
Enhanced Nitrate Production via Electrocatalytic Nitric Oxide Oxidation Reaction over MnO2 with Different Crystal Facets



Abstract
1. Introduction
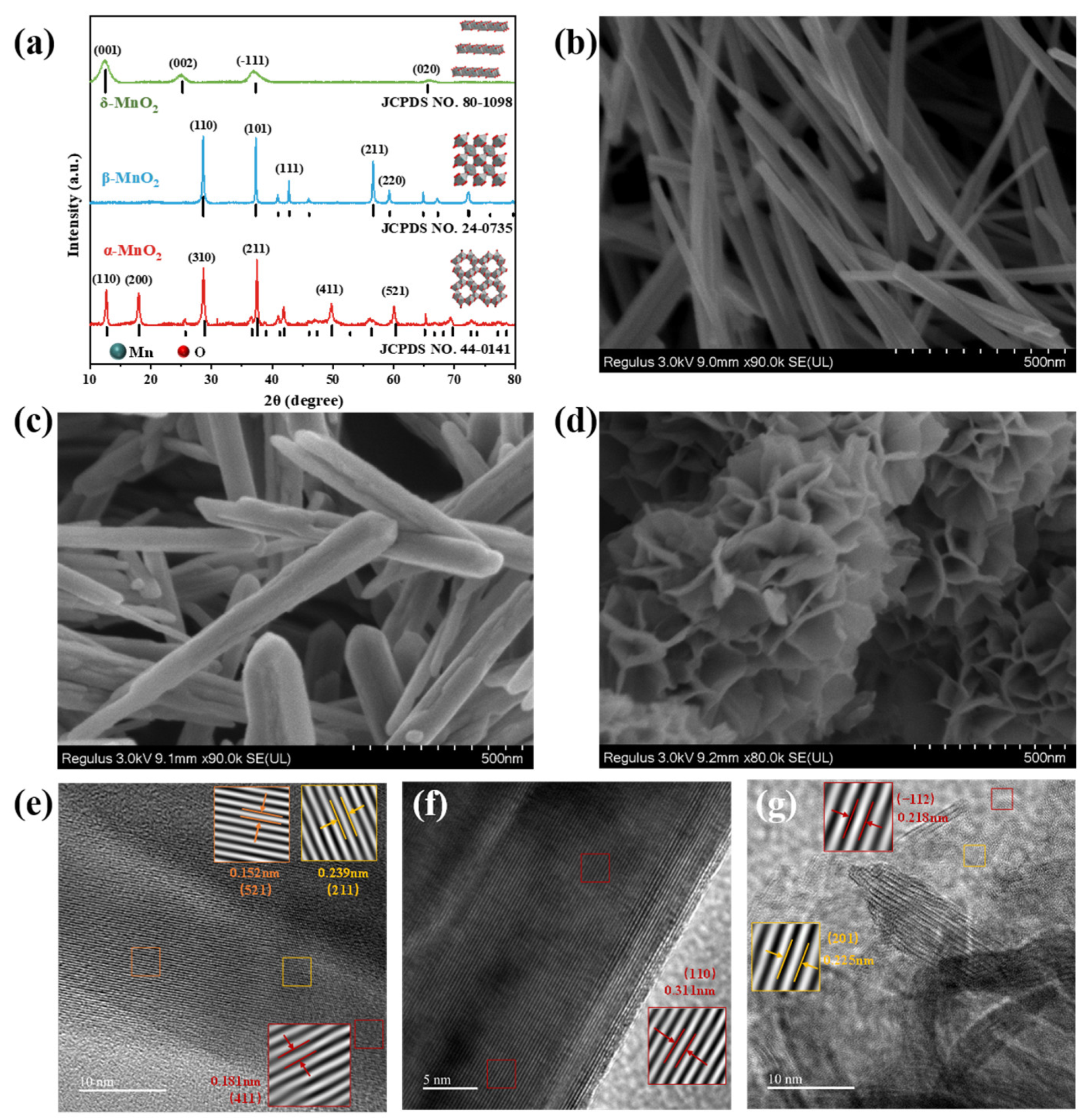
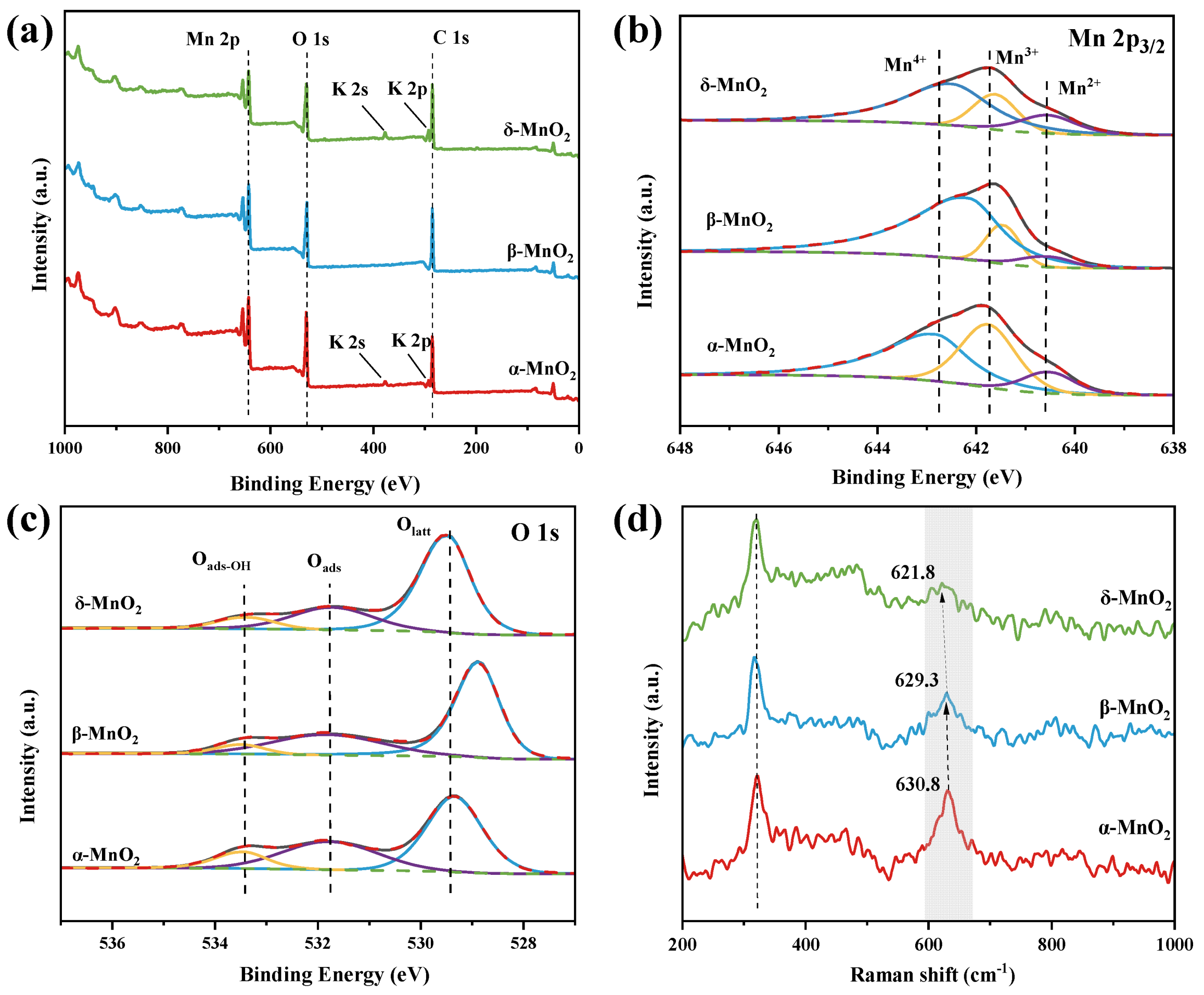
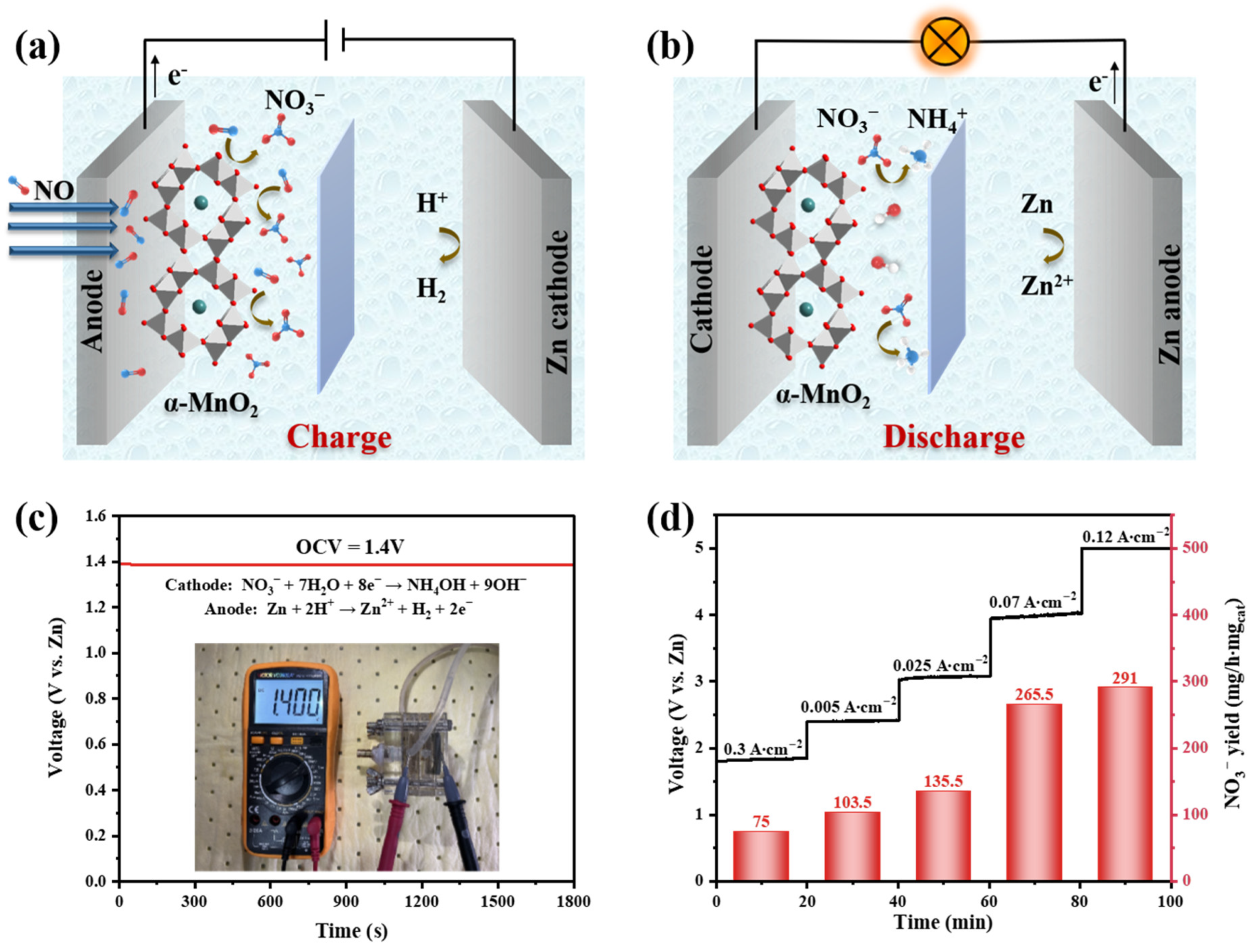
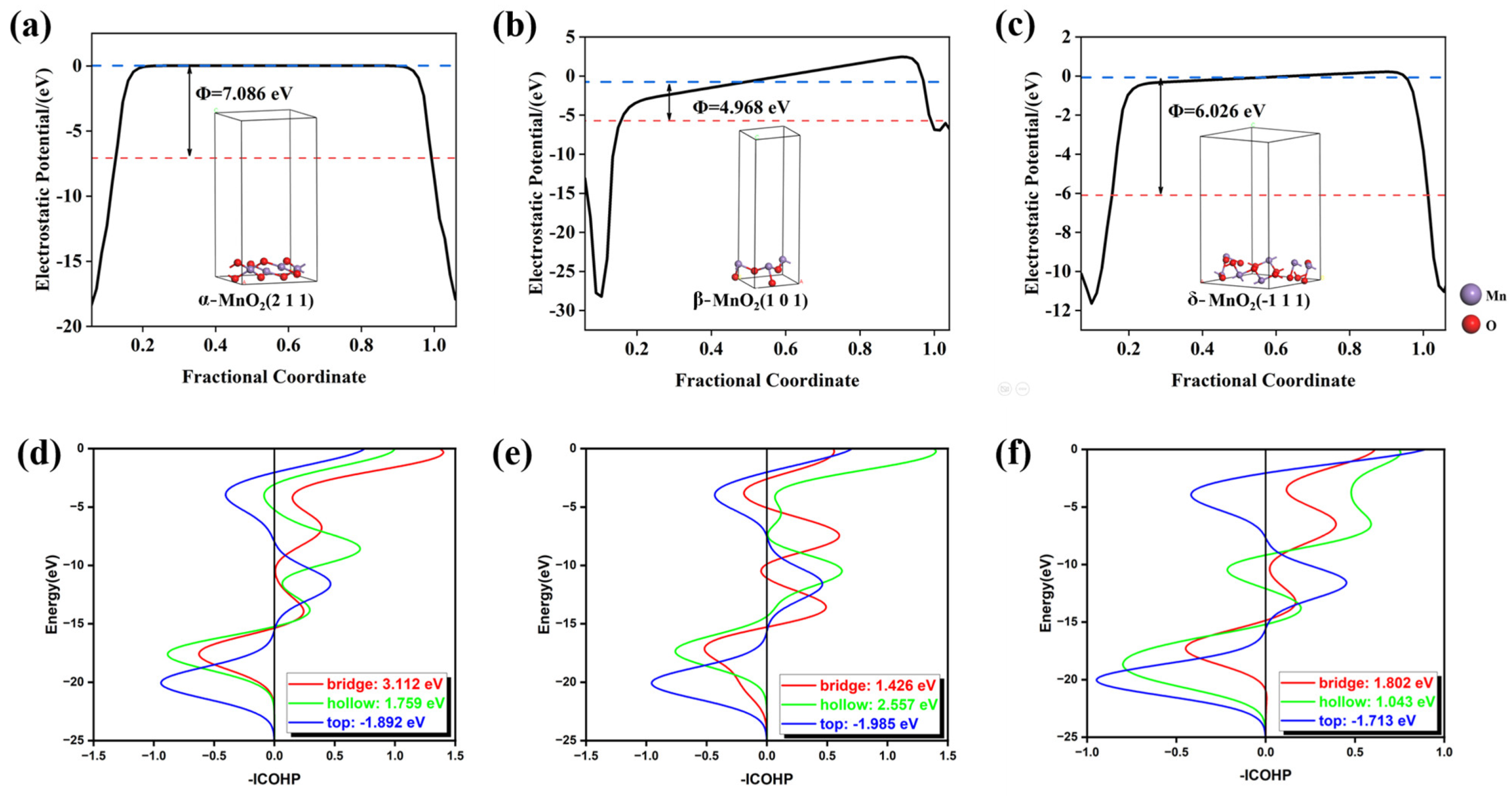
2. Results and Discussion
3. Experiments
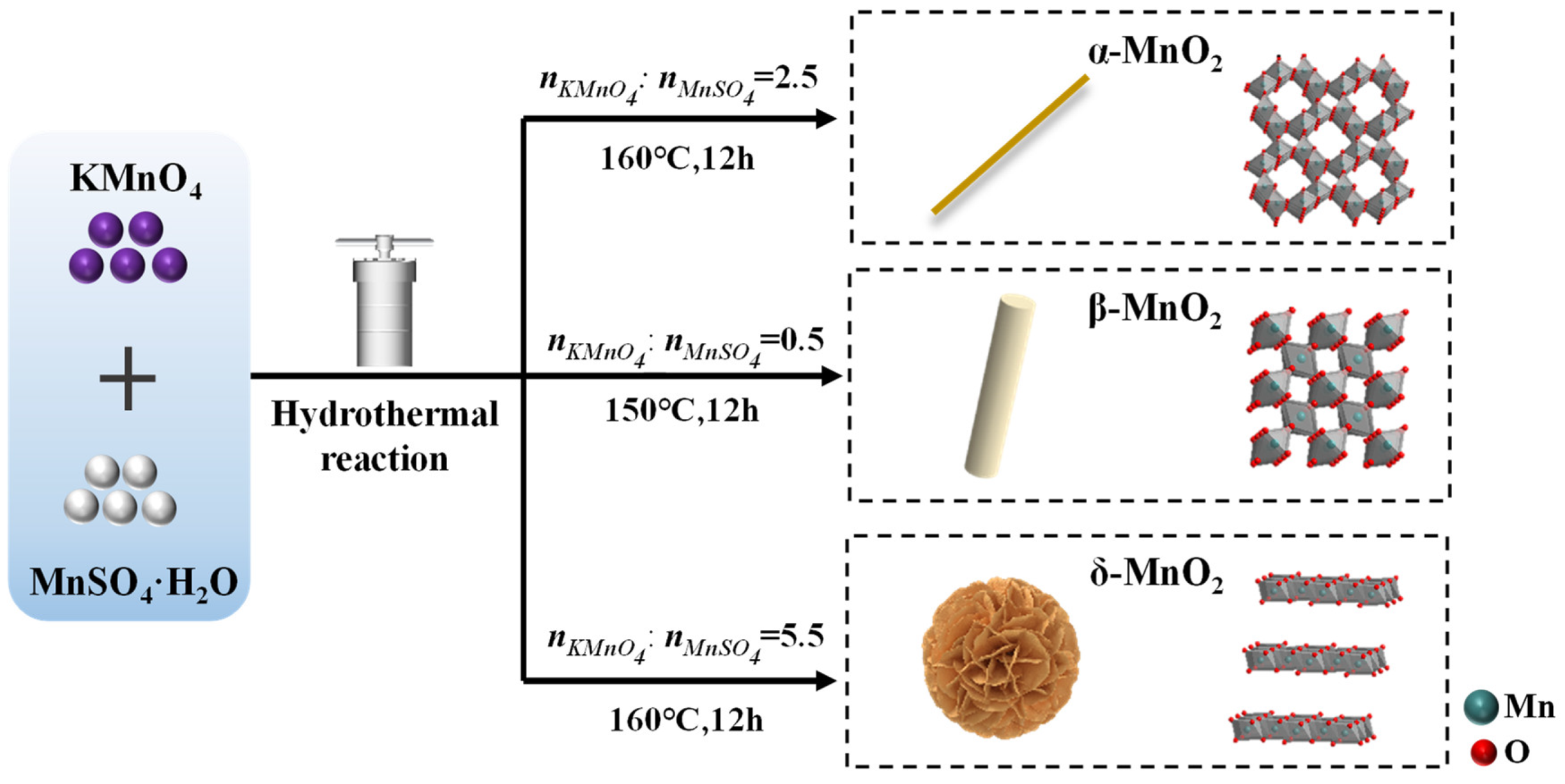
3.1. Synthesis Methods
3.2. Material Characterization
3.3. Electrochemical Measurement
3.4. Catalyst Activity Evaluation
3.5. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wang, D.; He, N.; Xiao, L.; Dong, F.; Chen, W.; Zhou, Y.; Chen, C.; Wang, S. Coupling Electrocatalytic Nitric Oxide Oxidation over Carbon Cloth with Hydrogen Evolution Reaction for Nitrate Synthesis. Angew. Chem. Int. Ed. 2021, 60, 24605–24611. [Google Scholar] [CrossRef]
- Li, S.; Shang, H.; Tao, Y.; Li, P.; Pan, H.; Wang, Q.; Zhang, S.; Jia, H.; Zhang, H.; Cao, J.; et al. Hydroxyl Radical-Mediated Efficient Photoelectrocatalytic NO Oxidation with Simultaneous Nitrate Storage Using a Flow Photoanode Reactor. Angew. Chem. Int. Ed. 2023, 62, e202305538. [Google Scholar]
- Long, J.; Chen, S.; Zhang, Y.; Guo, C.; Fu, X.; Deng, D.; Xiao, J. Direct Electrochemical Ammonia Synthesis from Nitric Oxide. Angew. Chem. Int. Ed. 2020, 59, 9711–9718. [Google Scholar] [CrossRef]
- Adalder, A.; Paul, S.; Ghorai, U.K. Progress of Electrochemical Synthesis of Nitric Acid: Catalyst Design, Mechanistic Insights, Protocol and Challenges. J. Mater. Chem. A 2023, 11, 10125–10148. [Google Scholar] [CrossRef]
- Yan, C.; Tham, Y.J.; Nie, W.; Xia, M.; Wang, H.; Guo, Y.; Ma, W.; Zhan, J.; Hua, C.; Li, Y.; et al. Increasing Contribution of Nighttime Nitrogen Chemistry to Wintertime Haze Formation in Beijing Observed during COVID-19 Lockdowns. Nat. Geosci. 2023, 16, 975–981. [Google Scholar] [CrossRef]
- Sumathi, S.; Bhatia, S.; Lee, K.T.; Mohamed, A.R. Selection of Best Impregnated Palm Shell Activated Carbon (PSAC) for Simultaneous Removal of SO2 and NOx. J. Hazard. Mater. 2010, 176, 1093–1096. [Google Scholar] [CrossRef]
- Xiao, S.; Wan, Z.; Zhou, J.; Li, H.; Zhang, H.; Su, C.; Chen, W.; Li, G.; Zhang, D.; Li, H. Gas-Phase Photoelectrocatalysis for Breaking Down Nitric Oxide. Environ. Sci. Technol. 2019, 53, 7145–7154. [Google Scholar] [CrossRef]
- Li, B.; Shen, D.; Xiao, Z.; Li, Q.; Yao, S.; Wang, W.; Liu, L. The In Situ Decoration of Ti3C2 Quantum Dots on Cu Nanowires for Highly Efficient Electrocatalytic Reduction of Nitric Oxide to Ammonia. Inorg. Chem. Front. 2023, 10, 5927–5936. [Google Scholar] [CrossRef]
- Luo, R.; Li, B.-J.; Wang, Z.-P.; Chen, M.-G.; Zhuang, G.-L.; Li, Q.; Tong, J.-P.; Wang, W.-T.; Fan, Y.-H.; Shao, F. Two-Dimensional MOF Constructed by a Binuclear-Copper Motif for High-Performance Electrocatalytic NO Reduction to NH3. JACS Au 2024, 4, 3823–3832. [Google Scholar] [CrossRef]
- Hao, M.; Shen, D.; Li, Q.; Xiao, Z.; Liu, L.; Li, C.; Wang, W. The Combination of Hydrogen Evolution, Nitric Oxide Oxidation and Zn-Nitrate Battery for Energy Conversion and Storage by an Efficient Nitrogen-Dopped CoOX Electrocatalyst with Turing Structure. J. Colloid Interface Sci. 2025, 683, 477–488. [Google Scholar] [CrossRef]
- Chen, J.G.; Crooks, R.M.; Seefeldt, L.C.; Bren, K.L.; Bullock, R.M.; Darensbourg, M.Y.; Holland, P.L.; Hoffman, B.; Janik, M.J.; Jones, A.K.; et al. Beyond Fossil Fuel–Driven Nitrogen Transformations. Science 2018, 360, eaar6611. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Fang, L.; Zhang, L.; Li, M.; Cong, M.; Guan, X.; Shi, C.; Gu, C.; Liu, X.; Wang, Y.; et al. Pulsed Electrocatalysis Enabling High Overall Nitrogen Fixation Performance for Atomically Dispersed Fe on TiO2. Angew. Chem. Int. Ed. 2023, 135, e202217635. [Google Scholar] [CrossRef]
- Miao, R.; Chen, D.; Guo, Z.; Zhou, Y.; Chen, C.; Wang, S. Recent Advances in Electrocatalytic Upgrading of Nitric Oxide and Beyond. Appl. Catal. B Environ. 2024, 344, 123662. [Google Scholar] [CrossRef]
- Zheng, H.; Ma, Z.; Liu, Y.; Zhang, Y.; Ye, J.; Debroye, E.; Zhang, L.; Liu, T.; Xie, Y. Perovskite Oxide as A New Platform for Efficient Electrocatalytic Nitrogen Oxidation. Angew. Chem. Int. Ed. 2024, 63, e202316097. [Google Scholar] [CrossRef]
- Zhang, L.; Liang, J.; He, X.; Yang, Q.; Luo, Y.; Zheng, D.; Sun, S.; Zhang, J.; Yan, H.; Ying, B.; et al. Integrating RuO2@TiO2 Catalyzed Electrochemical Chlorine Evolution with a NO Oxidation Reaction for Nitrate Synthesis. Inorg. Chem. Front. 2023, 10, 2100–2106. [Google Scholar] [CrossRef]
- Wu, F.; Huang, Y.; Wang, D. Experimental Study on Supported MnO2-Based Catalysts for NO Oxidation. React. Kinet. Mech. Cat. 2023, 136, 251–266. [Google Scholar] [CrossRef]
- Liang, J.; Zhang, L.; He, X.; Wang, Y.; Luo, Y.; Zheng, D.; Sun, S.; Cai, Z.; Zhang, J.; Ma, K.; et al. Redox Mediators Promote Electrochemical Oxidation of Nitric Oxide toward Ambient Nitrate Synthesis. J. Mater. Chem. A 2023, 11, 1098–1107. [Google Scholar] [CrossRef]
- Boningari, T.; Pappas, D.K.; Smirniotis, P.G. Metal Oxide-Confined Interweaved Titania Nanotubes M/TNT (M = Mn, Cu, Ce, Fe, V, Cr, and Co) for the Selective Catalytic Reduction of NOx in the Presence of Excess Oxygen. J. Catal. 2018, 365, 320–333. [Google Scholar] [CrossRef]
- Wang, H.; Chen, H.; Wang, Y.; Lyu, Y.-K. Performance and Mechanism Comparison of Manganese Oxides at Different Valence States for Catalytic Oxidation of NO. Chem. Eng. J. 2019, 361, 1161–1172. [Google Scholar] [CrossRef]
- Li, Y. The Exploration and Comparison of Adsorption Mechanisms in MnO2 with Different Crystal Structures for Capacitive Deionization. Desalination 2024, 577, 117387. [Google Scholar] [CrossRef]
- Meng, Y.; Song, W.; Huang, H.; Ren, Z.; Chen, S.-Y.; Suib, S.L. Structure-Property Relationship of Bifunctional MnO2 Nanostructures: Highly Efficient, Ultra-Stable Electrochemical Water Oxidation and Oxygen Reduction Reaction Catalysts Identified in Alkaline Media. J. Am. Chem. Soc. 2014, 136, 11452–11464. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Guo, Z.; Cai, L.; Zhu, R.; Fan, Y.; Zhang, Y.; Han, P.; Zhang, W.; Zhu, X.; Zhao, Q.; et al. Investigation into the Phase–Activity Relationship of MnO2 Nanomaterials toward Ozone-Assisted Catalytic Oxidation of Toluene. Small 2021, 17, 2103052. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ren, S.; Xing, X.; Yang, J.; Li, J.; Yang, J.; Liu, Q. Effect of MnO2 Crystal Types on CeO2@MnO2 Oxides Catalysts for Low-Temperature NH3-SCR. J. Environ. Chem. 2022, 10, 108239. [Google Scholar] [CrossRef]
- Liang, S.; Teng, F.; Bulgan, G.; Zong, R.; Zhu, Y. effect of phase structure of MnO2 Nanorod Catalyst on the Activity for CO Oxidation. J. Phys. Chem. C 2008, 112, 5307–5315. [Google Scholar] [CrossRef]
- Yang, W.; Su, Z.; Xu, Z.; Yang, W.; Peng, Y.; Li, J. Comparative Study of α-, β-, γ- and δ-MnO2 on Toluene Oxidation: Oxygen Vacancies and Reaction Intermediates. Appl. Catal. B Environ. 2020, 260, 118150. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, C.; Li, Y.; Chang, C.-R.; He, C.; Lu, Q.; Yu, Y.; Duan, P.; Zhang, Z.; Luque, R. Hierarchically Hollow MnO2 @CeO2 Heterostructures for NO Oxidation: Remarkably Promoted Activity and SO2 Tolerance. ACS Catal. 2021, 11, 10988–10996. [Google Scholar] [CrossRef]
- Yang, R.; Fan, Y.; Ye, R.; Tang, Y.; Cao, X.; Yin, Z.; Zeng, Z. MnO2—Based Materials for Environmental Applications. Adv. Mater. 2021, 33, 2004862. [Google Scholar] [CrossRef]
- Yang, J.; Ren, S.; Zhou, Y.; Hu, G.; Jiang, L.; Cao, J.; Liu, W.; Yao, L.; Kong, M.; Yang, J.; et al. Insight into N2O Formation Over Different Crystal Phases of MnO2 During Low-Temperature NH3–SCR of NO. Catal. Lett. 2021, 151, 2964–2971. [Google Scholar] [CrossRef]
- Su, H.; Liu, J.; Hu, Y.; Ai, T.; Gong, C.; Lu, J.; Luo, Y. Comparative Study of α- and β-MnO2 on Methyl Mercaptan Decomposition: The Role of Oxygen Vacancies. Nanomaterials 2023, 13, 775. [Google Scholar] [CrossRef]
- Xu, J.; Wu, Z.; Gao, E.; Zhu, J.; Yao, S.; Li, J. Revealing the Role of Oxygen Vacancies on α-MnO2 of Different Morphologies in CO Oxidation Using Operando DRIFTS-MS. Appl. Surf. Sci. 2023, 618, 156643. [Google Scholar] [CrossRef]
- Fu, Z.; Wang, D.; Yao, Y.; Gao, X.; Liu, X.; Wang, S.; Yao, S.; Wang, X.; Chi, X.; Zhang, K.; et al. Local Electric Field Induced by Atomic-Level Donor–Acceptor Couple of O Vacancies and Mn Atoms Enables Efficient Hybrid Capacitive Deionization. Small 2023, 19, 2205666. [Google Scholar] [CrossRef]
- Zhu, G.; Zhu, W.; Lou, Y.; Ma, J.; Yao, W.; Zong, R.; Zhu, Y. Encapsulate α-MnO2 Nanofiber within Graphene Layer to Tune Surface Electronic Structure for Efficient Ozone Decomposition. Nat. Commun. 2021, 12, 4152. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zhan, C.; He, K.; Chen, H.; Yao, W.; Sharifi-Asl, S.; Song, B.; Yang, Z.; Nie, A.; Luo, X.; et al. The Influence of Large Cations on the Electrochemical Properties of Tunnel-Structured Metal Oxides. Nat. Commun. 2016, 7, 13374. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Zhu, J.; Li, W.; Yao, W.; Zong, R.; Zhu, Y.; Zhang, Q. Tuning the K+ Concentration in the Tunnels of α-MnO2 to Increase the Content of Oxygen Vacancy for Ozone Elimination. Environ. Sci. Technol. 2018, 52, 8684–8692. [Google Scholar] [CrossRef]
- Zhang, T. Electrospun YMn2O5 Nanofibers: A Highly Catalytic Activity for NO Oxidation. Appl. Catal. B Environ. 2019, 247, 133141. [Google Scholar] [CrossRef]
- Wang, F.; Dai, H.; Deng, J.; Bai, G.; Ji, K.; Liu, Y. Manganese Oxides with Rod-, Wire-, Tube-, and Flower-Like Morphologies: Highly Effective Catalysts for the Removal of Toluene. Environ. Sci. Technol. 2012, 46, 4034–4041. [Google Scholar] [CrossRef]
- Ma, L.; Chen, S.; Yan, W.; Zhang, G.; Ying, Y.; Huang, H.; Ho, D.; Huang, W.; Zhi, C. A High-Energy Aqueous Zn‖NO2 Electrochemical Cell: A New Strategy for NO2 Fixation and Electric Power Generation. Energy Environ. Sci. 2023, 16, 1125–1134. [Google Scholar] [CrossRef]
- Li, Q.; Xiao, Z.; Jia, W.; Li, Q.; Li, X.; Wang, W. Copper Nanowires Decorated with TiO2−x from MXene for Enhanced Electrocatalytic Nitrogen Oxidation into Nitrate under Vacuum Assistance. Nano Res. 2023, 16, 12357–12362. [Google Scholar] [CrossRef]
- Greiner, M.T.; Chai, L.; Helander, M.G.; Tang, W.; Lu, Z. Transition Metal Oxide Work Functions: The Influence of Cation Oxidation State and Oxygen Vacancies. Adv. Funct. Mater. 2012, 22, 4557–4568. [Google Scholar] [CrossRef]
- Guo, L.; Li, R.; Jiang, J.; Zou, J.-J.; Mi, W. Electrocatalytic Performance of Mn-Adsorbed g-C3N4: A First-Principles Study. J. Mater. Chem. A 2021, 9, 26266–26276. [Google Scholar] [CrossRef]
- Ye, X.; Jiang, X.; Chen, L.; Jiang, W.; Wang, H.; Cen, W.; Ma, S. Effect of Manganese Dioxide Crystal Structure on Adsorption of SO2 by DFT and Experimental Study. Appl. Surf. Sci. 2020, 521, 146477. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, H.B.; Zhou, D.; Liu, B. Adsorption Energy in Oxygen Electrocatalysis. Chem. Rev. 2022, 122, 17028–17072. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qiu, W.; Song, E.; Gu, F.; Zheng, Z.; Zhao, X.; Zhao, Y.; Liu, J.; Zhang, W. Adsorption-Energy-Based Activity Descriptors for Electrocatalysts in Energy Storage Applications. Natl. Sci. Rev. 2018, 5, 327–341. [Google Scholar] [CrossRef]
- Ong, Y.-P.; Ho, L.-N.; Ong, S.-A.; Banjuraizah, J.; Ibrahim, A.H.; Thor, S.-H.; Yap, K.-L. A Highly Sustainable Hydrothermal Synthesized MnO2 as Cathodic Catalyst in Solar Photocatalytic Fuel Cell. Chemosphere 2021, 263, 128212. [Google Scholar] [CrossRef]
- Wu, A.; Lv, J.; Xuan, X.; Zhang, J.; Cao, A.; Wang, M.; Wu, X.; Liu, Q.; Zhong, Y.; Sun, W.; et al. Electrocatalytic Disproportionation of Nitric Oxide Toward Efficient Nitrogen Fixation. Adv. Energy Mater. 2023, 13, 2204231. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
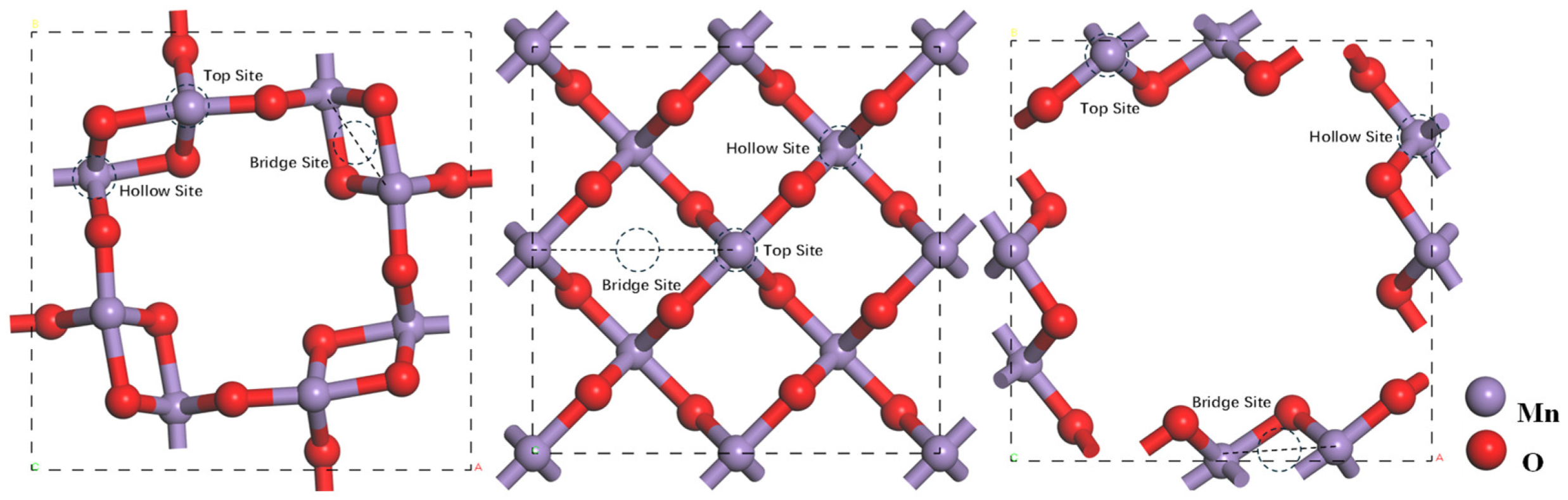
| Catalysts | Top Site | Bridge Site | Hollow Site |
|---|---|---|---|
| α-MnO2 | −10.417 | −1.1455 | −0.8948 |
| β-MnO2 | 0.8184 | 7.9489 | 11.1268 |
| δ-MnO2 | 0.8494 | 0.8494 | 13.1298 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, X.; Shen, D.; Li, Q.; Liu, X.; Wu, M.; Wang, W. Enhanced Nitrate Production via Electrocatalytic Nitric Oxide Oxidation Reaction over MnO2 with Different Crystal Facets. Catalysts 2025, 15, 342. https://doi.org/10.3390/catal15040342
Qin X, Shen D, Li Q, Liu X, Wu M, Wang W. Enhanced Nitrate Production via Electrocatalytic Nitric Oxide Oxidation Reaction over MnO2 with Different Crystal Facets. Catalysts. 2025; 15(4):342. https://doi.org/10.3390/catal15040342
Chicago/Turabian StyleQin, Xiaoyu, Dongcai Shen, Quan Li, Xin Liu, Mingrui Wu, and Wentai Wang. 2025. "Enhanced Nitrate Production via Electrocatalytic Nitric Oxide Oxidation Reaction over MnO2 with Different Crystal Facets" Catalysts 15, no. 4: 342. https://doi.org/10.3390/catal15040342
APA StyleQin, X., Shen, D., Li, Q., Liu, X., Wu, M., & Wang, W. (2025). Enhanced Nitrate Production via Electrocatalytic Nitric Oxide Oxidation Reaction over MnO2 with Different Crystal Facets. Catalysts, 15(4), 342. https://doi.org/10.3390/catal15040342