
Machine Learning-Assisted Catalysts for Advanced Oxidation Processes: Progress, Challenges, and Prospects

 , ,
, , 

Abstract
1. Introduction
2. Advanced Oxidation Process of Wastewater Treatment
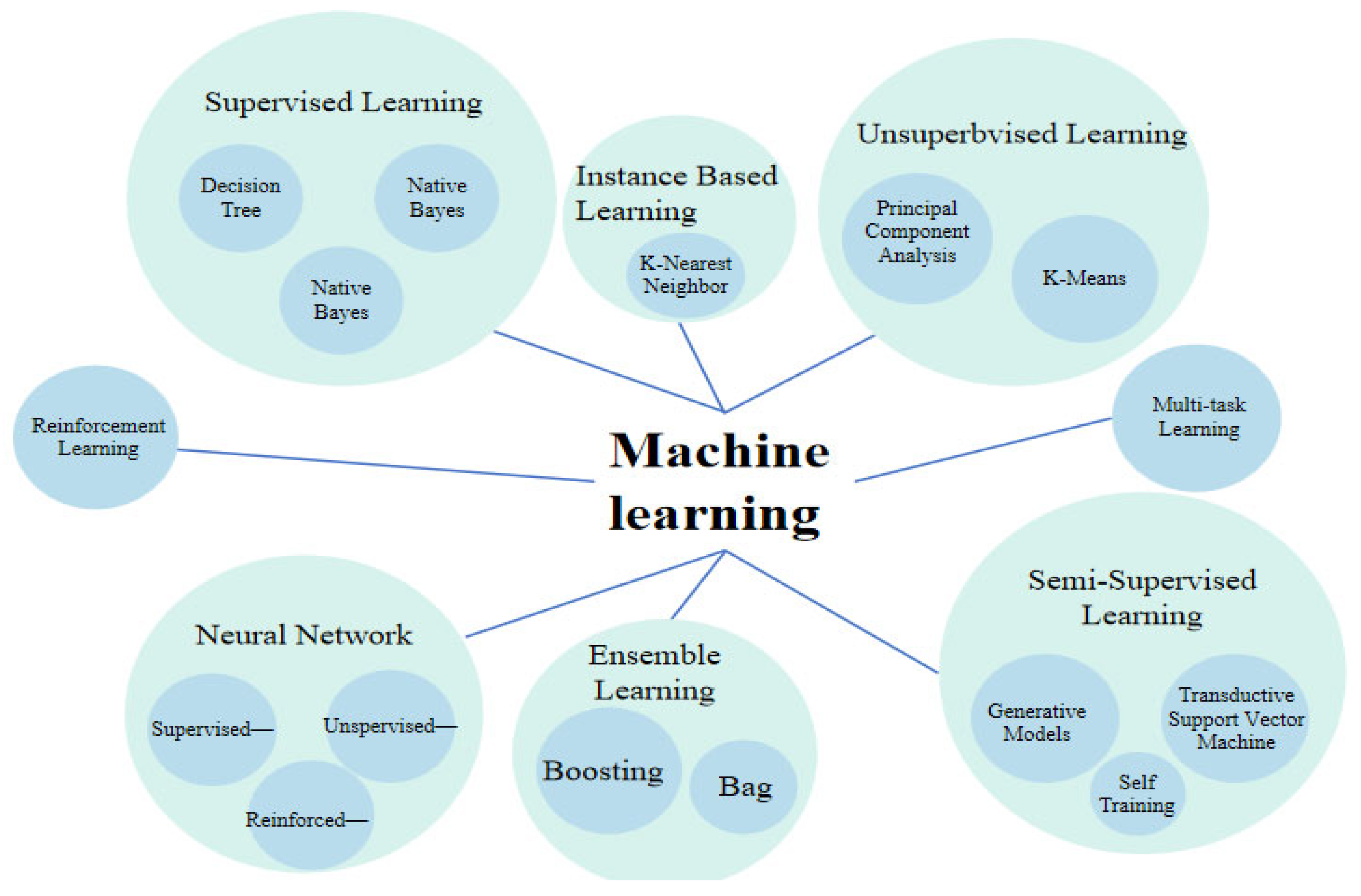
3. Machine Learning Algorithms
4. Catalyst Development for ML-Assisted Advanced Oxidation Water Treatment Processes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Industrial Wastewater Category | Typical Pollutants | AOPs | ML Models/Algorithms | Selected Catalyst Types | ML Implementation Mechanism | Advantages of ML-Driven Catalyst Screening | References |
|---|---|---|---|---|---|---|---|
| Pharmaceutical/ Textile Industry Effluent | Antibiotics (e.g., sulfamethoxazole) pharmaceutical residue, Azo dyes, recalcitrant organic compounds | Fenton/Photo-Fenton systems | Artificial neural network (ANN), deep neural network (DNN), random forest (RF) | Fe-based catalysts | Using RF and ANN to predict the reaction characteristics of AOPs and then optimize the screening of catalysts. DNN predicts the best catalytic outcome. | 1. Resolves nonlinear relationships between catalyst properties and activity; 2. Minimizes experimental redundancy; 3. Optimizes multivariate reaction parameters. | [70,71,72] |
| Electroplating/ Metallurgical Effluent | Heavy metals (Cr6+, Cu2+) with co-existing organics | Electro-Fenton processes, photocatalytic oxidation | Convolutional neural networks (CNN), genetic algorithms (GA), gradient boosting regression (GBR) | Pt-nanoparticles, TiO2-based nanocomposite | CNN is used for image analysis and the extraction of material features. GA detects the binding energy on the Pt surface to enable the catalyst to achieve maximum activity. The powerful search capability of the GBR model can be employed to identify target catalysts. | 1. Optimize the relationship between structure and performance; 2. Improve the accuracy of screening. | [73,74,75,76] |
| Agrochemical Effluent | Organochlorines (e.g., DDT), Coronated hydrocarbons | Ozone oxidation, Fenton, UV | Random forest (RF), Bayesian optimization (BO), CNN, ANN | TiO2-based nanocomposite | The generalized RF model has good predictive ability and is prospective to predict the photocatalytic degradation performance of organic pollutants with the experimental data of BO. | 1. Improve the efficiency of catalyst prediction; 2. Uncovers synergistic interfacial effects. | [77,78,79,80] |
4.1. Active Site Determination and Catalyst Screening
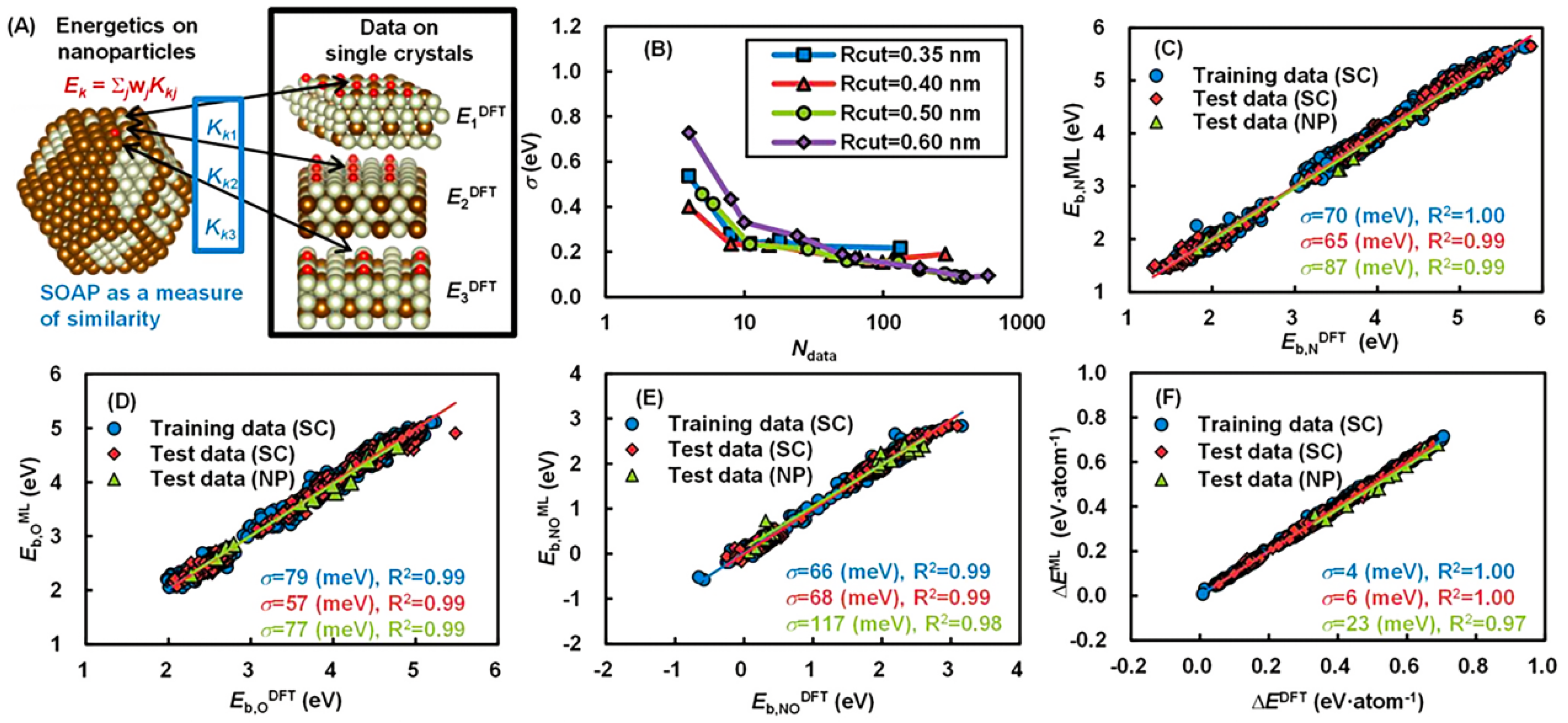
4.2. Finding Descriptors and Patterns in Catalysis Data
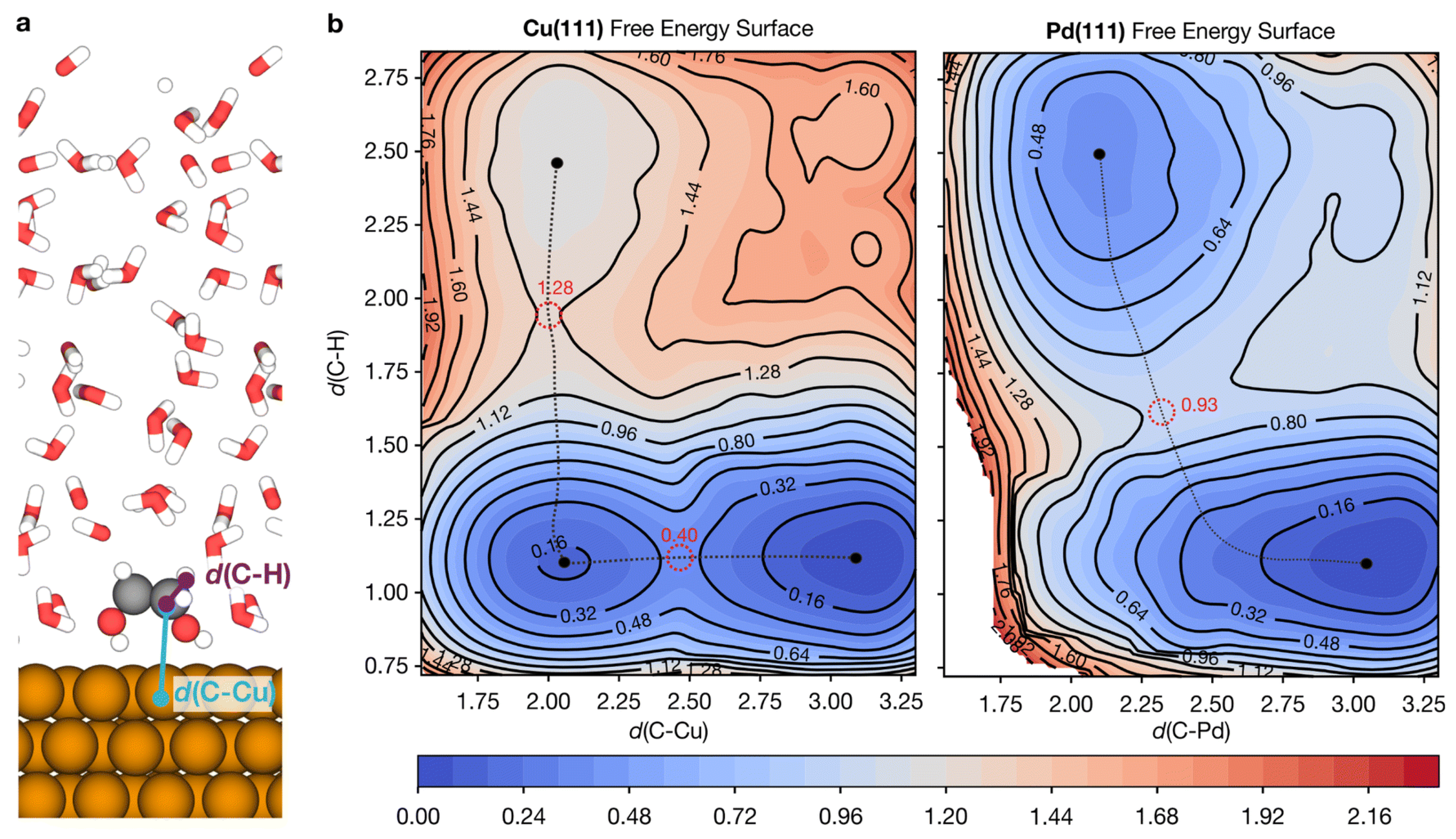
4.3. Machine-Learned Interatomic Potentials for Catalyst Simulation
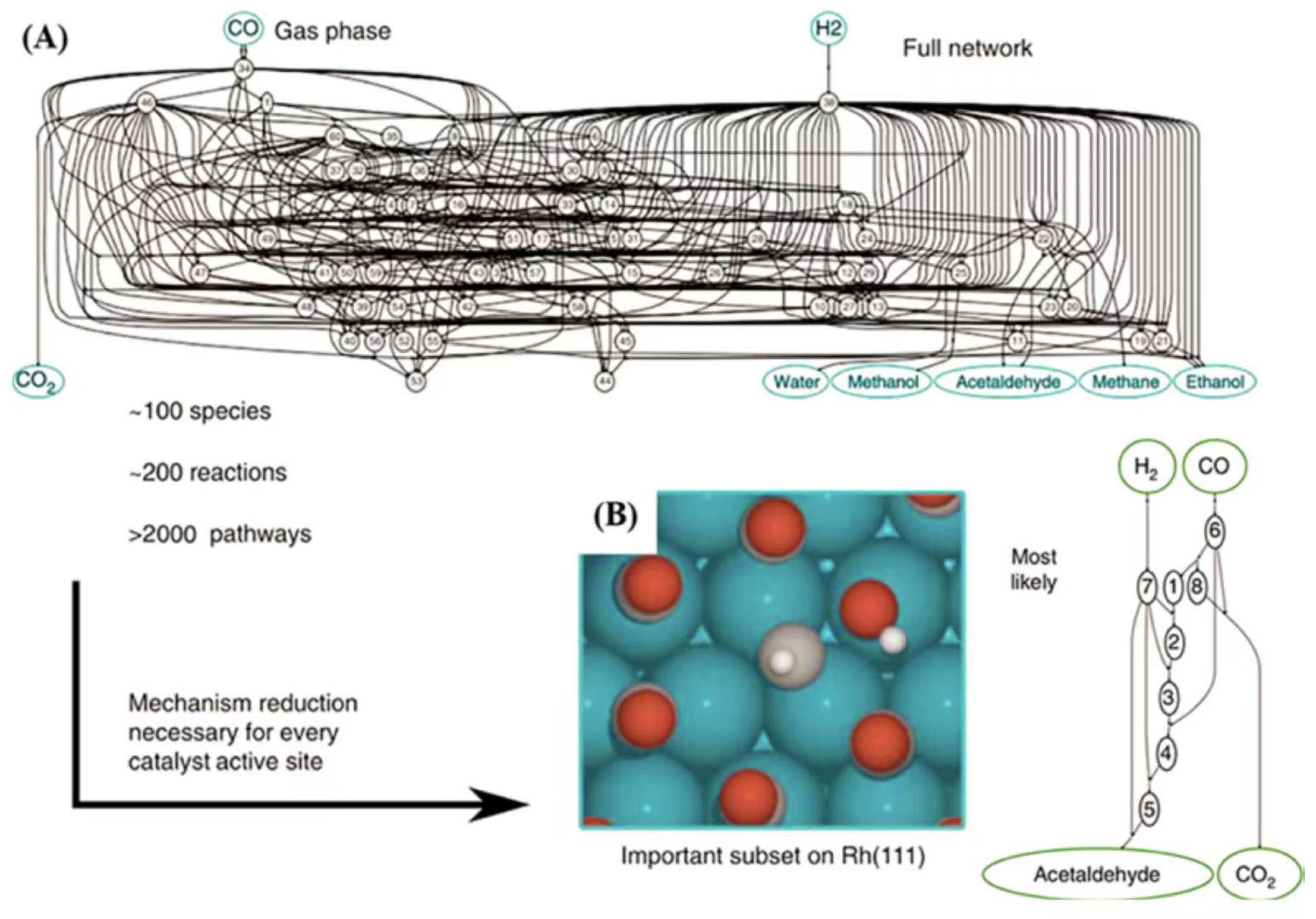
4.4. Accelerating the Discovery of Catalytic Mechanisms
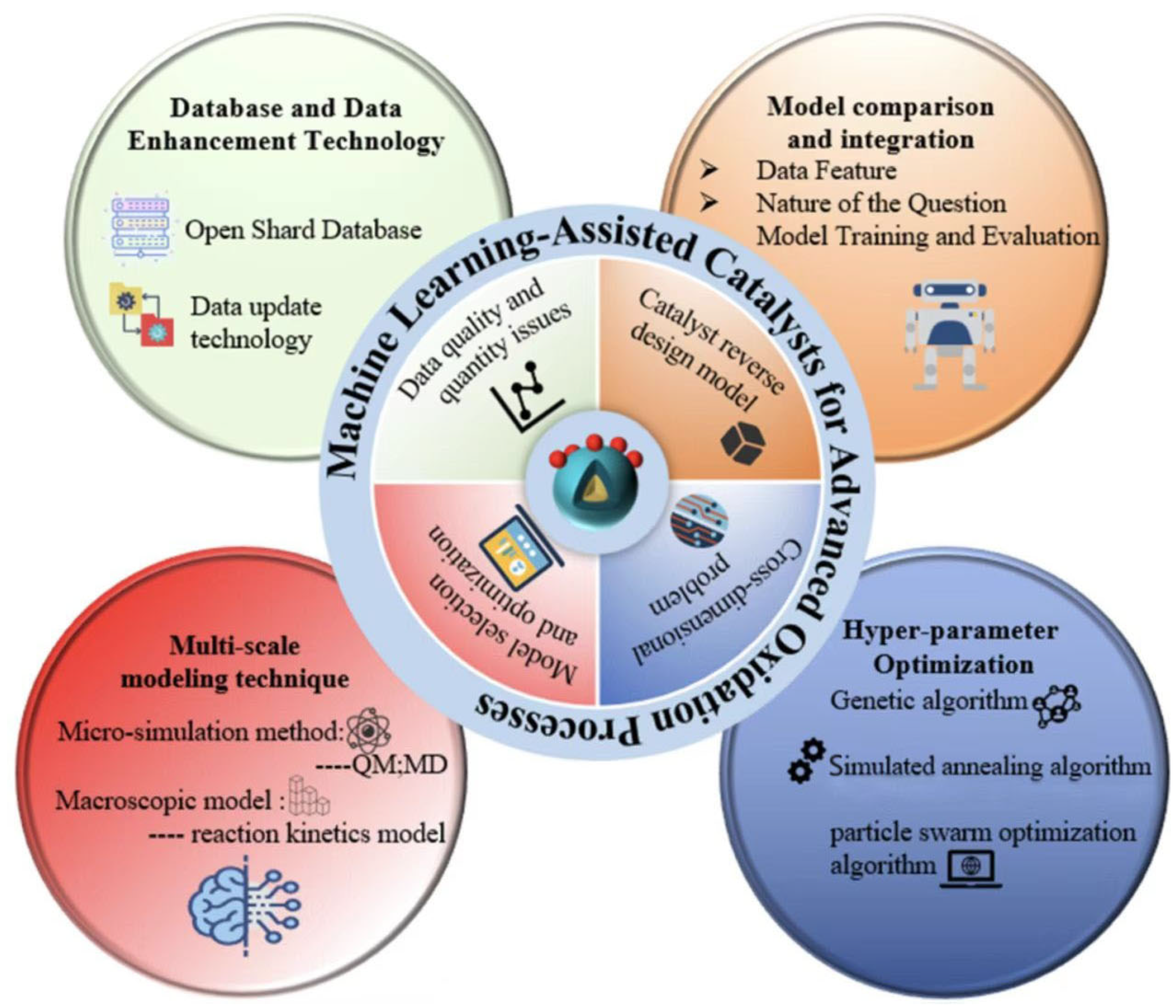
5. Future Development Directions and Challenges
5.1. Shared Database Establishment
5.2. Training and Development of Model
5.3. Cross-Latitude Fusion
5.4. Reverse Design of Catalysts
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Saravanan, A.; Deivayanai, V.; Kumar, P.S.; Rangasamy, G.; Hemavathy, R.; Harshana, T.; Gayathri, N.; Alagumalai, K. A detailed review on advanced oxidation process in treatment of wastewater: Mechanism, challenges and future outlook. Chemosphere 2022, 308, 136524. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhang, H.; Wang, F.F.; Xiong, X.G.Y.; Tian, K.; Sun, Y.B.; Yu, T.T. Application of heterogeneous catalytic ozonation for refractory organics in wastewater. Catalysts 2019, 9, 241. [Google Scholar] [CrossRef]
- Ma, D.S.; Yi, H.; Lai, C.; Liu, X.G.; Huo, X.Q.; An, Z.W.; Li, L.; Fu, Y.K.; Li, B.S.; Zhang, M.M.; et al. Critical review of advanced oxidation processes in organic wastewater treatment. Chemosphere 2021, 275, 130104. [Google Scholar] [CrossRef] [PubMed]
- Fraiha, O.; Hadoudi, N.; Najlae, Z.A.; Salhi, A.; Amhamdi, H.; Mourabit, F.; Ahari, M.H. Comprehensive review on the adsorption of pharmaceutical products from wastewater by clay materials. Desalination Water Treat. 2024, 317, 100114. [Google Scholar] [CrossRef]
- Li, L.X.; Han, J.Z.; Huang, X.H.; Qiu, S.; Liu, X.H.; Liu, L.L.; Zhao, M.J.; Qu, J.W.; Zou, J.L.; Zhang, J. Organic pollutants removal from aqueous solutions using metal-organic frameworks (MOFs) as adsorbents: A review. J. Environ. Chem. Eng. 2023, 6, 111217. [Google Scholar] [CrossRef]
- Chen, H.J.; Xiao, R.H.; Huang, D.L.; Deng, R.; Li, R.J.; Chen, Y.S.; Zhou, W. Three kinds of apatite adsorbents prepared by co-precipitation for Pb (II) and Cd (II) removal from wastewater: Performance, competitive effects and mechanisms. J. Mol. Liq. 2024, 400, 124478. [Google Scholar] [CrossRef]
- Kavitha, E.; Poonguzhali, E.; Nanditha, D.; Kapoor, A.; Arthanareeswaran, G.; Prabhakar, S. Current status and future prospects of membrane separation processes for value recovery from wastewater. Chemosphere 2022, 291, 132690. [Google Scholar] [CrossRef]
- Öcal, Z.B.; Öncel, M.S.; Keskinler, B.; Khataee, A.; Karagündüz, A. Sustainable treatment of boron industry wastewater with precipitation-adsorption hybrid process and recovery of boron species. Process Saf. Environ. Prot. 2024, 182, 719–726. [Google Scholar] [CrossRef]
- Bartolomeu, M.; Neves, M.G.; Faustino, M.A.; Almeida, A. Wastewater chemical contaminants: Remediation by advanced oxidation processes. Photochem. Photobiol. Sci. 2018, 17, 1573–1598. [Google Scholar] [CrossRef]
- Li, L.X.; He, Z.M.; Liang, T.J.; Sheng, T.; Zhang, F.G.; Wu, D.; Ma, F. Colonization of biofilm in wastewater treatment: A review. Environ. Pollut. 2022, 293, 118514. [Google Scholar] [CrossRef]
- Li, L.X.; Han, J.Z.; Huang, L.L.; Liu, L.L.; Qiu, S.; Ding, J.; Liu, X.H.; Zhang, J. Activation of PMS by MIL-53 (Fe)@ AC composites contributes to tetracycline degradation: Properties and mechanisms. Surf. Interfaces 2024, 51, 104521. [Google Scholar] [CrossRef]
- Li, L.X.; Chai, W.; Sun, C.Y.; Huang, L.L.; Sheng, T.; Song, Z.W.; Ma, F. Role of microalgae-bacterial consortium in wastewater treatment: A review. J. Environ. 2024, 360, 121226. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhou, M.W.; Zhang, Y.M.; Li, D.P.; Hou, N.; Zhao, X.Y. Novel strategies for enhancing energy metabolism and wastewater treatment in algae-bacteria symbiotic system through carbon dots-induced photogenerated electrons: The definitive role of accelerated electron transport. Chem. Eng. J. 2024, 500, 157016. [Google Scholar] [CrossRef]
- Crini, G.; Lichtfouse, E. Advantages and disadvantages of techniques used for wastewater treatment. Environ. Chem. Lett. 2019, 17, 145–155. [Google Scholar] [CrossRef]
- Ahmed, M.B.; Zhou, J.L.; Ngo, H.H.; Guo, W.; Thomaidis, N.S.; Xu, J. Progress in the biological and chemical treatment technologies for emerging contaminant removal from wastewater: A critical review. J. Hazard. Mater. 2017, 323, 274–298. [Google Scholar] [CrossRef]
- Ponnusami, A.B.; Sinha, S.; Ashokan, H.; Paul, M.V.; Hariharan, S.P.; Arun, J.; Gopinath, K.P.; Le, Q.H.; Pugazhendhi, A. Advanced oxidation process (AOP) combined biological process for wastewater treatment: A review on advancements, feasibility and practicability of combined techniques. Environ. Res. 2023, 237, 116944. [Google Scholar] [CrossRef]
- Zhang, S.Z.; Chen, S.; Jiang, H. A new tool to predict the advanced oxidation process efficiency: Using machine learning methods to predict the degradation of organic pollutants with Fe-carbon catalyst as a sample. Chem. Eng. Sci. 2023, 280, 119069. [Google Scholar] [CrossRef]
- Liu, S.Q.; Long, Z.Q.; Liu, H.Z.; Zhang, J.; Zhang, G.M.; Liang, J.S. Machine learning predict the degradation efficiency of aqueous refractory organic pollutants by ultrasound-based advanced oxidation processes. J. Water Process. Eng. 2024, 66, 106022. [Google Scholar] [CrossRef]
- Li, L.X.; Xu, H.B.; Zhang, Q.; Zhan, Z.S.; Liang, X.W.; Xing, J. Estimation methods of wetland carbon sink and factors influencing wetland carbon cycle: A review. Carbon Res. 2024, 3, 50. [Google Scholar] [CrossRef]
- Zeng, Q.; Gao, Y.; Guan, K.; Liu, J.; Feng, Z. Machine learning and a computational fluid dynamic approach to estimate phase composition of chemical vapor deposition boron carbide. J. Adv. Ceram. 2021, 10, 537–550. [Google Scholar] [CrossRef]
- Latif, J.; Chen, N.; Saleem, A.; Li, K.; Qin, J.J.; Yang, H.Q.; Jia, H.Z. Machine learning for persistent free radicals in biochar: Dual prediction of contents and types using regression and classification models. Carbon Res. 2024, 3, 39. [Google Scholar] [CrossRef]
- Huang, X.Y.; Wang, H.; Xue, W.H.; Ullah, A.; Xiang, S.; Huang, H.L.; Meng, L.; Ma, G.; Zhang, G.Z. A combined machine learning model for the prediction of time-temperature-transformation diagrams of high-alloy steels. J. Alloys Compd. 2020, 823, 153694. [Google Scholar] [CrossRef]
- Ma, X.; Chen, P.; Chen, C.; Feng, W.; Wang, Y. Prediction of surrounding rock stability of coal roadway based on machine learning and its application. J. Min. Sci. Technol. 2023, 8, 156–165. [Google Scholar]
- Wang, K.; Li, K.; Du, F.; Zhang, X.; Wang, Y.; Zhou, J. Prediction of coal-gas compound dynamic disaster based on convolutional neural network. J. Min. Sci. Technol. 2023, 8, 613–622. [Google Scholar]
- Zhang, X.L.; Zhu, T.; Yuan, B.; Wang, M.D.; Zhang, B.X. Combustion performance of sub-nanometer Pd/S-1 catalyst for ventilation air methane. J. Min. Sci. Technol. 2023, 8, 339–347. [Google Scholar] [CrossRef]
- Fernández-Castro, P.; Vallejo, M.; San Román, M.F.; Ortiz, I. Insight on the fundamentals of advanced oxidation processes. Role and review of the determination methods of reactive oxygen species. J. Chem. Technol. Biotechnol. 2015, 90, 796–820. [Google Scholar] [CrossRef]
- Clennan, E.L.; Pace, A. Advances in singlet oxygen chemistry. Tetrahedron 2005, 61, 6665–6691. [Google Scholar] [CrossRef]
- Battistella, B.; Ray, K. O2 and H2O2 activations at dinuclear Mn and Fe active sites. Coord. Chem. Rev. 2020, 408, 213176. [Google Scholar] [CrossRef]
- Ding, Y.B.; Wang, X.R.; Fu, L.B.; Peng, X.Q.; Pan, C.; Mao, Q.H.; Wang, C.J.; Yan, J.C. Nonradicals induced degradation of organic pollutants by peroxydisulfate (PDS) and peroxymonosulfate (PMS): Recent advances and perspective. Sci. Total Environ. 2021, 765, 142794. [Google Scholar] [CrossRef]
- Wang, J.L.; Wang, S.Z. Reactive species in advanced oxidation processes: Formation, identification and reaction mechanism. Chem. Eng. J. 2020, 401, 1385–8947. [Google Scholar] [CrossRef]
- Pignatello, J.J.; Oliveros, E.; MacKay, A. Advanced oxidation processes for organic contaminant destruction based on the Fenton reaction and related chemistry. Crit. Rev. Environ. Sci. Technol. 2006, 36, 1–84. [Google Scholar] [CrossRef]
- Rachmilovich-Calis, S.; Masarwa, A.; Meyerstein, N.; Meyerstein, D.; Van Eldik, R. New mechanistic aspects of the Fenton reaction. Chem. Eur. J. 2009, 15, 8303–8309. [Google Scholar] [CrossRef] [PubMed]
- Brillas, E.; Sirés, I.; Oturan, M.A. Electro-Fenton process and related electrochemical technologies based on Fenton’s reaction chemistry. Chem. Rev. 2009, 109, 6570–6631. [Google Scholar] [CrossRef]
- Wu, M.Q.; Guo, X.; Cao, Y.D.; Yu, H.C.; Hu, Z.R.; Yang, Y.; Yao, T.J.; Wu, J. Cascading H2O2 photosynthesis and fenton reaction for self-sufficient photo-fenton reactions: A review of recent advances. Chem. Eng. J. 2024, 489, 151091. [Google Scholar] [CrossRef]
- Dai, C.L.; Feng, Z.H.; Hu, Q.M.; Qiu, J.Y.; You, J.H.; Guo, R.; Liu, X.W.; Zhang, H.Z. Recent progress in modification and composite strategies of graphitic carbon nitride as catalysts for heterogeneous photo-Fenton reaction. Mater Sci. Semicond. Process 2023, 167, 107807. [Google Scholar] [CrossRef]
- Dong, C.C.; Fang, W.Z.; Yi, Q.Y.; Zhang, J.L. A comprehensive review on reactive oxygen species (ROS) in advanced oxidation processes (AOPs). Chemosphere 2022, 308, 136205. [Google Scholar] [CrossRef]
- Pandis, P.K.; Kalogirou, C.; Kanellou, E.; Vaitsis, C.; Savvidou, M.G.; Sourkouni, G.; Zorpas, A.A.; Argirusis, C. Key points of advanced oxidation processes (AOPs) for wastewater, organic pollutants and pharmaceutical waste treatment: A mini review. Chem. Eng. 2022, 6, 8. [Google Scholar] [CrossRef]
- Wang, X.C.; Jiang, J.N.; Zhou, M.H.; Dewil, R. Recent advances in H2O2-based advanced oxidation processes for removal of antibiotics from wastewater. Chin. Chem. Lett. 2023, 34, 107621. [Google Scholar] [CrossRef]
- Tian, F.X.; Ye, W.K.; Xu, B.; Hu, X.J.; Ma, S.X.; Lai, F.; Gao, Y.Q.; Xing, H.B.; Xia, W.H.; Wang, B. Comparison of UV-induced AOPs (UV/Cl2, UV/NH2Cl, UV/ClO2 and UV/H2O2) in the degradation of iopamidol: Kinetics, energy requirements and DBPs-related toxicity in sequential disinfection processes. Chem. Eng. J. 2020, 398, 125570. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, S.Y.; He, X.W.; Yang, Y.Y.; Yang, X.T.; Van Hulle, S.W. Comparison of macro and micro-pollutants abatement from biotreated landfill leachate by single ozonation, O3/H2O2, and catalytic ozonation processes. Chem. Eng. J. 2023, 452, 139503. [Google Scholar] [CrossRef]
- Yuan, F.; Hu, C.; Hu, X.X.; Qu, J.H.; Yang, M. Degradation of selected pharmaceuticals in aqueous solution with UV and UV/H2O2. Water Res. 2009, 43, 1766–1774. [Google Scholar] [CrossRef]
- Jung, Y.J.; Kim, W.G.; Yoon, Y.; Kang, J.W.; Hong, M.Y.; Kim, H.W. Removal of amoxicillin by UV and UV/H2O2 processes. Sci. Total Environ. 2012, 420, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Al-Mamun, M.R.; Kader, S.; Islam, M.S.; Khan, M.Z. Photocatalytic activity improvement and application of UV-TiO2 photocatalysis in textile wastewater treatment: A review. J. Environ. Chem. Eng. 2019, 7, 103248. [Google Scholar] [CrossRef]
- ES, E. Photocatalytic degradation of amoxicillin, ampicillin and cloxacillin antibiotics in aqueous solution using UV/TiO2 and UV/H2O2/TiO2 photocatalysis. Desalination 2010, 252, 46–52. [Google Scholar]
- Hua, I.; Hoffmann, M.R. Optimization of ultrasonic irradiation as an advanced oxidation technology. Environ. Sci. Technol. 1997, 31, 2237–2243. [Google Scholar] [CrossRef]
- Lin, C.C.; Hsiao, H.H. Degradation of Rhodamine B in water by heat/persulfate process. J. Taiwan Inst. Chem. Eng. 2022, 132, 104190. [Google Scholar] [CrossRef]
- Xu, Q.H.; Shi, F.; You, H.; Wang, S.T. Integrated remediation for organic-contaminated site by forcing running-water to modify alkali-heat/persulfate via oxidation process transfer. Chemosphere 2021, 262, 128352. [Google Scholar] [CrossRef]
- Yang, Y.; Lu, X.L.; Jiang, J.; Ma, J.; Liu, G.; Cao, Y.; Liu, W.L.; Li, J.; Pang, S.Y.; Kong, X.J.; et al. Degradation of sulfamethoxazole by UV, UV/H2O2 and UV/persulfate (PDS): Formation of oxidation products and effect of bicarbonate. Water Res. 2017, 118, 196–207. [Google Scholar] [CrossRef]
- Xiao, Y.J.; Zhang, L.F.; Zhang, W.; Lim, K.Y.; Webster, R.D.; Lim, T.T. Comparative evaluation of iodoacids removal by UV/persulfate and UV/H2O2 processes. Water Res. 2016, 102, 629–639. [Google Scholar] [CrossRef]
- Chen, G.Y.; Yu, Y.; Liang, L.; Duan, X.G.; Li, R.; Lu, X.K.; Yan, B.B.; Li, N.; Wang, S.B. Remediation of antibiotic wastewater by coupled photocatalytic and persulfate oxidation system: A critical review. J. Hazard. Mater. 2021, 408, 124461. [Google Scholar] [CrossRef]
- Mahesh, B. Machine learning algorithms-a review. Int. J. Sci. Res. 2020, 9, 381–386. [Google Scholar] [CrossRef]
- Mahmoodzadeh, A.; Fakhri, D.; Mohammed, A.H.; Mohammed, A.S.; Ibrahim, H.H.; Rashidi, S. Estimating the effective fracture toughness of a variety of materials using several machine learning models. Eng. Fract. Mech. 2023, 286, 109321. [Google Scholar] [CrossRef]
- Shinde, P.P.; Shah, S. A review of machine learning and deep learning applications. In Proceedings of the 2018 Fourth International Conference on Computing Communication Control and Automation (ICCUBEA), Pune, India, 16–18 August 2018; pp. 1–6. [Google Scholar]
- Feng, Y.; Wang, Q.H.; Wu, D.; Luo, Z.; Chen, X.J.; Zhang, T.Y.; Gao, W. Machine learning aided phase field method for fracture mechanics. Int. J. Eng. Sci. 2021, 169, 103587. [Google Scholar] [CrossRef]
- Sharma, N.; Sharma, R.; Jindal, N. Machine learning and deep learning applications-a vision. Glob. Transit. Proc. 2021, 2, 24–28. [Google Scholar] [CrossRef]
- Zhang, W.Y.; Su, Y.M.; Jiang, Y.F.; Hu, Z.Q.; Bi, J.T.; He, W.T. Data-driven fatigue crack propagation and life prediction of tubular T-joint: A fracture mechanics based machine learning surrogate model. Eng. Fract. Mech. 2024, 311, 110556. [Google Scholar] [CrossRef]
- Wu, Y.; Feng, J. Development and application of artificial neural network. Wirel. Pers. Commun. 2018, 102, 1645–1656. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Meng, X.W.; Li, Q.L.; Ho, S.H. Nitrogen metabolic responses of non-rhizosphere and rhizosphere microbial communities in constructed wetlands under nanoplastics disturbance. J. Hazard. Mater. 2025, 484, 136777. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Guo, M.R.; Wang, Y.N.; Jin, M.; Hou, N.; Wu, H.M. Toxic effects of nanoplastics on biological nitrogen removal in constructed wetlands: Evidence from iron utilization and metabolism. Water Res. 2024, 256, 121577. [Google Scholar] [CrossRef]
- Gu, W.; Yang, B.; Li, D.; Shang, X.; Zhou, Z.; Guo, J. Accelerated design of lead-free high-performance piezoelectric ceramics with high accuracy via machine learning. J. Adv. Ceram. 2023, 12, 1389–1405. [Google Scholar] [CrossRef]
- He, J.; Wang, C.; Li, J.; Liu, C.; Xue, D.; Cao, J.; Bai, Y. Machine learning assisted prediction of dielectric temperature spectrum of ferroelectrics. J. Adv. Ceram. 2023, 12, 1793–1804. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, L.; Ma, F.; Yang, J.X. Effects of arbuscular mycorrhizal fungi on CH4 emissions from rice paddies. Int. J. Phytorem. 2017, 19, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, D.G.; Ma, F.; Wang, G.; You, Y.Q. Recent advances in responses of arbuscular mycorrhizal fungi-Plant symbiosis to engineered nanoparticles. Chemosphere 2022, 286, 131644. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, L.; Ma, F.; Yang, D.G.; You, Y.Q. Earthworm and arbuscular mycorrhiza interactions: Strategies to motivate antioxidant responses and improve soil functionality. Environ. Pollut. 2021, 272, 115980. [Google Scholar] [CrossRef] [PubMed]
- Zahrt, A.F.; Henle, J.J.; Rose, B.T.; Wang, Y.; Darrow, W.T.; Denmark, S.E. Prediction of higher-selectivity catalysts by computer-driven workflow and machine learning. Science 2019, 363, 6424. [Google Scholar] [CrossRef]
- Sun, M.; Dougherty, A.W.; Huang, B.; Li, Y.; Yan, C.H. Accelerating atomic catalyst discovery by theoretical calculations-machine learning strategy. Adv. Energy Mater. 2020, 10, 1903949. [Google Scholar] [CrossRef]
- Fang, Z.L.; Li, S.Y.; Zhang, Y.J.; Wang, Y.X.; Meng, K.; Huang, C.Y.; Sun, S.R. The DFT and machine learning method accelerated the discovery of DMSCs with high ORR and OER catalytic activities. J. Phys. Chem. Lett. 2024, 15, 281–289. [Google Scholar] [CrossRef]
- Duan, X.; Li, Y.; Zhao, J.; Zhang, M.Y.; Wang, X.P.; Zhang, L.; Ma, X.X.; Qu, Y.; Zhang, P.F. Machine Learning Accelerated Discovery of Entropy-Stabilized Oxide Catalysts for Catalytic Oxidation. J. Am. Chem. Soc. 2024, 147, 651–661. [Google Scholar] [CrossRef]
- Liu, J.Y.; Guo, H.Q.; Xiong, Y.L.; Chen, X.; Yu, Y.F.; Wang, C.H. Rational design of Pt-anchored single-atom alloy electrocatalysts for NO-to-NH3 conversion by density functional theory and machine learning. Chin. J. Catal. 2024, 62, 243–253. [Google Scholar] [CrossRef]
- Acosta-Angulo, B.; Lara-Ramos, J.; Niño-Vargas, A.; Diaz-Angulo, J.; Benavides-Guerrero, J.; Bhattacharya, A.; Cloutier, S.; Machuca-Martínez, F. Unveiling the potential of machine learning in cost-effective degradation of pharmaceutically active compounds: A stirred photo-reactor study. Chemosphere 2024, 358, 142222. [Google Scholar] [CrossRef]
- Xie, Y.; Yang, F.; Liu, J.; Ren, M.J. Prediction of the degradation of organic pollutants by metal-activated peracetic acid using machine learning. J. Environ. Chem. Eng. 2024, 12, 113700. [Google Scholar] [CrossRef]
- Yadav, K.K.; Elboughdiri, N.; Fetimi, A.; Bhutto, J.K.; Merouani, S.; Tamam, N.; Alreshidi, M.A.; Rodríguez-Díaz, J.M.; Benguerba, Y. Enhanced wastewater treatment by catalytic persulfate activation with protonated hydroxylamine-assisted iron: Insights from a deep learning-based numerical investigation. Chemosphere 2024, 360, 142367. [Google Scholar] [CrossRef] [PubMed]
- Back, S.; Yoon, J.; Tian, N.; Zhong, W.; Tran, K.; Ulissi, Z.W. Convolutional Neural Network of Atomic Surface Structures to Predict Binding Energies for High-Throughput Screening of Catalysts. J. Phys. Chem. Lett. 2019, 10, 4401–4408. [Google Scholar] [CrossRef] [PubMed]
- Ooka, H.; Wintzer, M.E.; Nakamura, R. Non-Zero Binding Enhances Kinetics of Catalysis: Machine Learning Analysis on the Experimental Hydrogen Binding Energy of Platinum. ACS Catal. 2021, 11, 6298–6303. [Google Scholar] [CrossRef]
- Bai, Y.; Wilbraham, L.; Slater, B.J.; Zwijnenburg, M.A.; Sprick, R.S.; Cooper, A.I. Accelerated Discovery of Organic Polymer Photocatalysts for Hydrogen Evolution from Water through the Integration of Experiment and Theory. J. Am. Chem. Soc. 2019, 141, 9063–9071. [Google Scholar] [CrossRef]
- Mai, H.; Le, T.C.; Chen, D.; Chen, D.H.; David, A.W.; Rachel, A.C. Machine learning for electrocatalyst and photocatalyst design and discovery. Chem. Rev. 2022, 122, 13478–13515. [Google Scholar] [CrossRef]
- Jiang, Z.; Hu, J.; Zhang, X.; Zhao, Y.; Fan, X.; Zhong, S.; Zhang, H.; Yu, X. A generalized predictive model for TiO2-Catalyzed photo-degradation rate constants of water contaminants through artificial neural network. Environ. Res. 2020, 187, 109697. [Google Scholar] [CrossRef]
- Jiang, Z.; Hu, J.; Samia, A.; Yu, X. Predicting active sites in photocatalytic degradation process using an interpretable molecular-image combined convolutional neural network. Catalysts 2022, 12, 746. [Google Scholar] [CrossRef]
- Shang, Q.; Liu, X.; Zhang, M.; Zhang, P.; Ling, Y.; Cui, G.; Liu, W.; Shi, X.; Yue, J.; Tang, B. Photocatalytic degradation of ofloxacin antibiotic wastewater using TS-1/C3N4 composite photocatalyst: Reaction performance optimisation and estimation of wastewater component synergistic effect by artificial neural network and genetic algorithm. Chem. Eng. J. 2022, 443, 136354. [Google Scholar] [CrossRef]
- Alshehri, F.; Shahfahad; Rahman, A. Optimizing Machine Learning Models with Bayesian Techniques for Prediction of Groundwater Quality Index in Southwest Saudi Arabia. Earth Syst. Environ. 2024, 8, 1417–1436. [Google Scholar] [CrossRef]
- Biswas, P.; Wu, C.Y. Nanoparticles and the environment. J. Air Waste Manag. 2005, 55, 708–746. [Google Scholar] [CrossRef]
- Lauher, J.W.; Fowler, F.W.; Goroff, N.S. Single-crystal-to-single-crystal topochemical polymerizations by design. Acc. Chem. Res. 2008, 41, 1215–1229. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Fryling, M.A.; McCreery, R.L. Electron transfer kinetics at modified carbon electrode surfaces: The role of specific surface sites. Anal. Chem. 1995, 67, 3115–3122. [Google Scholar] [CrossRef]
- Jinnouchi, R.; Asahi, R. Predicting catalytic activity of nanoparticles by a DFT-aided machine-learning algorithm. J. Phys. Chem. Lett. 2017, 8, 4279–4283. [Google Scholar] [CrossRef]
- Li, Z.; Wang, S.W.; Chin, W.S.; Achenie, L.E.; Xin, H.L. High-throughput screening of bimetallic catalysts enabled by machine learning. J. Mater. Chem. A 2017, 5, 24131–24138. [Google Scholar] [CrossRef]
- Tamtaji, M.; Kazemeini, M.; Abdi, J. DFT and machine learning studies on a multi-functional single-atom catalyst for enhanced oxygen and hydrogen evolution as well as CO2 reduction reactions. Int. J. Hydrogen Energy 2024, 80, 1075–1083. [Google Scholar] [CrossRef]
- Deng, C.F.; Su, Y.; Li, F.H.; Shen, W.F.; Chen, Z.F.; Tang, Q. Understanding activity origin for the oxygen reduction reaction on bi-atom catalysts by DFT studies and machine-learning. J. Mater. Chem. A 2020, 8, 24563–24571. [Google Scholar] [CrossRef]
- Ishioka, S.; Fujiwara, A.; Nakanowatari, S.; Takahashi, L.; Taniike, T.; Takahashi, K. Designing catalyst descriptors for machine learning in oxidative coupling of methane. ACS Catal. 2022, 12, 11541–11546. [Google Scholar] [CrossRef]
- Liu, W.X.; Zhu, Y.; Wu, Y.Q.; Chen, C.; Hong, Y.; Yue, Y.N.; Zhang, J.C.; Hou, B. Molecular dynamics and machine learning in catalysts. Catalysts 2021, 11, 1129. [Google Scholar] [CrossRef]
- Mortazavi, B.; Zhuang, X.; Rabczuk, T.; Shapeev, A.V. Atomistic modeling of the mechanical properties: The rise of machine learning interatomic potentials. Mater. Horiz. 2023, 10, 1956–1968. [Google Scholar] [CrossRef]
- Chen, B.W.J.; Zhang, X.L.; Zhang, J. Accelerating explicit solvent models of heterogeneous catalysts with machine learning interatomic potentials. Chem. Sci. 2023, 14, 8338–8354. [Google Scholar] [CrossRef]
- Chan, H.; Narayanan, B.; Cherukara, M.J.; Sen, F.G.; Sasikumar, K.; Gray, S.K.; Chan, M.K. Sankaranarayanan SK. Machine learning classical interatomic potentials for molecular dynamics from first-principles training data. J. Phys. Chem. C 2019, 123, 6941–6957. [Google Scholar] [CrossRef]
- Yu, W.; Ji, C.Y.; Wan, X.H.; Zhang, Z.F.; Robertson, J.; Liu, S.; Guo, Y.Z. Machine-learning-based interatomic potentials for advanced manufacturing. IJMSD 2021, 1, 159–172. [Google Scholar] [CrossRef]
- Sheppard, D.; Terrell, R.; Henkelman, G. Optimization methods for finding minimum energy paths. J. Chem. Phys. 2008, 128, 13. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, B.R.; Esterhuizen, J.; Liu, J.X.; Bartel, C.J.; Sutton, C. Machine learning for heterogeneous catalyst design and discovery. AIChE J. 2018, 64, 2311–2323. [Google Scholar] [CrossRef]
- Ma, S.; Liu, Z.P. Machine learning for atomic simulation and activity prediction in heterogeneous catalysis: Current status and future. ACS Catal. 2020, 10, 13213–13226. [Google Scholar] [CrossRef]
- Mohan, O.; Choksi, T.S.; Lapkin, A.A. The design and optimization of heterogeneous catalysts using computational methods. Catal. Sci. Technol. 2024, 14, 515–532. [Google Scholar]
- Li, H.B.; Jiao, Y.; Davey, K.; Qiao, S.Z. Data-driven machine learning for understanding surface structures of heterogeneous catalysts. Angew. Chem. Int. Ed. 2023, 62, e202216383. [Google Scholar]
- Guan, Y.N.; Chaffart, D.; Liu, G.H.; Tan, Z.Y.; Zhang, D.S.; Wang, Y.J.; Li, J.D.; Ricardez-Sandoval, L. Machine learning in solid heterogeneous catalysis: Recent developments, challenges and perspectives. Chem. Eng. Sci. 2022, 248, 117224. [Google Scholar] [CrossRef]
- Yang, W.; Fidelis, T.T.; Sun, W.H. Machine learning in catalysis, from proposal to practicing. ACS Omega 2019, 5, 83–88. [Google Scholar] [CrossRef]
- Zhang, T.S.; Shi, Y.B.; Liu, Y.; Yang, J.Y.; Guo, M.R.; Bai, S.W.; Hou, N.; Zhao, X.Y. A study on microbial mechanism in response to different nanoplastics concentrations in constructed wetland and its carbon footprints analysis. Chem. Eng. J. 2024, 480, 148023. [Google Scholar]
- Wang, R.P.; Chen, H.L.; He, Z.X.; Zhang, S.Y.; Wang, K.; Ren, N.Q.; Ho, S.H. Discovery of an end-to-end pattern for contaminant-oriented advanced oxidation processes catalyzed by biochar with explainable machine learning. Environ. Sci. Technol. 2024, 58, 16867–16876. [Google Scholar] [CrossRef] [PubMed]
- Pascacio, P.; Vicente, D.J.; Salazar, F.; Guerra-Rodríguez, S.; Rodríguez-Chueca, J. Predictive modeling of Enterococcus sp. removal with limited data from different advanced oxidation processes: A machine learning approach. J. Environ. Chem. Eng. 2024, 12, 112530. [Google Scholar] [CrossRef]
- Liu, Y.H.; Li, Z.Y.; Cao, C.Q.; Zhang, X.Z.; Meng, S.Q.; Davari, M.D.; Xu, H.J.; Ji, Y.; Schwaneberg, U.; Liu, L. Engineering of Substrate Tunnel of P450 CYP116B3 though Machine Learning. Catalysts 2023, 13, 1228. [Google Scholar] [CrossRef]
- Jaffari, Z.H.; Abbas, A.; Lam, S.M.; Park, S.; Chon, K.; Kim, E.S.; Cho, K.H. Machine learning approaches to predict the photocatalytic performance of bismuth ferrite-based materials in the removal of malachite green. J. Hazard. Mater. 2023, 442, 130031. [Google Scholar] [CrossRef]
- Oviedo, L.R.; Durzian, D.M.; Montagner, G.E.; Ruiz, Y.P.; Galembeck, A.; Pavoski, G.; Espinosa, D.C.; Nora, L.D.D.; da Silva, W.L. Supported heterogeneous catalyst of the copper oxide nanoparticles and nanozeolite for binary dyes mixture degradation: Machine learning and experimental design. J. Mol. Liq. 2024, 402, 124763. [Google Scholar] [CrossRef]
- Liu, M.H.; Li, N.; Meng, S.Y.; Yang, S.L.; Jing, B.J.; Zhang, J.Y.; Jiang, J.Z.; Qiu, S.; Deng, F.X. Bio-inspired Cu2O cathode for O2 capturing and oxidation boosting in electro-Fenton for sulfathiazole decay. J. Hazard. Mater. 2024, 478, 135484. [Google Scholar] [CrossRef]
- Li, L.X.; Liang, T.J.; Zhao, M.J.; Lv, Y.; Song, Z.W.; Sheng, T.; Ma, F. A review on mycelial pellets as biological carriers: Wastewater treatment and recovery for resource and energy. Bioresour. Technol. 2022, 355, 127200. [Google Scholar] [CrossRef]
- Yazdi, A.A.; Pirbazari, A.E.; Saraei, F.E.; Esmaeili, A.; Pirbazari, A.E.; Kohnehsari, A.A.; Derakhshesh, A. Design of 2D/2D β-Ni (OH)2/ZnO heterostructures via photocatalytic deposition of nickel for sonophotocatalytic degradation of tetracycline and modeling with three supervised machine learning algorithms. Chemosphere 2024, 352, 141328. [Google Scholar]
- Cüce, H.; Özçelik, D. Application of machine learning (ML) and artificial intelligence (AI)-based tools for modelling and enhancing sustainable optimization of the classical/photo-Fenton processes for the landfill leachate treatment. SDGs 2022, 14, 11261. [Google Scholar] [CrossRef]
- Yu, G.F.; Wu, Y.Q.; Cao, H.B.; Ge, Q.F.; Dai, Q.; Sun, S.H.; Xie, Y.B. Insights into the mechanism of ozone activation and singlet oxygen generation on N-doped defective nanocarbons: A DFT and machine learning study. Environ. Sci. Technol. 2022, 56, 7853–7863. [Google Scholar] [CrossRef]
- Liu, J.X.; Jia, H.; Mei, M.; Wang, T.; Chen, S.; Li, J.P. Efficient degradation of diclofenac by digestate-derived biochar catalyzed peroxymonosulfate oxidation: Performance, machine learning prediction, and mechanism. Process Saf. Environ. Prot. 2022, 167, 77–88. [Google Scholar] [CrossRef]
- Wang, L.J.; Yang, T.Y.; Wei, M.J.; Guan, R.Q.; Wei, W.; Jiang, J.Z. Machine learning and DFT dual-guidance of carbon dots implanted SrTiO3 hollow nanosphere for efficient all-pH-value photocatalysis. J Mater. Sci. Technol. 2025, 217, 169–181. [Google Scholar] [CrossRef]
- Bergstra, J.; Komer, B.; Eliasmith, C.; Yamins, D.; Cox, D.D. Hyperopt: A python library for model selection and hyperparameter optimization. CSD 2015, 8, 014008. [Google Scholar] [CrossRef]
- Liu, X.J.; Xu, P.C.; Zhao, J.J.; Lu, W.C.; Li, M.J.; Wang, G. Material machine learning for alloys: Applications, challenges and perspectives. J. Alloys Compd. 2022, 921, 165984. [Google Scholar] [CrossRef]
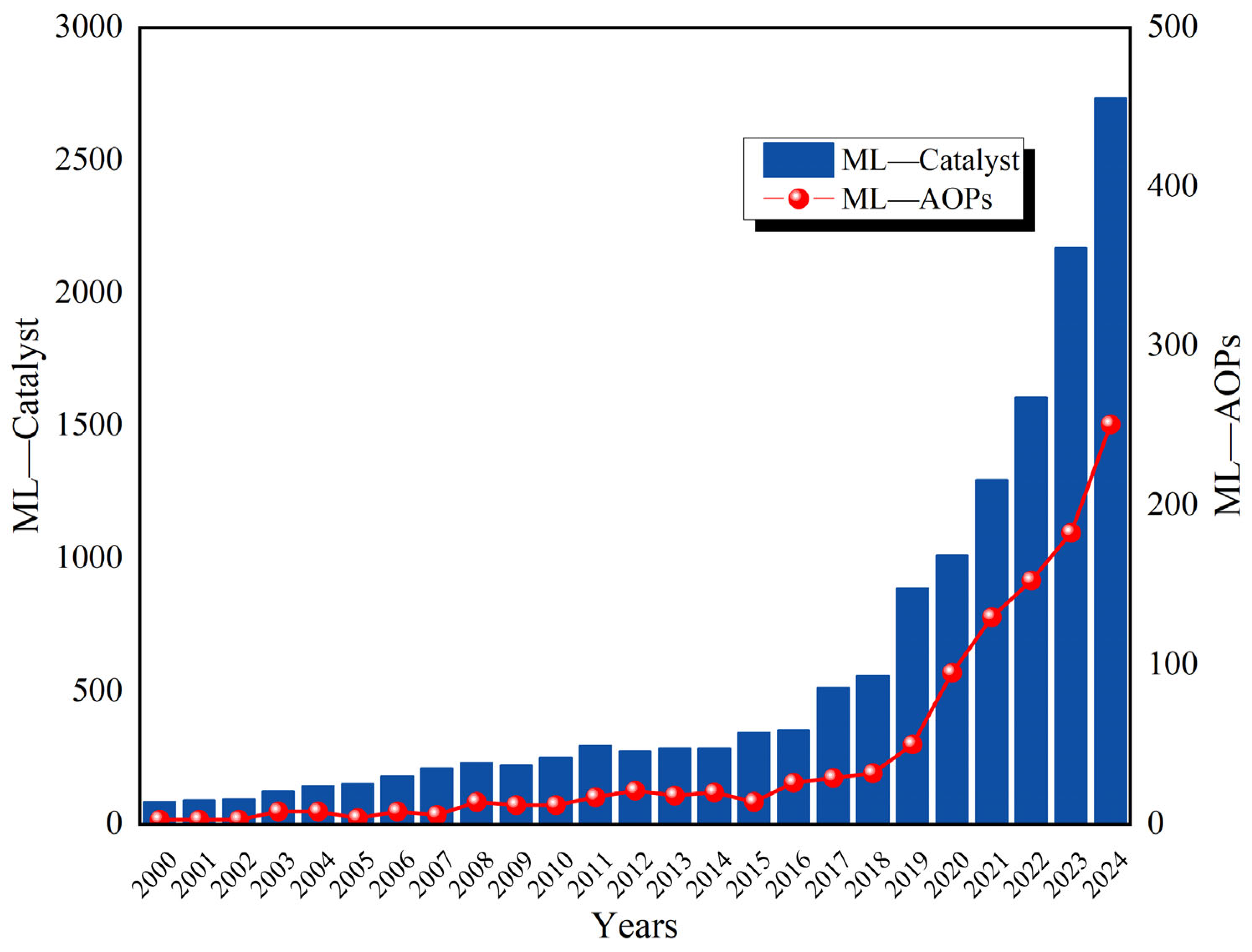
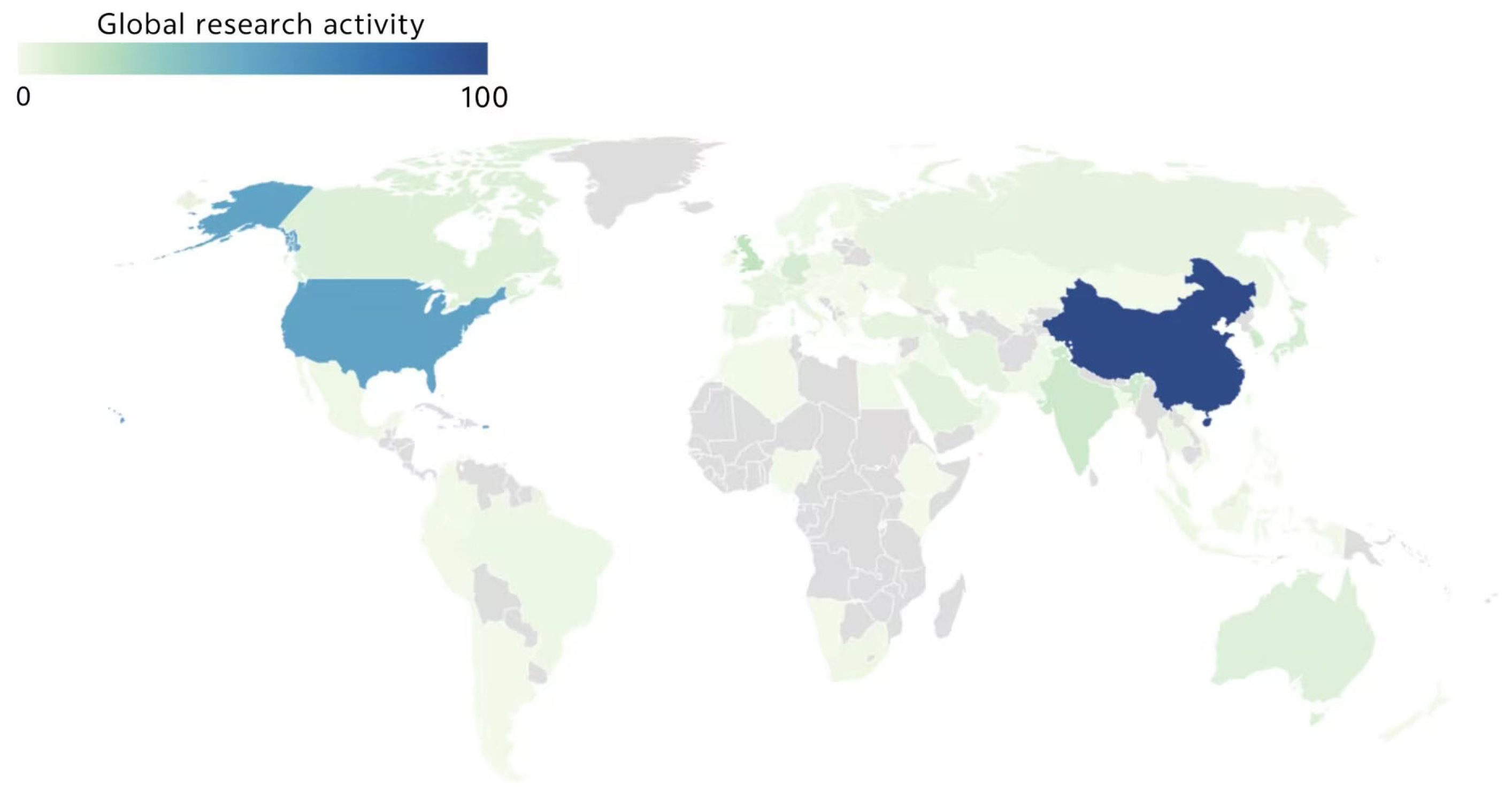
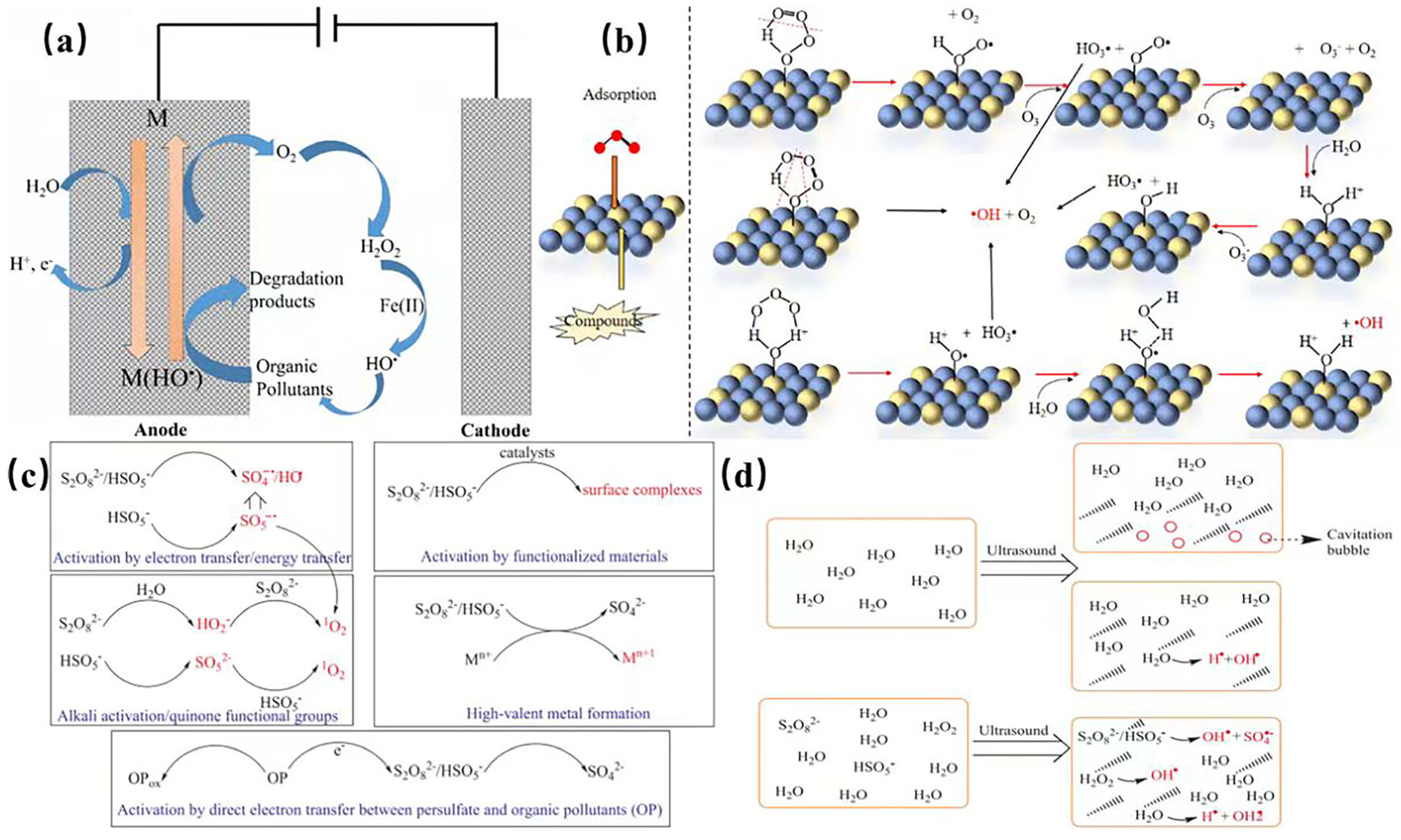

| AOP Types | ROS Types | Other Occurring Mechanisms | Reference |
|---|---|---|---|
| Fenton reaction | •OH | Iron coagulation Iron sludge-induced adsorption | [31,32,33] |
| Photo-Fenton reaction | •OH | Iron coagulation Iron sludge-induced adsorption UV photolysis | [34,35] |
| O3 | •OH | Direct O3 oxidation | [36,37] |
| O3/H2O2 | •OH | Direct O3 oxidation H2O2oxidation | [38,39,40] |
| O3/UV | •OH | UV photolysis | [39,40] |
| UV/H2O2 | •OH | UV photolysis H2O2oxidation | [41,42] |
| UV/TiO2 | •OH | UV photolysis | [43,44] |
| Ultrasonic irradiation | •OH | Acoustic cavitation generates transient high temperatures (≥5000 K) and pressures (≥1000 atm) and produce H+ and HO2−, besides OH− | [45] |
| Heat/persulfate | SO4− | Persulfate oxidation | [46,47] |
| UV/persulfate | SO4− | Persulfate oxidation UV photolysis | [48] |
| Fe(II)/persulfate | SO4− | Persulfate oxidation Iron coagulation Iron sludge-induced adsorption | [49,50] |
| OH−/persulfate | SO4−/•OH | Persulfate oxidation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, Q.; Wang, X.; Xu, D.; Liu, H.; Zhang, H.; Yu, Q.; Bi, Y.; Li, L. Machine Learning-Assisted Catalysts for Advanced Oxidation Processes: Progress, Challenges, and Prospects. Catalysts 2025, 15, 282. https://doi.org/10.3390/catal15030282
Yuan Q, Wang X, Xu D, Liu H, Zhang H, Yu Q, Bi Y, Li L. Machine Learning-Assisted Catalysts for Advanced Oxidation Processes: Progress, Challenges, and Prospects. Catalysts. 2025; 15(3):282. https://doi.org/10.3390/catal15030282
Chicago/Turabian StyleYuan, Qinghui, Xiaobei Wang, Dongdong Xu, Hongyan Liu, Hanwen Zhang, Qian Yu, Yanliang Bi, and Lixin Li. 2025. "Machine Learning-Assisted Catalysts for Advanced Oxidation Processes: Progress, Challenges, and Prospects" Catalysts 15, no. 3: 282. https://doi.org/10.3390/catal15030282
APA StyleYuan, Q., Wang, X., Xu, D., Liu, H., Zhang, H., Yu, Q., Bi, Y., & Li, L. (2025). Machine Learning-Assisted Catalysts for Advanced Oxidation Processes: Progress, Challenges, and Prospects. Catalysts, 15(3), 282. https://doi.org/10.3390/catal15030282