
Computational Search for a Novel Effective Ligand for Ni-Catalyzed Asymmetric Hydrogenation

 , ,
, , 
Abstract

1. Introduction
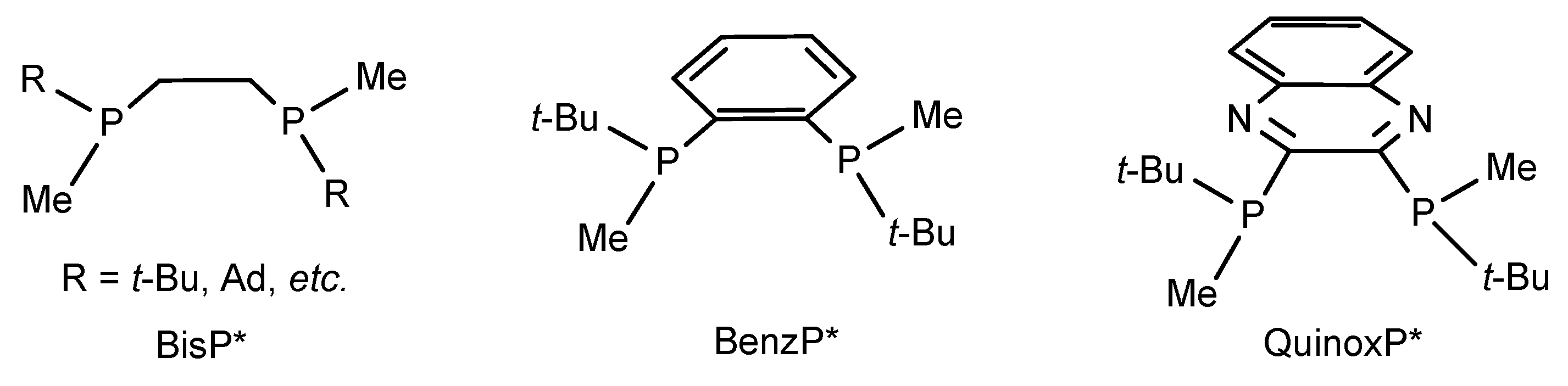
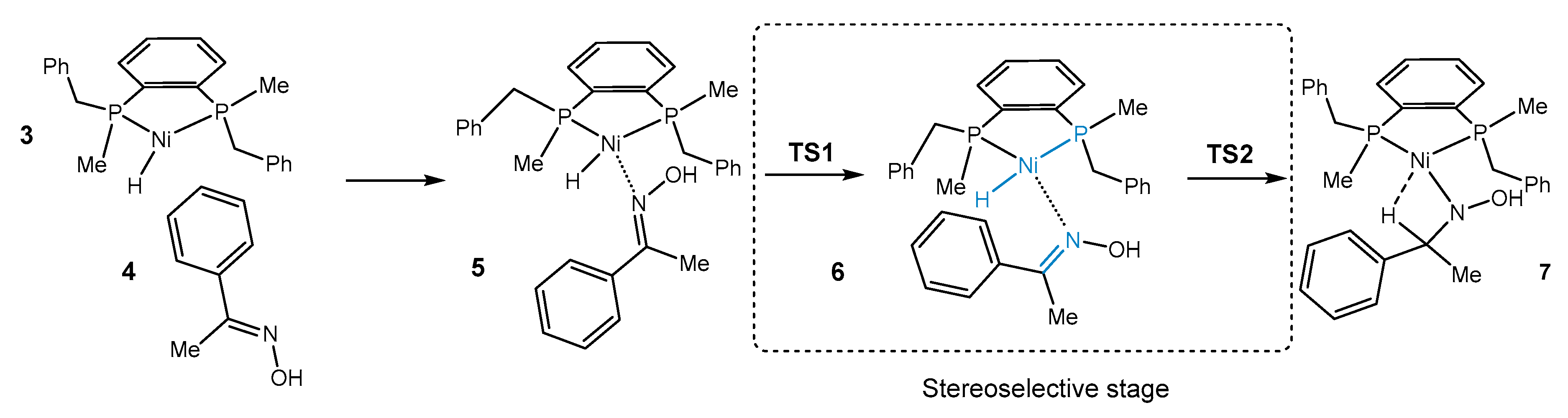
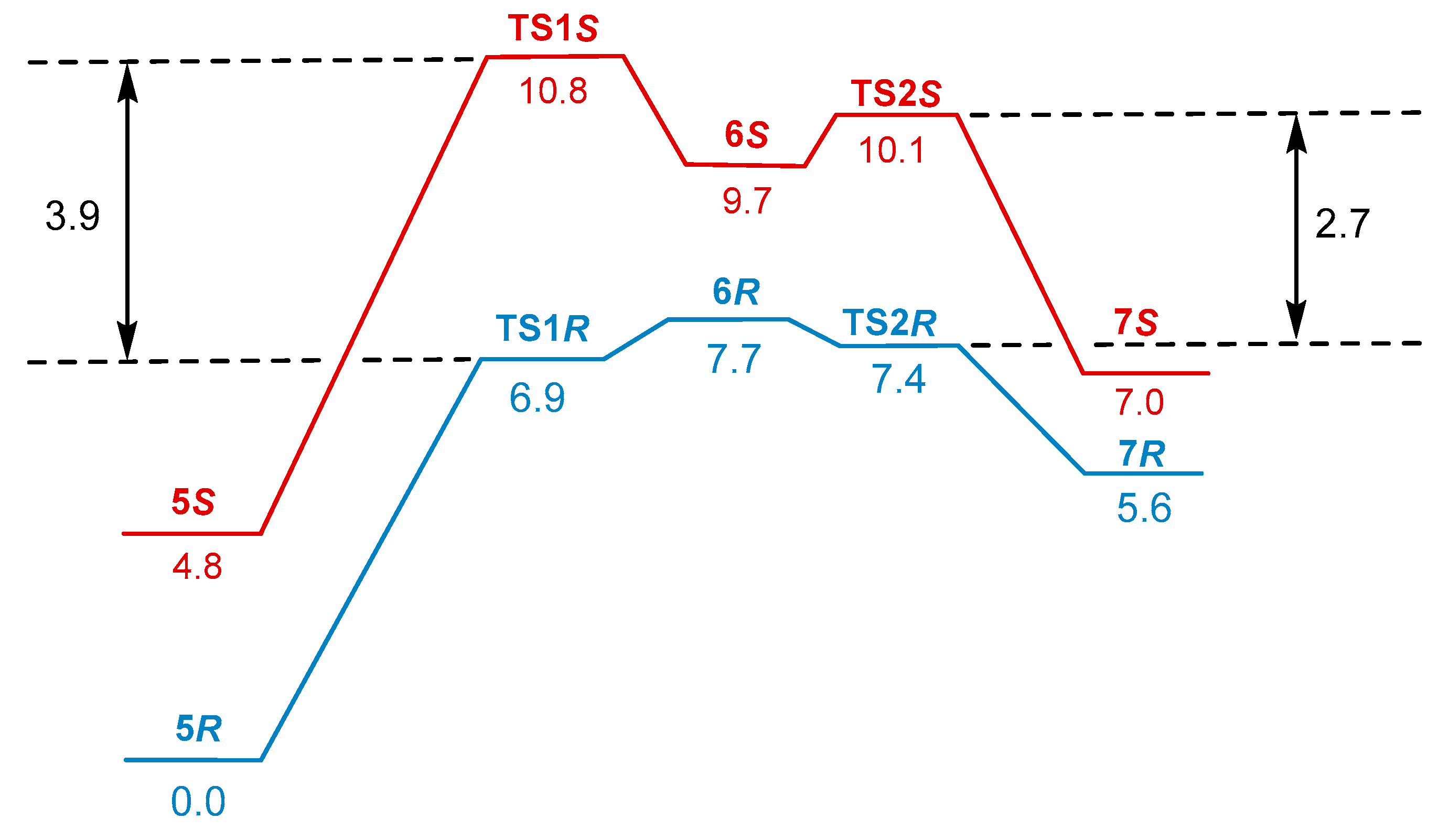
2. Results and Discussion
3. Materials and Methods
Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sun, Y.; Krska, S.; Shultz, C.S.; Tellers, D.M. Enabling asymmetric hydrogenation for the design of efficient synthesis of drug substances. In Asymmetric Catalysis on Industrial Scale: Challenges, Approaches and Solutions, 2nd ed.; Blaser, H.U., Fesersel, H.J., Eds.; Wiley-VCH: Weinheim, Germany, 2010; pp. 331–376. [Google Scholar]
- Kuremoto, T.; Yasukawa, T.; Kobayashi, S. Hydrogenation for the synthesis of an (S)-Metolachlor intermediate using catalysts immobilized on a core/shell-type support. Adv. Synth. Cat. 2024, 366, 757–761. [Google Scholar] [CrossRef]
- Biosca, M.; Diégues, M.; Zanotti-Gerosa, A. Asymmetric hydrogenation in industry. Adv. Cat. 2021, 68, 341–383. [Google Scholar]
- Gridnev, I.D.; Dub, P.A. Enantioselection in Asymmetric Catalysis; Boca Raton CRC Press: London, UK; New York, NY, USA, 2017. [Google Scholar]
- Bayat, P.; Karami, K.; Gallagher, J.F.; Sillanpää, M. Factors controlling asymmetric hydrogenation reactions catalyzed by complexes of phospholanes, and investigation in to the correlation of molecular and electronic structure from 1991 until 2020. J. Mol. Struct. 2023, 1286, 135538. [Google Scholar] [CrossRef]
- Rojo, P.; Riera, A.; Verdaguer, X. Bulky P-stereogenic ligands. A success story in asymmetric catalysis. Coord. Chem. Rev. 2023, 489, 215192. [Google Scholar] [CrossRef]
- Imamoto, T. Development of P-Chirogenic Phosphine Ligands Based on Chemistry of Phosphine–Boranes: Searching for Novelty and Utility in Synthetic Organic Chemistry. TCImail 2017. Available online: https://www.tcichemicals.com/assets/cms-pdfs/174drE_1.pdf (accessed on 25 March 2025).
- Burke, A.J.; Federsel, H.J.; Hermann, G.J. Recent Advances in Asymmetric Hydrogenation Catalysis Utilizing Spiro and Other Rigid C-Stereogenic Phosphine Ligands. J. Org. Chem. 2021, 87, 1898–1924. [Google Scholar] [CrossRef] [PubMed]
- Dobereiner, G.E.; Zhang, X.; Wang, H. Phosphine ligand development for homogeneous asymmetric hydrogenation. In Comprehensive Organometallic Chemistry IV, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2022; Volume 13, pp. 1–31. [Google Scholar]
- Imamoto, T. P-Stereogenic phosphorus ligands in asymmetric catalysis. Chem. Rev. 2024, 124, 8657–8739. [Google Scholar] [CrossRef]
- Imamoto, T.; Watanabe, J.; Wada, Y.; Masuda, H.; Yamada, H.; Tsuruta, H.; Natsukawa, S.; Yamaguchi, K. P-Chiral bis(trialkylphosphine) ligands and their use in highly enantioselective hydrogenation reactions. J. Am. Chem. Soc. 1998, 120, 1635–1636. [Google Scholar] [CrossRef]
- Gridnev, I.D.; Yamanoi, Y.; Higashi, N.; Tsuruta, H.; Yasutake, M.; Imamoto, T. Asymmetric hydrogenation catalyzed by (S,S)-bisp*-Rh and (R,R)-miniphos-Rh complexes: Scope, limitations and mechanism. Adv. Synth. Cat. 2001, 343, 118–136. [Google Scholar] [CrossRef]
- Yasutake, M.; Gridnev, I.D.; Higashi, N.; Imamoto, T. Highly enantioselective hydrogenationof (E)-β-(acylamio)acrylates catalyzed by Rh(I)-complexes of electron-rich P-chirogenic diphosphines. Org. Lett. 2001, 3, 1701–1704. [Google Scholar] [CrossRef]
- Gridnev, I.D.; Yasutake, M.; Imamoto, T.; Beletskaya, I.P. Asymmetric Hydrogenation of α,β-umsaturated phosphonates with Rh-BisP* and Rh-MiniPHOS catalysts: Scope and mechanism of the reaction. Proc. Nat. Acad. Sci. USA 2004, 101, 5385–5390. [Google Scholar] [CrossRef]
- Imamoto, T.; Sugita, K.; Yoshida, K. An air-stable P-chiral phosphine ligand for highly enantioselective transition-metal-catalyzed reactions. J. Am. Chem. Soc. 2005, 127, 11934–11935. [Google Scholar] [CrossRef] [PubMed]
- Imamoto, T.; Tamura, K.; Zhang, Z.; Horiuchi, Y.; Sugiya, M.; Yoshida, K.; Yanagisawa, A.; Gridnev, I.D. Rigid P-chiral phosphine ligands with tert-butylmethylphosphino groups for rhodium-catalyzed asymmetric hydrogenation of functionalized alkenes. J. Am. Chem. Soc. 2012, 134, 1754–1769. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Sugiya, M.; Yoshida, K.; Yanagisawa, A.; Imamoto, T. Enantiopure 1,2-bis(tert-butylmethylphosphino)benzene as a highly efficient ligand in rhodium-catalyzed asymmetric hydrogenation. Org. Lett. 2010, 13, 4400–4403. [Google Scholar] [CrossRef] [PubMed]
- Imamoto, T.; Nishimura, M.; Koide, A.; Yoshida, K. t-Bu-QuinoxP* ligand: Applications in asymmetric Pd-catalyzed allylic substitution and Ru-catalyzed hydrogenation. J. Org. Chem. 2019, 72, 7413–7416. [Google Scholar] [CrossRef]
- Li, B.; Chen, J.; Zhang, Z.; Gridnev, I.D.; Zhang, W. Nickel-catalyzed asymmetric hydrogenation of N-sulfonyl imines. Angew. Chem. Int. Ed. 2019, 58, 7329–7334. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Xie, C.; Guo, Q.; Zi, G.; Hou, G.; Huang, Y. Nickel-catalyzed asymmetric hydrogenation ofγ-keto acids, esters, and amides to chiralγ-lactones andγ-hydroxy acid derivatives. Org. Lett. 2022, 24, 2722–2727. [Google Scholar] [CrossRef] [PubMed]
- Muci, A.R.; Campos, K.R.; Evans, D.A. Enantioselective Deprotonation as a Vehicle for the Asymmetric Synthesis of C2-Symmetric P-Chiral Diphosphines. J. Am. Chem. Soc. 1995, 117, 9075–9076. [Google Scholar] [CrossRef]
- Maienza, F.; Spindler, F.; Thommen, M.; Pugin, B.; Malan, C.; Mezzetti, A. Exploring stereogenic phosphorus: Synthetic strategies for diphosphines containing bulky highly symmetric substituents. J. Org. Chem. 2002, 67, 5239–5249. [Google Scholar] [CrossRef]
- Gridnev, I.D.; Higashi, N.; Imamoto, T. Formation of a stable rhodium(I) dihydride complex and its reactions with prochiral substrates of asymmetric hydrogenation. Organometallics 2001, 21, 4542–4553. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Gaussian 16; Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar]
- Gordon, M.S.; Binkley, J.S.; Pople, J.A.; Pietro, W.J.; Hehre, W.J. Self-consistent molecular-orbital methods. Small split-valence basis sets for second-row elements. J. Am. Chem. Soc. 1982, 104, 2797–2803. [Google Scholar] [CrossRef]
- Rassolov, V.A.; Pople, J.A.; Ratner, M.A.; Windus, T.L. 6-31G * basis set for atoms K through Zn. J. Chem. Phys. 1998, 109, 1223–1229. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Chen, J.; Liu, D.; Gridnev, I.D.; Zhang, W. Nickel-catalyzed asymmetric hydrogenation of oximes. Nat. Chem. 2022, 14, 920–927. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, J.; Li, B.; Zhang, Z.; Gridnev, I.D.; Zhang, W. Nickel-Catalyzed Asymmetric Hydrogenation of 2-Amidoacrylates. Angew. Chem. Int. Ed. 2020, 59, 5371–5375. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Chen, H.; Chen, J.; Gridnev, I.D.; Zhang, W. Nickel-Catalyzed Asymmetric Hydrogenation of alpha-Substituted Vinylphosphonates and Diarylvinylphosphine Oxides. Angew. Chem. Int. Ed. 2023, 62, e202214990. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Luo, Y.; Li, J.; Chen, J.; Gridnev, I.D.; Zhang, W. Enantioselective Synthesis of Chiral β2-Amino Phosphorus Derivatives via Nickel-Catalyzed Asymmetric Hydrogenation. J. Am. Chem. Soc. 2025, 147, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wei, H.; Gridnev, I.D.; Zhang, W. Weak Attractive Non-Covalent Interactions in Metal-Catalyzed Asymmetric Hydrogenation. Angew. Chem. Int. Ed. 2025, 64, e202425589. [Google Scholar]
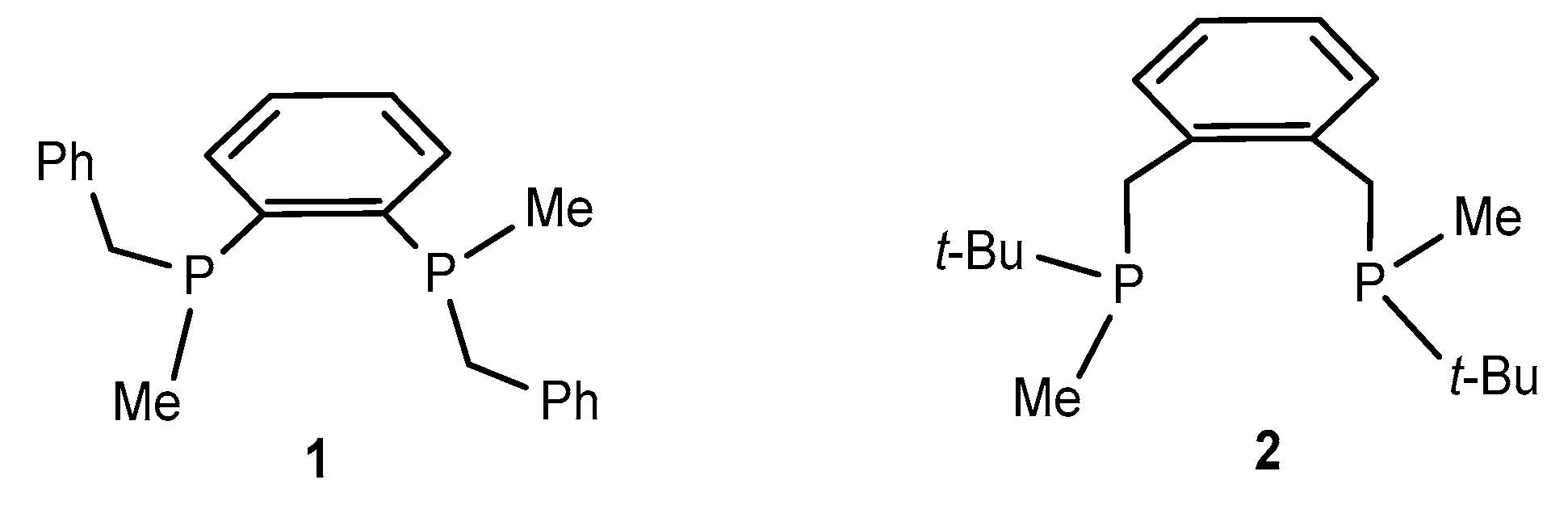

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
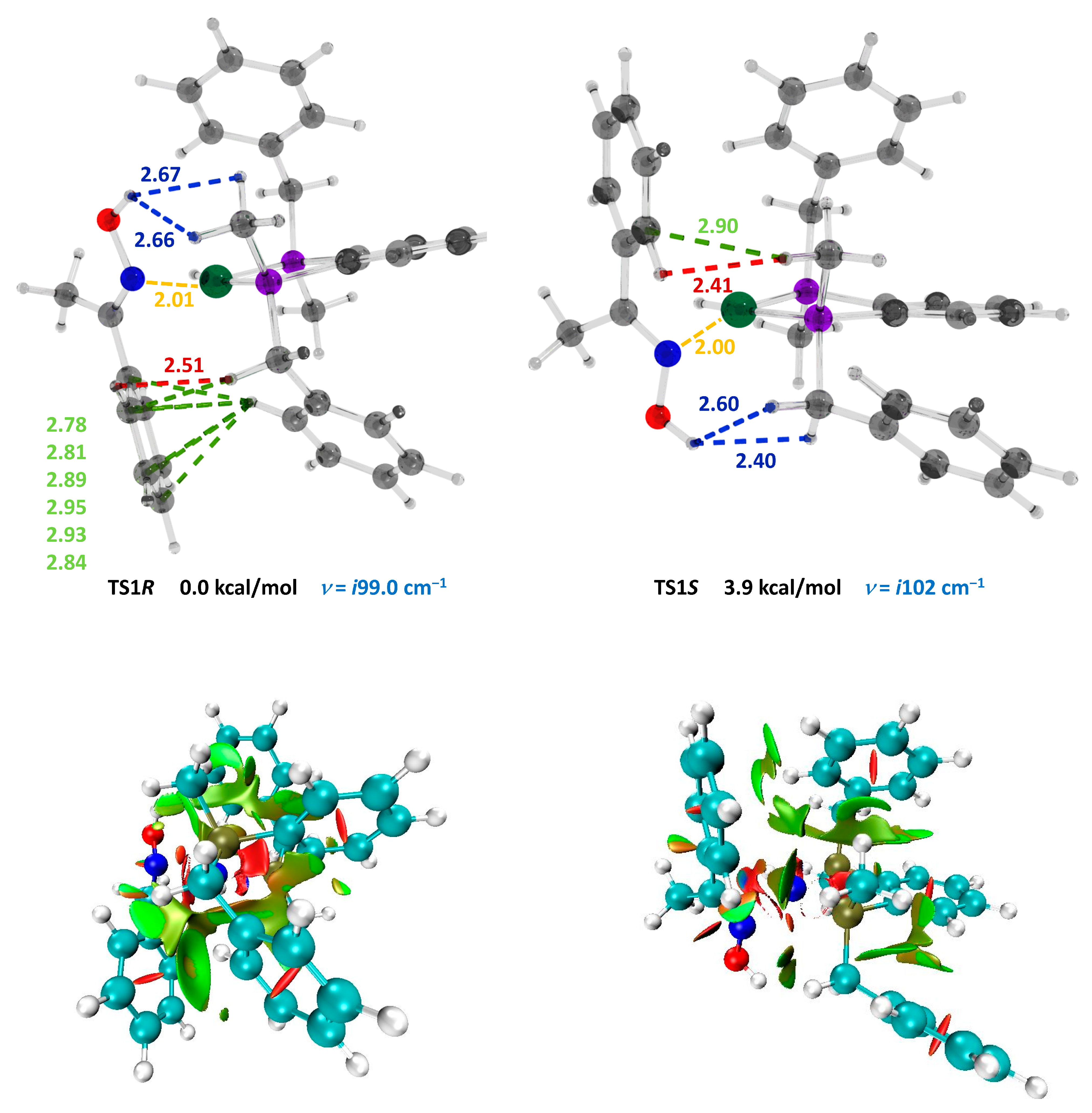
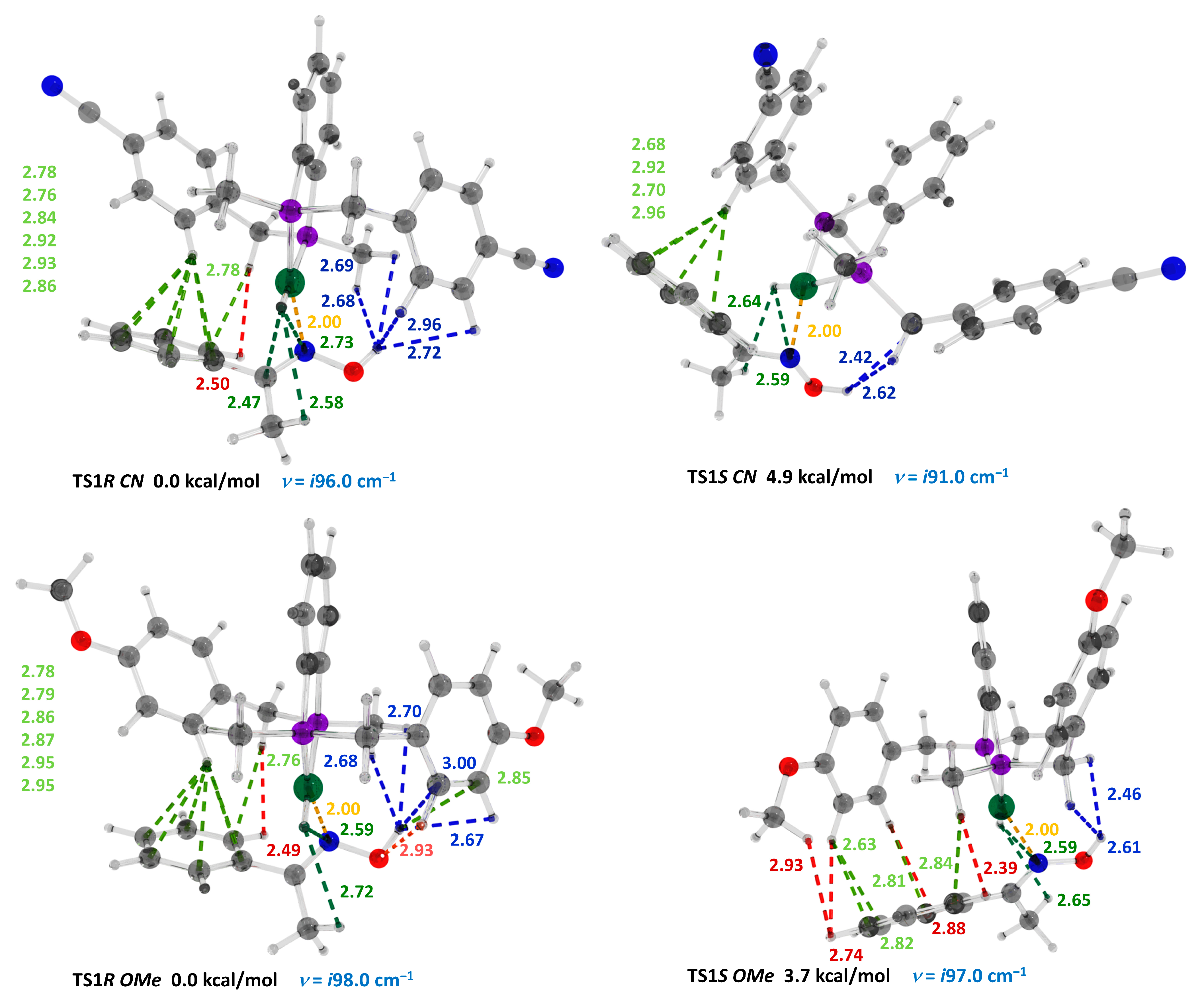
 | Transition State | Number of Contacts in the Interval 2.0–3.0 Å | ΔN (Relative Number of Stabilizing NCI) | Nɩ–N Distance (Å) | ΔΔG wb97xd/6-31g(d,p) (Kcal/mol) |
|---|---|---|---|---|---|
| Ar = 4-CNC6H4 | TS1R CN | 15 | 7 | 2.00 | |
| TS1S CN | 8 | 2.00 | +4.9 | ||
| Ar = C6H5 | TS1R | 9 | 5 | 2.01 | |
| TS1S | 4 | 2.00 | +3.9 | ||
| Ar = 4-(OMe)C6H4 | TS1R OMe | 16 | 4 | 2.00 | |
| TS1S OMe | 12 | 2.00 | +3.7 | ||
| Ar = 4-CNC6H4 | TS2R CN | 8 | 0 | 1.93 | |
| TS2S CN | 8 | 1.94 | +2.1 | ||
| Ar = C6H5 | TS2R | 9 | 0 | 1.93 | |
| TS2S | 9 | 1.94 | +2.7 | ||
| Ar = 4-(OMe)C6H4 | TS2R OMe | 8 | 2 | 1.93 | |
| TS2S OMe | 6 | 1.94 | +1.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pospelov, E.V.; Golovanov, I.S.; Chen, J.; Zhang, W.; Gridnev, I.D. Computational Search for a Novel Effective Ligand for Ni-Catalyzed Asymmetric Hydrogenation. Catalysts 2025, 15, 352. https://doi.org/10.3390/catal15040352
Pospelov EV, Golovanov IS, Chen J, Zhang W, Gridnev ID. Computational Search for a Novel Effective Ligand for Ni-Catalyzed Asymmetric Hydrogenation. Catalysts. 2025; 15(4):352. https://doi.org/10.3390/catal15040352
Chicago/Turabian StylePospelov, Evgeny V., Ivan S. Golovanov, Jianzhong Chen, Wanbin Zhang, and Ilya D. Gridnev. 2025. "Computational Search for a Novel Effective Ligand for Ni-Catalyzed Asymmetric Hydrogenation" Catalysts 15, no. 4: 352. https://doi.org/10.3390/catal15040352
APA StylePospelov, E. V., Golovanov, I. S., Chen, J., Zhang, W., & Gridnev, I. D. (2025). Computational Search for a Novel Effective Ligand for Ni-Catalyzed Asymmetric Hydrogenation. Catalysts, 15(4), 352. https://doi.org/10.3390/catal15040352