined with reduced costs, makes cobalt boride catalysts a viable choice for practical applications.
3.2. High Efficiency and Scalability
Under optimized conditions, cobalt boride catalysts demonstrate a high HGR, making them suitable for a wide range of applications, from small-scale portable hydrogen generators to large-scale industrial systems. The highest HGR observed for cobalt boride (CO4) was 353.5 mL min−1 (15 mg cat.)−1 (i.e., 23.56 L min−1 g cat.−1), achieved under optimized synthesis conditions (monomer-to-metal nitrate ratio of 4:1, APS content of 4 wt.%, and calcination at 600 °C for 2 h). This HGR is competitive with Ru-based systems, showing the high efficiency of cobalt boride catalysts. The tunable synthesis parameters (e.g., monomer-to-metal nitrate ratio, APS concentration, and calcination conditions) allow for the precise control of structural and functional properties, enabling adaptation for different reactor designs and operational scales.
3.3. Industrial Relevance
The ability to fine-tune cobalt boride catalyst properties offers significant potential for integrating these materials into industrial hydrogen production systems. The following advantages make cobalt boride catalysts particularly relevant for industrial applications:
Structural Control: This study demonstrated that cobalt boride’s particle size and surface area could be effectively controlled through synthesis parameters. For example, CO4, which has the smallest particle size (16 ± 5 nm), achieved the highest HGR due to its greater active surface area and dense active sites.
Thermal Stability: The optimal calcination temperature (600 °C) ensured the formation of stable crystalline phases without excessive grain growth, highlighting the robustness of these catalysts under operational conditions.
4. Materials and Methods
4.1. Preparation of Cobalt Oxide Nanopowders
Cobalt oxide nanopowders were synthesized using an aqueous gel-casting technique. Cobalt nitrate hexahydrate (Co(NO3)2·6H2O (99%)) served as the cobalt source, acrylamide (AM) served as the monomer, N,N′-methylenebis (acrylamide) (MBAM) served as the crosslinker, ammonium persulphate (APS) acted as the initiator, and deionized water functioned as the solvent. Together, these components were utilized as starting materials. In this work, all chemicals were purchased from Sigma-Aldrich (Saint Louis, MI, SUA) and used as received without further purification. Initially, cobalt nitrate and the appropriate amounts of AM and MBAM were dissolved in deionized water under continuous stirring for approximately 30 min to achieve a homogeneous solution. Subsequently, the APS solution was added to the pre-mixed homogeneous solution, which was further stirred briefly to ensure uniform distribution. The solution was then transferred to an oven and heated at approximately 80–90 °C, resulting in polymerization and gel formation.
After solidification, the fragmented gel underwent a preliminary heating step on a corrosion-resistant ceramic-coated digital hotplate at 380 ± 10 °C. This step included auto-ignition, followed by a self-propagating combustion process. This highly exothermic reaction rapidly converted the precursor materials into ultrafine precursor powders, characterized by their dark brown appearance. The rapid combustion process facilitated the formation of amorphous or partially crystalline cobalt oxide precursors. Subsequently, the resulting powders were subjected to controlled calcination at high temperature for 2 h in air to yield crystalline cobalt oxide nanoparticles. Details of the process and procedure for synthesizing nanopowders via the aqueous gel-casting technique can be found in the literature. The synthesis and calcination conditions are summarized in
Table 2. The cobalt oxide nanopowders are designated as COx (x ranges from 1 to 10) based on the synthesis conditions. Pristine poly(acrylamide) (PAM) was prepared by polymerizing acrylamide with the crosslinker MBAM, using APS as the initiator, followed by heating the mixture in an oven at 85 °C for 30 min to form a gel.
4.2. Characterization
Thermogravimetric analysis (TGA) of the gel-cast cobalt oxide nanopowders, calcined at different temperatures, and of PAM was carried out using a 2950 thermal analyzer (TA Instruments, Inc., New Castle, DE, USA) at a heating rate of 10 °C/min with an air flow rate of 100 sccm. Specific surface area measurements were conducted using the Brunauer–Emmett–Teller (BET) method on a Micromeritics ASAP Tristar II 3020 (Micromeritics Instrument Corp., Norcross, GA, USA). The phase characteristics of the cobalt oxide powders were determined by X-ray diffraction (XRD, Philips MPD 1880, Oxford, UK) using Cu Kα radiation at room temperature. X-ray scans were performed over a 2θ spectrum of 20–80° at a scan rate of 0.02° per step and 4°/min. The morphology of the synthesized powders was examined using a transmission electron microscope (TEM, JEOL 360, JEOL Ltd., Akishima, Tokyo, Japan).
4.3. Catalyst Performance Evaluation
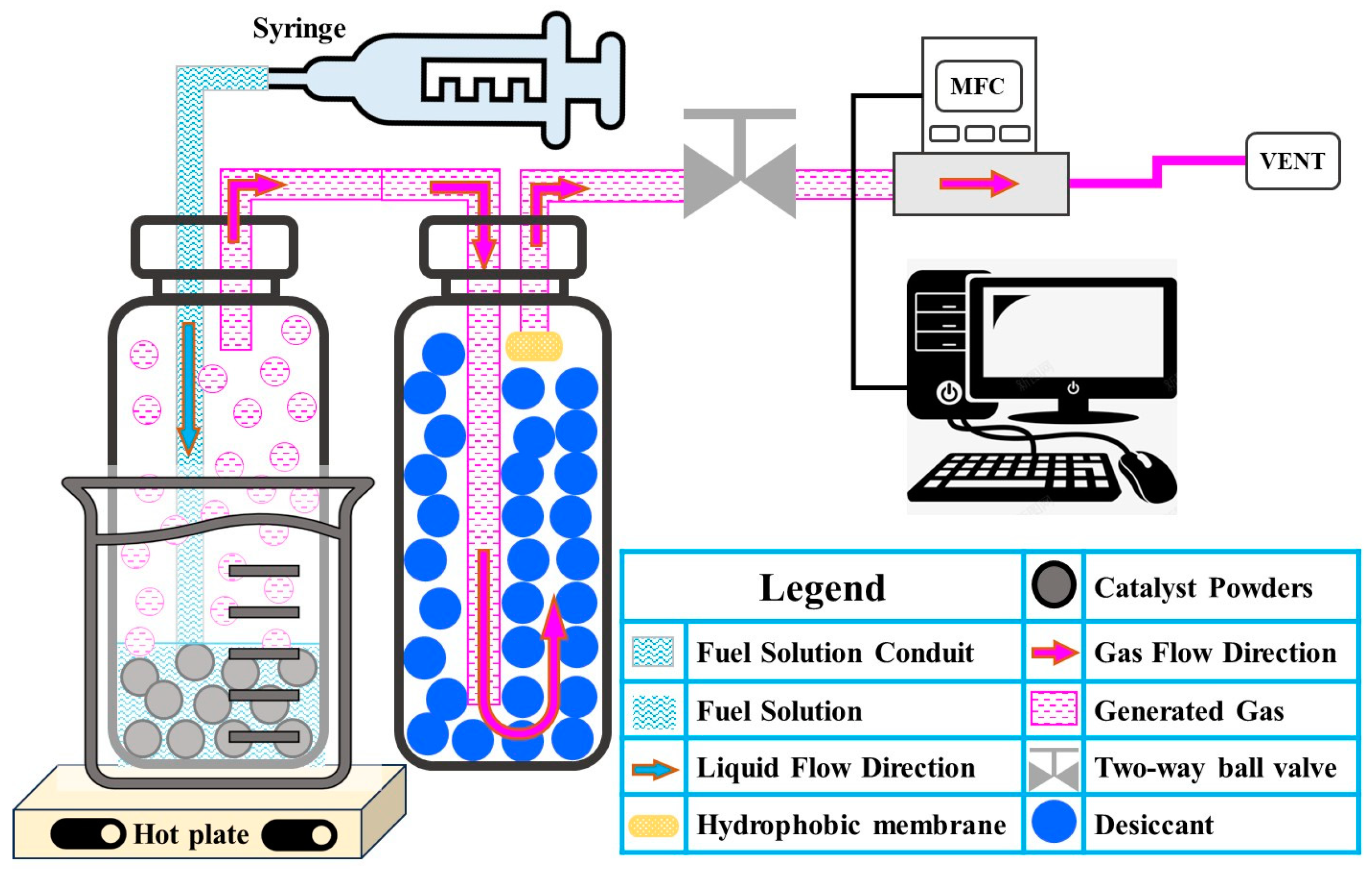
In a typical hydrogen generation experiment, 6 g of a NaOH (0.5 wt.%)-stabilized NaBH
4 (4 wt.%) solution was used in the presence of as-synthesized cobalt oxide nanopowders, where the catalyst loading was 6.25 wt.% relative to the weight of NaBH
4 (15 mg net Co
3O
4 weight). The experiment was conducted at room temperature (25~28 °C). The nanopowders were weighed at around 15 mg~16.7 mg (actual weight dependent on TGA results) and placed into a centrifuge tube (Oak Ridge Centrifuge Tube, 50 mL, FEP, Oak Ridge, TN, USA), followed by the injection of 6 g of the NaOH-stabilized NaBH
4 solution (see
Figure 6).
The hydrogen generation rate (HGR) was recorded using the Flow Vision 3 program (Alicat Scientific, Inc., Tucson, AZ, USA) at normal temperature and pressure. Since the flow meter is highly sensitive to moisture, the hydrogen was passed through a drying agent to remove moisture before measurement. For activation energy determination, the stabilized NaBH4 solution was injected into a centrifuge tube maintained at a specific temperature using a water bath.
5. Conclusions and Future Work
In summary, nano-spherical Co3O4 powders were successfully synthesized using an aqueous gel-casting technique. The crystalline Co3O4 phase formed as early as 400 °C, with calcination at 600 °C yielding optimal catalytic properties. A systematic study of synthesis parameters revealed that increasing the monomer-to-metal nitrate ratio and initiator (APS) concentration led to significant reductions in particle size, from approximately 40 nm down to 16 nm, resulting in improved catalytic efficiency. Among all synthesized powders, the sample CO4 calcined at 600 °C exhibited the highest hydrogen generation rate (HGR), reaching 422 mL min−1 (15 mg cat.)−1, or 28.13 L min−1 g cat.−1).
The extracted activation energy of 51.81 kJ mol−1 is comparable to that of Ru-based catalysts, demonstrating the potential of Co3O4 as an efficient and cost-effective alternative to precious metal-based catalysts. The short synthesis cycle, combined with the active morphology achieved through the gel-casting technique, could have a significant impact on metal hydride storage systems by enabling the rapid and economical production of catalyst precursors, offering a viable replacement for precious metals.
Looking forward, further enhancement of catalyst performance may be achieved through nanostructure refinement, such as the development of hollow or core-shell architectures, and surface modifications via doping with elements like Ni or Mo. These adjustments could further improve active site accessibility and electron transfer kinetics. Additionally, in situ characterization techniques are essential to elucidate the dynamic changes in structure and surface chemistry during catalysis, providing critical insights for rational catalyst design.
Overall, this study highlights the potential of gel-cast Co3O4 catalysts for hydrogen generation applications and lays a solid foundation for future innovations in catalyst design and development.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}