
Charge Transfer and Synergy on Mn-Mn Dimer Sites in Manganese Oxides: Activity for NO Oxidation


 and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
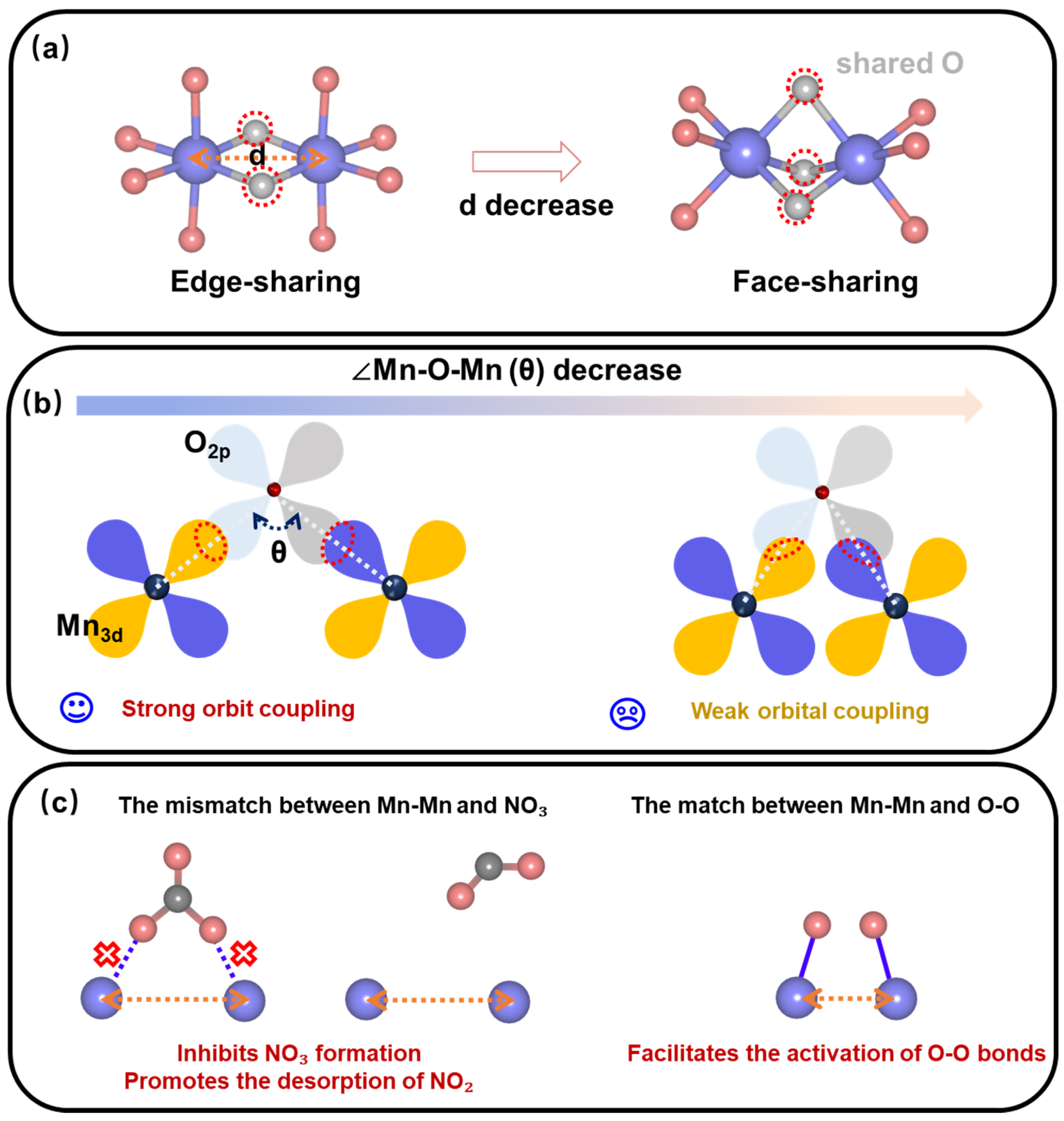
2.1. The Influence of Mn-Mn Bond Length on Geometric and Electronic Structure in Different Manganese Oxides
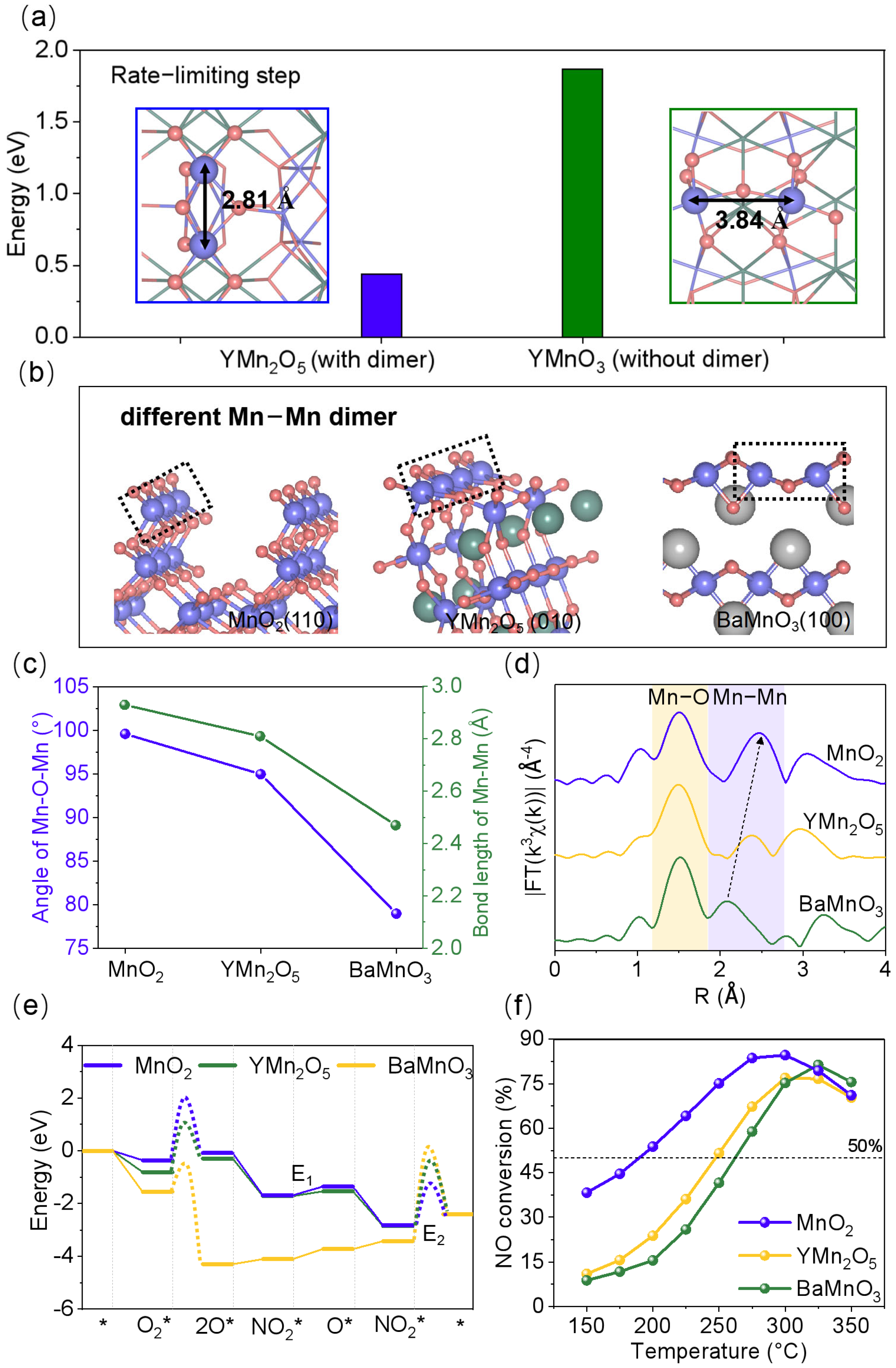
2.2. Enhanced NO Oxidation Activity of Mn-Mn Dimers in Mullite YMn2O5 Compared to Single Mn Sites in Perovskite YMnO3
2.3. Geometric Structure Characterization and NO Oxidation Activity of Mn-Based Oxides with Mn-Mn Dimers of Varying Bond Lengths
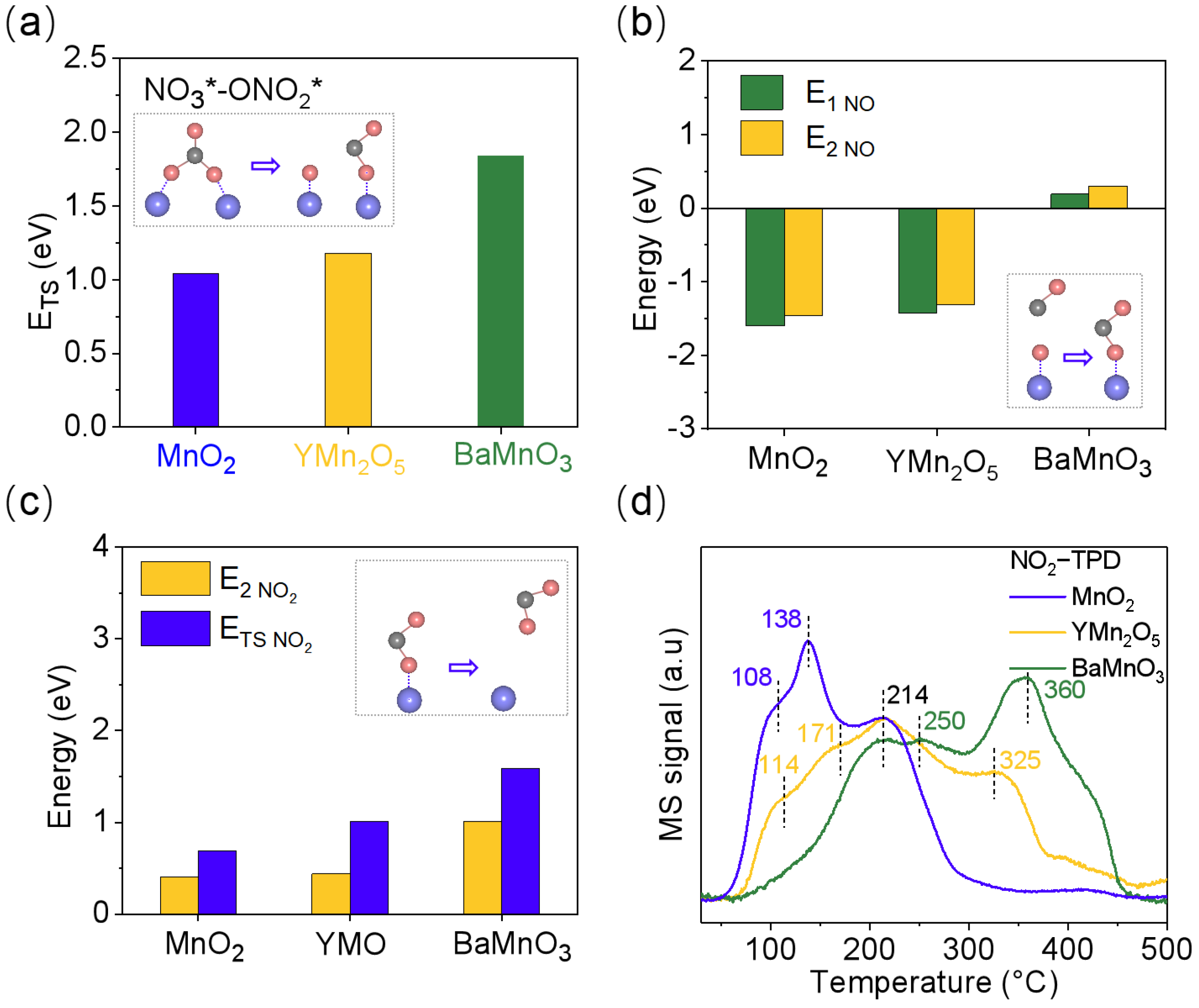
2.4. Dynamic Regulation of Adsorbed Intermediates by Different Bond Lengths in Mn-Mn Dimers
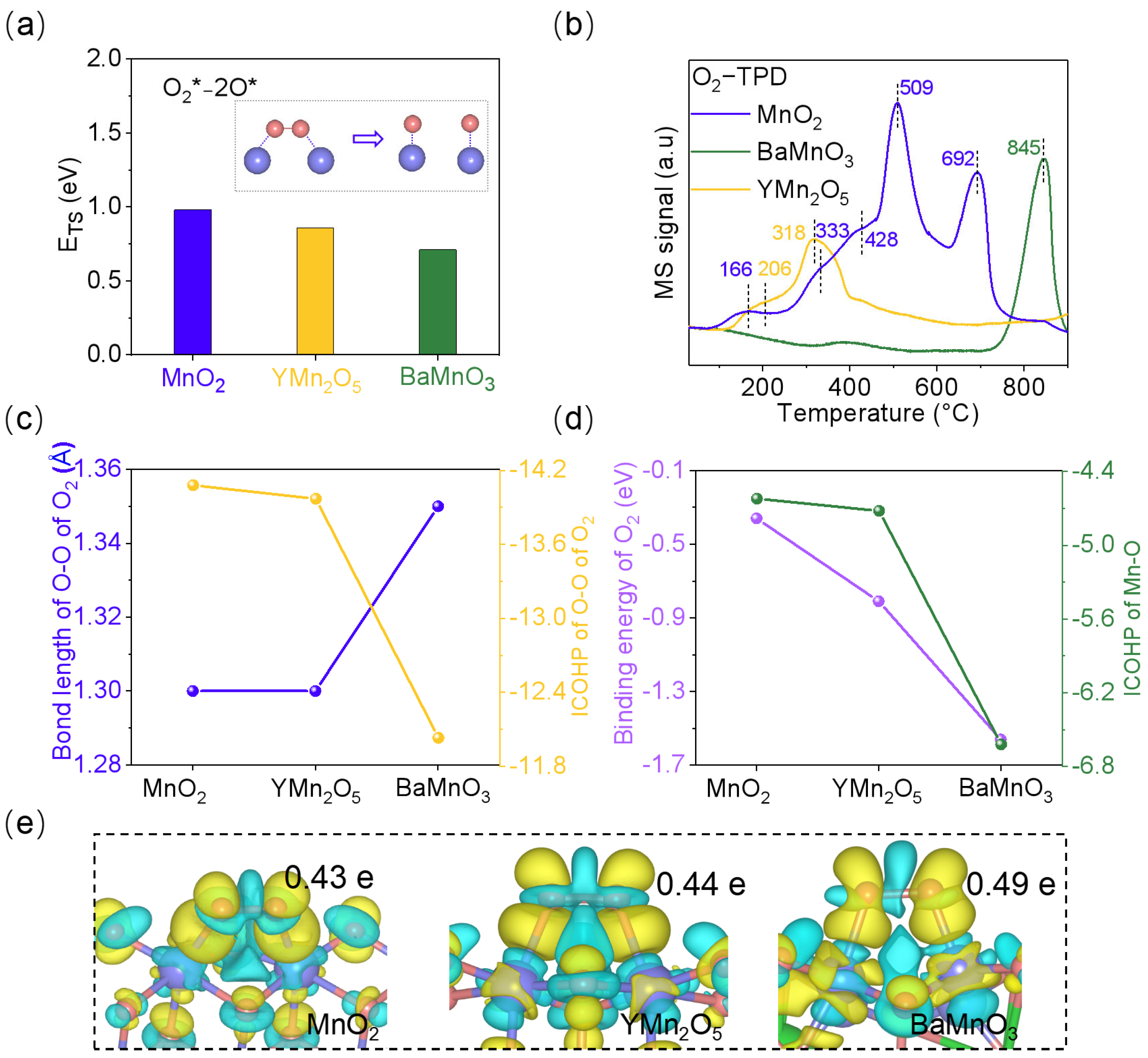
2.5. The Influence of Mn-Mn Bond Length on O2 Activation Efficiency in NO Oxidation Catalysis
2.6. The Role of Mn-Mn Bond Length in Mechanisms of NO Oxidation Catalysis
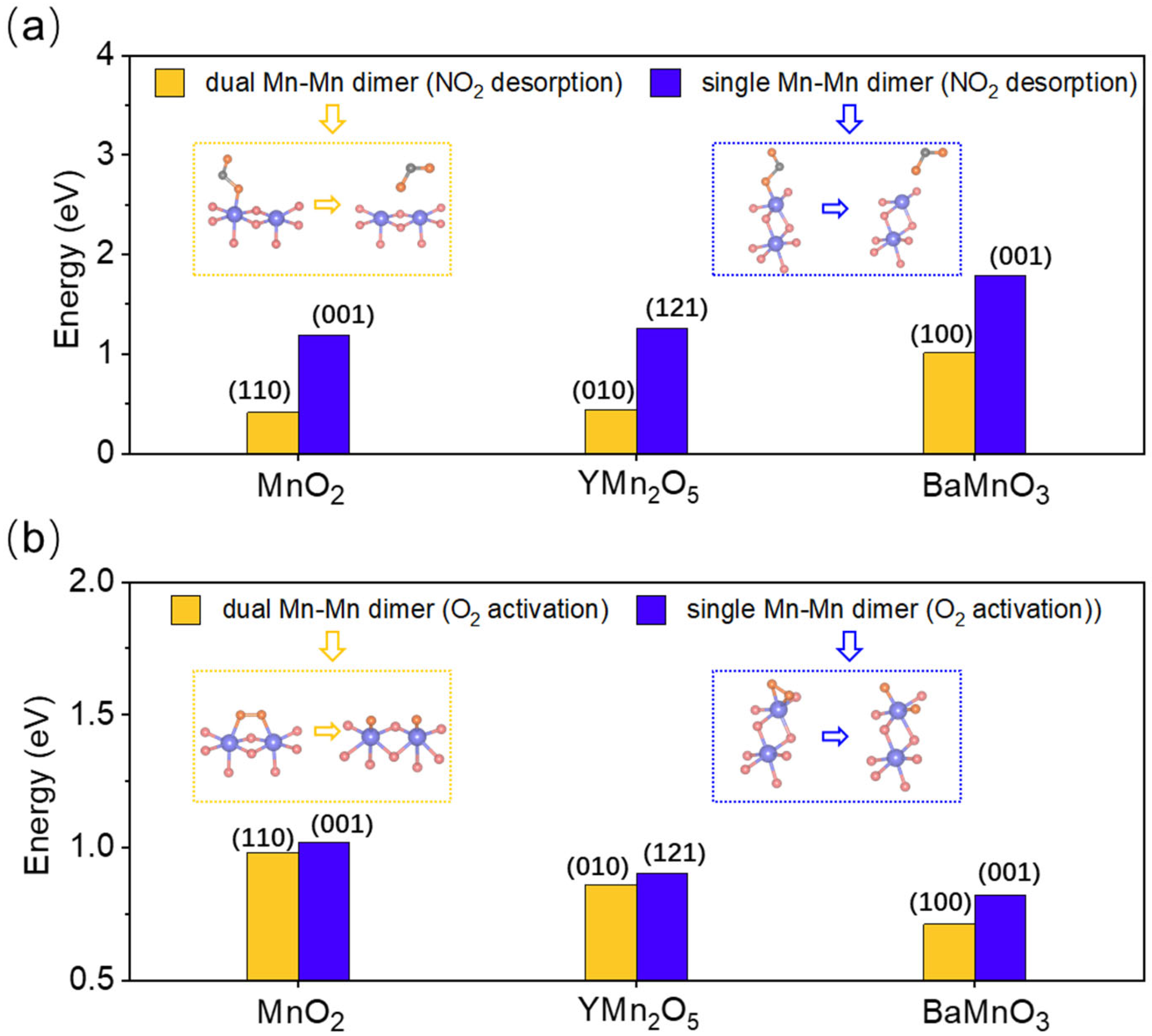
2.7. Catalytic Advantage of Dual-Site Mn-Mn Dimer Exposure over Single-Site Exposure in NO Oxidation
3. Materials and Methods
3.1. Catalyst Synthesis
3.2. Characterization
3.3. Catalytic Activity Measurements
3.4. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhou, X.; Min, Y.; Zhao, C.; Chen, C.; Ke, M.-K.; Xu, S.-L.; Chen, J.-J.; Wu, Y.; Yu, H.-Q. Constructing sulfur and oxygen super-coordinated main-group electrocatalysts for selective and cumulative H2O2 production. Nat. Commun. 2024, 15, 193. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Wang, Z.; Wu, H.; Xiao, M.; Liu, C.; Xing, W. High-density Ir single sites from rapid ligand transformation for efficient water electrolysis. Chin. J. Catal. 2024, 66, 223–232. [Google Scholar] [CrossRef]
- Li, W.; Hong, J.; Shang, J.; Yamashita, H.; Wei, C.; Hu, Y. In situ construction of CuBi-MOF derived heterojunctions with electron-rich effects enhances localized CO2 enrichment integrated with Si photocathodes for CO2 reduction. Appl. Catal. B Environ. 2025, 365, 124890. [Google Scholar] [CrossRef]
- Chang, J.; Shi, Y.; Wu, H.; Yu, J.; Jing, W.; Wang, S.; Waterhouse, G.I.N.; Tang, Z.; Lu, S. Oxygen radical coupling on short-range ordered Ru atom arrays enables exceptional activity and stability for acidic water oxidation. J. Am. Chem. Soc. 2024, 146, 12958–12968. [Google Scholar] [CrossRef]
- Fukui, K. Role of frontier orbitals in chemical reactions. Science 1982, 218, 747–754. [Google Scholar] [CrossRef]
- Fukui, K.; Inagaki, S. Orbital interaction rationale for the role of catalysts. J. Am. Chem. Soc. 1975, 97, 4445–4452. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Rossmeisl, J.; Christensen, C.H. Towards the computational design of solid catalysts. Nat. Chem. 2009, 1, 37–46. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Vojvodic, A.; Nørskov, J.K.; Abild-Pedersen, F. Electronic structure effects in transition metal surface chemistry. Top. Catal. 2014, 57, 25–32. [Google Scholar] [CrossRef]
- Greeley, J.; Stephens, I.E.L.; Bondarenko, A.S.; Johansson, T.P.; Hansen, H.A.; Jaramillo, T.F.; Rossmeisl, I.; Chorkendorff, J.; Nørskov, J.K. Alloys of platinum and early transition metals as oxygen reduction electrocatalysts. Nat. Chem. 2009, 1, 552–556. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Yoneyama, H.; Tamura, H. Catalytic activity for electrochemical reduction of oxygen of lanthanum nickel oxide and related oxides. J. Electroanal. Chem. Interfacial Electrochem. 1977, 79, 319–326. [Google Scholar] [CrossRef]
- Wei, C.; Feng, Z.; Scherer, G.G.; Barber, J.; Shao-Horn, Y.; Xu, Z.J. Cations in octahedral sites: A descriptor for oxygen electrocatalysis on transition-metal spinels. Adv. Mater. 2017, 29, 1606800. [Google Scholar] [CrossRef] [PubMed]
- Suntivich, J.; May, K.J.; Gasteiger, H.A.; Goodenough, J.B.; Shao-Horn, Y. A perovskite oxide optimized for oxygen evolution catalysis from molecular orbital principles. Science 2011, 334, 1383–1385. [Google Scholar] [CrossRef]
- Grimaud, A.; May, K.J.; Carlton, C.E.; Lee, Y.-L.; Risch, M.; Hong, W.T.; Zhou, J.; Shao-Horn, Y. Double perovskites as a family of highly active catalysts for oxygen evolution in alkaline solution. Nat. Commun. 2013, 4, 2439. [Google Scholar] [CrossRef]
- Lee, Y.-L.; Kleis, J.; Rossmeisl, J.; Shao-Horn, Y.; Morgan, D. Prediction of solid oxide fuel cell cathode activity with first-principles descriptors. Energy Environ. Sci. 2011, 4, 3966–3970. [Google Scholar] [CrossRef]
- Liu, J.; Liu, H.; Chen, H.; Du, X.; Zhang, B.; Hong, Z.; Sun, S.; Wang, W. Progress and challenges toward the rational design of oxygen electrocatalysts based on a descriptor approach. Adv. Sci. 2020, 7, 1901614. [Google Scholar] [CrossRef]
- Li, R.; Hu, A.; Zhao, C.; Zhou, B.; He, M.; Fan, Y.; Chen, J.; Yan, Z.; Pan, Y.; Long, J. Tailoring mixed geometrical configurations in amorphous catalysts to activate oxygen electrode reactions of lithium-oxygen batteries. Chem. Eng. J. 2023, 452, 139162. [Google Scholar]
- Chen, S.; Guan, J. Structural regulation strategies of nitrogen reduction electrocatalysts. Chin. J. Catal. 2024, 66, 20–52. [Google Scholar] [CrossRef]
- Wu, J.; Wang, X.; Zheng, W.; Sun, Y.; Xie, Y.; Ma, K.; Zhang, Z.; Liao, Q.; Tian, Z.; Kang, Z.; et al. Identifying and interpreting geometric configuration-dependent activity of spinel catalysts for water reduction. J. Am. Chem. Soc. 2022, 144, 19163–19172. [Google Scholar] [CrossRef]
- Wang, L.; Hua, W.; Wan, X.; Feng, Z.; Hu, Z.; Li, H.; Niu, J.; Wang, L.; Wang, A.; Liu, J.; et al. Design rules of a sulfur redox electrocatalyst for lithium-sulfur batteries. Adv. Mater. 2022, 34, 2110279. [Google Scholar] [CrossRef]
- Wang, W.; McCool, G.; Kapur, N.; Yuan, G.; Shan, B.; Nguyen, M.; Graham, U.M.; Davis, B.H.; Jacobs, G.; Cho, K.; et al. Mixed-phase oxide catalyst based on Mn-mullite (Sm, Gd)Mn2O5 for NO oxidation in diesel exhaust. Science 2012, 337, 832–835. [Google Scholar]
- Zheng, Y.; Thampy, S.; Ashburn, N.; Dillon, S.; Wang, L.; Jangjou, Y.; Tan, K.; Kong, F.; Nie, Y.; Kim, M.J.; et al. Stable and active oxidation catalysis by cooperative lattice oxygen redox on SmMn2O5 mullite surface. J. Am. Chem. Soc. 2019, 141, 10722–10728. [Google Scholar] [PubMed]
- Ma, S.-J.; Wang, X.-W.; Chen, T.; Yuan, Z.-H. Effect of surface morphology on catalytic activity for NO oxidation of SmMn2O5 nanocrystals. Chem. Eng. J. 2018, 354, 191–196. [Google Scholar]
- Thampy, S.; Zheng, Y.; Dillon, S.; Liu, C.; Jangjou, Y.; Lee, Y.-J.; Epling, W.S.; Xiong, K.; Chabal, Y.J.; Cho, K.; et al. Superior catalytic performance of Mn-Mullite over Mn-Perovskite for NO oxidation. Catal. Today 2018, 310, 195–201. [Google Scholar]
- Yang, W.; Su, Z.A.; Xu, Z.; Yang, W.; Peng, Y.; Li, J. Comparative study of α-, β-, γ- and δ-MnO2 on toluene oxidation: Oxygen vacancies and reaction intermediates. Appl. Catal. B Environ. 2020, 260, 118150. [Google Scholar]
- Zhang, T.; Li, H.; Yang, Z.; Cao, F.; Li, L.; Chen, H.; Liu, H.; Xiong, K.; Wu, J.; Hong, Z.; et al. Electrospun YMn2O5 nanofibers: A highly catalytic activity for NO oxidation. Appl. Catal. B Environ. 2019, 247, 133–141. [Google Scholar]
- Ao, R.; Ma, L.; Guo, Z.; Yang, J.; Mu, L.; Yang, J.; Wei, Y. NO oxidation performance and kinetics analysis of BaMO3 (M=Mn, Co) perovskite catalysts. Environ. Sci. Pollut. Res. 2021, 28, 6929–6940. [Google Scholar]
- Stueben, B.L.; Cantrelle, B.; Sneddon, J.; Beck, J.N. Manganese K-edge XANES studies of Mn speciation in Lac des Allemands as a function of depth. Microchem. J. 2004, 76, 113–120. [Google Scholar]
- Celorrio, V.; Calvillo, L.; Granozzi, G.; Russell, A.E.; Fermin, D.J. AMnO3 (A = Sr, La, Ca, Y) perovskite oxides as oxygen reduction electrocatalysts. Top. Catal. 2018, 61, 154–161. [Google Scholar]
- Chaboy, J.; Prieto, C.; Hernando, M.; Parras, M.; González-Calbet, J. Ab initio X-ray absorption study of the manganese K-edge XANES spectra in Mn- and Zn-related hexagonal perovskites. Phys. Rev. B 2006, 74, 174433. [Google Scholar]
- Kim, K.; Jeong, J.; Azad, A.K.; Jin, S.B.; Kim, J.H. X-ray photoelectron spectroscopic study of direct reforming catalysts Ln0.5Sr0.5Ti0.5Mn0.5O3±d (Ln = La, Nd, and Sm) for high temperature-operating solid oxide fuel cell. Appl. Surf. Sci. 2016, 365, 38–46. [Google Scholar]
- Zheng, Y.; Liu, Q.; Shan, C.; Su, Y.; Fu, K.; Lu, S.; Han, R.; Song, C.; Ji, N.; Ma, D. Defective ultrafine MnOx nanoparticles confined within a carbon matrix for low-temperature oxidation of volatile organic compounds. Environ. Sci. Technol. 2021, 55, 5403–5411. [Google Scholar] [PubMed]
- Xiong, T.; Yu, Z.G.; Wu, H.; Du, Y.; Xie, Q.; Chen, J.; Zhang, Y.-W.; Pennycook, S.J.; Lee, W.S.V.; Xue, J. Defect engineering of oxygen-deficient manganese oxide to achieve high-performing aqueous zinc ion battery. Adv. Energy Mater. 2019, 9, 1803815. [Google Scholar]
- Chen, X.; Li, Y.; Fu, W.; Tian, S.; Yang, Y.; Yang, K.; Xu, X.; Zhang, X. Oriented generation of singlet oxygen in H2O2 activation for water decontamination: Regulation of oxygen vacancies over α-MnO2 nanocatalysts. Environ. Sci. Nano 2023, 10, 1428–1440. [Google Scholar] [CrossRef]
- Litvinchuk, A.P. Lattice dynamics and spin-phonon interactions in multiferroic RMn2O5: Shell model calculations. J. Magn. Magn. Mater. 2009, 321, 2373–2377. [Google Scholar]
- Zhao, C.; Zhu, A.; Gao, S.; Wang, L.; Wan, X.; Wang, A.; Wang, W.-H.; Xue, T.; Yang, S.; Sun, D.; et al. Phonon resonance catalysis in NO oxidation on Mn-based mullite. ACS Catal. 2022, 12, 12113–12122. [Google Scholar] [CrossRef]
- Zhao, C.; Zhang, X.; Yu, M.; Wang, A.; Wang, L.; Xue, L.; Liu, J.; Yang, Z.; Wang, W. Cooperative catalysis toward oxygen reduction reaction under dual coordination environments on intrinsic AMnO3-type perovskites via regulating stacking configurations of coordination units. Adv. Mater. 2020, 32, 2006145. [Google Scholar]
- Sacchetti, A.; Baldini, M.; Postorino, P.; Martin, C.; Maignan, A. Raman spectroscopy on cubic and hexagonal SrMnO3. J. Raman Spectrosc. 2006, 37, 591–596. [Google Scholar]
- Joly, J.P.; Méhier, C.; Béré, K.E.; Abon, M. TPD study of labile oxygen on a (VO)2P2O7 catalyst active in n-butane partial oxidation. Appl. Catal. A Gen. 1998, 169, 55–63. [Google Scholar]
- Li, B.; Yang, Q.; Peng, Y.; Chen, J.; Deng, L.; Wang, D.; Hong, X.; Li, J. Enhanced low-temperature activity of LaMnO3 for toluene oxidation: The effect of treatment with an acidic KMnO4. Chem. Eng. J. 2019, 366, 92–99. [Google Scholar]
- Yang, J.; Zhang, J.; Liu, X.; Duan, X.; Wen, Y.; Chen, R.; Shan, B. Origin of the superior activity of surface doped SmMn2O5 mullites for NO oxidation: A first-principles based microkinetic study. J. Catal. 2018, 359, 122–129. [Google Scholar] [CrossRef]
- Zhang, S.; Li, H.; Cai, G.; Wang, Y.; Gao, Z.; Hao, R.; Bao, X.; Zhao, C.; Wang, W. Spatial geometric effect driven by the different [MnO6] octahedra entity stacking configurations to facilitate the catalytic decomposition of H2O2 in wastewater. Appl. Surf. Sci. 2024, 669, 160589. [Google Scholar] [CrossRef]
- Yang, R.; Guo, Z.; Cai, L.; Zhu, R.; Fan, Y.; Zhang, Y.; Han, P.; Zhang, W.; Zhu, X.; Zhao, Q.; et al. Investigation into the phase-activity relationship of MnO2 nanomaterials toward ozone-assisted catalytic oxidation of toluene. Small 2021, 17, 2103052. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. 1996, 6, 15–50. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Wen, R.; Dong, A.; Wang, W.; Xu, J.; Zhang, S.; Chen, G.; Wang, W.; Hao, R.; Wan, X.; et al. Charge Transfer and Synergy on Mn-Mn Dimer Sites in Manganese Oxides: Activity for NO Oxidation. Catalysts 2025, 15, 307. https://doi.org/10.3390/catal15040307
Li H, Wen R, Dong A, Wang W, Xu J, Zhang S, Chen G, Wang W, Hao R, Wan X, et al. Charge Transfer and Synergy on Mn-Mn Dimer Sites in Manganese Oxides: Activity for NO Oxidation. Catalysts. 2025; 15(4):307. https://doi.org/10.3390/catal15040307
Chicago/Turabian StyleLi, Huan, Rui Wen, Anqi Dong, Wanying Wang, Jinchao Xu, Shen Zhang, Gang Chen, Wen Wang, Ruiting Hao, Xiang Wan, and et al. 2025. "Charge Transfer and Synergy on Mn-Mn Dimer Sites in Manganese Oxides: Activity for NO Oxidation" Catalysts 15, no. 4: 307. https://doi.org/10.3390/catal15040307
APA StyleLi, H., Wen, R., Dong, A., Wang, W., Xu, J., Zhang, S., Chen, G., Wang, W., Hao, R., Wan, X., Zhao, C., & Wang, W. (2025). Charge Transfer and Synergy on Mn-Mn Dimer Sites in Manganese Oxides: Activity for NO Oxidation. Catalysts, 15(4), 307. https://doi.org/10.3390/catal15040307