1. Introduction
A ProTide, a prodrug and nucleotide that contains a phosphoramidate or a phosphonate group, is considered a breakthrough design for delivering nucleoside analogs into cells [
1,
2]. The phosphate moiety acts as a masked group improving formation of the active triphosphate metabolites, increasing the oral bioavailability and breaking the limit of resistance to transporter downregulation [
2]. FDA-approved ProTide prodrugs including tenofovir alafinamide, sofosbuvir, and remdesivir have become essential medications for the treatment of viral infections such as human immunodeficiency Virus (HIV) and hepatitis B virus (HBV), hepatitis C virus (HCV), and coronavirus, respectively [
3,
4]. These ProTide are a chiral molecule which has two diastereomers which require separation. For example, the case of sofosbuvir found that the (
Sp)-diastereomer is more potent than the (
Rp)-diastereomer for treatment due to (
Sp)-diastereomer exhibiting a 35-fold better substrate for CatA in liver cells [
5] and being 18-fold better against hepatitis C [
6]. Similarly to tenofovir alafinamide, (
Sp)-diastereomer is 12-fold more active against HIV than (
Rp)-diastereomer [
7]. In addition, (
Sp)-remdesivir exhibits higher selectivity and an appropriate therapeutic window that is suitable for antiviral therapy [
8].
To address the separation of the two diastereoisomers, there are several ways such as column chromatography [
1], crystallization [
9], and synthetic procedures with stereocontrol of the phosphorus chirality of the ProTides [
10,
11]. Recently, an alternative way of production for high purities ProTide has been reported using biocatalysts (phosphotriesterase, PTE) [
12,
13,
14]. A mutant library of PTE from
Brevundimonas diminuta (formerly
Pseudomonas diminuta) was constructed and screened for its ability to selectively hydrolyze a
p-nitrophenyl-containing ProTide: sofosbuvir precursor [
12,
15]. The mutant G60A-PTE exhibits a 165-fold preference for hydrolysis of the (
Rp)-diastereomer, while the mutant In1W-PTE exhibits a 1400-fold preference for hydrolysis of the (
Sp)-diastereomer. Moreover, their experimental investigation demonstrated the irreversibility of the reaction from (
Sp)- to (
Rp)- or (
Rp)- to (
Sp)-diastereomer [
12]. Based on this published data, the mutants In1W-PTE and G60A-PTE were selected for additional research in the diastereomeric separation of the remdesivir precursor. The mutant In1W-PTE demonstrated consistent specific hydrolysis of the (
Sp)-diastereomer, aligning with prior findings, whereas the mutant G60A-PTE did not show measurable enantioselective hydrolysis properties for the (
Rp)-diastereomer of remdesivir precursor [
13]. However, the selective hydrolysis of the (
Rp)-diastereomer was improved in our previous work, where site-directed mutagenesis was rationally designed and led to the successful preparation of the pure (
Sp)-diastereomer of remdesivir and sofosbuvir precursors within a remarkably short hydrolysis time [
14]. This suggests that the mutated PTE holds the potential for enhancing the production of diastereomerically pure nucleotide phosphoramidate prodrugs and can be an alternative method for enantioseparation.
Despite its effectiveness, the overall production cost remains a burden of this technology due to using free enzymes. Compared to free enzymes, immobilized enzymes are more robust and more resistant to environmental changes including temperature, pH, and organic solvents. In addition, the immobilized enzymes allow for the easy recovery of both enzymes and products, the multiple reuses of enzymes, the continuous operation of enzymatic processes, the rapid termination of reactions, and a greater variety of bioreactor designs [
16,
17]. There have been reports of PTE immobilization onto cotton fabric [
18], polyether sulphone (PES) and polyvinylidene fluoride (PVDF) membranes, polyhydroxyalkanoate (PHA) nano-granules [
19], chitosan beads containing glutaraldehyde [
20], Fuller’s earth [
21], polyamide nanofibrous membrane [
22], modified cellulose microfibers [
23], and hybrid copper nanoflowers [
24]. The immobilized PTEs exhibited remaining higher activity even though they were used in several consecutive reactions, a wider temperature and pH application range, and higher catalytic activity [
16,
18,
22,
24]. However, the choice of supporting material and immobilization technique is controversial, with advantages and disadvantages depending on the working conditions. For example, nanomaterials provide a large surface area for enzyme attachment and can disperse evenly in the reaction medium, promoting their catalytic performance. However, reusing such small particles involves high-energy, high-cost separation processes like centrifugation or nanofiltration. Thus, our research was interested in an alternative material, polymeric macroporous beads with a hierarchical structure. These beads are synthesized in sizes ranging from tens to hundreds of microns, with micro- and nanosized pores, which are ideal for handling while providing ample space for protein loading.
In this work, the mutated PTE (W131M-PTE), which demonstrated high potential for isolating the desired isomer of the ProTide precursor [
14], was selected to enhance enzyme recyclability and facilitate its recovery from chemical reactions. The immobilization of the W131M-PTE on polymeric macroporous beads, including commercial beads, modified commercial beads, and synthetic beads, was demonstrated. The commercial beads (Immobead 150P; IB) were modified to introduce amine functionality (IB-EDA) and coated with maltodextrin (IB-MTD). Additionally, synthetic polyacrylamide beads (PAM) were prepared and functionalized with two coupling agents, resulting in PAM-GA and PAM-EDC. All five types of beads were characterized and evaluated for their immobilization efficiency, with the most effective bead selected for isolating the pure diastereomer of the sofosbuvir precursor.
2. Results and Discussion
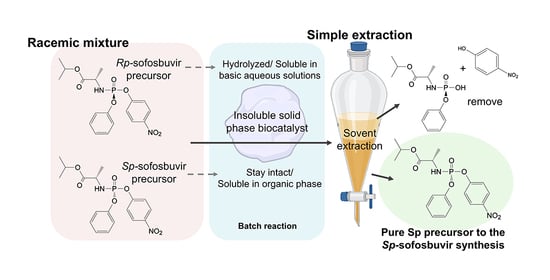
Chiral phosphate-based antiviral drugs, such as sofosbuvir, are initially synthesized as a racemic mixture of (
Sp and
Rp)-diastereomers (
Scheme 1). The (
Sp)-enantiomer is typically isolated using chiral preparative HPLC or recrystallization. As an alternative approach, engineered PTEs have been explored for enantioselective separation. The catalytic efficiency (
kcat/K
m) of W131M-PTE for the (
Rp)-sofosbuvir precursor was determined to be (2.97 ± 0.13) × 10
5 M
−1s
−1, which is 187-fold higher than its efficiency for the (
Sp)-sofosbuvir precursor (1.58 ± 0.10) × 10
3 M
−1s
−1. In comparison, the catalytic efficiency of G60A-PTE for the (
Rp)-sofosbuvir precursor was (3.91 ± 0.36) × 10
2 M
−1s
−1, only 13-fold higher than for the
Sp-sofosbuvir precursor (2.90 ± 0.05) × 10
1 M
−1s
−1 [
14]. These results strongly indicate that W131M-PTE exhibits significantly higher selectivity and efficiency in hydrolyzing the (
Rp)-diastereomer compared to G60A-PTE, making it a more suitable biocatalyst for enantioselective hydrolysis of sofosbuvir precursors. Its practical application in large-scale synthesis remains challenging due to enzyme instability and high production costs. Free enzymes are often susceptible to degradation and require frequent replenishment, which limits their reusability and economic feasibility. To overcome these limitations, enzyme immobilization offers a promising strategy by enhancing enzyme stability, allowing for multiple reuses and facilitating efficient separation of the biocatalyst from the reaction mixture. Therefore, this study explores the immobilization of W131M-PTE onto various polymeric macroporous beads to optimize its stability and reusability in diastereomer separation.
2.1. Bead Preparation for Covalent Binding to Enzyme
Macroporous beads were selected as supporting materials due to their favorable physical properties, including an easily separable size range and a large surface area per volume for enzyme attachment. The interaction between the bead and enzyme during immobilization is determined by surface chemistry. In this context, the commercial bead IB was expected to bind to PTE through covalent interactions via nucleophilic substitution at the epoxide ring (
Figure 1a). While covalent binding is the primary mechanism, adsorption may also occur, resulting in a combination of these interactions. The modified commercial bead, IB-EDA, was prepared by immersing the beads in ethylenediamine at an elevated temperature, a harsher condition compared to the immobilization process. As a result, the epoxide group would be consumed and converted to an amine functional group during this step. Consequently, IB-EDA would covalently bond with PTE with the aid of coupling agent GA (
Figure 1b). For IB-MTD, the bead’s surface was covered with maltodextrin, and the interaction is expected to rely on biorecognition between maltodextrin and the maltose-binding protein fused PTE (
Figure 1d). For synthetic beads, PAM bead coupling agents including GA and EDC were utilized to facilitate covalent bonding. The interactions are expected to involve the formation of imine bonds in the case of GA and amide bonds when using EDC (
Figure 1b,c).
2.2. Bead Characterization
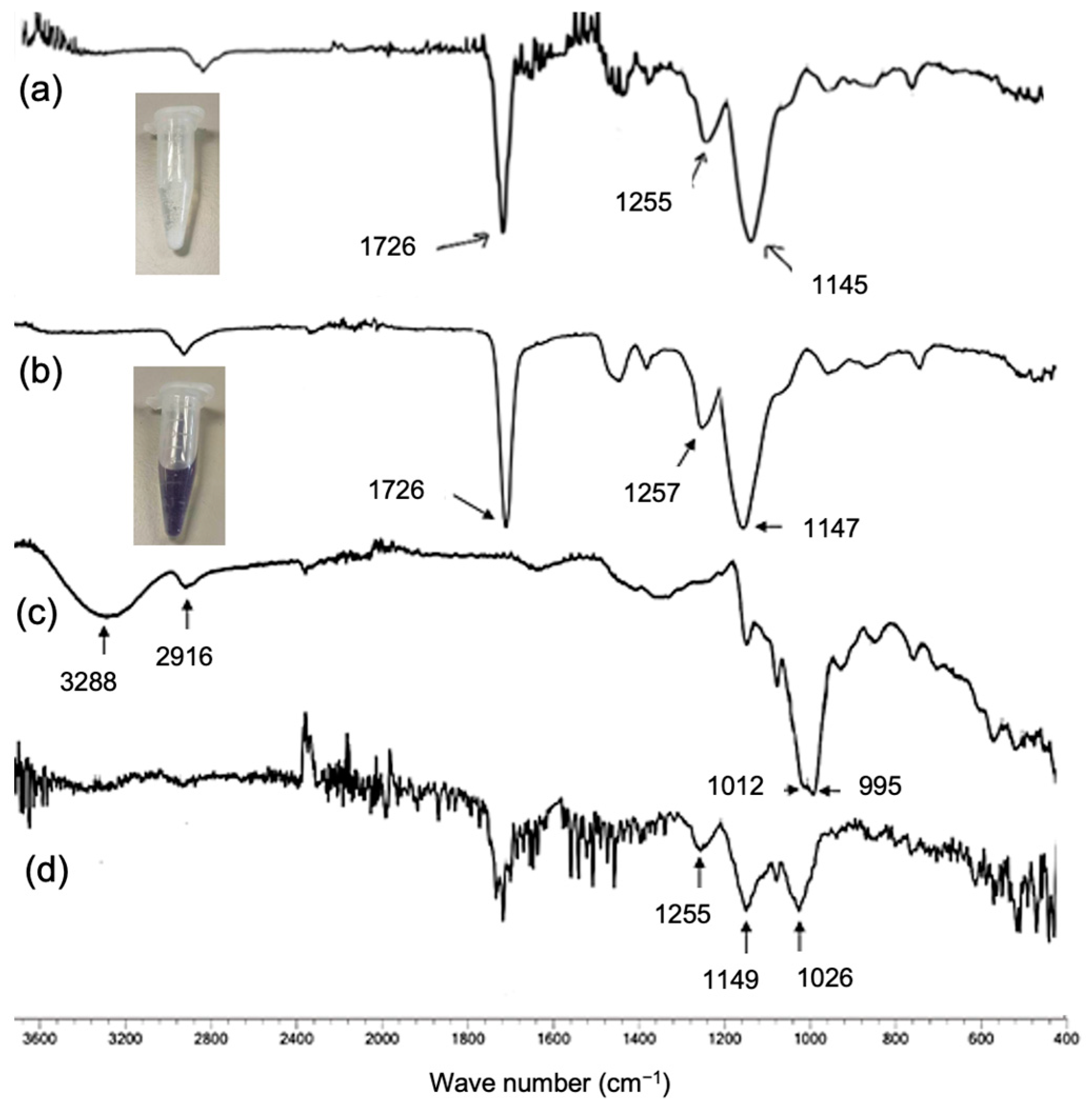
Prior to immobilization, the modified beads were characterized. They were subjected to ATR-FTIR to observe changes in functional groups. As shown in
Figure 2a, IB exhibited absorption at 1726 cm
−1, corresponding to the C=O stretching of the acrylate group. The epoxide characteristic of IB was observed at 1255 cm
−1. The spectrum of IB-EDA was similar to that of IB (
Figure 2b). This similarity is likely due to the bulk properties of the beads dominating the observed signals as the beads were ground prior to analysis with the ATR-FTIR probe. The beads were further examined. The IB-EDA beads were subjected to the ninhydrin test, which showed a vivid purple color, indicating the presence of amine functionality, while IB showed a negative result and remained white (
Figure 2a,b). In addition to the FTIR and ninhydrin tests, the immobilization properties, including protein loading and enzymatic activity of IB-EDA, were significantly altered, which will be discussed in more detail later. The oxidized maltodextrin showed absorption of O-H bending as a broad peak around 1012 cm
−1 and 995 cm
−1, indicating primary and secondary OH groups on maltodextrin (
Figure 2c). This characteristic was also found in the FTIR results for IB-MTD (wave number of 1026 cm
−1), confirming the success of the modification (
Figure 2d). The characteristics of PAM amination were referenced from previous work [
25].
In the SEM analysis of physical appearance, IB has a spherical shape with a rough and porous surface (
Figure 3a,b). After amination, IB-EDA showed a smoother surface, likely due to the attachment of amine pendant groups (
Figure 3c,d). This modification resulted in a decrease in surface area from 331.99 to 283.29 m
2/g. The SEM images of IB-MTD revealed full coverage of maltodextrin on the bead surface, leading to a complete transformation in its appearance (
Figure 3e,f). The surface area significantly decreased to 63.02 m
2/g. For a PAM bead, a surface area of 43.93 m
2/g was observed (
Figure 3g,h). The surface appeared rough, and the shape was irregular rather than perfectly circular, with a tail-like feature resulting from its droplet formation.
2.3. The Results of PTE Immobilization
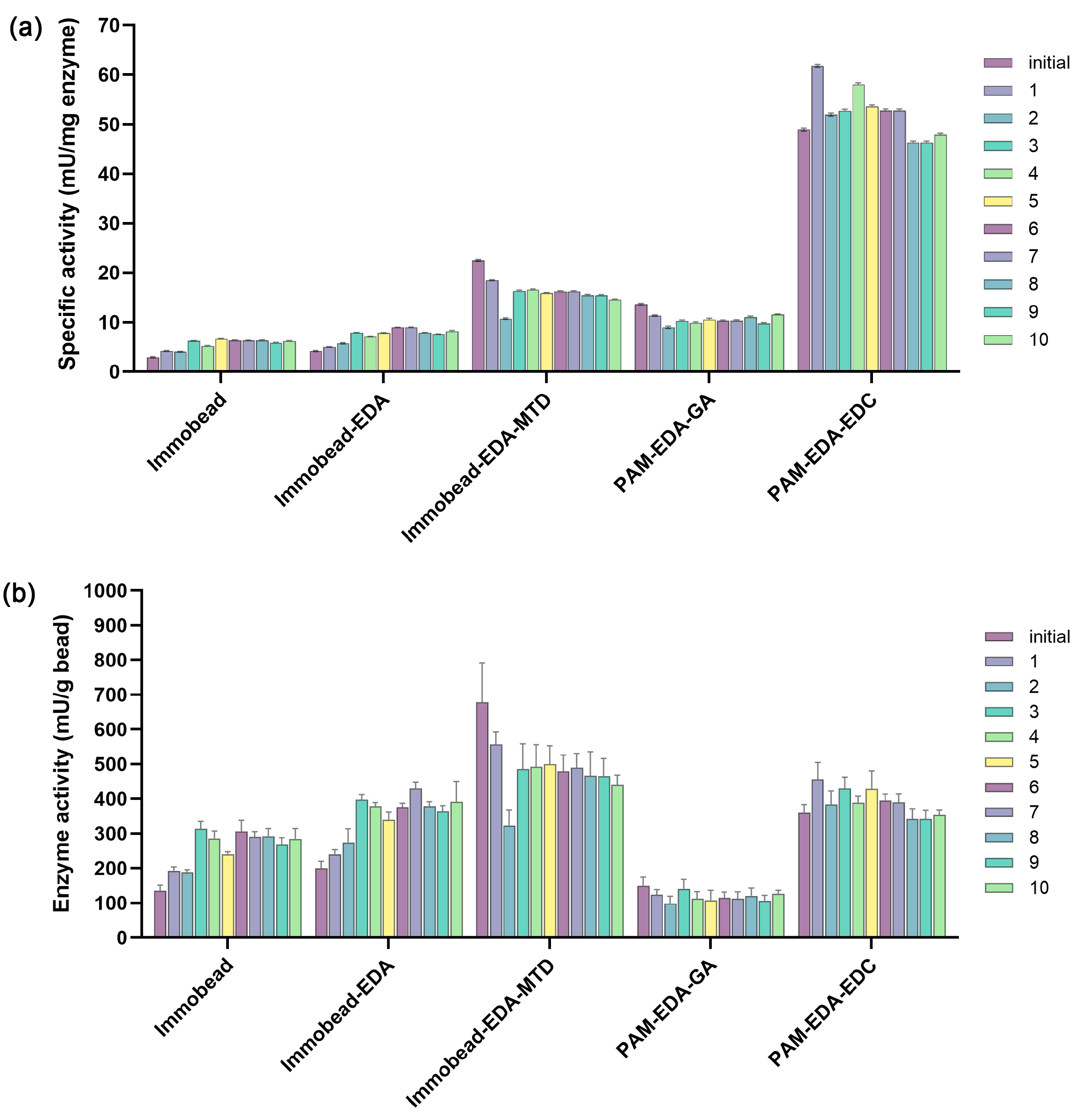
2.3.1. Protein Loading and Enzyme Activity
The protein was initially introduced at 1 mg per 20 mg of beads for all five bead types. However, the immobilization efficiency varied due to differences in surface area, pore structure, and surface chemistry, as detailed in
Table 1. In general, IB had the largest surface area and pore volume (0.37 cm
3/g), indicating micro- and mesoporous structures that allowed both covalent attachment via epoxide ring opening and adsorption within the pores [
26]. In contrast, IB-EDA exhibited a smaller surface area and a smoother surface after amination (
Figure 3d), suggesting reduced pore accessibility. For IB-MTD and PAM, no visible pores were observed on the SEM images (
Figure 3f,h) but they exhibited rougher surfaces. Despite IB having the largest surface area and highest theoretical capacity for enzyme attachment, IB-EDA showed unexpectedly high enzyme adsorption (47.8 ± 0.2 mg/g), even with a lower surface area and reduced pore accessibility than IB. This suggests that factors beyond surface area and porosity influence enzyme loading. As noted by Mateo C. [
27], amine-functionalized surfaces can enhance enzyme adsorption through electrostatic interactions and hydrogen bonding, significantly contributing to protein attachment. However, excessive enzyme loading does not necessarily translate to higher activity. Structural crowding and steric hindrance can restrict substrate accessibility, potentially reducing specific activity rather than enhancing it. For IB-MTD, protein loading decreased to 30.1 ± 1.7 mg/g bead, likely due to the reduction in the porous structure after coating with maltodextrin, a large polysaccharide. Despite this, the enzymatic activity of IB-MTD increased to 22.5 ± 0.2 mU/mg protein, likely due to specific binding interactions between maltodextrin and maltose-binding protein.
For PAM beads, both PAM-GA and PAM-EDC exhibited lower enzyme loadings than the IBs, consistent with their lower surface areas. However, they showed relatively high enzymatic activity, with PAM-EDC demonstrating the highest activity among the tested materials at 48.9 ± 0.3 mU/mg protein. The higher activity observed for PAM compared to IB is noteworthy and likely influenced by differences in bonding characteristics. PAM-GA, with its imine bonds, and PAM-EDC, with its amide bonds, both exhibited higher activities than IB, where covalent bonding was the primary interaction. Additionally, the hydrophilic nature of PAM may contribute to its enhanced activity by providing a better hydration environment which improves substrate accessibility and enzyme mobility.
However, the specific activity of the free W131M enzyme was notably higher than that of the immobilized form, with values of 2338 ± 492 mU/mg. This difference could be attributed to mass transfer limitations affecting enzyme–substrate interactions. In the immobilized state, substrate diffusion into the bead matrix may be restricted, limiting enzyme access. In contrast, the free enzyme remains fully available in solution, allowing unrestricted interaction with the substrate. However, the improvement of reusability was prioritized due to the practical and economic advantages of immobilized biocatalysts. Free enzymes, despite their higher activity, suffer from limited separation from the reaction. Whereas immobilization enhances enzyme recovery, enabling multiple reaction cycles with minimal loss of activity.
2.3.2. Reusability
A reusability test was conducted to evaluate the stability and operational durability of immobilized PTE on various solid supports during repeated incubation cycles in the reaction medium. The activity of the immobilized enzyme was measured after each incubation to assess changes over multiple rounds of use. The IB-MTD exhibited the highest value at 678.28 ± 113.6 mU/g bead, attributed to its combination of relatively high enzyme loading and specific activity. However, after incubation in the reaction medium, the activity dropped significantly. This decline suggests partial detachment of PTE during the initial incubation rounds. Notably, after the fourth round the activity stabilized (
Figure 4).
For IB and IB-EDA, an increase in activity was observed during the first three incubation rounds (
Figure 4). This phenomenon is partly explained by IB’s partial adsorption of the analytical product,
p-nitrophenol, on its surface, which led to false-negative results from enzyme activity assays (
Supplementary Table S1). After the fourth incubation, the activities of IB and IB-EDA plateaued.
In contrast, the synthetic PAM bead demonstrated relatively stable activity throughout ten incubation rounds (
Figure 4), retaining a relative activity as high as 97% by the tenth round. Compared to the literature, the residual enzymatic activity of organophosphorous hydrolase (OPH) immobilized on a cellulose-based system was approximately 59% after ten cycles of batch operation [
23]. Similarly, OPH immobilized on chitosan beads exhibited less than 40% residual activity after five usage cycles [
20]. In another study, Cu-PTE hybrid nanoflowers maintained 72.3% relative activity after ten consecutive reactions [
24]. This high retention suggests strong enzyme–support interactions, minimizing enzyme leaching. Previous studies have reported that enzyme leaching is a significant challenge in non-covalent immobilization systems, leading to a gradual loss of activity over repeated cycles [
20,
23,
24,
25]. The superior performance of PAM-EDC in this study likely results from the covalent amide bond formation between the enzyme and support, ensuring stronger attachment and reducing enzyme desorption. Furthermore, this stability is advantageous from a user perspective as it minimizes variability and facilitates process control during production. Therefore, the PAM-EDC with an activity of 361.2 ± 22.72 mU/g bead was selected for isolation of the (
Rp/
Sp)-sofosbuvir precursor.
2.4. The Utilization of Immobilized PTE in Diastereomer Isolation
To hydrolyze the (
Rp/
Sp)-sofosbuvir precursor, the substrate-to-bead ratio and substrate concentration were adjusted, as detailed in
Table 2.
The reaction was conducted by mixing hydrated PAM-EDC, a racemic mixture of the sofosbuvir precursor, CoCl
2, and MeOH, followed by incubation at 30 °C. The remaining amount of (
Rp)-diastereomer in the reaction was measured using HPLC and plotted (
Figure 5).
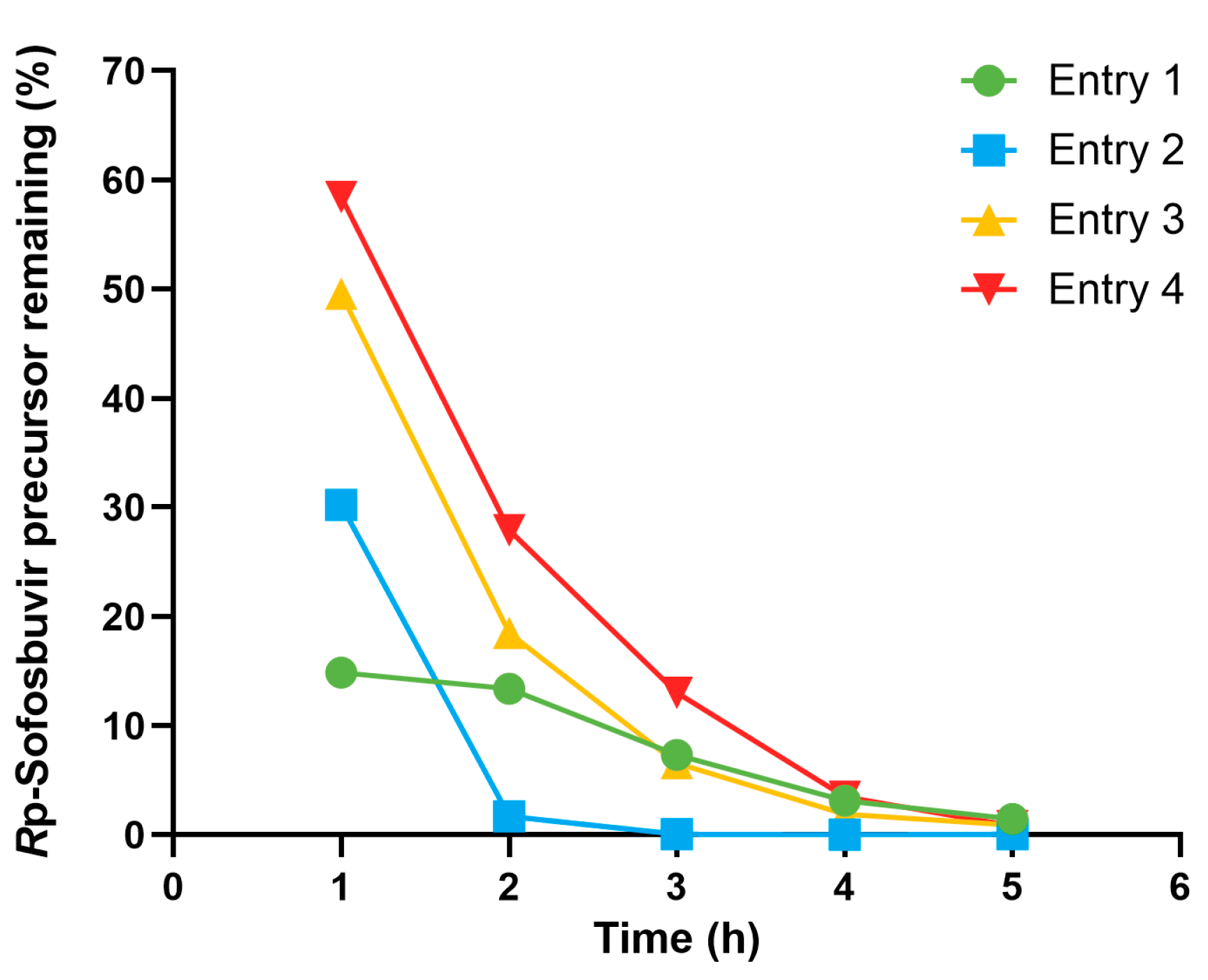
For entry 1, in which the substrate-to-bead ratio is 1:5, the (Rp)-diastereomer was not completely hydrolyzed within 5 h. The (Rp)-diastereomer remained in the reaction for 13.38% at the second hour and 1.45% at the fifth hour. The incomplete hydrolysis of the (Rp)-diastereomer was likely due to the low catalytic activity of the added PTE. Therefore, in entry 2, the number of beads was doubled. As a result, the (Rp)-diastereomer was reduced to 1.68% by 2 h and completely hydrolyzed by 4 h. In addition, the reaction volume was found to have a significant effect on the hydrolysis reaction. In entries 3 and 4, the reaction volume was increased to 2.5 and 5 mL/mg substrate, respectively. A significant decrease in the hydrolysis rate was observed, emphasizing that the reaction prefers concentrated substrate.
Entry 2 was selected as the optimal condition for the yield of (Sp)-diastereomer isolation. The obtained mixture was filtered to remove the insoluble beads from the reaction. Additionally, the beads were washed with MeOH to recover any precipitated Sp-precursor remaining on the beads. The combined filtrates were evaporated to afford a crude product containing both the hydrolyzed (Rp)-diastereomer and the unhydrolyzed (Sp)-diastereomer. The isolation of the crude product was performed using a simple solvent extraction based on solubility differences. The intact (Sp)-diastereomer was extracted with DCM, while the hydrolyzed (Rp)-diastereomer was removed by washing with saturated NaHCO3 aqueous solution. Finally, the DCM extract was evaporated, and the weight was measured to determine the (Sp)-diastereomer isolation yield, which was 92%.
Compared with the use of free enzymes, the actual weight of W131-PTE was calculated. The protein adsorption on 1 g of dry beads was 8.74 ± 1.73 mg. However, during use, the beads were in a hydrated state to maintain enzymatic activity, resulting in additional weight due to absorbed water. The beads were found to swell to five times their original weight, yielding a value of 1.748 mg protein per gram of hydrated beads. This corresponded to enzyme-to-substrate weight ratios of 1:114 and 1:57 for entries 1 and 2, respectively. For the free enzymes, reactions were carried out using enzyme-to-substrate weight ratios of 1:20, 1:40, 1:100, and 1:200. The (
Rp)-sofosbuvir precursor was hydrolyzed to 1%, 2%, 4%, and 17% at the fourth hour, respectively, at the aforementioned enzyme/substrate ratios. These results suggest that using the immobilized enzyme would use an enzyme dosage less than the free enzyme required to achieve complete hydrolysis of the (
Rp)-sofosbuvir precursor within 4 h (
Table 3). This suggests that the immobilized system enables efficient substrate conversion under optimized conditions. Furthermore, in the reusability test, the immobilized enzyme retained full catalytic activity over 10 reaction cycles (
Figure 4). This stability, combined with sustained activity, demonstrates that the immobilized enzyme can efficiently hydrolyze the substrate over multiple rounds without loss of performance. These results highlight that enzyme immobilization on beads offers significant advantages in terms of hydrolysis efficiency, enzyme stability, and cost-effectiveness, making it an attractive option for large-scale enzymatic hydrolysis applications such as the production of chiral precursors.
3. Materials and Methods
3.1. Plasmid Construction
The phosphotriesterase gene carrying a mutation (W131M-PTE) of
Brevundimonas diminuta was obtained from the previous study [
14]. The target gene was synthesized by GenScript with His-tag at C-terminal and inserted at
NdeI and
HindIII site into pMAL.c5x vector. The plasmid was transformed into
E. coli BL21(DE3) for protein expression. The single colony obtained from electroporation of the mutagenic plasmid was picked, the colony PCR was checked, and it was inoculated in Luria broth (LB) supplement with an antibiotic at 37 °C overnight with a shaking condition. The culture sample was kept in 30% (
v/
v) glycerol at −20 °C to be the seed culture for enzyme production.
3.2. Protein Expression and Purification
The W131M-PTE in E. coli BL21(DE3) was produced in shake flasks with 1 L LB medium supplemented with 50 µg/mL ampicillin. Bacterial cells were grown at 37 °C, 200 rpm until OD600 reached 0.6 and then induced with isopropyl thio-ß-D-galactoside (IPTG) at a final concentration of 0.2 mM. After adding IPTG, cells were cultured at 16 °C for 16–20 h and cells were harvested by centrifugation (10 min, 8500 rpm, 4 °C). The harvested cells were resuspended in 50 mM Tris-HCl buffer pH 8.0 before sonication. The sample was centrifuged at 4 °C, 17,000 rpm for 15 min for pellet separation. The crude enzyme extract in the supernatant was loaded onto a HisPrep FF 16/10 column (Cytiva, Marlborough, MA, USA), equilibrated with the equilibration buffer (20 mM Tris-HCl buffer pH 8.0 containing 20 mM imidazole and 0.1 M NaCl). The column was washed with 5-times volumes of the same buffer at a flow rate of 5 mL/min and the bound protein was eluted with 0.2 M imidazole in the equilibration buffer. The eluted protein fractions had their purity confirmed by Coomassie-stained sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The target protein fractions were pooled, dialyzed in 50 mM sodium phosphate buffer pH 8.0, and concentrated by ultrafiltration (Amicon Ultra-15 centrifugal filter unit, Darmstadt, Germany). The concentrated protein was kept at 4 °C containing 0.5 mM CoCl2 for long-term stability.
3.3. Bead Preparation
3.3.1. Commercial Beads Modification
Amination using ethylene diamine (EDA) was conducted to convert epoxide to amine. Briefly, Immobead 150P (commercial bead from Sigma-Aldrich, Burlington, MA, USA), which was named in this study as IB, was rehydrated using deionized water. After the bead was fully swollen, the excess water was removed. Then, it was immersed in EDA and heated to 70 °C for 24 h. The obtained bead was cooled to room temperature and washed thrice with deionized water, followed by 95% ethanol. The beads were dried at 70 °C overnight in an oven and named IB-EDA. This bead was activated using glutaraldehyde (GA) before the immobilization. Twenty mg of IB-EDA was placed in a 1.5 mL microcentrifuge tube, followed by the addition of 1 mL of a 0.5% (v/v) GA solution. The sample was mixed in a rotating shaker (10 rpm) for 30 min at room temperature. The bead was washed with 1 mL of distilled water 3 times to remove the unreacted GA. The water was removed. The activated IB-EDA was utilized as a wet-form material support for enzyme immobilization.
IB-EDA was coated with maltodextrin, which can link to the maltose-binding protein fused W131M-PTE. The procedure was adapted from a previous study [
21]. A stock solution of maltodextrin was prepared by dissolving 19 g of maltodextrin in 133 mL of 100 mM phosphate buffer pH 7.4. The prepared bead IB-EDA (20 mg) was added to 10 mL of the maltodextrin stock solution. Then, 275 µL of 0.467 M NaIO
4 was added to the mixture to oxidize the maltodextrin, generating aldehyde functional groups that subsequently covalently linked to the amine groups on IB-EDA. The reaction was carried out at room temperature with agitation at 150 rpm for 4 h. The beads were rinsed three times with deionized water, followed by a rinse with 95% (
v/
v) ethanol, and then dried overnight at 70 °C in an oven. The resulting beads were named IB-MTD and were used as the material support for enzyme immobilization.
3.3.2. Polyacrylamide (PAM) Beads Preparation
PAM beads were prepared as described in our previous work [
25]. Briefly, 10 mL of an aqueous solution containing 10% (
w/
v) acrylamide monomer, 4% (
w/
v) N’N methylenebisacrylamide, 1% (
w/
v) sodium alginate, and 0.4%
w/v ammonium was loaded in a syringe with a blunt 25 G needle. The 8 kV positive voltage was applied to the needle tip. The solution in the syringe was dropped into a collecting bath containing 3% (
w/
v) calcium chloride dihydrate and 0.4% (
v/
v) tetramethylenediamine (TEMED) with the flow rate fixed at 20 mL/h. The beads were removed by being poured through filter paper (Whatman No. 1, Cytiva, Marlborough, MA USA), rinsed 3 times with deionized water, and washed with 95% (
v/
v) ethanol before being dried in an oven.
The amination process introduced an amine functionality onto the PAM beads using ethylene diamine. The PAM beads were rehydrated using deionized water. After the beads were fully swollen, the excess water was removed. Then, the beads were immersed in ethylene diamine and heated to 70 °C for 3 days to convert amide groups to amine groups. The obtained beads were cooled to room temperature and washed thrice with deionized water, followed by 95% ethanol. The beads were dried at 70 °C overnight in an oven.
The generated amine groups on PAM would be coupled to the enzyme with the aid of crosslinking agents glutaraldehyde (GA) and 1-Ethyl-3-(3-dimethyl aminopropyl) carbodiimide (EDC). They would be named PAM-GA and PAM-EDC, respectively. In the case of PAM-GA, the GA activation was performed prior to the addition of the enzyme. Twenty mg of aminated PAM beads were placed in a 1.5 mL microcentrifuge tube, followed by the addition of 1 mL of a 0.5% (v/v) GA solution. The sample was mixed in a rotating shaker (10 rpm) for 30 min at room temperature. The beads were washed with 1 mL of distilled water 3 times to remove the unreacted GA. The water was removed from the beads, and the PAM-GA beads were ready for enzyme immobilization. In the case of PAM-EDC, the crosslinking agent EDC/NHS was mixed with the enzyme solution in one pot and described in PTE immobilization section.
3.4. PTE Immobilization
The beads IB, IB-EDA, and IB-MTD with a dry weight of 20 mg were placed in 1.5 mL microcentrifuge tubes. The beads were rehydrated in 0.5 mL of water for 5 min, then the water was removed. After that, 1 mL of 1 mg/mL W131M-PTE in 50 mM sodium phosphate buffer pH 8.0 in the presence of 0.5 mM CoCl2 was gently mixed with the beads in a 1.5 mL microcentrifuge tube at 10 rpm in a rotating shaker over 4 h at room temperature. After the immobilization process, any unbound enzyme was then washed out with 1 mL of distilled water 3 times. The immobilized enzyme was kept in a hydrated form without the excess water at 4 °C for further studies.
The beads PAM-GA 20 mg were mixed with 1 mL of 1 mg/mL W131M-PTE in 50 mM sodium phosphate buffer pH 8.0 in the presence of 0.5 mM CoCl2 in the same manner as IB, IB-EDA, and IB-MTD immobilization.
The beads PAM-EDC 20 mg, and the crosslinking agent EDC/NHS were mixed with the enzyme solution in one pot. In addition to the W131M-PTE and CoCl2 in the immobilization procedure, 12 mg of EDC and 18 mg of N-Hydroxysuccinimide (NHS) were added. The rest of the procedure was performed in the same manner as IB, IB-EDA, and IB-MTD immobilization.
3.5. Protein Loading Evaluation
In the step of enzyme immobilization, the remaining protein content in the supernatant was measured using Bradford’s assay. Protein loading refers to the amount of protein initially introduced or immobilized onto a support material. Protein loading was calculated from the equation below, which is as follows:
3.6. Enzymatic Activity Evaluation
The activity of immobilized W131M-PTE was measured using racemic sofosbuvir precursor as the substrate. The reaction mixture (1 mL) contained an appropriate amount of immobilized PTE (approximately 3 mg equivalent to dry bead), 50 mM CHES buffer (pH 9.0), 0.5 mM CoCl2, and 0.12 mM substrate. The mixture was incubated at 30 °C for 10 min. Enzyme activity was determined using a colorimetric assay method by monitoring the release of free p-nitrophenol at an absorbance of 400 nm. One unit of hydrolysis activity is defined as one micromole of p-nitrophenol per minute. The activity of immobilized PTE was reported in milliunits per gram of dry bead (mU/g). It is noted that the immobilized PTE beads were in wet form. The dry weight of the immobilized PTE was measured separately by oven-drying the beads. The weight ratio of the hydrated beads to dry beads was used for the activity evaluation.
3.7. Reusability Evaluation
In the reusability test, the initial activity of immobilized PTE was measured. Then, the immobilized PTE was incubated to the operating conditions (50 mM CHES pH 9.0, 0.5 mM CoCl2, and 10% (v/v) methanol) at 30 °C for 2 h. After that, the solution was removed, and the activity of immobilized PTE was remeasured. This process was repeated ten times, and the experiment was performed in triplicate.
3.8. Enzymatic Resolution of Sofosbuvir Precursor
The diastereomers of the sofosbuvir precursor were synthesized following the previous work [
14]. The reaction mixture (25 mL) containing 25 mg of (
Rp/
Sp)-sofosbuvir precursor, 50 mM CHES pH 9.0, 125–250 mg of hydrated PAM-EDC, 6.25 nmol CoCl
2, and 10% MeOH was gently stirred at 30 °C. To monitor the reaction progress, 100 µL of the sample was taken and diluted with 400 µL of methanol at interval times. The (
Rp/
Sp)-diastereomers were evaluated by a Dionex™ UltiMate™ 3000 UHPLC (Thermo Fisher Scientific Inc., Waltham, MA USA) using a CHIRALPAK IG-U (100 mm × 3 mm i.d., 1.6 um) (Daicel Chiral Technologies Pvt Ltd., Telangana, India). The column temperature, the sample injected volume, and the mobile phase’s flow rate were 40 °C, 50 µL, and 0.4 mL/min, respectively. Mobile phase A was 95% (
v/
v) methanol, and mobile phase B was 100% acetonitrile. The elution was initially performed with 100% of mobile phase A for 3 min, then was then programmed to use gradient solvent systems with 0 to 35% of mobile phase B for 3–10 min. The HPLC peaks were detected at the wavelength of 270 nm. The (
Rp/
Sp)-sofosbuvir precursor was calculated from the peak area compared to the standard calibration curve extrapolated from the known concentration of racemic sofosbuvir precursor. The retention time of (
Rp)- and (
Sp)-diastereomer were 4.44 and 5.62 min, respectively. To evaluate the isolated yield, the obtained product was collected by evaporation to remove methanol, extracted with DCM, washed by saturated NaHCO
3 5–8 times, and evaporated again. The isolation yield was calculated from an equation below:
4. Conclusions
This study demonstrated the immobilization of W131M-PTE onto various polymeric macroporous beads, with modifications significantly influencing enzyme loading, stability, and catalytic performance. Among the tested material supports, commercial beads (IB) and amine-modified IB (IB-EDA) exhibited the highest number of enzymes immobilized on beads. However, their specific activity was lower due to excessive enzyme crowding, which led to structural overlapping and restricted substrate accessibility. This limitation ultimately reduced catalytic efficiency. In contrast, IB-MTD exhibited the highest activity per gram of bead due to specific interactions between the maltose-binding protein (MBP) fusion enzyme and the maltodextrin-coated surface. These interactions may have promoted optimal enzyme orientation, enhancing catalytic activity. However, the enzyme’s stability on IB-MTD was limited, as a significant drop in activity was observed after the second reuse cycle. Among all immobilization supports, the synthetic bead PAM-EDC proved to be the most effective. It offered high enzymatic activity, strong immobilization stability, and excellent reusability over multiple reaction cycles. Additionally, the application of immobilized W131M-PTE for the enantioselective hydrolysis of sofosbuvir precursors in pharmaceutical processes required fewer enzymes than the free enzyme system.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}