
Intramolecular [2+2+2] Cyclotrimerization of a Model Triyne to [7]Helical Indeno[2,1-c]Fluorene with Air-Stable Ni(0) and Other Precatalysts



Abstract

1. Introduction
2. Results and Discussion

{kind=link}
{kind=link}
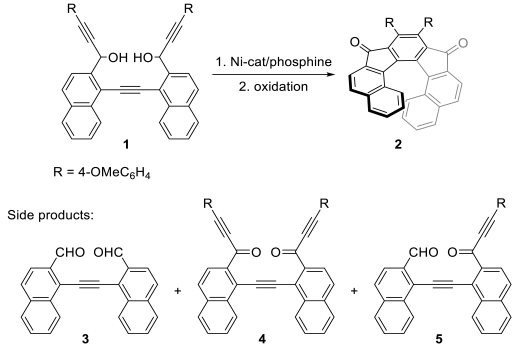 | |||
|---|---|---|---|
| Entry | Ligand (20 mol%) | Bite Angle (°) 2 | 2 (%) 3 |
| 1 | PPh3 | - | 33 (19) 4,5 |
| 2 | PCy3 | - | ~10 (n.i.) 5 |
| 3 | dppm | 71 | 70 (65) |
| 4 | dppe | 83 | 40 (23) 4,5 |
| 5 | dppBz | 82 | 77 (55) |
| 6 | dppp | 91 | 50 (47) |
| 7 | dppb | 97 | 56 (44) |
| 8 | dppf | 99 | n.i. |
| 9 | BINAP | 93 | 32 (18) 4,5 |
| 10 | DPEPhos | 101 | 43 (39) |
| 11 | Xantphos | 105 | 42 (37) |
| 12 | Bipy | - | 33 (26) 4,5 |
| Entry | Precatalyst | Ligand 2 | Solvent | T (°) | 2 (%) 3 | e.r. |
|---|---|---|---|---|---|---|
| 1 | Ni(COD)(DQ) | (S)-DIFLUORPHOS | toluene | 100 | 46 (19) 4 | 56:44 |
| 2 | (S)-SEGPHOS | toluene | 100 | 40 (26) 4 | 50:50 | |
| 3 | (S)-BINAP | toluene | 100 | 30 (19) 4,5 | 52:48 | |
| 4 | (R)-SEGPHOS | toluene | 100 | 46 (20) | 52:48 | |
| 5 | (R)-Xyl-Garphos | toluene | 100 | 23 (18) 4,5 | 54:46 | |
| 6 | (R)-DMM-Garphos | toluene | 100 | 38 (17) 4,5 | 52:48 | |
| 7 | Me-MeoBiphep | toluene | 100 | n.i 5 | ||
| 8 | t-Bu-MeoBiphep | toluene | 100 | n.i 5 | ||
| 9 | (R)-PROPHOS | toluene | 100 | 100 (60) | 52:48 | |
| 10 | Ni(4-tBustb)3 | (R)-PROPHOS | toluene | 80 | n.i. 5 | |
| 11 | (R)-PROPHOS | THF | 80 | 27 (13) 5 | 55:45 | |
| 12 | (R)-PROPHOS | HFIP | 80 | 13 (12) 4,5 | 19:81 |
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| COD | 1,5-cyclooctadiene |
| DQ | duroquinone |
| 4-tBustb | (E)-1,2-bis(4-(tert-butyl)phenyl)ethene |
| PPh3 | triphenylphosphine |
| PCy3 | tricyclohexylphosphine |
| dppm | bis(diphenylphosphino)methane |
| dppe | 1,2-bis(diphenylphosphino)ethane |
| dppBz | 1,2-bis(diphenylphosphino)benzene |
| dppp | 1,3-bis(diphenylphosphino)propane |
| dppb | 1,4-bis(diphenylphosphino)butane |
| dppf | 1,1′-bis(diphenylphosphino)ferrocene |
| BINAP | 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl |
| DPEPhos | bis[(2-diphenylphosphino)phenyl] ether |
| Xantphos | 9,9-dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphine) |
| Bipy | 2,2′-bipyridine |
| (S)-DIFLUORPHOS | (S)-(+)-2,2,2′,2′-tetrafluoro-4,4′-bibenzo[d][1,3]dioxole-5,5′-diyl)bis(diphenylphosphine) |
| (S)-SEGPHOS | (S)-(−)-5,5′-bis(diphenylphosphino)-4,4′-bi-1,3-benzodioxole |
| (S)-BINAP | (S)-(−)-2,2′-bis(diphenylphosphino)-1,1′-binaphthalene |
| (R)-SEGPHOS | (R)-(+)-5,5′-bis(diphenylphosphino)-4,4′-bi-1,3-benzodioxole |
| (R)-Xyl-Garphos | (R)-(4,4′,6,6′-tetramethoxybiphenyl-2,2′-diyl)bis(bis(3,5-dimethylphenyl)phosphine) |
| (R)-DMM-Garphos | (R)-(4,4′,6,6′-tetramethoxybiphenyl-2,2′-diyl) bis[bis(4-methoxy-3,5-dimethylphenyl)phosphine] |
| Me-MeoBiphep | (R)-(+)-2,2′-bis[di(3,5-xylyl)phosphino]-6,6′-dimethoxy-1,1′-biphenyl |
| t-Bu-MeoBiphep | (R)-(+)-2,2′-bis[bis(3,5-di-tert-butyl)phosphino]-6,6′-dimethoxy-1,1′-biphenyl |
| (R)-Prophos | (R)-(+)-1,2-bis(diphenylphosphino)propane |
| HFIP | hexafluoroisopropanol |
References
- Berthelot, M. Ueber die Einwirkung der Hitze auf einige Kohlenwasserstoffe. Justus Liebigs Ann. Chem. 1866, 139, 272–282. [Google Scholar] [CrossRef]
- Berthelot, M.C.R. Action de la chauler sur quelques carbures d’hydrogene. Hebd. Seances Acad. Sci. 1866, 62, 905–909. [Google Scholar]
- Reppe, W.; Schweckendiek, W.J. Cyclisierende Polymerisation von Acetylen. III Benzol, Benzolderivate und hydroaromatische Verbindungen. Justus Liebigs Ann. Chem. 1948, 560, 104. [Google Scholar]
- Pla-Quintana, A.; Roglans, A. [2+2+2] Cycloaddition Reactions of Macrocyclic Systems Catalyzed by Transition Metals. A Review. Molecules 2010, 15, 9230–9251. [Google Scholar] [CrossRef]
- Matton, P.; Huvelle, S.; Haddad, M.; Ohansavatrh, P.; Ratovelomanana-Vidal, V. Recent Progress in Metal-Catalyzed [2+2+2] Cycloaddition Reactions. Synthesis 2022, 54, 4–32. [Google Scholar]
- Shaaban, M.R.; El-Sayed, R.; Elwahy, A.H.M. Construction of fused heterocycles by metal-mediated [2þ2þ2] cyclotrimerization of alkynes and/or nitriles. Tetrahedron 2011, 67, 6095–6130. [Google Scholar] [CrossRef]
- Trotus, I.-T.; Zimmermann, T.; Schüth, F. Catalytic Reactions of Acetylene: A Feedstock for the Chemical Industry Revisited. Chem. Rev. 2014, 114, 1761–1782. [Google Scholar] [CrossRef]
- Zweig, J.E.; Kim, D.E.; Newhouse, T.R. Methods Utilizing First-Row Transition Metals in Natural Product Total Synthesis. Chem. Rev. 2017, 117, 11680–11752. [Google Scholar] [CrossRef]
- Kotha, S.; Lahiri, K.; Sreevani, G. Design and Synthesis of Aromatics through [2+2+2] Cyclotrimerization. Synlett 2018, 29, 2342–2361. [Google Scholar] [CrossRef]
- Zhu, X.-Q.; Li, Y.-C.; Ye, L.-W. Enantioselective Reaction of Diynes and Multiynes for the Synthesis of Axially Chiral Compounds. Asian J. Org. Chem. 2024, 13, e202300554. [Google Scholar] [CrossRef]
- Stará, I.G.; Starý, I. Helically Chiral Aromatics: The Synthesis of Helicenes by [2 + 2 + 2] Cycloisomerization of π-Electron Systems. Acc. Chem. Res. 2020, 53, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Morita, F. Enantioselective Helicene Synthesis by Rhodium─Catalyzed [2+2+2] Cycloaddition. Bull. Chem. Soc. Jpn. 2022, 80, 1019–1027. [Google Scholar] [CrossRef]
- Cadart, T.; Nečas, D.; Kaiser, R.P.; Favereau, L.; Císařová, I.; Gyepes, R.; Hodačová, J.; Kalíková, K.; Bednárová, L.; Crassous, J.; et al. Rhodium Catalyzed Enantioselective Synthesis of Highly Fluorescent and CPL Active Dispiroindeno [2,1-c]fluorenes. Chem. Eur. J. 2021, 27, 11279–11284. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, R.P.; Nečas, D.; Cadart, T.; Gyepes, R.; Císařová, I.; Mosinger, J.; Pospíšil, L.; Kotora, M. Straightforward Synthesis and Properties of Highly Fluorescent [5]- and [7]-Helical Dispiroindeno [2,1-c]fluorenes . Angew. Chem. Int. Ed. 2019, 58, 17169–17174. [Google Scholar] [CrossRef]
- Alexandrová, Z.; Sehnal, P.; Stará, I.G.; Starý, I.; Šaman, D.; Urquhart, S.G.; Otero, E. Modified Synthesis of Heptahelicene and Its Resolution into Single Enantiomers. Collect. Czech. Chem. Commun. 2006, 71, 1256–1264. [Google Scholar] [CrossRef]
- Stará, I.G.; Andronova, A.; Kollárovič, A.; Vyskočil, Š.; Jugé, S.; Lloyd-Jones, G.C.; Guiry, P.; Starý, I. Enantioselective [2+2+2] Cycloisomerization of Alkynes in the Synthesis of Helicenes: The Search for Effective Chiral Ligands. Collect. Czechoslov. Chem. Commun. 2011, 76, 2005–2022. [Google Scholar] [CrossRef]
- Kiss, A.; Feriancová, L.; Gyepes, R.; Cadart, T.; Kotora, M. Substituted indeno [2,1-c]fluorene-5,8-diones as a platform for synthesis of polyaromatic hydrocarbons possessing extended π-conjugated systems with butterfly-like frameworks. Appl. Organomet. Chem. 2024, 38, e7621. [Google Scholar]
- Seino, K.; Okano, T.; Oya, K.; Katagiri, H.; Murase, T. Helix-to-Disc Conversion of Thia [6]helicenes into Coronenes Facilitated by Sulfur Oxidation and Fluorination. Chem. Eur. J. 2024, 30, e202402445. [Google Scholar] [CrossRef]
- Shen, C.; Zhang, G.; Ding, Y.; Yang, N.; Gan, F.; Crassous, J.; Qiu, H. Oxidative cyclo-rearrangement of helicenes into chiral nanographenes. Nat. Commun. 2021, 12, 2786. [Google Scholar] [CrossRef]
- Uematsu, K.; Hayasaka, C.; Takase, K.; Noguchi, K.; Nakano, K. Transformation of Thia [7]helicene to Aza [7]helicenes and [7]Helicene-like Compounds via Aromatic Metamorphosis. Molecules 2022, 27, 606. [Google Scholar] [CrossRef]
- Turek, P.; Novák, P.; Pohl, R.; Hocek, M.; Kotora, M. Preparation of Highly Substituted 6-Arylpurine Ribonucleosides by Ni-Catalyzed Cyclotrimerization. Scope of the Reaction. J. Org. Chem. 2006, 71, 8978–8980. [Google Scholar] [CrossRef] [PubMed]
- Tran, V.T.; Li, Z.-Q.; Apolinar, O.; Derosa, J.; Joannou, M.V.; Wisniewski, S.R.; Eastgate, M.D.; Engle, K.M. Ni(COD)(DQ): An Air-Stable 18-Electron Nickel(0)–Olefin Precatalyst. Angew. Chem. Int. Ed. 2020, 59, 7409–7413. [Google Scholar] [CrossRef] [PubMed]
- Nattmann, L.; Cornella, J. Ni(4-tBustb)3: A Robust 16-Electron Ni(0) Olefin Complex for Catalysis. Organometallics 2020, 39, 3295–3300. [Google Scholar] [CrossRef]
- Gou, T.; Wang, B.-Q.; Shi, Z.-J. Catalytic activations of unstrained C–C bond involving organometallic intermediates. Chem. Soc. Rev. 2018, 47, 7078–7115. [Google Scholar] [CrossRef]
- Dierkes, P.; van Leeuwen, P.W.N.M. The bite angle makes the difference: A practical ligand parameter for diphosphine ligands. J. Chem. Soc. Dalton Trans. 1999, 1519–1529. [Google Scholar] [CrossRef]
- Heller, B.; Hapke, M.; Fischer, C.; Andronova, A.; Starý, I.; Stará, I.G. Chiral cobaltI and nickel0 complexes in the synthesis of nonracemic helicenes through the enantioselective [2 + 2 + 2] cyclotrimerisation of alkynes. J. Organomet. Chem. 2013, 723, 98–102. [Google Scholar] [CrossRef]
- Stará, I.G.; Starý, I.; Kollárovič, A.; Teplý, F.; Vyskočil, Š.; Šaman, D. Transition Metal Catalysed Synthesis of Tetrahydro Derivatives of [5]-, [6]- and [7]Helicene. Tetrahedron Lett. 1999, 40, 1993–1996. [Google Scholar] [CrossRef]
- Teplý, F.; Stará, I.G.; Starý, I.; Kollárovič, A.; Šaman, D.; Vyskočil, Š.; Fiedler, P. Synthesis of 3-Hexahelicenol and Its Transformation to 3-Hexahelicenylamines, Diphenylphosphine, Methyl Carboxylate, and Dimethylthiocarbamate. J. Org. Chem. 2003, 68, 5193–5197. [Google Scholar] [CrossRef]
- Motiwala, H.F.; Armaly, A.M.; Cacioppo, J.G.; Coombs, T.C.; Koehn, K.R.K.; Norwood IV, V.M.; Aube, J. HFIP in Organic Synthesis. Chem. Rev. 2022, 122, 12544–12747. [Google Scholar] [CrossRef]
- Li, K.; Wei, L.; Sun, M.; Li, B.; Liu, M.; Li, C. Enantioselective Synthesis of Pyridines with All-Carbon Quaternary Carbon Centers via Cobalt-Catalyzed Desymmetric [2+2+2]Cycloaddition. Angew. Chem. Int. Ed. 2021, 60, 20204–20209. [Google Scholar] [CrossRef]
- Jeulin, S.; Duprat de Paule, S.; Ratovelomanana-Vidal, V.; Genet, J.-P.; Champion, N.; Dellis, P. Chiral biphenyl diphosphines for asymmetric catalysis: Stereoelectronic design and industrial perspectives. Proc. Natl. Acad. Sci. USA 2004, 101, 5799–5804. [Google Scholar] [CrossRef] [PubMed]
- Brun, S.; Garcia, L.; Gonzáles, I.; Torrent, A.; Dachs, A.; Pla-Quitana, A.; Parella, T.; Roglans, A. Fused tetracycles with a benzene or cyclohexadiene core: [2 + 2 + 2] cycloadditions on macrocyclic systems. Chem. Commun. 2005, 36, 4339–4341. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, T.; Jia, J.-W.; Yamaguchi, Y.; Tanimoto, H.; Kakiuchi, K. Cationic Rhodium(I)-Catalyzed Carbonylative [2+2+1] Cycloaddition of Diynes. Asian J. Org. Chem. 2020, 9, 1778–1782. [Google Scholar] [CrossRef]
- Lynch, L.; Sherwood, J.; McElroy, C.R.; Murray, J.; Shimizu, S. Dichloromethane replacement: Towards greener chromatography via Kirkwood–Buff integrals. Anal. Methods 2023, 15, 596–605. [Google Scholar] [CrossRef]
- Thiel, I.; Spannenberg, A.; Hapke, M. Synthesis of Air-Stable and Recyclable CpCoI-Complexes. ChemCatChem 2013, 5, 2865–2868. [Google Scholar] [CrossRef]
- Fischer, F.; Hapke, M. Air-Stable CpCoI–Phosphite–Fumarate Precatalyst in Cyclization Reactions: Comparing Different Methods of Energy Supply. Eur. J. Org. Chem. 2018, 2018, 3193–3201. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Degač, M.; Kotora, M. Intramolecular [2+2+2] Cyclotrimerization of a Model Triyne to [7]Helical Indeno[2,1-c]Fluorene with Air-Stable Ni(0) and Other Precatalysts. Catalysts 2025, 15, 150. https://doi.org/10.3390/catal15020150
Degač M, Kotora M. Intramolecular [2+2+2] Cyclotrimerization of a Model Triyne to [7]Helical Indeno[2,1-c]Fluorene with Air-Stable Ni(0) and Other Precatalysts. Catalysts. 2025; 15(2):150. https://doi.org/10.3390/catal15020150
Chicago/Turabian StyleDegač, Marina, and Martin Kotora. 2025. "Intramolecular [2+2+2] Cyclotrimerization of a Model Triyne to [7]Helical Indeno[2,1-c]Fluorene with Air-Stable Ni(0) and Other Precatalysts" Catalysts 15, no. 2: 150. https://doi.org/10.3390/catal15020150
APA StyleDegač, M., & Kotora, M. (2025). Intramolecular [2+2+2] Cyclotrimerization of a Model Triyne to [7]Helical Indeno[2,1-c]Fluorene with Air-Stable Ni(0) and Other Precatalysts. Catalysts, 15(2), 150. https://doi.org/10.3390/catal15020150