


Assessing the CO2 Capture and Electro-Reduction in Imidazolium-Based Ionic Liquids: Role of the Ion Exchange Membrane



Abstract

1. Introduction
2. Results
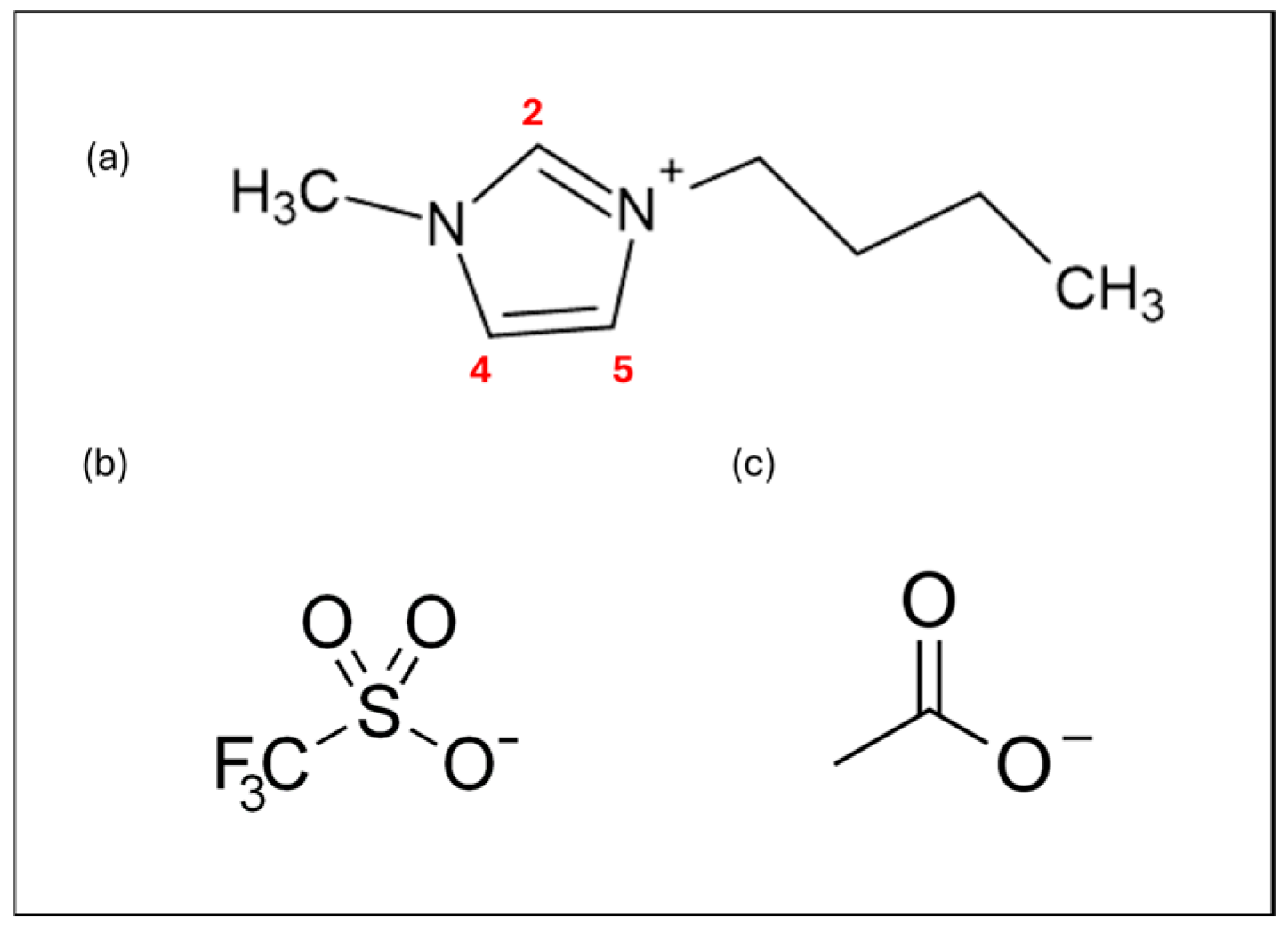
2.1. Electrolyte Preparation
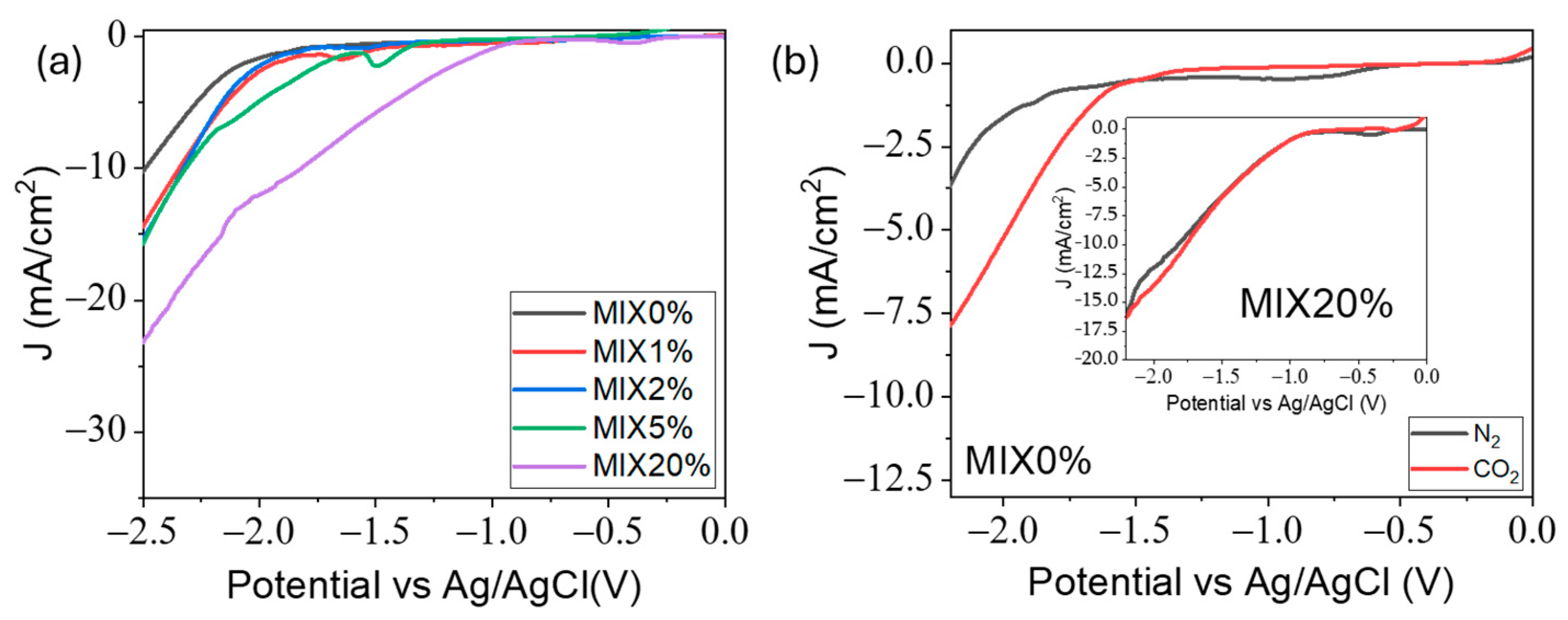
2.2. RTIL-Based Electrolyte Electrochemical Characterization
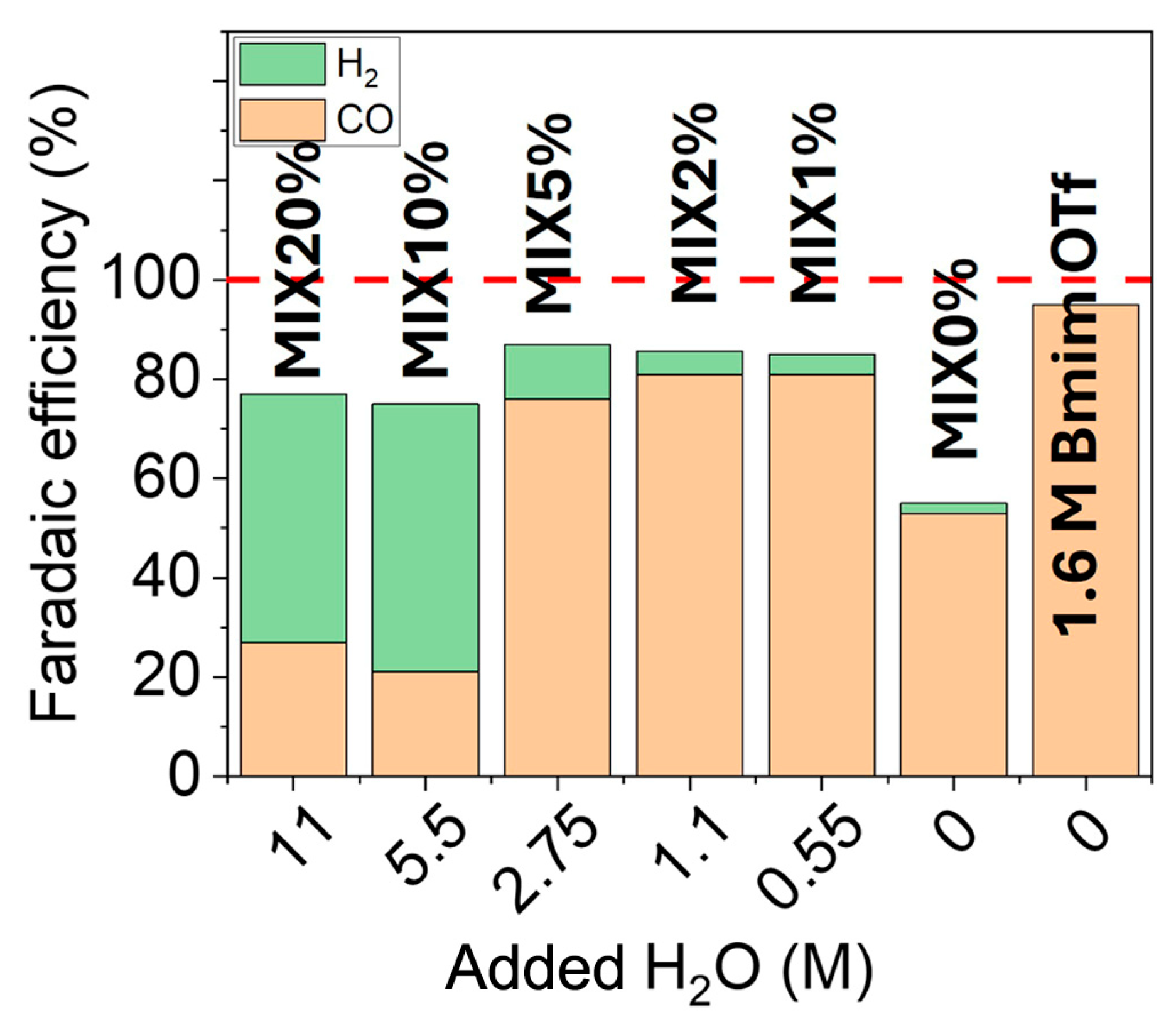
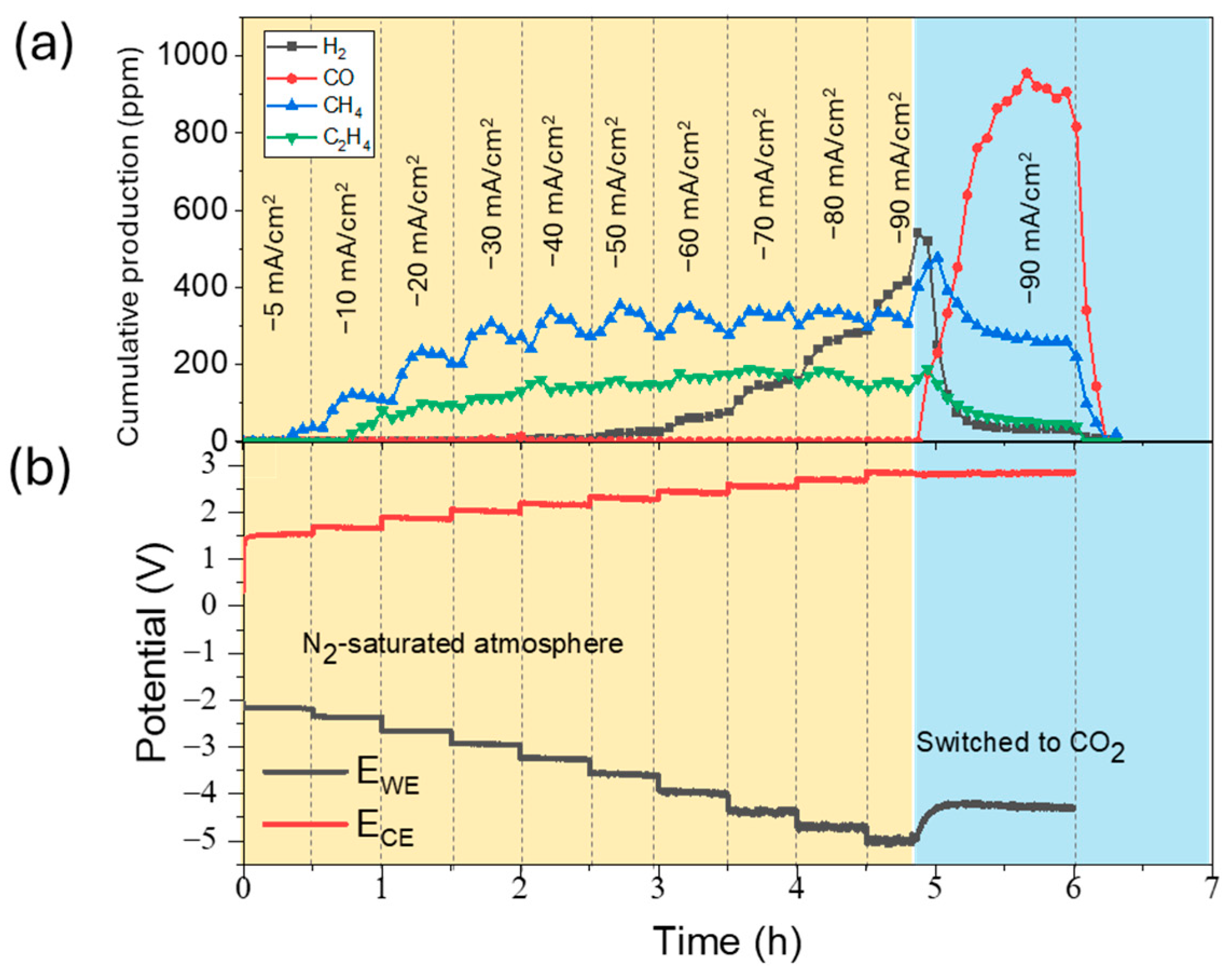
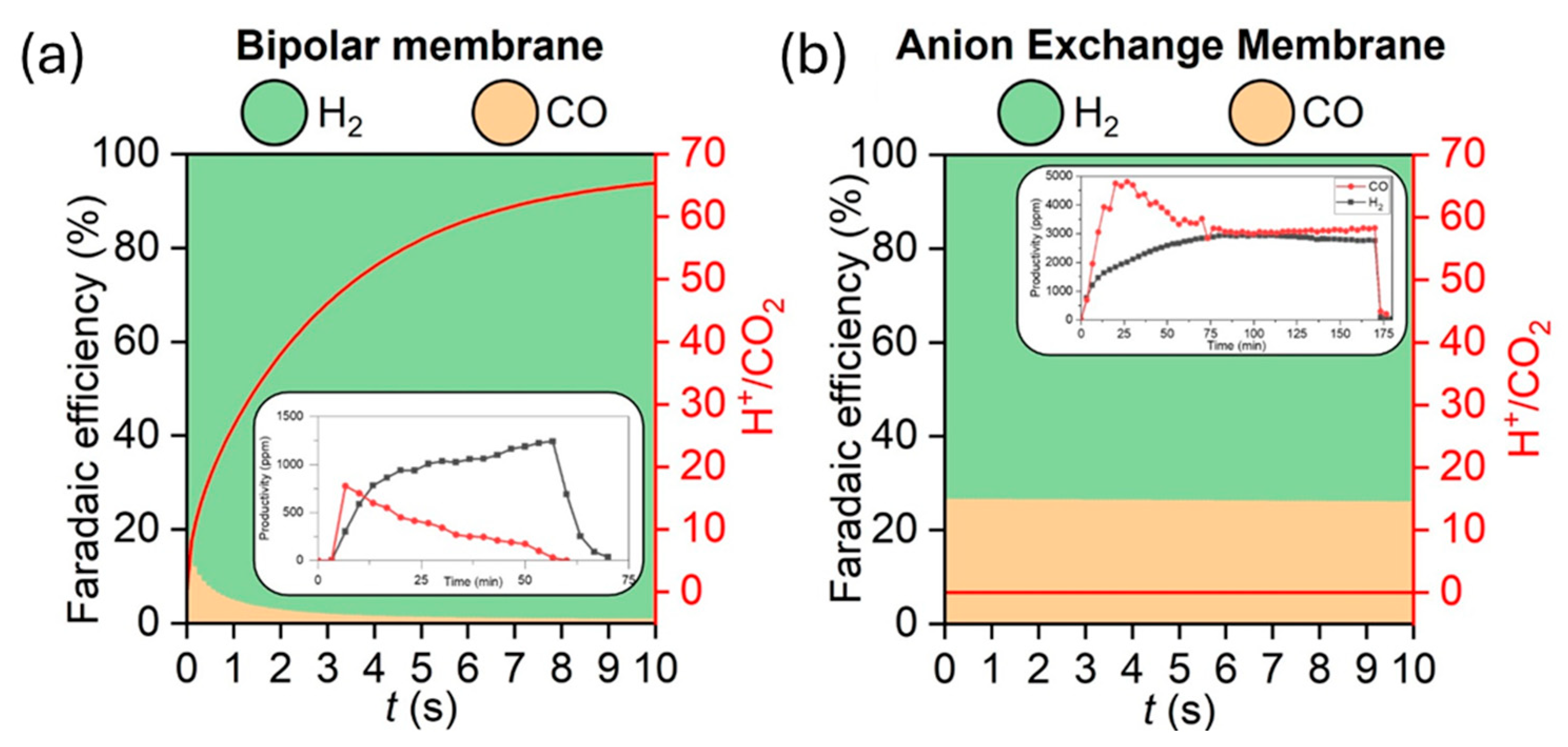
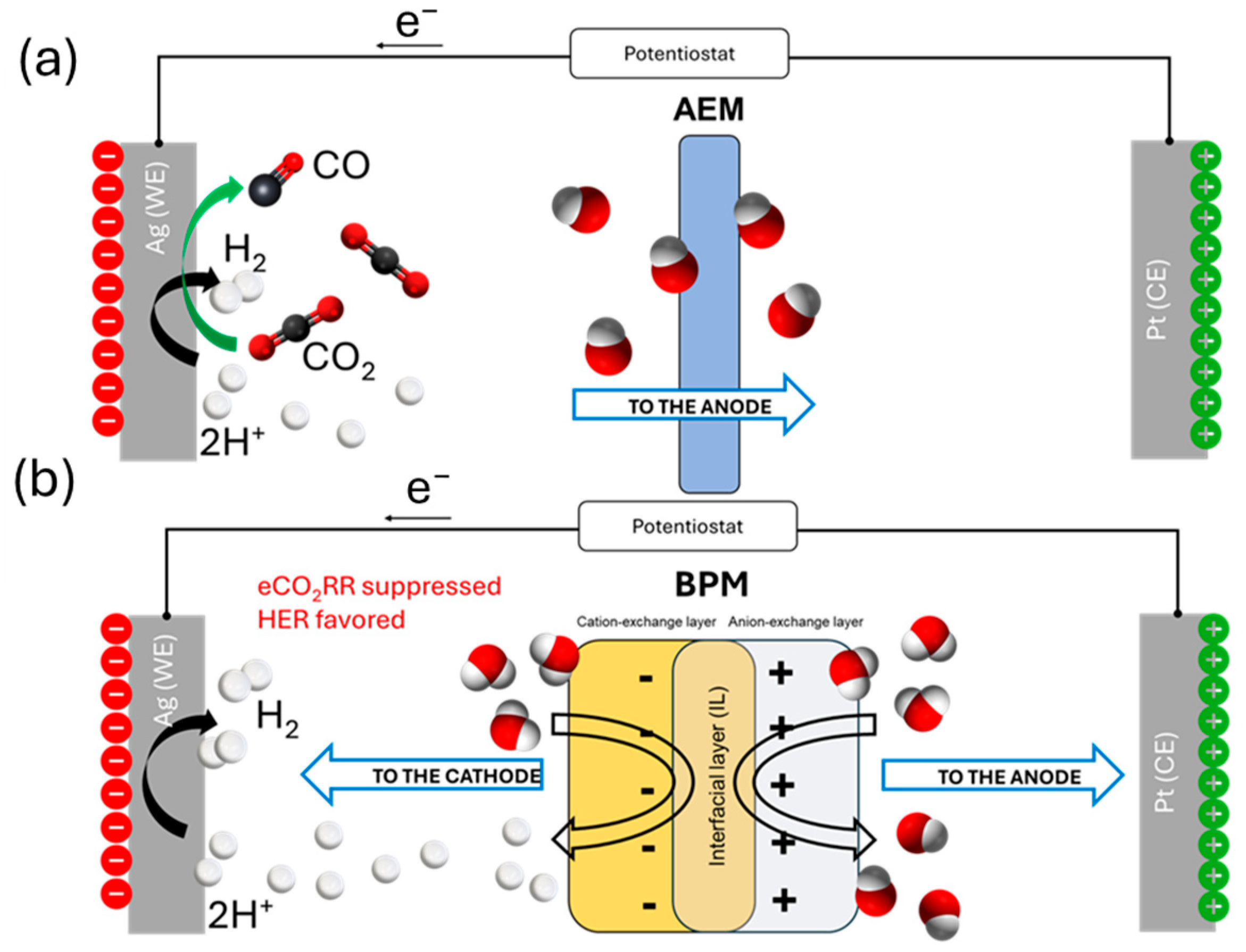
2.3. CO2 Electrolysis Tests
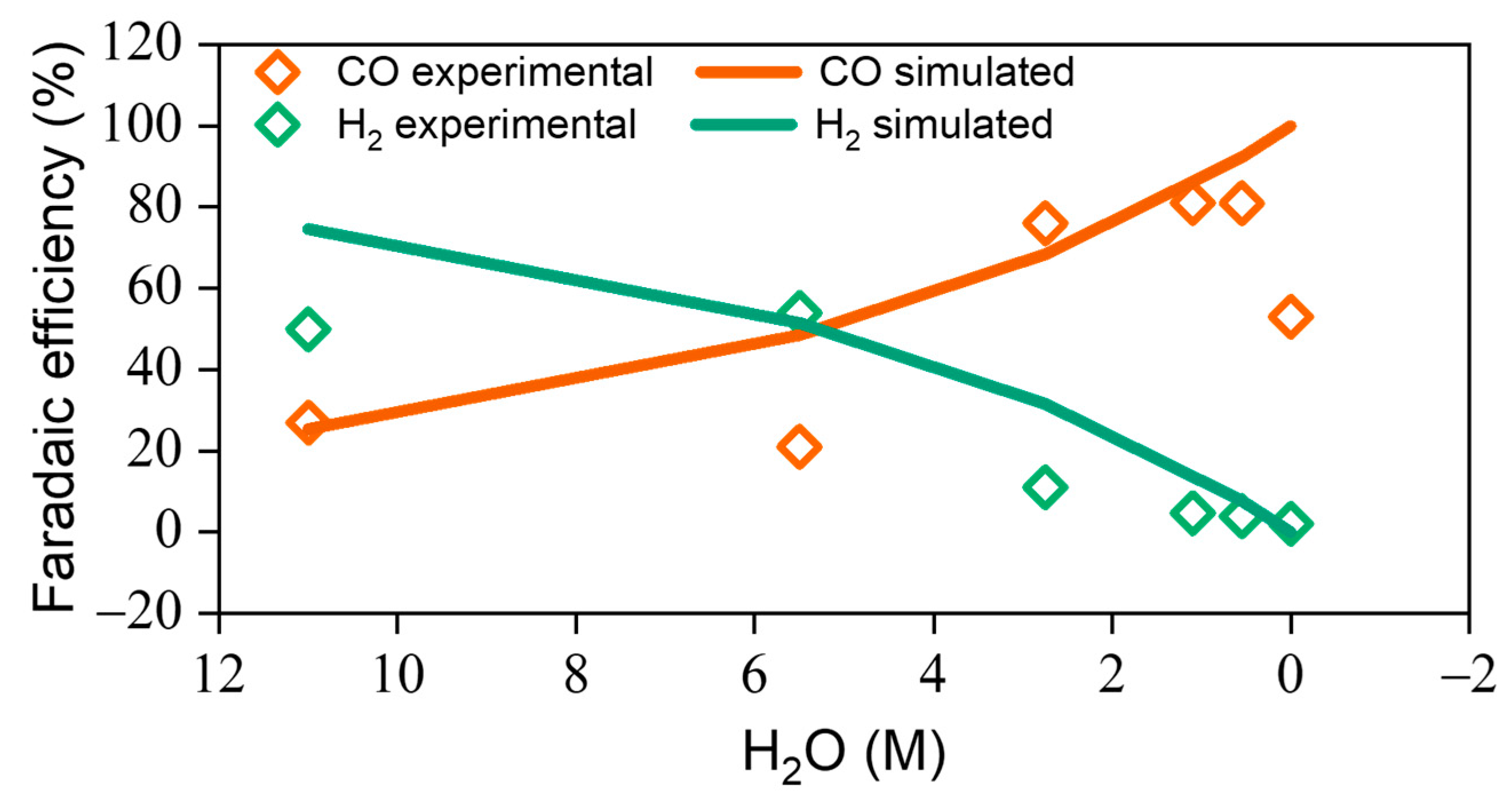
2.4. Computational Modeling
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Characterizations
4.3. Electrochemical Tests
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Monteiro, M.C.O.; Philips, M.F.; Schouten, K.J.P.; Koper, M.T.M. Efficiency and selectivity of CO2 reduction to CO on gold gas diffusion electrodes in acidic media. Nat. Commun. 2021, 12, 4943. [Google Scholar] [CrossRef] [PubMed]
- Marcandalli, G.; Goyal, A.; Koper, M.T.M. Electrolyte effects on the faradaic efficiency of CO2 reduction to co on a gold electrode. ACS Catal. 2021, 11, 4936–4945. [Google Scholar] [CrossRef] [PubMed]
- Cuatto, G.; Zoli, M.; Gallone, M.; Guzmán, H.; Castellino, M.; Hernández, S. Standardization of Cu2O nanocubes synthesis: Role of precipitation process parameters on physico-chemical and photo-electrocatalytic properties. Chem. Eng. Res. Des. 2023, 199, 384–398. [Google Scholar] [CrossRef]
- Rotundo, L.; Garino, C.; Priola, E.; Sassone, D.; Rao, H.; Ma, B.; Robert, M.; Fiedler, J.; Gobetto, R.; Nervi, C. Electrochemical and Photochemical Reduction of CO2 Catalyzed by Re(I) Complexes Carrying Local Proton Sources. Organometallics 2019, 38, 1351–1360. [Google Scholar] [CrossRef]
- Franco, F.; Cometto, C.; Nencini, L.; Barolo, C.; Sordello, F.; Minero, C.; Fiedler, J.; Robert, M.; Gobetto, R.; Nervi, C. Local Proton Source in Electrocatalytic CO2 Reduction with [Mn(bpy–R)(CO)3Br] Complexes. Chem. A Eur. J. 2017, 23, 4782–4793. [Google Scholar] [CrossRef]
- Franco, F.; Cometto, C.; Vallana, F.F.; Sordello, F.; Priola, E.; Minero, C.; Nervi, C.; Gobetto, R. A local proton source in a [Mn(bpy-R)(CO)3Br]-type redox catalyst enables CO2 reduction even in the absence of Brønsted acids. Chem. Commun. 2014, 50, 14670–14673. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Z.; Han, P.; Du, Y.; Gu, Z.; Xu, X.; Zheng, G. Single-Atomic Cu with Multiple Oxygen Vacancies on Ceria for Electrocatalytic CO2 Reduction to CH4. ACS Catal. 2018, 8, 7113–7119. [Google Scholar] [CrossRef]
- Shen, J.; Kortlever, R.; Kas, R.; Birdja, Y.Y.; Diaz-Morales, O.; Kwon, Y.; Ledezma-Yanez, I.; Schouten, K.J.P.; Mul, G.; Koper, M.T.M. Electrocatalytic reduction of carbon dioxide to carbon monoxide and methane at an immobilized cobalt protoporphyrin. Nat. Commun. 2015, 6, 8177. [Google Scholar] [CrossRef]
- Weng, Z.; Jiang, J.; Wu, Y.; Wu, Z.; Guo, X.; Materna, K.L.; Liu, W.; Batista, V.S.; Brudvig, G.W.; Wang, H. Electrochemical CO2 Reduction to Hydrocarbons on a Heterogeneous Molecular Cu Catalyst in Aqueous Solution. J. Am. Chem. Soc. 2016, 138, 8076–8079. [Google Scholar] [CrossRef]
- Li, F.; Thevenon, A.; Rosas-Hernández, A.; Wang, Z.; Li, Y.; Gabardo, C.M.; Ozden, A.; Dinh, C.T.; Li, J.; Wang, Y.; et al. Molecular tuning of CO2-to-ethylene conversion. Nature 2020, 577, 509–513. [Google Scholar] [CrossRef]
- Hori, Y.; Murata, A.; Takahashi, R.; Suzuki, S. Enhanced formation of ethylene and alcohols at ambient temperature and pressure in electrochemical reduction of carbon dioxide at a copper electrode. ChemInform 1988, 19, 17–19. [Google Scholar] [CrossRef]
- Dinh, C.-T.; Burdyny, T.; Kibria, M.G.; Seifitokaldani, A.; Gabardo, C.M.; de Arquer, F.P.G.; Kiani, A.; Edwards, J.P.; De Luna, P.; Bushuyev, O.S.; et al. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 2018, 360, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Zhan, C.; Dattila, F.; Rettenmaier, C.; Bergmann, A.; Kühl, S.; García-Muelas, R.; López, N.; Cuenya, B.R. Revealing the CO coverage-driven C–C coupling mechanism for electrochemical CO2 reduction on Cu2O nanocubes via Operando Raman spectroscopy. ACS Catal. 2021, 11, 7694–7701. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wu, Y.; Li, G.; Lin, Q.; Hu, Q.; Zhang, Q.; Liu, J.; He, C. Scalable Production of Efficient Single-Atom Copper Decorated Carbon Membranes for CO2 Electroreduction to Methanol. J. Am. Chem. Soc. 2019, 141, 12717–12723. [Google Scholar] [CrossRef]
- Lu, L.; Sun, X.; Ma, J.; Yang, D.; Wu, H.; Zhang, B.; Zhang, J.; Han, B. Highly Efficient Electroreduction of CO2 to Methanol on Palladium–Copper Bimetallic Aerogels. Angew. Chem. Int. Ed. Engl. 2018, 57, 14149–14153. [Google Scholar] [CrossRef]
- Albo, J.; Sáez, A.; Solla-Gullón, J.; Montiel, V.; Irabien, A. Production of methanol from CO2 electroreduction at Cu2O and Cu2O/ZnO-based electrodes in aqueous solution. Appl. Catal. B 2015, 176–177, 709–717. [Google Scholar] [CrossRef]
- Bohra, D.; Chaudhry, J.H.; Burdyny, T.; Pidko, E.A.; Smith, W.A. Modeling the electrical double layer to understand the reaction environment in a CO2 electrocatalytic system. Energy Environ. Sci. 2019, 12, 3380–3389. [Google Scholar] [CrossRef]
- Siritanaratkul, B.; Eagle, C.; Cowan, A.J. Manganese Carbonyl Complexes as Selective Electrocatalysts for CO2 Reduction in Water and Organic Solvents. Acc. Chem. Res. 2022, 55, 955–965. [Google Scholar] [CrossRef]
- Cadena, C.; Anthony, J.L.; Shah, J.K.; Morrow, T.I.; Brennecke, J.F.; Maginn, E.J. Why Is CO2 So Soluble in Imidazolium-Based Ionic Liquids? J. Am. Chem. Soc. 2004, 126, 5300–5308. [Google Scholar] [CrossRef]
- Feng, J.; Zeng, S.; Feng, J.; Dong, H.; Zhang, X. CO2 Electroreduction in Ionic Liquids: A Review. Chin. J. Chem. 2018, 36, 961–970. [Google Scholar] [CrossRef]
- Sun, L.; Ramesha, G.K.; Kamat, P.V.; Brennecke, J.F. Switching the reaction course of electrochemical CO2 reduction with ionic liquids. Langmuir 2014, 30, 6302–6308. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, F.; Laaksonen, A.; Wang, C.; Cobden, P.; Boden, P.; Liu, Y.; Zhang, X.; Ji, X. Electrochemical CO2reduction with ionic liquids: Review and evaluation. Ind. Chem. Mater. 2023, 1, 410–430. [Google Scholar] [CrossRef]
- Parada, W.A.; Vasilyev, D.V.; Mayrhofer, K.J.J.; Katsounaros, I. CO2 Electroreduction on Silver Foams Modified by Ionic Liquids with Different Cation Side Chain Length. ACS Appl. Mater. Interfaces 2022, 14, 14193–14201. [Google Scholar] [CrossRef] [PubMed]
- Coskun, O.K.; Dongare, S.; Doherty, B.; Klemm, A.; Tuckerman, M.; Gurkan, B. Tailoring Electrochemical CO2 Reduction on Copper by Reactive Ionic Liquid and Native Hydrogen Bond Donors. Angew. Chem. Int. Ed. Engl. 2023, 63, e202312163. [Google Scholar] [CrossRef]
- Brennecke, J.F.; Gurkan, B.E. Ionic liquids for CO2 capture and emission reduction. J. Phys. Chem. Lett. 2010, 1, 3459–3464. [Google Scholar] [CrossRef]
- Anderson, J.L.; Dixon, J.K.; Brennecke, J.F. Solubility of CO2, CH4, C2H6, C2H4, O2, and N2 in 1-hexyl-3-methylpyridinium bis(trifluoromethylsulfonyl)imide: Comparison to other ionic liquids. Acc. Chem. Res. 2007, 40, 1208–1216. [Google Scholar] [CrossRef]
- Papasizza, M.; Yang, X.; Cheng, J.; Cuesta, A. Electrocatalytic reduction of CO2 in neat and water-containing imidazolium-based ionic liquids. Curr. Opin. Electrochem. 2020, 23, 80–88. [Google Scholar] [CrossRef]
- Reche, I.; Gallardo, I.; Guirado, G. Electrochemical studies of CO2 in imidazolium ionic liquids using silver as a working electrode: A suitable approach for determining diffusion coefficients, solubility values, and electrocatalytic effects. RSC Adv. 2014, 4, 65176–65183. [Google Scholar] [CrossRef]
- Dattila, F.; Fortunati, A.; Zammillo, F.; Guzmán, H.; López, N.; Hernández, S. Descriptors for Electrochemical CO2 Reduction in Imidazolium-Based Electrolytes. ACS Catal. 2024, 14, 16166–16174. [Google Scholar] [CrossRef]
- Fortunati, A.; Risplendi, F.; Fiorentin, M.R.; Cicero, G.; Parisi, E.; Castellino, M.; Simone, E.; Iliev, B.; Schubert, T.J.S.; Russo, N.; et al. Understanding the role of imidazolium-based ionic liquids in the electrochemical CO2 reduction reaction. Commun. Chem. 2023, 6, 84. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, S.-X.; Gandionco, K.A.; Bond, A.M.; Zhang, J. Electrocatalytic carbon dioxide reduction: From fundamental principles to catalyst design. Mater. Today Adv. 2020, 7, 100074. [Google Scholar] [CrossRef]
- Singh, M.R.; Clark, E.L.; Bell, A.T. Effects of electrolyte, catalyst, and membrane composition and operating conditions on the performance of solar-driven electrochemical reduction of carbon dioxide. Phys. Chem. Chem. Phys. 2015, 17, 18924–18936. [Google Scholar] [CrossRef] [PubMed]
- Marcandalli, G.; Monteiro, M.C.O.; Goyal, A.; Koper, M.T.M. Electrolyte Effects on CO2 Electrochemical Reduction to CO. Acc. Chem. Res. 2022, 55, 1900–1911. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Altaf, N.; Huang, L.; Gao, Y.; Wang, Q. Electrolytic cell design for electrochemical CO2 reduction. J. CO2 Util. 2020, 35, 90–105. [Google Scholar] [CrossRef]
- Xi, W.; Xi, W.; Yang, P.; Yang, P.; Jiang, M.; Jiang, M.; Wang, X.; Wang, X.; Zhou, H.; Zhou, H.; et al. Electrochemical CO2 reduction coupled with alternative oxidation reactions: Electrocatalysts, electrolytes, and electrolyzers. Appl. Catal. B Environ. 2024, 341, 123291. [Google Scholar] [CrossRef]
- Jiang, N.; Zhu, Z.; Xue, W.; Xia, B.Y.; You, B. Emerging Electrocatalysts for Water Oxidation under Near-Neutral CO2 Reduction Conditions. Adv. Mater. 2022, 34, 2105852. [Google Scholar] [CrossRef]
- Tufa, R.A.; Chanda, D.; Ma, M.; Aili, D.; Demissie, T.B.; Vaes, J.; Li, Q.; Liu, S.; Pant, D. Towards highly efficient electrochemical CO2 reduction: Cell designs, membranes and electrocatalysts. Appl. Energy 2020, 277, 115557. [Google Scholar] [CrossRef]
- Hernández, S.; Farkhondehfal, M.A.; Sastre, F.; Makkee, M.; Saracco, G.; Russo, N. Syngas production from electrochemical reduction of CO2: Current status and prospective implementation. Green Chem. 2017, 19, 2326–2346. [Google Scholar] [CrossRef]
- Rosen, J.; Hutchings, G.S.; Lu, Q.; Rivera, S.; Zhou, Y.; Vlachos, D.G.; Jiao, F. Mechanistic Insights into the Electrochemical Reduction of CO2 to CO on Nanostructured Ag Surfaces. ACS Catal. 2015, 5, 4293–4299. [Google Scholar] [CrossRef]
- Hoshi, N.; Kato, M.; Hori, Y. Electrochemical reduction of CO, on single crystal electrodes of silver Ag(111), Ag(100) and Agi(110). J. Electroanal. Chem. 1997, 44, 283–286. [Google Scholar]
- Hori, Y. Electrochemical CO2 Reduction on Metal Electrodes. In Modern Aspects of Electrochemistry. Modern Aspects of Electrochemistry; Springer: New York, NY, USA, 2008; pp. 89–189. [Google Scholar] [CrossRef]
- Mahyoub, S.A.; Qaraah, F.; Chen, C.; Zhang, F.; Yan, S.; Cheng, Z. An overview on the recent developments of Ag-based electrodes in the electrochemical reduction of CO2 to CO. Sustain. Energy Fuels 2019, 4, 50–67. [Google Scholar] [CrossRef]
- Jensen, M.T.; Rønne, M.H.; Ravn, A.K.; Juhl, R.W.; Nielsen, D.U.; Hu, X.-M.; Pedersen, S.U.; Daasbjerg, K.; Skrydstrup, T. Scalable carbon dioxide electroreduction coupled to carbonylation chemistry. Nat. Commun. 2017, 8, 489. [Google Scholar] [CrossRef] [PubMed]
- Ponsard, L.; Nicolas, E.; Tran, N.H.; Lamaison, S.; Wakerley, D.; Cantat, T.; Fontecave, M. Coupling Electrocatalytic CO2 Reduction with Thermocatalysis Enables the Formation of a Lactone Monomer. ChemSusChem 2021, 14, 2198–2204. [Google Scholar] [CrossRef]
- Miró, R.; Fernández-Llamazares, E.; Godard, C.; Bernardos, M.D.d.L.; Gual, A. Synergism between iron porphyrin and dicationic ionic liquids: Tandem CO2 electroreduction–carbonylation reactions. Chem. Commun. 2022, 58, 10552–10555. [Google Scholar] [CrossRef]
- Biswas, A.N.; Xie, Z.; Xia, R.; Overa, S.; Jiao, F.; Chen, J.G. Tandem Electrocatalytic–Thermocatalytic Reaction Scheme for CO2 Conversion to C3 Oxygenates. ACS Energy Lett. 2022, 7, 2904–2910. [Google Scholar] [CrossRef]
- Kroon, M.C.; Buijs, W.; Peters, C.J.; Witkamp, G.-J. Decomposition of ionic liquids in electrochemical processing. Green Chem. 2006, 8, 241–245. [Google Scholar] [CrossRef]
- Dutta, A.; Morstein, C.E.; Rahaman, M.; López, A.C.; Broekmann, P. Beyond Copper in CO2 Electrolysis: Effective Hydrocarbon Production on Silver-Nanofoam Catalysts. ACS Catal. 2018, 8, 8357–8368. [Google Scholar] [CrossRef]
- Shiratsuchi, R.; Nogami, G. Pulsed Electroreduction of CO2 on Silver Electrodes. J. Electrochem. Soc. 1996, 143, 582–586. [Google Scholar] [CrossRef]
- Kumełan, J.; Kamps, Á.P.-S.; Tuma, D.; Maurer, G. Solubility of CO2 in the ionic liquids [bmim][CH3SO4] and [bmim][PF6]. J. Chem. Eng. Data 2006, 51, 1802–1807. [Google Scholar] [CrossRef]
- Bui, J.C.; Kim, C.; Weber, A.Z.; Bell, A.T. Dynamic boundary layer simulation of pulsed CO2 electrolysis on a copper catalyst. ACS Energy Lett. 2021, 6, 1181–1188. [Google Scholar] [CrossRef]
- Wang, J.H. Self-Diffusion Coefficients of Water. J. Phys. Chem. 1965, 69, 4412. [Google Scholar]
- Moya, C.; Palomar, J.; Gonzalez-Miquel, M.; Bedia, J.; Rodriguez, F. Diffusion coefficients of CO2 in ionic liquids estimated by gravimetry. Ind. Eng. Chem. Res. 2014, 53, 13782–13789. [Google Scholar] [CrossRef]
- Liu, S.; Govindarajan, N.; Prats, H.; Chan, K. Understanding the reaction mechanism of Kolbe electrolysis on Pt anodes. Chem Catal. 2022, 2, 1100–1113. [Google Scholar] [CrossRef]
- Habibzadeh, F.; Mardle, P.; Zhao, N.; Riley, H.D.; Salvatore, D.A.; Berlinguette, C.P.; Holdcroft, S.; Shi, Z. Ion Exchange Membranes in Electrochemical CO2 Reduction Processes. Electrochem. Energy Rev. 2023, 6, 26. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | H2O (%v/v) | H2O (mL) | H2O (mmol) | Added H2O (M) | [SO₃CF₃] (mmol) | [SO₃CF₃] (M) | [CO2CH₃] (mmol) | [CO2CH₃] (M) | [Anion] (mmol) | Conductivity (mS/cm) |
|---|---|---|---|---|---|---|---|---|---|---|
| MIX20% | 20 | 10 | 550.0 | 11.0 | 48 | 0.96 | 24 | 0.48 | 72 | 21.7 |
| MIX10% | 10 | 5 | 275.0 | 5.5 | 54 | 1.08 | 27 | 0.54 | 81 | 18.8 |
| MIX5% | 5 | 2.5 | 137.5 | 2.75 | 57 | 1.14 | 28 | 0.57 | 85 | 16.1 |
| MIX1% | 1 | 0.5 | 27.5 | 0.55 | 59 | 1.19 | 29 | 0.59 | 88 | 12.7 |
| MIX0% | ~0.2 | 0 | ~3 | 0 | 59 | 1.20 | 30 | 0.60 | 89 | 11.6 |
| j0 (mA/cm2) | α | E0/V vs. RHE | Reference | |
|---|---|---|---|---|
| H2 | 6.36 10−2 | 0.14 | 0 | [51] |
| CO | 2.01 10−3 | 0.35 | −0.11 | [51] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallone, M.; Fortunati, A.; Hernández, S. Assessing the CO2 Capture and Electro-Reduction in Imidazolium-Based Ionic Liquids: Role of the Ion Exchange Membrane. Catalysts 2025, 15, 318. https://doi.org/10.3390/catal15040318
Gallone M, Fortunati A, Hernández S. Assessing the CO2 Capture and Electro-Reduction in Imidazolium-Based Ionic Liquids: Role of the Ion Exchange Membrane. Catalysts. 2025; 15(4):318. https://doi.org/10.3390/catal15040318
Chicago/Turabian StyleGallone, Mario, Alessia Fortunati, and Simelys Hernández. 2025. "Assessing the CO2 Capture and Electro-Reduction in Imidazolium-Based Ionic Liquids: Role of the Ion Exchange Membrane" Catalysts 15, no. 4: 318. https://doi.org/10.3390/catal15040318
APA StyleGallone, M., Fortunati, A., & Hernández, S. (2025). Assessing the CO2 Capture and Electro-Reduction in Imidazolium-Based Ionic Liquids: Role of the Ion Exchange Membrane. Catalysts, 15(4), 318. https://doi.org/10.3390/catal15040318