
Enantioselectivity Enhancement of a Geobacillus thermoleovorans CCR11 Lipase by Rational Design

 ,
,  , , , and
, , , and 
Abstract

1. Introduction
2. Results and Discussion
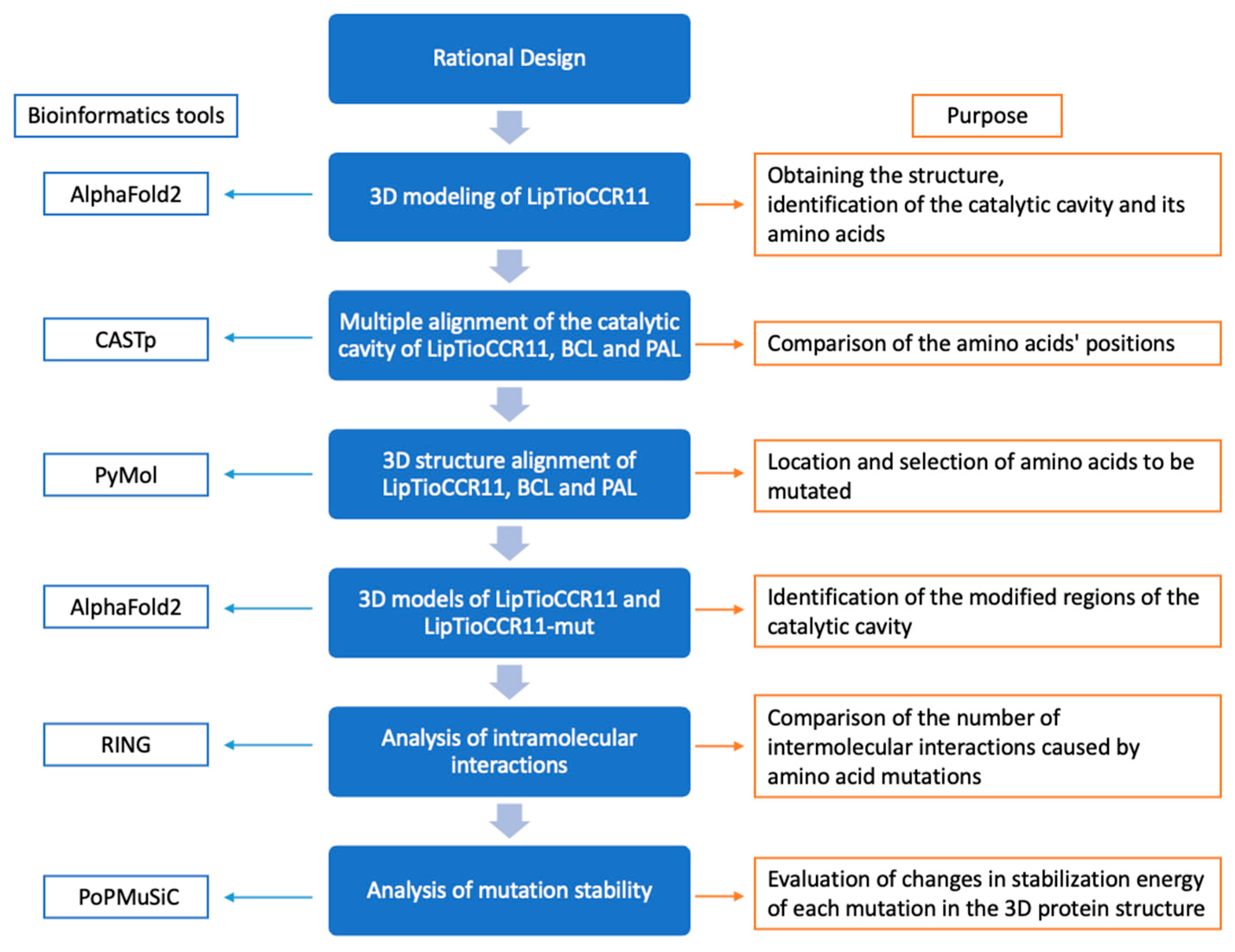
2.1. Rational Design of LipTioCCR11 Lipase to Increase Its Enantioselectivity
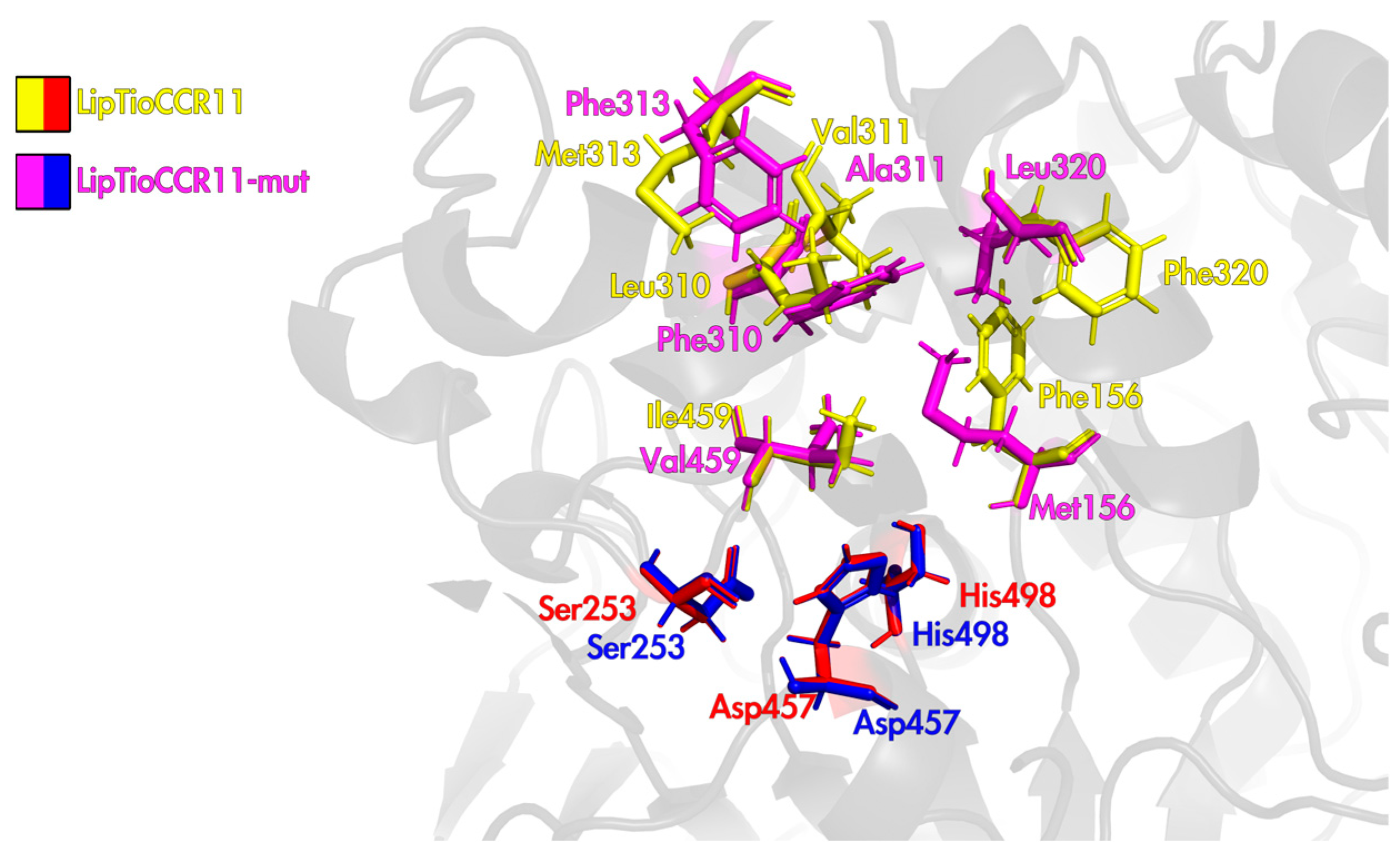
2.2. Three-Dimensional Structure of LipTioCCR11 Lipase
2.3. Three-Dimensional Structure LipTioCCR11-Mut Lipase
2.4. Cloning and Expression of LipTioCCR11 and LipTioCCR11-Mut Lipases
2.5. Biochemical Characteristics of LipTioCCR11 and LipTioCCR11-Mut Activity
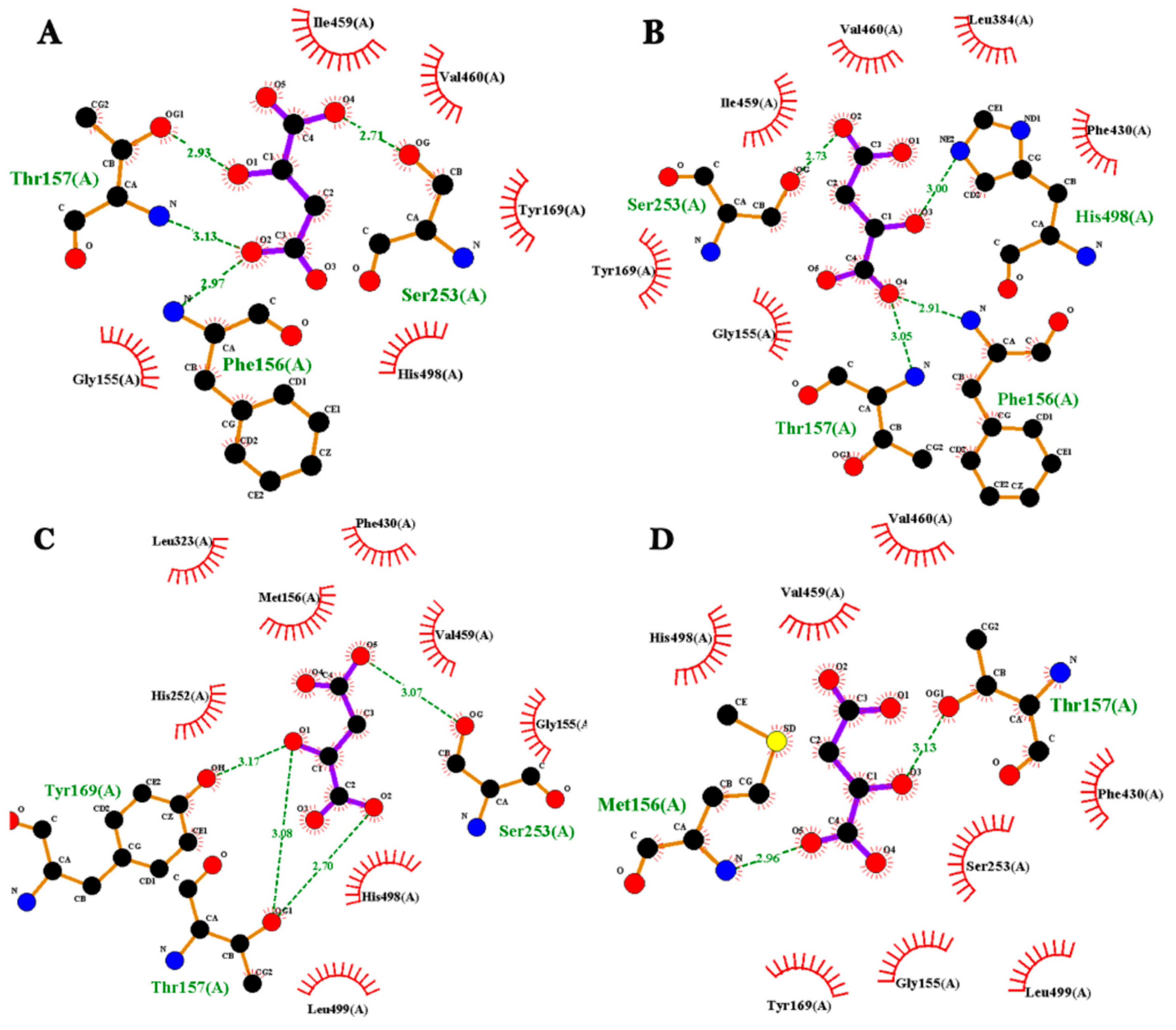
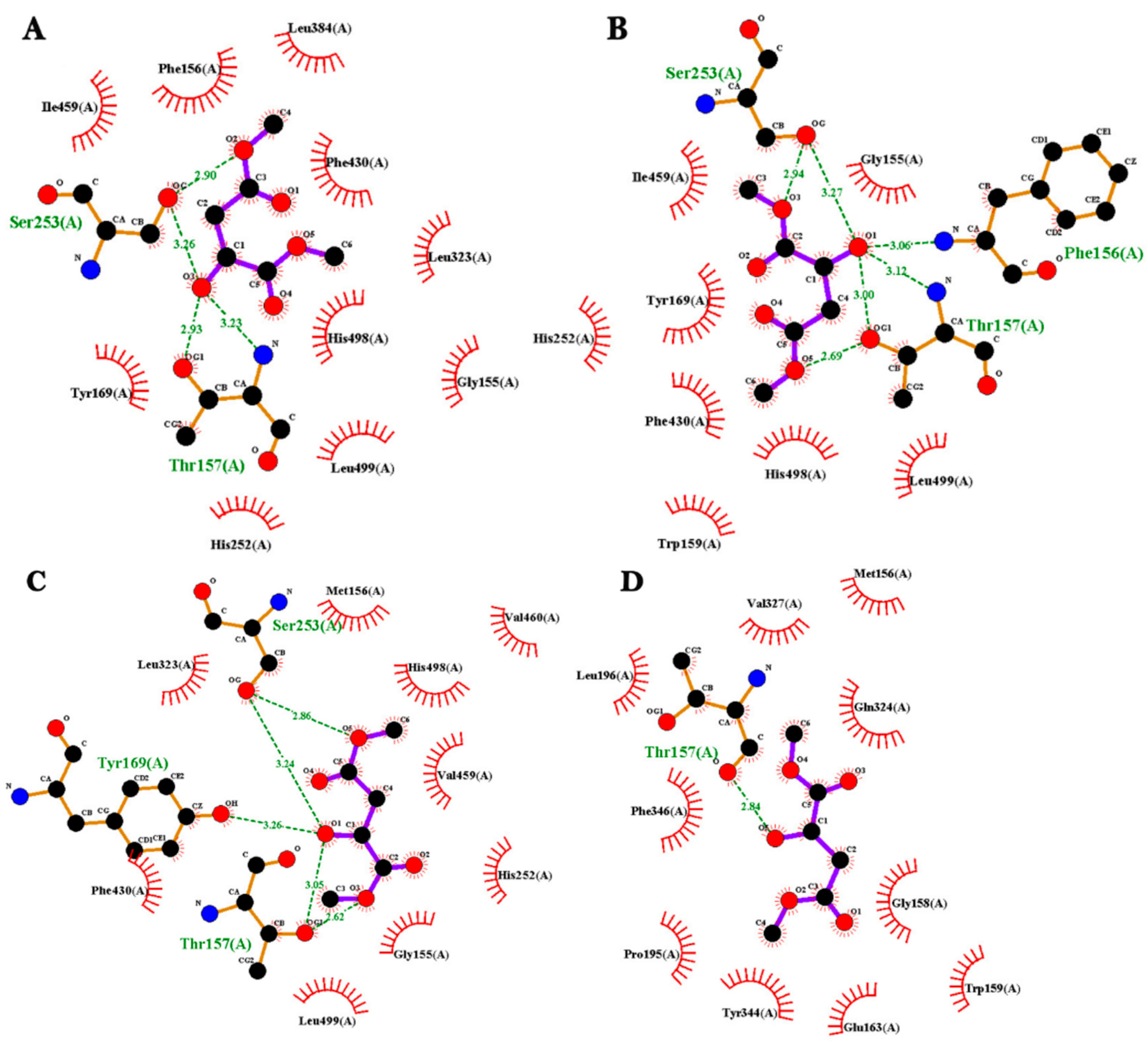
2.6. Molecular Docking Analysis
2.7. Analysis of the Enantioselective Synthesis of (R)- and (S)-Dimethyl Malate
3. Materials and Methods
3.1. Bacterial Strain, Plasmids and Culture Medium
3.2. Rational Design of LipTioCCR11 Amino Acid Sequence
3.3. Construction of Expression Systems for LipTioCCR11 and LipTioCCR11-Mut
3.4. Expression and Collection of LipTioCCR11 and LipTioCCR11-Mut Lipases
3.4.1. Purification of LipTioCCR11 and LipTioCCR11-Mut Lipases
3.4.2. Protein Quantification
3.4.3. Quantification of Lipolytic Activity
3.4.4. Polyacrylamide Gel Electrophoresis (SDS-PAGE)
3.4.5. Zymography
3.5. Characterization of LipTioCCR11 and LipTioCCR11-Mut Lipases
3.6. Molecular Docking Studies
3.7. Analysis of the Enantioselective Production of (RS)-Dimethyl Malate
3.8. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Casas-Godoy, L.; Gasteazoro, F.; Duquesne, S.; Bordes, F.; Marty, A.; Sandoval, G. Lipases: An overview. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2018; Volume 1835, pp. 3–38. [Google Scholar] [CrossRef]
- Treichel, H.; de Oliveira, D.; Mazutti, M.A.; Di Luccio, M.; Oliveira, J.V. A Review on microbial lipases production. Food Bioprocess Technol. 2010, 3, 182–196. [Google Scholar] [CrossRef]
- Xu, X. Production of specific-structured triacylglycerols by lipase-catalyzed reactions: A review. Eur. J. Lip. Sci. Technol. 2000, 102, 287–303. [Google Scholar] [CrossRef]
- Park, J.-Y.; Park, K.-M. Lipase and Its Unique Selectivity: A Mini-Review. J. Chem. 2022, 2022, 7609019. [Google Scholar] [CrossRef]
- Maldonado, M.R.; Alnoch, R.C.; de Almeida, J.M.; dos Santos, L.A.; Andretta, A.T.; Ropaín, R.d.P.C.; de Souza, E.M.; Mitchell, D.A.; Krieger, N. Key mutation sites for improvement of the enantioselectivity of lipases through protein engineering. Biochem. Eng. J. 2021, 172, 108047. [Google Scholar] [CrossRef]
- Chen, H.; Meng, X.; Xu, X.; Liu, W.; Li, S. The molecular basis for lipase stereoselectivity. Appl. Microbiol. Biotechnol. 2018, 102, 3487–3495. [Google Scholar] [CrossRef]
- Carvalho, A.C.L.d.M.; Fonseca, T.D.S.; de Mattos, M.C.; Oliveira, M.D.C.F.d.; de Lemos, T.L.G.; Molinari, F.; Romano, D.; Serra, I. Recent advances in lipase-mediated preparation of pharmaceuticals and their intermediates. Int. J. Mol. Sci. 2015, 16, 29682–29716. [Google Scholar] [CrossRef]
- Bhardwaj, K.K.; Gupta, R. Synthesis of chirally pure enantiomers by lipase. J. Oleo Sci. 2017, 66, 1073–1084. [Google Scholar] [CrossRef]
- Ferraccioli, R. Progress on the stereoselective synthesis of chiral molecules based on metal-catalyzed dynamic kinetic resolu-tion of alcohols with lipases. Symmetry 2021, 13, 1744. [Google Scholar] [CrossRef]
- Soto, M.; Sanz-Machín, I.; Rodríguez-Solla, H.; Gotor-Fernández, V. Chemoenzymatic stereoselective synthesis of trans-flavan-4-ols via lipase-catalyzed kinetic resolutions. Catalysts 2021, 11, 1296. [Google Scholar] [CrossRef]
- Watts, O.F.B.; Berreur, J.; Collins, B.S.L.; Clayden, J. Biocatalytic enantioselective synthesis of atropisomers. Acc. Chem. Res. 2022, 55, 3362–3375. [Google Scholar] [CrossRef]
- Ferrario, V.; Ebert, C.; Nitti, P.; Pitacco, G.; Gardossi, L. Modelling and predicting enzyme enantioselectivity: The aid of computational methods for the rational use of lipase B from Candida antarctica. Curr. Biotechnol. 2015, 4, 87–99. [Google Scholar] [CrossRef]
- Yang, H.; Yu, H.; Stolarzewicz, I.A.; Tang, W. Enantioselective transformations in the synthesis of therapeutic agents. Chem. Rev. 2023, 123, 9397–9446. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, S.P.; Schuenck-Rodrigues, R.A. Lipases in the Enantioselective Biocatalysis Process to Obtain Enantiomerically Pure Racemic Products. J. Phys. Chem. Res. 2022, 4, 146. [Google Scholar]
- Vivek, K.; Sandhia, G.S.; Subramaniyan, S.J.B.A. Extremophilic lipases for industrial applications: A general review. Biotechnol. Adv. 2022, 60, 108002. [Google Scholar] [CrossRef]
- Choi, Y.; Park, J.-Y.; Chang, P.-S. Integral Stereoselectivity of lipase based on the chromatographic resolution of enantiomeric/regioisomeric diacylglycerols. J. Agric. Food Chem. 2021, 69, 325–331. [Google Scholar] [CrossRef]
- Cen, Y.; Li, D.; Xu, J.; Wu, Q.; Wu, Q.; Lin, X. Highly Focused Library-Based Engineering of Candida antarctica Lipase B with (S)-Selectivity Towards sec-Alcohols. Adv. Synth. Catal. 2019, 361, 126–134. [Google Scholar] [CrossRef]
- Li, D.; Chen, X.; Chen, Z.; Lin, X.; Xu, J.; Wu, Q. Directed evolution of lipase A from Bacillus subtilis for the preparation of enantiocomplementary sec-alcohols. Green Synth. Catal. 2021, 2, 290–294. [Google Scholar] [CrossRef]
- McLoughlin, E.C.; Twamley, B.; O’Boyle, N.M. Candida antarctica Lipase B mediated kinetic resolution: A sustainable method for chiral synthesis of antiproliferative β-lactams. Eur. J. Med. Chem. 2024, 276, 116692. [Google Scholar] [CrossRef]
- Wu, Q.; Soni, P.; Reetz, M.T. Laboratory evolution of enantiocomplementary Candida antarctica lipase B mutants with broad substrate scope. J. Am. Chem. Soc. 2013, 135, 1872–1881. [Google Scholar] [CrossRef]
- Qin, Z.; Wang, Y.; Li, S.; Wu, Q.; Zhu, D. Enhancing the enantioselectivity of Candida antarctica lipase B for secondary alcohols by engineering the entrance to the binding pocket. J. Mol. Catal. B Enzym. 2013, 85–86, 100–106. [Google Scholar]
- Shen, J.-W.; Qi, J.-M.; Zhang, X.-J.; Liu, Z.-Q.; Zheng, Y.-G. Significantly increased catalytic activity of Candida antarctica lipase B for the resolution of cis-(±)-dimethyl 1-acetylpiperidine-2,3-dicarboxylate. Catal. Sci. Technol. 2018, 8, 4718–4725. [Google Scholar] [CrossRef]
- Engström, K.; Nyhlén, J.; Sandström, A.G.; Bäckvall, J.-E. Directed evolution of an enantioselective lipase with broad substrate scope for hydrolysis of α-substituted esters. J. Am. Chem. Soc. 2010, 132, 7038–7042. [Google Scholar] [CrossRef] [PubMed]
- Sandström, A.G.; Wikmark, Y.; Engström, K.; Nyhlén, J.; Bäckvall, J.-E. Combinatorial reshaping of the Candida antarctica lipase A substrate pocket for enantioselectivity using an extremely condensed library. Proc. Natl. Acad. Sci. USA 2012, 109, 78–83. [Google Scholar] [CrossRef]
- Reetz, M.T. Witnessing the birth of directed evolution of stereoselective enzymes as catalysts in organic chemistry. Adv. Synth. Catal. 2022, 364, 3326–3335. [Google Scholar] [CrossRef]
- Reetz, M.T.; Wilensek, S.; Zha, D.; Jaeger, K.-E. Directed evolution of an enantioselective enzyme through combinatorial multiple-cassette mutagenesis. Angew. Chem. Int. Ed. Engl. 2001, 40, 3589–3591. [Google Scholar] [CrossRef]
- Prasad, S.; Bocola, M.; Reetz, M.T. Revisiting the lipase from Pseudomonas aeruginosa: Directed evolution of substrate ac-ceptance and enantioselectivity using iterative saturation mutagenesis. ChemPhysChem 2011, 12, 1550–1557. [Google Scholar] [CrossRef]
- Tian, M.; Yang, L.; Lv, P.; Wang, Z.; Fu, J.; Miao, C.; Li, Z.; Li, L.; Liu, T.; Du, W.; et al. Improvement of methanol tolerance and catalytic activity of Rhizomucor miehei lipase for one-step synthesis of biodiesel by semi-rational design. Bioresour. Technol. 2022, 348, 126769. [Google Scholar] [CrossRef]
- Song, Z.; Zhang, Q.; Wu, W.; Pu, Z.; Yu, H. Rational design of enzyme activity and enantioselectivity. Front. Bioeng. Biotechnol. 2023, 11, 1129149. [Google Scholar] [CrossRef]
- Sternke, M.; Tripp, K.W.; Barrick, D. Consensus sequence design as a general strategy to create hyperstable, biologically active proteins. Proc. Natl. Acad. Sci. USA 2019, 116, 11275–11284. [Google Scholar] [CrossRef]
- Zhang, Y.; Feng, T.; Ni, X.; Xia, J.; Suo, H.; Yan, L.; Zou, B. Immobilized lipase based on SBA-15 adsorption and gel embedding for catalytic synthesis of isoamyl acetate. Food Biosci. 2024, 60, 104427. [Google Scholar] [CrossRef]
- Kazlauskas, R.J.; Bornscheuer, U.T. Finding better protein engineering strategies. Nat. Chem. Biol. 2009, 5, 526–529. [Google Scholar] [CrossRef] [PubMed]
- Palomo, J.M. Semisynthetic Enzymes by Protein–Peptide Site-Directed Covalent Conjugation: Methods and Applications. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2017; Volume 590, pp. 305–316. [Google Scholar]
- Monteiro, R.R.; Virgen-Ortiz, J.J.; Berenguer-Murcia, Á.; da Rocha, T.N.; dos Santos, J.C.; Alcántara, A.R.; Fernandez-Lafuente, R. Biotechnological relevance of the lipase A from Candida antarctica. Catal. Today 2021, 362, 141–154. [Google Scholar] [CrossRef]
- Löfgren, J.; Görbe, T.; Oschmann, M.; Svedendahl Humble, M.; Bäckvall, J.E. Transesterification of a tertiary alcohol by en-gineered Candida antarctica Lipase A. ChemBioChem 2019, 20, 1438–1443. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.; Hummel, A.; Yang, J.; Horino, S.; Kanomata, K.; Akai, S.; Gröger, H. Protein engineering of lipase a from Candida antarctica to improve esterification of tertiary alcohols. ChemBioChem 2024, 25, e202400082. [Google Scholar] [CrossRef] [PubMed]
- Yi, S.; Park, S. Enhancing enantioselectivity of Candida antarctica lipase B towards chiral sec-alcohols bearing small substituents through hijacking sequence of A homolog. Tetrahedron Lett. 2021, 75, 153186. [Google Scholar] [CrossRef]
- Castro-Ochoa, L.D.; Rodríguez-Gómez, C.; Valerio-Alfaro, G.; Ros, R.O. Screening, purification and characterization of the thermoalkalophilic lipase produced by Bacillus thermoleovorans CCR11. Enzym. Microb. Technol. 2005, 37, 648–654. [Google Scholar] [CrossRef]
- Quintana-Castro, R.; Díaz, P.; Valerio-Alfaro, G.; García, H.S.; Oliart-Ros, R. Gene cloning, expression and characterization of the Geobacillus thermoleovorans CCR11 thermoalkaliphilic lipase. Mol. Biotechnol. 2009, 42, 75–83. [Google Scholar] [CrossRef]
- Espinosa-Luna, G.; Sánchez-Otero, M.G.; Quintana-Castro, R.; Matus-Toledo, R.E.; Oliart-Ros, R.M. Gene Cloning and Characterization of the Geobacillus thermoleovorans CCR11 Carboxylesterase CaesCCR11, a New Member of Family XV. Mol. Biotechnol. 2016, 58, 37–46. [Google Scholar] [CrossRef]
- Sánchez-Otero, M.G.; Quintana-Castro, R.; Mora-González, P.C.; Márquez-Molina, O.; Valerio-Alfaro, G.; Oliart-Ros, R. Enzymatic reactions and synthesis of n-butyl caproate: Esterification, transesterification and aminolysis using a recombinant lipase from Geobacillus thermoleovorans CCR11. Environ. Technol. 2010, 31, 1101–1106. [Google Scholar] [CrossRef]
- Badillo-Zeferino, G.; Ruiz-López, I.; Oliart-Ros, R.; Sánchez-Otero, M. Improved expression and immobilization of Geobacillus thermoleovorans CCR11 thermostable recombinant lipase. Biotechnol. Appl. Biochem. 2017, 64, 62–69. [Google Scholar] [CrossRef]
- Ema, T. Rational strategies for highly enantioselective lipase-catalyzed kinetic resolutions of very bulky chiral compounds: Substrate design and high-temperature biocatalysis. Tetrahedron Asymmetry 2004, 15, 2765–2770. [Google Scholar] [CrossRef]
- Mariani, V.; Biasini, M.; Barbato, A.; Schwede, T. lDDT: A local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics 2013, 29, 2722–2728. [Google Scholar] [CrossRef] [PubMed]
- Melnyk, I.; Lozano, A.; Das, P.; Chenthamarakshan, V. Alphafold distillation for improved inverse protein folding. arXiv 2022, arXiv:2210.03488. [Google Scholar] [CrossRef]
- Nealon, J.O.; Philomina, L.S.; McGuffin, L.J. Predictive and experimental approaches for elucidating protein–protein interactions and quaternary structures. Int. J. Mol. Sci. 2017, 18, 2623. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M. Open-access data: A cornerstone for artificial intelligence approaches to protein structure prediction. Structure 2021, 29, 515–520. [Google Scholar] [CrossRef]
- Shin, H.-D.; Kim, J.-H.; Kim, T.-K.; Kim, S.-H.; Lee, Y.-H. Esterification of hydrophobic substrates by lipase in the cyclodextrin induced emulsion reaction system. Enzym. Microb. Technol. 2002, 30, 835–842. [Google Scholar] [CrossRef]
- Kurcinski, M.; Oleniecki, T.; Ciemny, M.P.; Kuriata, A.; Kolinski, A.; Kmiecik, S. CABS-flex standalone: A simulation environment for fast modeling of protein flexibility. Bioinformatics 2018, 35, 694–695. [Google Scholar] [CrossRef]
- Clementel, D.; Del Conte, A.; Monzon, A.M.; Camagni, G.F.; Minervini, G.; Piovesan, D.; E Tosatto, S.C. RING 3.0: Fast generation of probabilistic residue interaction networks from structural ensembles. Nucleic Acids Res. 2022, 50, W651–W656. [Google Scholar] [CrossRef]
- Vahidi, S.H.; Monhemi, H.; Sabzevar, B.H.; Eftekhari, M. Electrostatic interactions of enzymes in non-aqueous conditions: Insights from molecular dynamics simulations. J. Biomol. Struct. Dyn. 2023, 43, 291–304. [Google Scholar] [CrossRef]
- Seiler, M.; Stock, S.; Drews, A. pH-dependent electrostatic interactions between enzymes and nanoparticles in pickering emulsions-influence on activity and droplet size. J. Biotechnol. 2024, 382, 28–36. [Google Scholar] [CrossRef]
- Dehouck, Y.; Grosfils, A.; Folch, B.; Gilis, D.; Bogaerts, P.; Rooman, M. Fast and accurate predictions of protein stability changes upon mutations using statistical potentials and neural networks: PoPMuSiC-2.0. Bioinformatics 2009, 25, 2537–2543. [Google Scholar] [CrossRef] [PubMed]
- McNiff, M.; Haynes, E.; Dixit, N.; Gao, F.; Laurence, J. Thioredoxin fusion construct enables high-yield production of soluble, active matrix metalloproteinase-8 (MMP-8) in Escherichia coli. Protein Expr. Purif. 2016, 122, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Kamal, Z.; Mohammad, T.A.S.; Krishnamoorthy, G.; Rao, N.M. Role of active site rigidity in activity: MD simulation and fluorescence study on a lipase mutant. PLoS ONE 2012, 7, e35188. [Google Scholar] [CrossRef] [PubMed]
- Poveda, J. Beneficial effects of microbial volatile organic compounds (MVOCs) in plants. Appl. Soil Ecol. 2021, 168, 104118. [Google Scholar] [CrossRef]
- Wang, Q.; Al Makishah, N.H.; Li, Q.; Li, Y.; Liu, W.; Sun, X.; Wen, Z.; Yang, S. Developing clostridia as cell factories for short-and medium-chain ester production. Front. Bioeng. Biotechnol. 2021, 9, 661694. [Google Scholar] [CrossRef]
- Toledo, M.; José, C.; Briand, L. Esterificación enzimática de antiinflamatorios no esteroideos con glicerol. Jorn. En Cienc. Apl. "Dr. Jorge Ronco" 2020, 2. Available online: https://revistas.unlp.edu.ar/CienciasAplicadas/article/view/9574 (accessed on 17 January 2024).
- Martín-Matute, B.; Edin, M.; Bäckvall, J. Highly Efficient Synthesis of Enantiopure Diacetylated C2-Symmetric Diols by Ruthenium- and Enzyme-Catalyzed Dynamic Kinetic Asymmetric Transformation (DYKAT). Chem. A Eur. J. 2006, 12, 6053–6061. [Google Scholar] [CrossRef]
- Ottosson, J.; Fransson, L.; Hult, K. Substrate entropy in enzyme enantioselectivity: An experimental and molecular modeling study of a lipase. Protein Sci. 2002, 11, 1462–1471. [Google Scholar] [CrossRef]
- Kumar, R.; Goomber, S.; Kaur, J. Engineering lipases for temperature adaptation: Structure function correlation. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2019, 1867, 140261. [Google Scholar] [CrossRef]
- Overbeeke, P.L.A.; Koops, B.C.; Verheij, H.M.; Slotboom, A.J.; Egmond, M.R.; Jongejan, J.A.; Heijnen, J.J. Activity and enantioselectivity of modified lipases in organic solvents. Biocatal. Biotransform. 2000, 18, 59–77. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Applying and improving AlphaFold at CASP14. Proteins Struct. Funct. Bioinform. 2021, 89, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Nawani, N.; Dosanjh, N.; Kaur, J. A novel thermostable lipase from a thermophilic Bacillus sp.: Characterization and esterification studies. Biotechnol. Lett. 1998, 20, 997–1000. [Google Scholar] [CrossRef]
- Prim, N.; Sánchez, M.; Ruiz, C.; Pastor, F.J.; Diaz, P. Use of methylumbeliferyl-derivative substrates for lipase activity characterization. J. Mol. Catal. B Enzym. 2003, 22, 339–346. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Reges, M.; Marinho, M.M.; Marinho, E.S. In silico characterization of hypoglycemic agent Phenformin using classical force field MMFF94. Int. J. Rec. Res. Rev. 2018, 11, 36–43. [Google Scholar]
- Chen, S.; Li, B.; Chen, L.; Jiang, H. Uncovering the mechanism of resveratrol in the treatment of diabetic kidney disease based on network pharmacology, molecular docking, and experimental validation. J. Transl. Med. 2023, 21, 380. [Google Scholar] [CrossRef]
- Abadi, M.H.J.N.; Abyaneh, F.A.; Zare, N.; Zamani, J.; Abdoli, A.; Aslanbeigi, F.; Hamblin, M.R.; Tarrahimofrad, H.; Rahimi, M.; Hashemian, S.M.; et al. In silico design and immunoinformatics analysis of a chimeric vaccine construct based on Salmonella pathogenesis factors. Microb. Pathog. 2023, 180, 106130. [Google Scholar] [CrossRef]
- Chiacchio, M.A.; Legnani, L.; Fassi, E.M.A.; Roda, G.; Grazioso, G. Development of AMBER Parameters for Molecular Simulations of Selected Boron-Based Covalent Ligands. Molecules 2023, 28, 2866. [Google Scholar] [CrossRef]
- Callil-Soares, P.H.; Biasi, L.C.K.; Filho, P.d.A.P. Effect of preprocessing and simulation parameters on the performance of molecular docking studies. J. Mol. Model. 2023, 29, 251. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipases | Amino Acids | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| PAL | Met16 | Phe108 | His109 | Ser112 | Thr114 | Ala115 | Phe117 | Leu231 | Leu232 |
| BCL | Leu17 | Phe113 | His114 | Ser117 | Phe119 | Ala120 | Phe122 | Val266 | Val267 |
| LipTioCCR11 | Phe156Met | Phe304 | His305 | Ser308 | Leu310Phe | Val311Ala | Met313Phe | Phe320Leu | Ile459Val |
| Mutation | ΔΔG (kcal/mol) |
|---|---|
| Phe156Met | −0.89 |
| Leu310Phe | −0.66 |
| Val311Ala | −0.13 |
| Met313Phe | −0.86 |
| Phe320Leu | −1.06 |
| Ile459Val | −0.36 |
| Biochemical Characteristic | LipTioCCR11 | LipTioCCR11-Mut |
|---|---|---|
| Optimal pH | 8 | 8 |
| Optimal temperature (°C) | 40 | 60 |
| Substrate preference | C16 | C12 |
| Specific activity | 3.2 kat/mg | 0.95 kat/mg |
| Effect of inhibitors and detergents (relative activity ± standard deviation, %) | None: 100 Triton-X100: 26.7 ± 2.3 Tween-20: 41.6 ± 1.6 Tween-80: 23.3 ± 1.9 EDTA: 66.5 ± 0.9 β-mercaptoethanol: 54.8 ± 0.7 SDS: 0 PMSF: 0 | None: 100 Triton-X100: 54.4 ± 0.7 Tween-20: 86.8 ± 1.5 Tween-80: 81.3 ± 2.1 EDTA: 59.5 ± 1.3 β-mercaptoethanol: 109.4 ± 1.8 SDS: 0 PMSF: 0 |
| Effect of metal ions (relative activity ± standard deviation, %) | None: 100 K+1: 79.7 ± 2.6 Na+1: 24.8 ± 0.6 Ca+2: 99.3 ± 2.3 Ba+2: 65.7 ± 2.7 Mn+2: 13.8 ± 1.1 Li+1: 37.8 ± 1.6 Hg+2: 6.3 ± 0.8 Sr+2: 39.3 ± 0.8 Cu+2: 25.5 ± 0.6 Mg+2: 55.1 ± 1.7 | None: 100 K+1: 115.3 ± 1.8 Na+1: 55.4 ± 0.5 Ca+2: 109.9 ± 2 Ba+2: 132.9 ± 2.3 Mn+2: 103.6 ± 1.4 Li+1: 101.7 ± 1.9 Hg+2: 52.1 ± 1 Sr+2: 114.8 ± 1.5 Cu+2: 103.8 ± 1.3 Mg+2: 125.7 ± 1.6 |
| Effect of exposure to organic solvents (relative activity ± standard deviation, %) | None: 100 Methanol: 37.2 ± 2.1 Ethanol: 54.5 ± 0.3 Acetone: 7.1 ± 0.6 2-propanol: 18.8 ± 1.4 Acetonitrile: 76.9 ± 0.7 Tertbutanol: 33.4 ± 1.2 Butanol: 6.4 ± 0.8 Octanol: 53.1 ± 1.7 Hexane: 86.9 ± 2.3 Heptane: 32.3 ± 2 | None: 100 Methanol: 103.7 ± 1.1 Ethanol: 128.2 ± 0.9 Acetone: 61.3 ± 2.1 2-propanol: 82.7 ± 2.3 Acetonitrile: 83.8 ± 0.4 Tertbutanol: 77.5 ± 1.8 Butanol: 25.6 ± 1.1 Octanol: 154.3 ± 1.7 Hexane: 118.4 ± 1.3 Heptane: 182 ± 1 |
| Enzyme | Methanol 25 °C | ||
|---|---|---|---|
| RAUC (AU) | SAUC (AU) | ee% | |
| LipTioCCR11 | 54 ± 1.8 | 37 ± 2.3 * | 59 R |
| LipTioCCR11-mut | 990 ± 0.7 | 58 ± 2.1 * | 94.5 R |
| CAL-B | 197 ± 1.4 | 935 ± 2.2 * | 83.5 S |
| Methanol 40 °C | |||
| LipTioCCR11 | 124 ± 0.9 | 107 ± 0.9 * | 53.5 R |
| LipTioCCR11-mut | 975 ± 0.7 | 181 ± 1.3 * | 84 R |
| CAL-B | 505 ± 0.55 | 950 ± 0.45 * | 65.5 S |
| Enzyme | Acetonitrile 25 °C | ||
|---|---|---|---|
| RAUC (AU) | SAUC (AU) | ee% | |
| LipTioCCR11 | 271 ± 1.6 | 320 ± 0.6 | 54.5 S |
| LipTioCCR11-mut | 909 ± 2.1 | 322 ± 1.6 | 80.5 R |
| CAL-B | 1030 ± 1.2 | 1001 ± 1.4 | 51 R |
| Acetonitrile 40 °C | |||
| LipTioCCR11 | 124 ± 0.9 | 107 ± 0.9 | 55.5 R |
| LipTioCCR11-mut | 1033 ± 1.6 | 220 ± 2.2 | 82.5 R |
| CAL-B | 406 ± 2.3 | 840 ± 1.8 | 67.5 S |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bustos-Baena, A.-S.; Quintana-Castro, R.; Sánchez-Otero, M.G.; Espinosa-Luna, G.; Mendoza-López, M.R.; Peña-Montes, C.; Oliart-Ros, R.M. Enantioselectivity Enhancement of a Geobacillus thermoleovorans CCR11 Lipase by Rational Design. Catalysts 2025, 15, 168. https://doi.org/10.3390/catal15020168
Bustos-Baena A-S, Quintana-Castro R, Sánchez-Otero MG, Espinosa-Luna G, Mendoza-López MR, Peña-Montes C, Oliart-Ros RM. Enantioselectivity Enhancement of a Geobacillus thermoleovorans CCR11 Lipase by Rational Design. Catalysts. 2025; 15(2):168. https://doi.org/10.3390/catal15020168
Chicago/Turabian StyleBustos-Baena, Aaron-Salvador, Rodolfo Quintana-Castro, María Guadalupe Sánchez-Otero, Graciela Espinosa-Luna, María Remedios Mendoza-López, Carolina Peña-Montes, and Rosa María Oliart-Ros. 2025. "Enantioselectivity Enhancement of a Geobacillus thermoleovorans CCR11 Lipase by Rational Design" Catalysts 15, no. 2: 168. https://doi.org/10.3390/catal15020168
APA StyleBustos-Baena, A.-S., Quintana-Castro, R., Sánchez-Otero, M. G., Espinosa-Luna, G., Mendoza-López, M. R., Peña-Montes, C., & Oliart-Ros, R. M. (2025). Enantioselectivity Enhancement of a Geobacillus thermoleovorans CCR11 Lipase by Rational Design. Catalysts, 15(2), 168. https://doi.org/10.3390/catal15020168