
Selective Catalytic Reduction of NO by NH3 and SOx Poisoning Mechanisms on Mn3O4 Catalysts: A Density Functional Investigation

 ,
, 
Abstract

1. Introduction
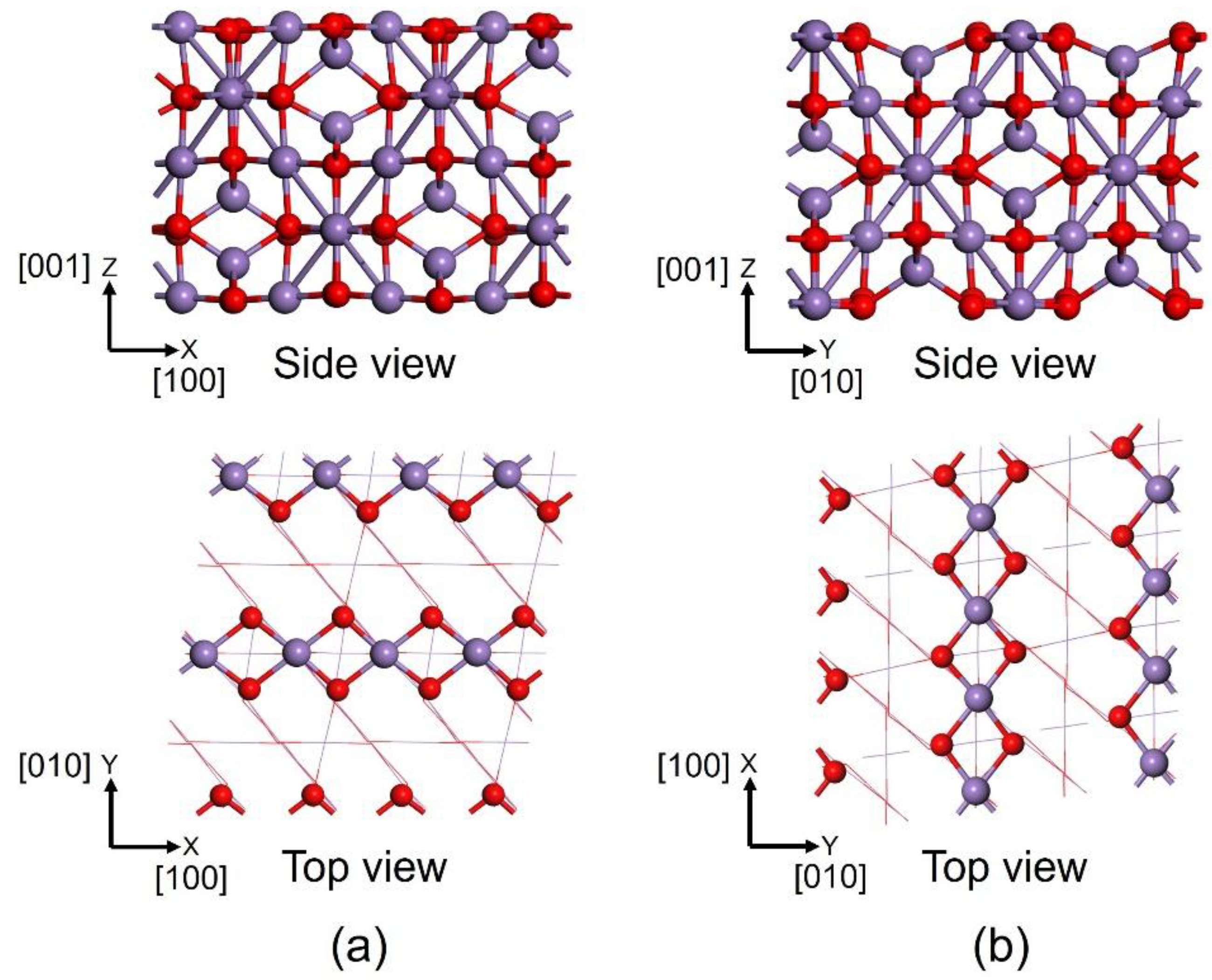
2. Results and Discussion
2.1. SCR Mechanism
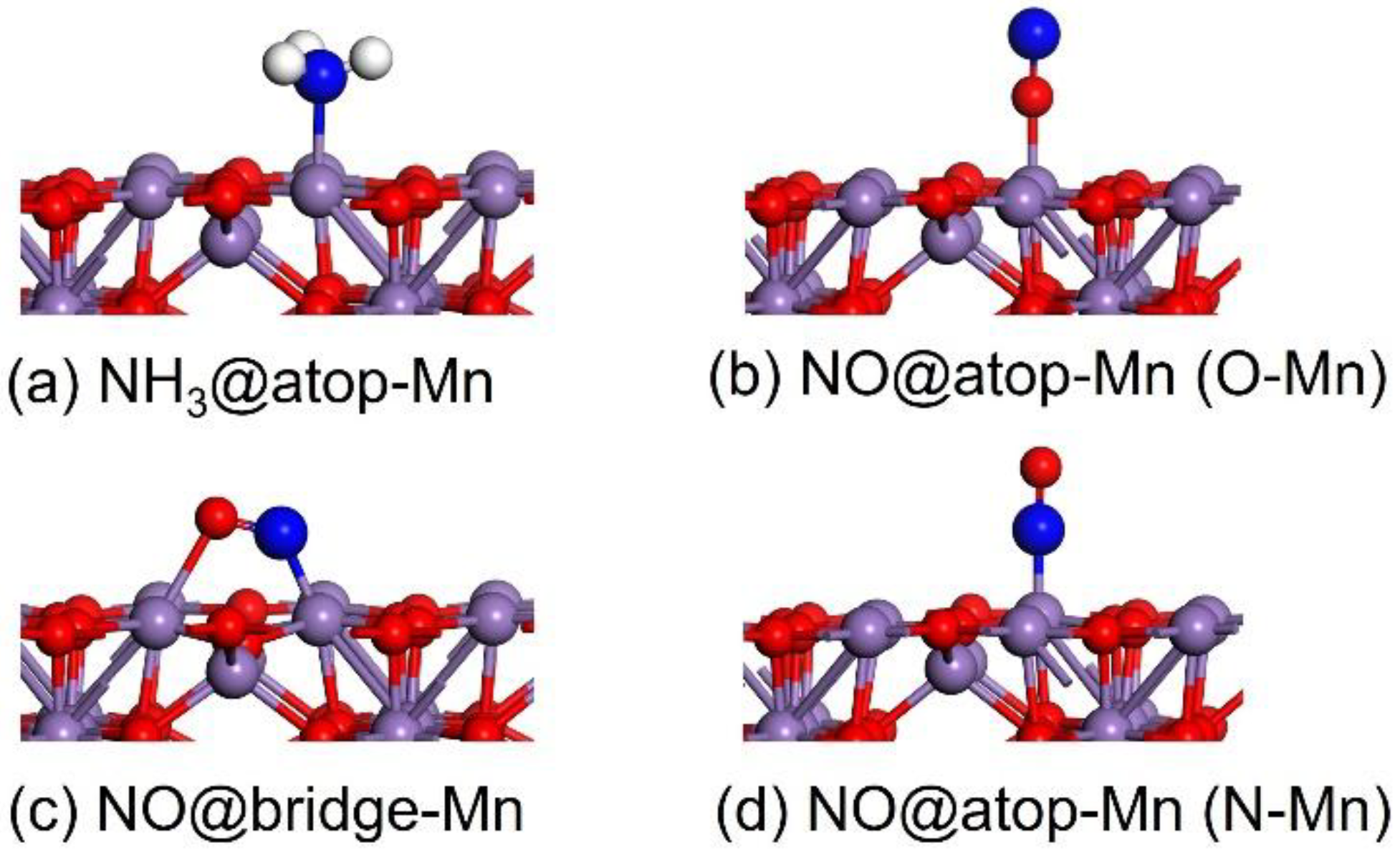
2.1.1. Adsorption Properties of NH3 and NO
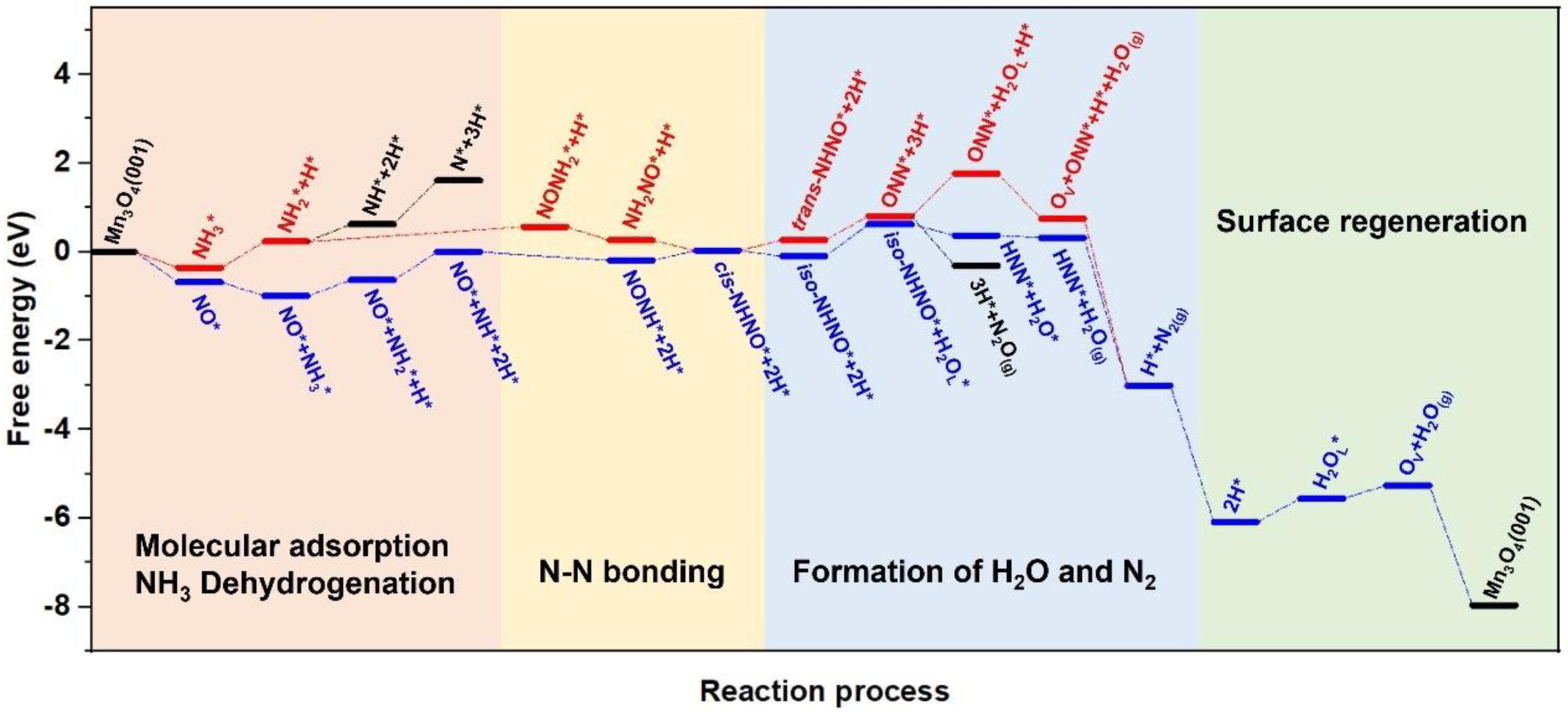
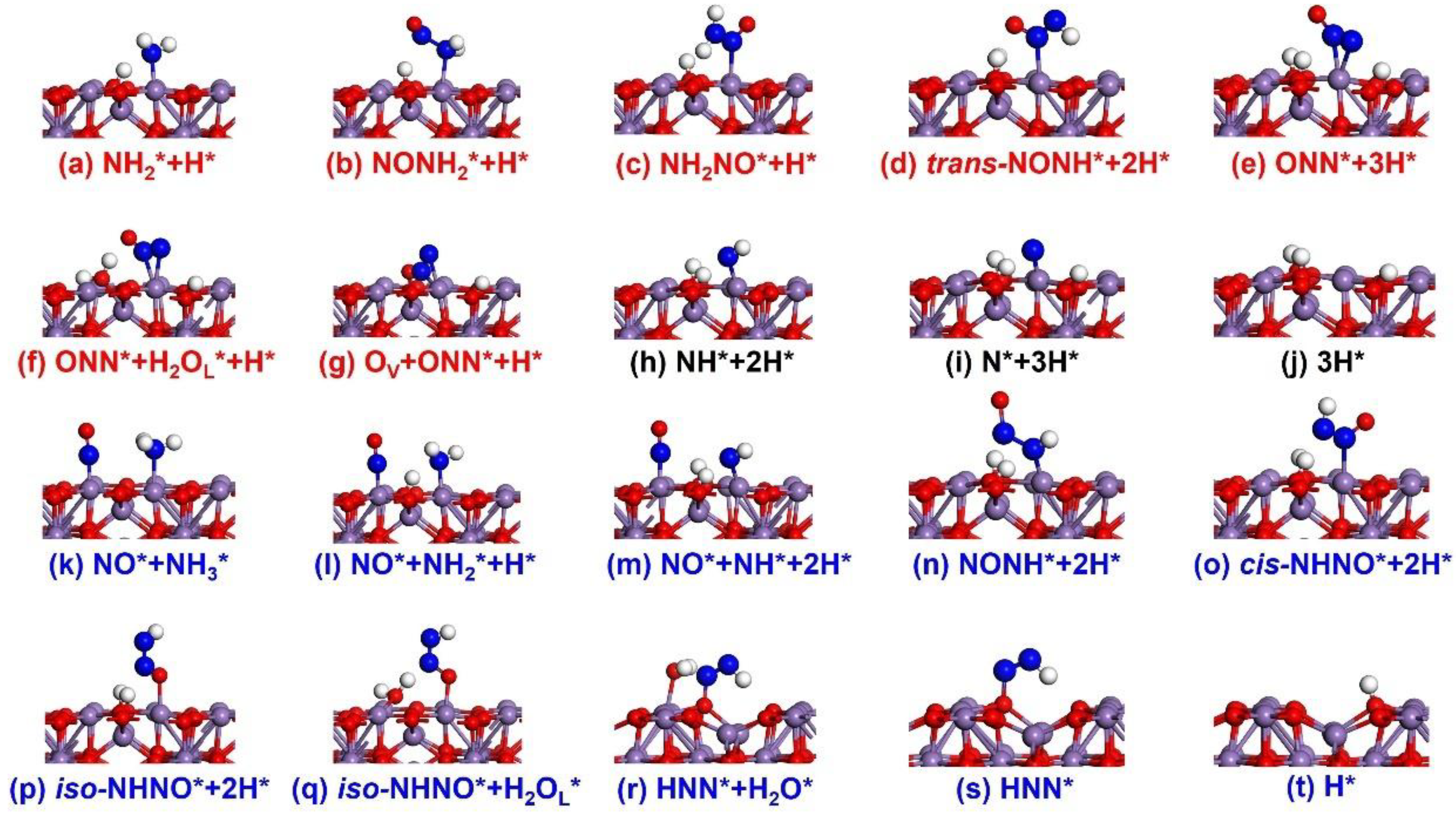
2.1.2. Reaction Pathways
- The E-R Mechanism
- The L-H Mechanism
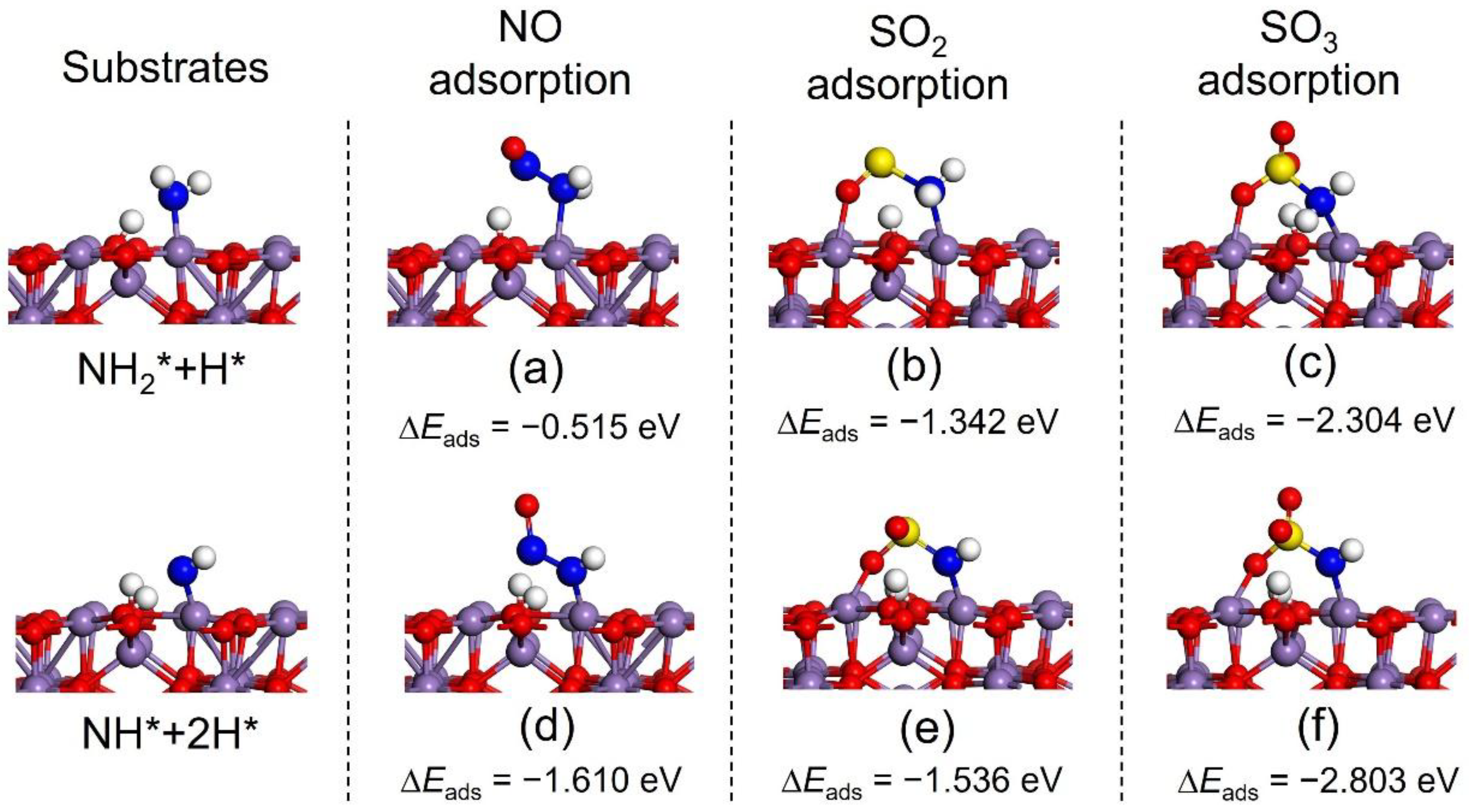
2.2. SOx Poisoning Mechanisms
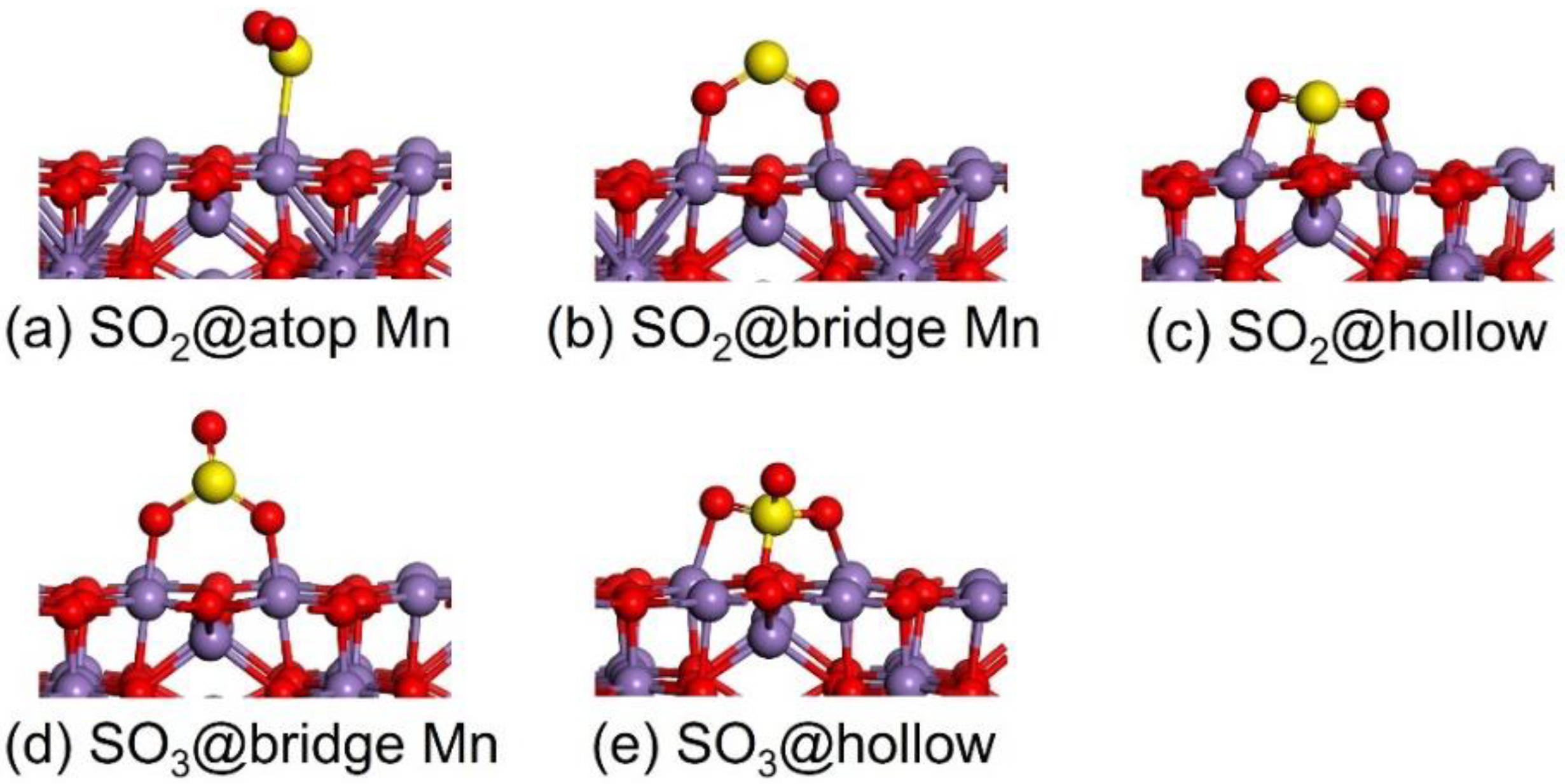
2.2.1. Adsorption Properties of SO2 and SO3
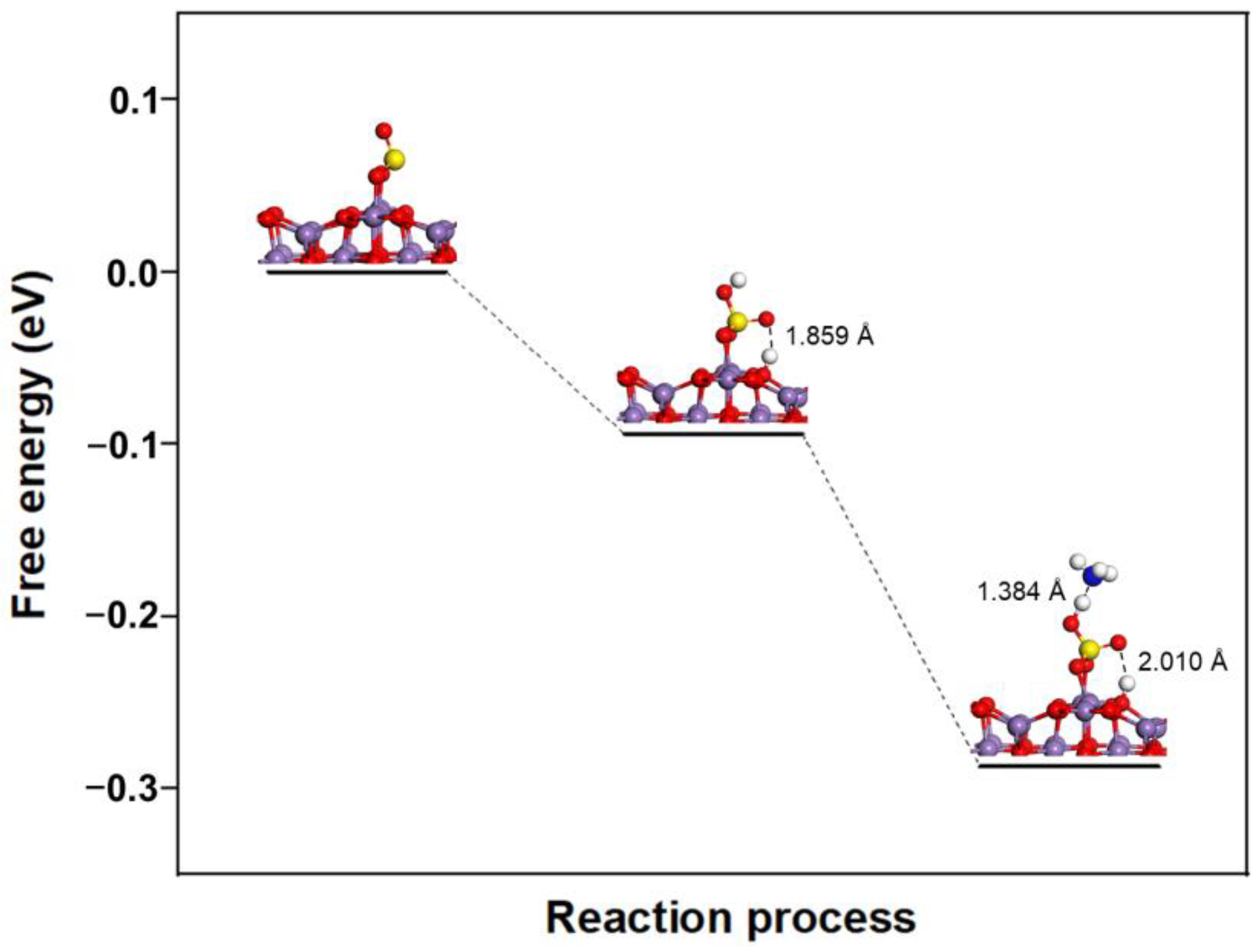
2.2.2. Formation of SOx-NH and SOx-NH2 Complexes
2.2.3. Formation of NH4HSO4
3. Computation Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Feng, X.; Zhu, J.; Song, K.; Zeng, J.; Zhou, X.; Guo, X.; Lin, K.; Zhang, C.; Xie, C.; Shi, J.-W. Insight into the Reasons for Enhanced NH3-SCR Activity and SO2 Tolerance of Mn-Co Layered Oxides. Sep. Purif. Technol. 2024, 336, 126285. [Google Scholar] [CrossRef]
- Liu, W.; Yang, Z.; Luan, X.; Zhai, Y.; Zhang, J.; Wang, L.; Wang, Z. Optimizing the Catalytic Performance of MnCo2O4 Spinel Catalysts for the NH3-SCR: Structure-Activity Relationships and Reaction Mechanism. Appl. Surf. Sci. 2024, 674, 160915. [Google Scholar] [CrossRef]
- Chen, W.; Zou, R.; Wang, X. Toward an Atomic-Level Understanding of the Catalytic Mechanism of Selective Catalytic Reduction of NOx with NH3. ACS Catal. 2022, 12, 14347–14375. [Google Scholar] [CrossRef]
- Shi, Z.; Peng, Q.; E, J.; Xie, B.; Wei, J.; Yin, R.; Fu, G. Mechanism, Performance and Modification Methods for NH3-SCR Catalysts: A Review. Fuel 2023, 331, 125885. [Google Scholar] [CrossRef]
- Park, E.D. Recent Progress on Low-Temperature Selective Catalytic Reduction of NOx with Ammonia. Molecules 2024, 29, 4506. [Google Scholar] [CrossRef]
- Guan, B.; Jiang, H.; Wei, Y.; Liu, Z.; Wu, X.; Lin, H.; Huang, Z. Density Functional Theory Researches for Atomic Structure, Properties Prediction, and Rational Design of Selective Catalytic Reduction Catalysts: Current Progresses and Future Perspectives. Mol. Catal. 2021, 510, 111704. [Google Scholar] [CrossRef]
- Liu, C.; Shi, J.-W.; Gao, C.; Niu, C. Manganese Oxide-Based Catalysts for Low-Temperature Selective Catalytic Reduction of NOx with NH3: A Review. Appl. Catal. Gen. 2016, 522, 54–69. [Google Scholar] [CrossRef]
- Han, L.; Cai, S.; Gao, M.; Hasegawa, J.; Wang, P.; Zhang, J.; Shi, L.; Zhang, D. Selective Catalytic Reduction of NOx with NH3 by Using Novel Catalysts: State of the Art and Future Prospects. Chem. Rev. 2019, 119, 10916–10976. [Google Scholar] [CrossRef]
- Paolucci, C.; Khurana, I.; Parekh, A.A.; Li, S.; Shih, A.J.; Li, H.; Di Iorio, J.R.; Albarracin-Caballero, J.D.; Yezerets, A.; Miller, J.T.; et al. Dynamic Multinuclear Sites Formed by Mobilized Copper Ions in NOxSelective Catalytic Reduction. Science 2017, 357, 898–903. [Google Scholar] [CrossRef]
- Wang, X.; Du, X.; Liu, S.; Yang, G.; Chen, Y.; Zhang, L.; Tu, X. Understanding the Deposition and Reaction Mechanism of Ammonium Bisulfate on a Vanadia SCR Catalyst: A Combined DFT and Experimental Study. Appl. Catal. B Environ. 2020, 260, 118168. [Google Scholar] [CrossRef]
- Zhu, M.; Lai, J.-K.; Tumuluri, U.; Wu, Z.; Wachs, I.E. Nature of Active Sites and Surface Intermediates during SCR of NO with NH3 by Supported V2O5–WO3 /TiO2 Catalysts. J. Am. Chem. Soc. 2017, 139, 15624–15627. [Google Scholar] [CrossRef]
- Lai, J.-K.; Wachs, I.E. A Perspective on the Selective Catalytic Reduction (SCR) of NO with NH3 by Supported V2O5–WO3/TiO2 Catalysts. ACS Catal. 2018, 8, 6537–6551. [Google Scholar] [CrossRef]
- Liu, J.; Wei, Y.; Li, P.-Z.; Zhang, P.; Su, W.; Sun, Y.; Zou, R.; Zhao, Y. Experimental and Theoretical Investigation of Mesoporous MnO2 Nanosheets with Oxygen Vacancies for High-Efficiency Catalytic DeNOx. ACS Catal. 2018, 8, 3865–3874. [Google Scholar] [CrossRef]
- Meng, D.; Zhan, W.; Guo, Y.; Guo, Y.; Wang, L.; Lu, G. A Highly Effective Catalyst of Sm-MnOx for the NH3-SCR of NOx at Low Temperature: Promotional Role of Sm and Its Catalytic Performance. ACS Catal. 2015, 5, 5973–5983. [Google Scholar] [CrossRef]
- Gao, F.; Tang, X.; Yi, H.; Zhao, S.; Li, C.; Li, J.; Shi, Y.; Meng, X. A Review on Selective Catalytic Reduction of NOx by NH3 over Mn–Based Catalysts at Low Temperatures: Catalysts, Mechanisms, Kinetics and DFT Calculations. Catalysts 2017, 7, 199. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, B.; Liu, B.; Sun, S. A Review of Mn-Containing Oxide Catalysts for Low Temperature Selective Catalytic Reduction of NOx with NH3: Reaction Mechanism and Catalyst Deactivation. RSC Adv. 2017, 7, 26226–26242. [Google Scholar] [CrossRef]
- Yang, Y.; Bian, X.; Xie, F.; Bai, Y.; Wang, J. Research Progress in the Composition and Performance of Mn-Based Low-Temperature Selective Catalytic Reduction Catalysts. Appl. Sci. 2024, 14, 10198. [Google Scholar] [CrossRef]
- Zhang, J.; Ma, Z.; Cao, A.; Yan, J.; Wang, Y.; Yu, M.; Hu, L.; Pan, S. Research Progress of Mn-Based Low-Temperature SCR Denitrification Catalysts. RSC Adv. 2024, 14, 32583–32601. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Shen, H.; Zhou, X.; Zheng, T.; Guo, F.; Zhang, Q.; Duan, M. Enhancement Low-Temperature NH3-SCR Activity of the Fe-Mn-Mo/TiO2 Catalyst and Its DFT Calculations and Kinetics. Mol. Catal. 2023, 551, 113657. [Google Scholar] [CrossRef]
- Guo, R.; Qin, B.; Wei, L.; Yin, T.; Zhou, J.; Pan, W. Recent Progress of Low-Temperature Selective Catalytic Reduction of NOx with NH3 over Manganese Oxide-Based Catalysts. Phys. Chem. Chem. Phys. 2022, 24, 6363–6382. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, J.; Tian, J.; Zhong, Y.; Zou, Z.; Dong, R.; Gao, S.; Xu, W.; Tan, D. The Effects of Mn-Based Catalysts on the Selective Catalytic Reduction of NOx with NH3 at Low Temperature: A Review. Fuel Process. Technol. 2022, 230, 107213. [Google Scholar] [CrossRef]
- Zhang, Y.; Han, B.; Chen, Y.; Xia, K.; Gao, Q.; Zhou, C. Understanding the Mechanism of Selective Catalytic Reduction on Spinel TiMn2O4(001) Surface. Mol. Catal. 2022, 518, 112070. [Google Scholar] [CrossRef]
- Qi, G.; Yang, R.T. Low-Temperature Selective Catalytic Reduction of NO with NH3 over Iron and Manganese Oxides Supported on Titania. Appl. Catal. B Environ. 2003, 44, 217–225. [Google Scholar] [CrossRef]
- Li, Y.; Li, Y.; Wang, P.; Hu, W.; Zhang, S.; Shi, Q.; Zhan, S. Low-Temperature Selective Catalytic Reduction of NOx with NH3 over MnFeOx Nanorods. Chem. Eng. J. 2017, 330, 213–222. [Google Scholar] [CrossRef]
- Yang, S.; Wang, C.; Li, J.; Yan, N.; Ma, L.; Chang, H. Low Temperature Selective Catalytic Reduction of NO with NH3 over Mn–Fe Spinel: Performance, Mechanism and Kinetic Study. Appl. Catal. B Environ. 2011, 110, 71–80. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, C.; Zhao, J.; Yang, L.; Sun, K.; Zeng, L.; Pan, Y.; Liu, Y.; Liu, C. Study on the NO2 Production Pathways and the Role of NO2 in Fast Selective Catalytic Reduction DeNOx at Low-Temperature over MnOx/TiO2 Catalyst. Chem. Eng. J. 2020, 379, 122288. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, M.; Liu, S.; Chen, Z.; Yang, L.; Sun, K.; Chen, Y.; Zeng, L.; Wang, W.; Zhao, J.; et al. Design of Assembled Composite of Mn3O4@Graphitic Carbon Porous Nano-Dandelions: A Catalyst for Low–Temperature Selective Catalytic Reduction of NOx with Remarkable SO2 Resistance. Appl. Catal. B Environ. 2020, 269, 118731. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, G.; Chen, C.; Sun, K.; Zeng, L.; Yang, L.; Chen, Y.; Wang, W.; Liu, B.; Lu, Y.; et al. Fe-Doped Mn3O4 Spinel Nanoparticles with Highly Exposed Feoct –O–Mntet Sites for Efficient Selective Catalytic Reduction (SCR) of NO with Ammonia at Low Temperatures. ACS Catal. 2020, 10, 6803–6809. [Google Scholar] [CrossRef]
- Tang, X.; Li, J.; Sun, L.; Hao, J. Origination of N2O from NO Reduction by NH3 over β-MnO2 and α-Mn2O3. Appl. Catal. B Environ. 2010, 99, 156–162. [Google Scholar] [CrossRef]
- Qi, G.; Yang, R.T.; Chang, R. MnOx-CeO2 Mixed Oxides Prepared by Co-Precipitation for Selective Catalytic Reduction of NO with NH3 at Low Temperatures. Appl. Catal. B Environ. 2004, 51, 93–106. [Google Scholar] [CrossRef]
- Meng, D.; Xu, Q.; Jiao, Y.; Guo, Y.; Guo, Y.; Wang, L.; Lu, G.; Zhan, W. Spinel Structured CoaMnbOx Mixed Oxide Catalyst for the Selective Catalytic Reduction of NOx with NH3. Appl. Catal. B Environ. 2018, 221, 652–663. [Google Scholar] [CrossRef]
- Wan, Y.; Zhao, W.; Tang, Y.; Li, L.; Wang, H.; Cui, Y.; Gu, J.; Li, Y.; Shi, J. Ni-Mn Bi-Metal Oxide Catalysts for the Low Temperature SCR Removal of NO with NH3. Appl. Catal. B Environ. 2014, 148–149, 114–122. [Google Scholar] [CrossRef]
- Fan, Z.; Shi, J.-W.; Gao, C.; Gao, G.; Wang, B.; Wang, Y.; He, C.; Niu, C. Gd-Modified MnOx for the Selective Catalytic Reduction of NO by NH3: The Promoting Effect of Gd on the Catalytic Performance and Sulfur Resistance. Chem. Eng. J. 2018, 348, 820–830. [Google Scholar] [CrossRef]
- Zhang, L.; Cui, S.; Guo, H.; Ma, X.; Luo, X. The Influence of K+ Cation on the MnOx-CeO2/TiO2 Catalysts for Selective Catalytic Reduction of NOx with NH3 at Low Temperature. J. Mol. Catal. Chem. 2014, 390, 14–21. [Google Scholar] [CrossRef]
- Liu, J.; Guo, R.; Li, M.; Sun, P.; Liu, S.; Pan, W.; Liu, S.; Sun, X. Enhancement of the SO2 Resistance of Mn/TiO2 SCR Catalyst by Eu Modification: A Mechanism Study. Fuel 2018, 223, 385–393. [Google Scholar] [CrossRef]
- Ye, B.; Lee, M.; Jeong, B.; Kim, J.; Lee, D.H.; Baik, J.M.; Kim, H.-D. Partially Reduced Graphene Oxide as a Support of Mn-Ce/TiO2 Catalyst for Selective Catalytic Reduction of NOx with NH3. Catal. Today 2019, 328, 300–306. [Google Scholar] [CrossRef]
- Li, J.; Chang, H.; Ma, L.; Hao, J.; Yang, R.T. Low-Temperature Selective Catalytic Reduction of NOx with NH3 over Metal Oxide and Zeolite Catalysts—A Review. Catal. Today 2011, 175, 147–156. [Google Scholar] [CrossRef]
- Fu, M.; Li, C.; Lu, P.; Qu, L.; Zhang, M.; Zhou, Y.; Yu, M.; Fang, Y. A Review on Selective Catalytic Reduction of NOx by Supported Catalysts at 100–300 °C—Catalysts, Mechanism, Kinetics. Catal. Sci. Technol. 2014, 4, 14–25. [Google Scholar] [CrossRef]
- Chen, C.; Wang, Y.; Li, J.; Tian, F.; Chen, W.; Feng, C.; Pan, Y.; Liu, Y. In Situ Construction of Heteroatom F-Doped Mn3O4 Spinel Catalysts with Robust Activity and SO2 Resistance for NH3-SCR at Low Temperature. Appl. Catal. B Environ. 2023, 338, 123086. [Google Scholar] [CrossRef]
- Chen, C.; Feng, C.; Wang, Y.; Li, J.; Liu, Z.; Wang, W.; Pan, Y.; Liu, Y. Design of Robust Co-Doped Mn3O4 Spinel Catalysts for Selective Catalytic Reduction of NO with NH3 at Low Temperatures. Appl. Surf. Sci. 2022, 602, 154384. [Google Scholar] [CrossRef]
- Yang, M.; Yuan, H.; Wang, H.; Hu, P. Insights into the Selective Catalytic Reduction of NO by NH3 over Mn3O4(110): A DFT Study Coupled with Microkinetic Analysis. Sci. China Chem. 2018, 61, 457–467. [Google Scholar] [CrossRef]
- Xiong, S.; Peng, Y.; Wang, D.; Huang, N.; Zhang, Q.; Yang, S.; Chen, J.; Li, J. The Role of the Cu Dopant on a Mn3O4 Spinel SCR Catalyst: Improvement of Low-Temperature Activity and Sulfur Resistance. Chem. Eng. J. 2020, 387, 124090. [Google Scholar] [CrossRef]
- Kapteijn, F.; Singoredjo, L.; Andreini, A.; Moulijn, J.A. Activity and Selectivity of Pure Manganese Oxides in the Selective Catalytic Reduction of Nitric Oxide with Ammonia. Appl. Catal. B Environ. 1994, 3, 173–189. [Google Scholar] [CrossRef]
- Kang, M.; Park, E.D.; Kim, J.M.; Yie, J.E. Manganese Oxide Catalysts for NOx Reduction with NH3 at Low Temperatures. Appl. Catal. Gen. 2007, 327, 261–269. [Google Scholar] [CrossRef]
- Xie, J.; Fang, D.; He, F.; Chen, J.; Fu, Z.; Chen, X. Performance and Mechanism about MnOx Species Included in MnOx/TiO2 Catalysts for SCR at Low Temperature. Catal. Commun. 2012, 28, 77–81. [Google Scholar] [CrossRef]
- Busca, G.; Lietti, L.; Ramis, G.; Berti, F. Chemical and Mechanistic Aspects of the Selective Catalytic Reduction of NOx by Ammonia over Oxide Catalysts: A Review. Appl. Catal. B Environ. 1998, 18, 1–36. [Google Scholar] [CrossRef]
- Forzatti, P. Present Status and Perspectives in De-NOx SCR Catalysis. Appl. Catal. Gen. 2001, 222, 221–236. [Google Scholar] [CrossRef]
- Marbán, G.; Fuertes, A.B. Kinetics of the Low-Temperature Selective Catalytic Reduction of NO with NH3 over Activated Carbon ®ber Composite-Supported Iron Oxides. Catal. Lett. 2002, 84, 13–19. [Google Scholar] [CrossRef]
- Marbán, G.; Valdés-Solís, T.; Fuertes, A.B. Mechanism of Low Temperature Selective Catalytic Reduction of NO with NH3 over Carbon-Supported Mn3O4: Active Phase and Role of Surface NO Species. Phys Chem Chem Phys 2004, 6, 453–464. [Google Scholar] [CrossRef]
- Marban, G. Mechanism of Low-Temperature Selective Catalytic Reduction of NO with BNH3 over Carbon-Supported Mn3O4Role of Surface NH3 Species: SCR Mechanism. J. Catal. 2004, 226, 138–155. [Google Scholar] [CrossRef]
- Wu, Z.; Jiang, B.; Liu, Y.; Wang, H.; Jin, R. DRIFT Study of Manganese/Titania-Based Catalysts for Low-Temperature Selective Catalytic Reduction of NO with NH3. Environ. Sci. Technol. 2007, 41, 5812–5817. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Cai, S.; Li, H.; Huang, L.; Shi, L.; Zhang, D. Mechanistic Aspects of deNO x Processing over TiO2 Supported Co–Mn Oxide Catalysts: Structure–Activity Relationships and In Situ DRIFTs Analysis. ACS Catal. 2015, 5, 6069–6077. [Google Scholar] [CrossRef]
- Hu, H.; Cai, S.; Li, H.; Huang, L.; Shi, L.; Zhang, D. In Situ DRIFTs Investigation of the Low-Temperature Reaction Mechanism over Mn-Doped Co3O4 for the Selective Catalytic Reduction of NOx with NH3. J. Phys. Chem. C 2015, 119, 22924–22933. [Google Scholar] [CrossRef]
- Suárez, S.; Martín, J.A.; Yates, M.; Avila, P.; Blanco, J. N2O Formation in the Selective Catalytic Reduction of NOx with NH3 at Low Temperature on CuO-Supported Monolithic Catalysts. J. Catal. 2005, 229, 227–236. [Google Scholar] [CrossRef]
- Ilchenko, N. Catalytic Oxidation of Ammonia, I. Reaction Kinetics and Mechanism. J. Catal. 1975, 39, 57–72. [Google Scholar] [CrossRef]
- Singoredjo, L.; Korver, R.; Kapteijn, F.; Moulijn, J. Alumina Supported Manganese Oxides for the Low-Temperature Selective Catalytic Reduction of Nitric Oxide with Ammonia. Appl. Catal. B Environ. 1992, 1, 297–316. [Google Scholar] [CrossRef]
- Madia, G.; Koebel, M.; Elsener, M.; Wokaun, A. Side Reactions in the Selective Catalytic Reduction of NOx with Various NO2 Fractions. Ind. Eng. Chem. Res. 2002, 41, 4008–4015. [Google Scholar] [CrossRef]
- Ciardelli, C.; Nova, I.; Tronconi, E.; Chatterjee, D.; Bandl-Konrad, B.; Weibel, M.; Krutzsch, B. Reactivity of NO/NO2–NH3 SCR System for Diesel Exhaust Aftertreatment: Identification of the Reaction Network as a Function of Temperature and NO2 Feed Content. Appl. Catal. B Environ. 2007, 70, 80–90. [Google Scholar] [CrossRef]
- He, Z.; Wang, Y.; Liu, Y.; Lian, L.; Kong, D.; Zhao, Y. Recent Advances in Sulfur Poisoning of Selective Catalytic Reduction (SCR) Denitration Catalysts. Fuel 2024, 365, 131126. [Google Scholar] [CrossRef]
- Yang, G.; Zhao, H.; Luo, X.; Shi, K.; Zhao, H.; Wang, W.; Chen, Q.; Fan, H.; Wu, T. Promotion Effect and Mechanism of the Addition of Mo on the Enhanced Low Temperature SCR of NOx by NH3 over MnOx/γ-Al2O3 Catalysts. Appl. Catal. B Environ. 2019, 245, 743–752. [Google Scholar] [CrossRef]
- Gao, F.; Tang, X.; Yi, H.; Li, J.; Zhao, S.; Wang, J.; Chu, C.; Li, C. Promotional Mechanisms of Activity and SO2 Tolerance of Co- or Ni-Doped MnOx-CeO2 Catalysts for SCR of NOx with NH3 at Low Temperature. Chem. Eng. J. 2017, 317, 20–31. [Google Scholar] [CrossRef]
- Yan, Q.; Chen, S.; Zhang, C.; Wang, Q.; Louis, B. Synthesis and Catalytic Performance of Cu1Mn0.5Ti0.5O Mixed Oxide as Low-Temperature NH3-SCR Catalyst with Enhanced SO2 Resistance. Appl. Catal. B Environ. 2018, 238, 236–247. [Google Scholar] [CrossRef]
- Deng, S.; Meng, T.; Xu, B.; Gao, F.; Ding, Y.; Yu, L.; Fan, Y. Advanced MnOx /TiO2 Catalyst with Preferentially Exposed Anatase {001} Facet for Low-Temperature SCR of NO. ACS Catal. 2016, 6, 5807–5815. [Google Scholar] [CrossRef]
- Jin, R.; Liu, Y.; Wang, Y.; Cen, W.; Wu, Z.; Wang, H.; Weng, X. The Role of Cerium in the Improved SO2 Tolerance for NO Reduction with NH3 over Mn-Ce/TiO2 Catalyst at Low Temperature. Appl. Catal. B Environ. 2014, 148–149, 582–588. [Google Scholar] [CrossRef]
- Yu, J.; Guo, F.; Wang, Y.; Zhu, J.; Liu, Y.; Su, F.; Gao, S.; Xu, G. Sulfur Poisoning Resistant Mesoporous Mn-Base Catalyst for Low-Temperature SCR of NO with NH3. Appl. Catal. B Environ. 2010, 95, 160–168. [Google Scholar] [CrossRef]
- Wei, L.; Cui, S.; Guo, H.; Ma, X.; Zhang, L. DRIFT and DFT Study of Cerium Addition on SO2 of Manganese-Based Catalysts for Low Temperature SCR. J. Mol. Catal. Chem. 2016, 421, 102–108. [Google Scholar] [CrossRef]
- Jarosch, D. Crystal Structure Refinement and Reflectance Measurements of Hausmannite, Mn3O4. Mineral. Petrol. 1987, 37, 15–23. [Google Scholar] [CrossRef]
- Vegge, T.; Rasmussen, T.; Leffers, T.; Pedersen, O.B.; Jacobsen, K.W. Atomistic Simulations of Cross-Slip of Jogged Screw Dislocations in Copper. Philos. Mag. Lett. 2001, 81, 137–144. [Google Scholar] [CrossRef]
- Howalt, J.G.; Bligaard, T.; Rossmeisl, J.; Vegge, T. DFT Based Study of Transition Metal Nano-Clusters for Electrochemical NH3 Production. Phys. Chem. Chem. Phys. 2013, 15, 7785. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, J.; Liu, F.; Wang, Z.; Ding, J.; Huang, H. Reaction Mechanism for NH3-SCR of NOx over CuMn2O4 Catalyst. Chem. Eng. J. 2019, 361, 578–587. [Google Scholar] [CrossRef]
- Zhang, L.; Cui, S.; Guo, H.; Ma, X.; Lu, W. Density Function Theoretical and Experimental Study of NH3+NO Adsorptions on MnO/TiO2 Surface. Comput. Mater. Sci. 2016, 112, 238–244. [Google Scholar] [CrossRef]
- Liu, B.; Liu, J.; Xin, L.; Zhang, T.; Xu, Y.; Jiang, F.; Liu, X. Unraveling Reactivity Descriptors and Structure Sensitivity in Low-Temperature NH3-SCR Reaction over CeTiOxCatalysts: A Combined Computational and Experimental Study. ACS Catal. 2021, 11, 7613–7636. [Google Scholar] [CrossRef]
- Alemany, L.J. Characterization and Composition of Commercial V2O5-WO3-TiO2 SCR Catalysts. Appl. Catal. B Environ. 1996, 10, 299–311. [Google Scholar] [CrossRef]
- Ramis, G.; Busca, G.; Bregani, F.; Forzatti*, P. Fourier Transform-Infrared Study of the Adsorption and Coadsorption of Nitric Oxide, Nitrogen Dioxide and Ammonia on Vanadia-Titania and Mechanism of Selective Catalytic Reduction. Appl. Catal. 1990, 64, 259–278. [Google Scholar] [CrossRef]
- Lietti, L.; Svachula, J.; Forzatti, P.; Busca, G.; Ramis, G.; Bregani, P. Surface and Catalytic Properties of Vanadia-Titania and Tungsta-Titania Systems in the Selective Catalytic Reduction of Nitrogen Oxides. Catal. Today 1993, 17, 131–139. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized Gradient Approximation for the Exchange-Correlation Hole of a Many-Electron System. Phys. Rev. B 1996, 54, 16533–16539. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Garcês Gonçalves, P.R., Jr.; De Abreu, H.A.; Duarte, H.A. Stability, Structural, and Electronic Properties of Hausmannite (Mn3O4) Surfaces and Their Interaction with Water. J. Phys. Chem. C 2018, 122, 20841–20849. [Google Scholar] [CrossRef]
- Liu, S.; Liu, L.; Cheng, Z.; Zhu, J.; Yu, R. Surface Structures of Mn3 O4 and the Partition of Oxidation States of Mn. J. Phys. Chem. Lett. 2021, 12, 5675–5681. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Liu, Y.; Wu, Y.; Zhang, H.; Zhang, D.; Zhang, Y.; Deng, H.; Sun, G.; Li, H.; Shan, W.; et al. Elucidating Atomic Structure and Reconstruction of Mn3O4(0 0 1) Surface. Appl. Surf. Sci. 2025, 680, 161339. [Google Scholar] [CrossRef]
- Johnson, R. NIST 101. Computational Chemistry Comparison and Benchmark Database. 1999. Available online: http://cccbdb.nist.gov (accessed on 22 January 2025).
- Wu, G.; Zelenay, P. Nanostructured Nonprecious Metal Catalysts for Oxygen Reduction Reaction. Acc. Chem. Res. 2013, 46, 1878–1889. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Site | Bond Length | ΔEads |
|---|---|---|---|
| NH3 | atop-Mn | dN-Mn = 2.088 | −0.971 |
| NO | atop-Mn (O-Mn) a | dO-Mn = 2.115 | −0.375 |
| bridge-Mn | dN-Mn = 1.758 dO-Mn = 2.241 | −1.431 | |
| atop-Mn (N-Mn) a | dN-Mn = 1.671 | −1.864 | |
| SO2 | atop-Mn | dS-Mn = 2.763 | −0.284 |
| bridge-Mn | dO-Mn = 1.955, 1.949 | −0.833 | |
| hollow | dS-O = 1.662 dO-Mn = 1.983, 2.021 | −1.451 | |
| SO3 | bridge-Mn | dO-Mn = 1.842, 1.839 | −1.424 |
| hollow | dS-O =1.597 dO-Mn = 2.005, 2.027 | −2.301 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, H.; Liu, Z.; Zhang, X.; Fan, Y.; Wang, X.; Liu, D.; Li, X.; Gong, X.; Guo, W.; Ren, H. Selective Catalytic Reduction of NO by NH3 and SOx Poisoning Mechanisms on Mn3O4 Catalysts: A Density Functional Investigation. Catalysts 2025, 15, 241. https://doi.org/10.3390/catal15030241
Zhu H, Liu Z, Zhang X, Fan Y, Wang X, Liu D, Li X, Gong X, Guo W, Ren H. Selective Catalytic Reduction of NO by NH3 and SOx Poisoning Mechanisms on Mn3O4 Catalysts: A Density Functional Investigation. Catalysts. 2025; 15(3):241. https://doi.org/10.3390/catal15030241
Chicago/Turabian StyleZhu, Houyu, Zhennan Liu, Xiaoxin Zhang, Yucheng Fan, Xin Wang, Dongyuan Liu, Xiaohan Li, Xiaoxiao Gong, Wenyue Guo, and Hao Ren. 2025. "Selective Catalytic Reduction of NO by NH3 and SOx Poisoning Mechanisms on Mn3O4 Catalysts: A Density Functional Investigation" Catalysts 15, no. 3: 241. https://doi.org/10.3390/catal15030241
APA StyleZhu, H., Liu, Z., Zhang, X., Fan, Y., Wang, X., Liu, D., Li, X., Gong, X., Guo, W., & Ren, H. (2025). Selective Catalytic Reduction of NO by NH3 and SOx Poisoning Mechanisms on Mn3O4 Catalysts: A Density Functional Investigation. Catalysts, 15(3), 241. https://doi.org/10.3390/catal15030241