
Electrocatalytic Pathways and Efficiency of Cuprous Oxide (Cu2O) Surfaces in CO2 Electrochemical Reduction (CO2ER) to Methanol: A Computational Approach

 , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
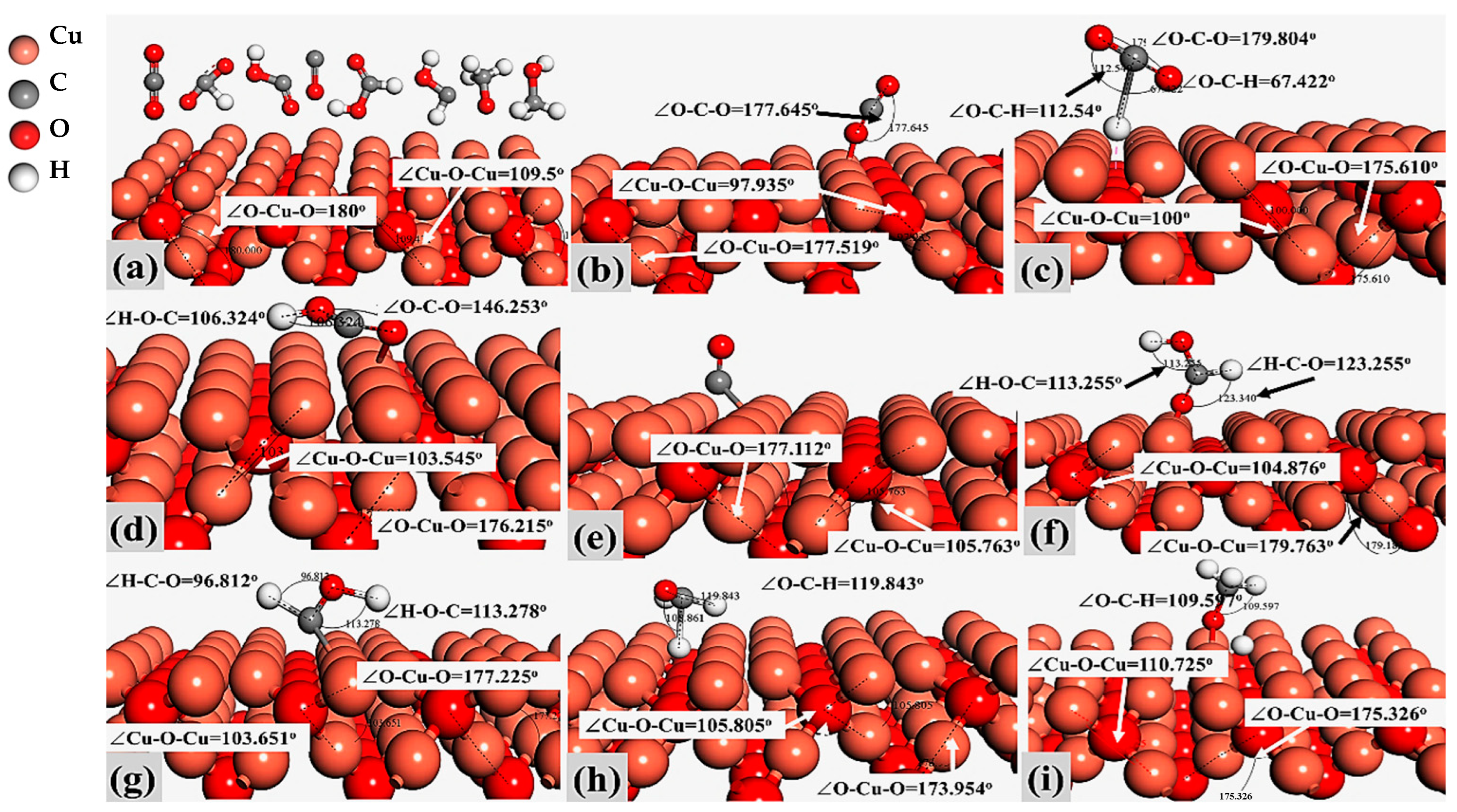
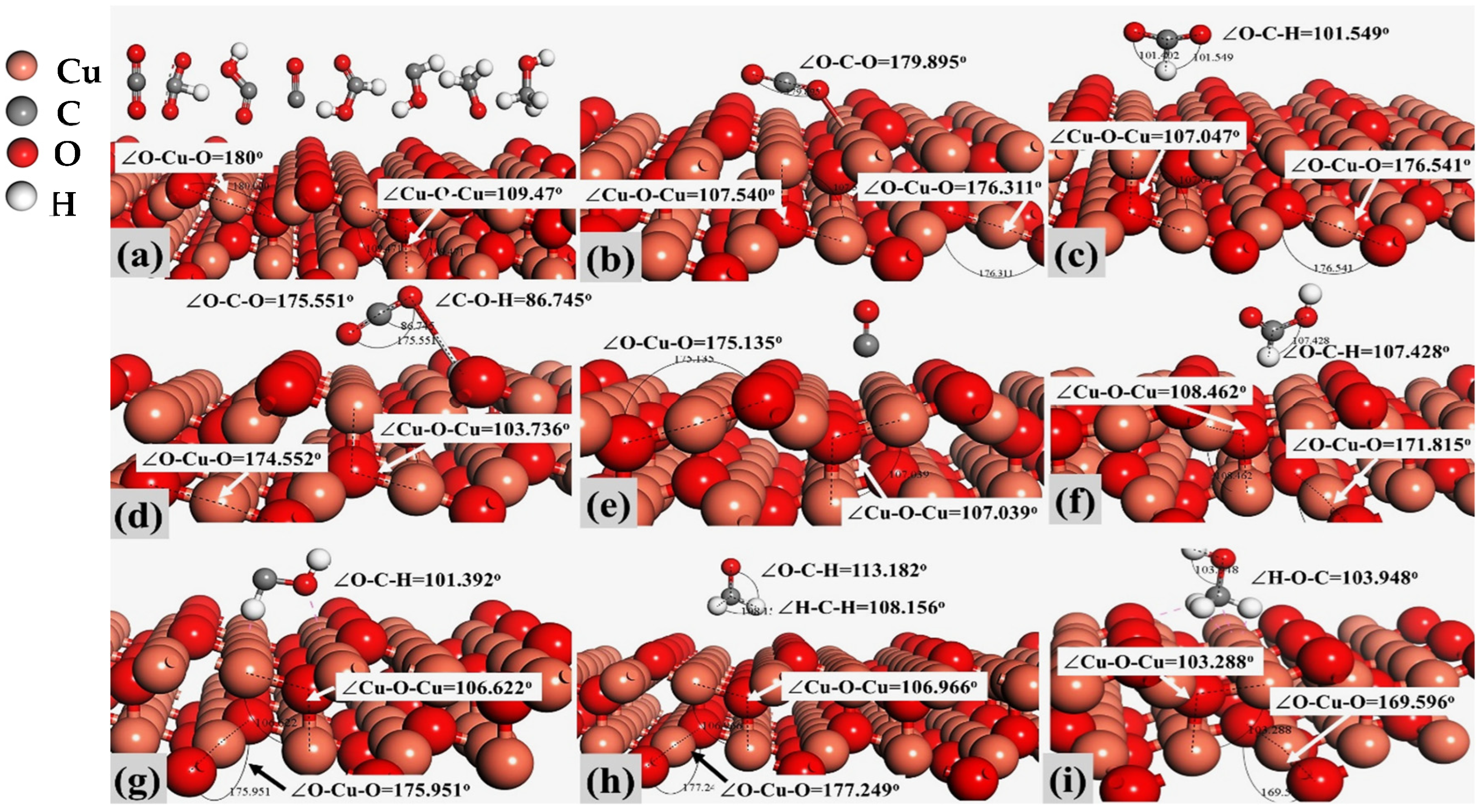
2.1. Models and Molecular Activation on Cu2O (100) and Cu2O (111) Sites
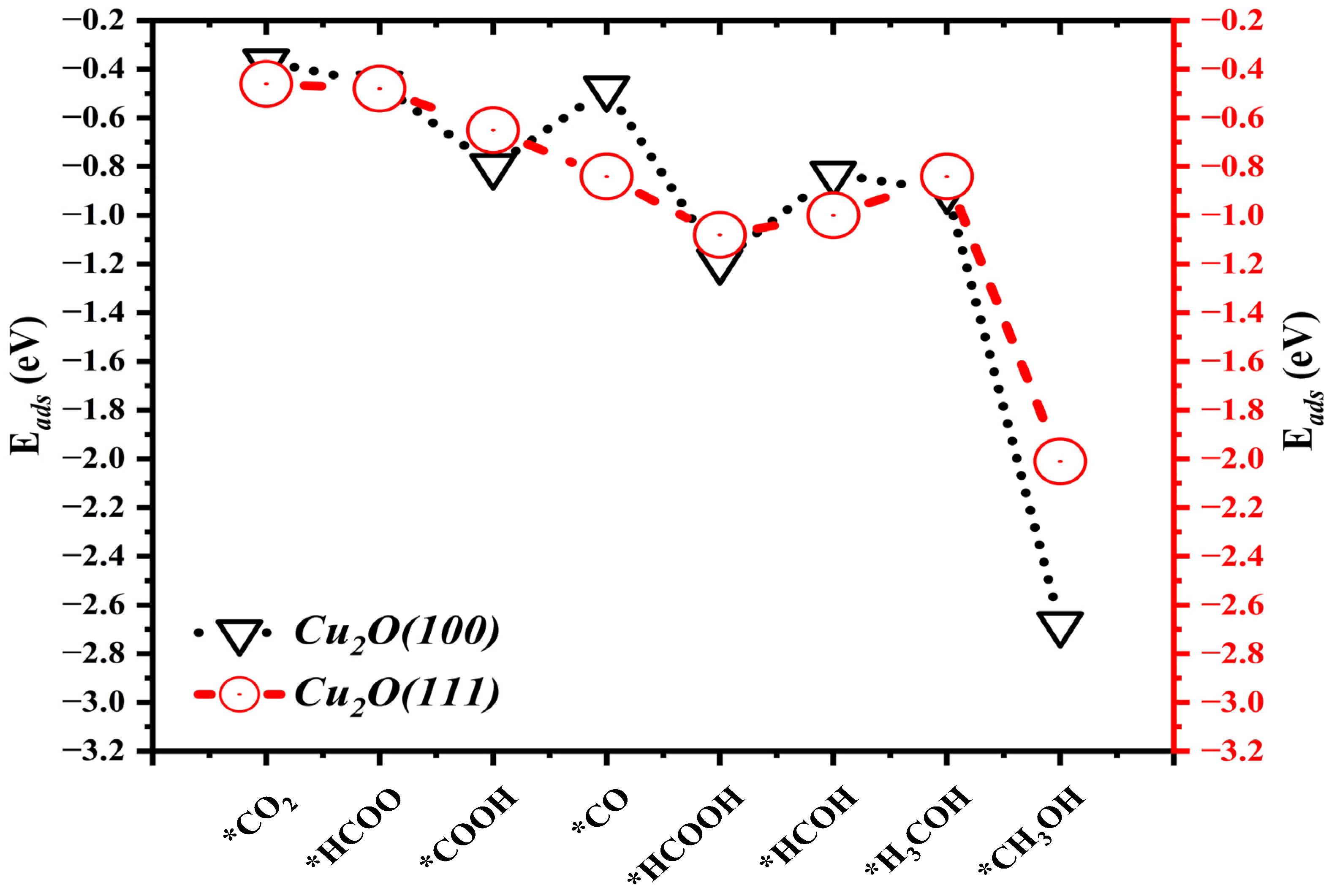
2.2. Adsorption Energies (Eads)
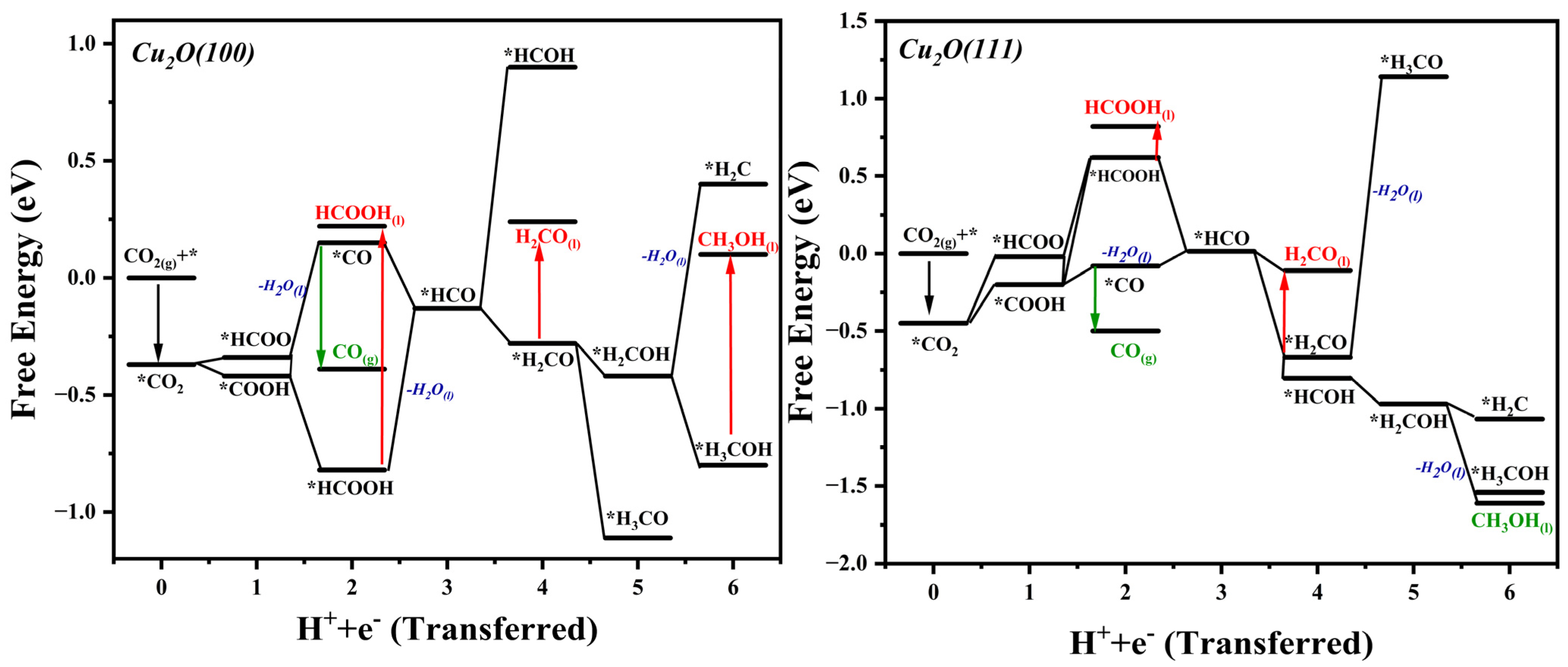
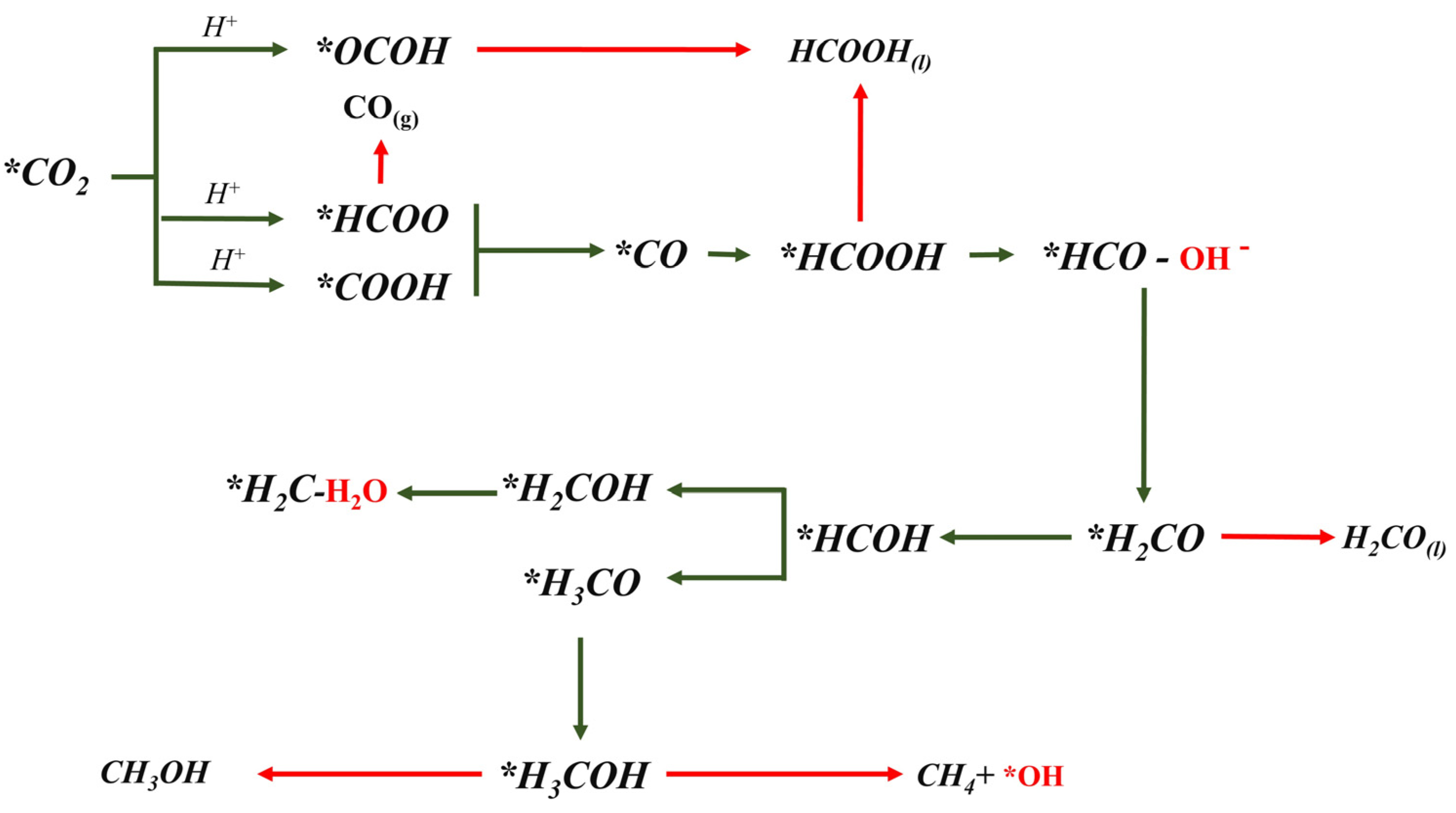
2.3. ΔG Profiles and Reduction Pathways of CO2ER
3. Model and Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kabir, M.; Habiba, U.E.; Khan, W.; Shah, A.; Rahim, S.; Rios-Escalante, P.R.D.l.; Farooqi, Z.-U.-R.; Ali, L.; Shafiq, M. Climate change due to increasing concentration of carbon dioxide and its impacts on environment in 21st century; a mini review. J. King Saud Univ.-Sci. 2023, 35, 102693. [Google Scholar] [CrossRef]
- Yang, M.; Chen, L.; Wang, J.; Msigwa, G.; Osman, A.I.; Fawzy, S.; Rooney, D.W.; Yap, P.-S. Circular economy strategies for combating climate change and other environmental issues. Environ. Chem. Lett. 2022, 21, 55–80. [Google Scholar] [CrossRef]
- Schleussner, C.-F.; Ganti, G.; Rogelj, J.; Gidden, M.J. An emission pathway classification reflecting the Paris Agreement climate objectives. Commun. Earth Environ. 2022, 3, 135. [Google Scholar] [CrossRef]
- Abdul-Latif, N.S.; Ong, M.Y.; Nomanbhay, S.; Salman, B.; Show, P.L. Estimation of carbon dioxide (CO2) reduction by utilization of algal biomass bioplastic in Malaysia using carbon emission pinch analysis (CEPA). Bioengineered 2020, 11, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Modak, A.; Bhanja, P.; Dutta, S.; Chowdhury, B.; Bhaumik, A. Catalytic reduction of CO2 into fuels and fine chemicals. Green Chem. 2020, 22, 4002–4033. [Google Scholar] [CrossRef]
- Fouda, A. Nano-porous Carbon-Based Electrodes for the Electrocatalytic Reduction of CO2 to Methanol. Doctoral Dissertation, Khalifa University of Science, Abu Dhabi, United Arab Emirates, 2023. [Google Scholar]
- Lu, S.; Lou, F.; Yu, Z. Recent progress in two-dimensional materials for electrocatalytic CO2 reduction. Catalysts 2022, 12, 228. [Google Scholar] [CrossRef]
- Albo, J.; Alvarez-Guerra, M.; Castaño, P.; Irabien, A. Towards the electrochemical conversion of carbon dioxide into methanol. Green Chem. 2015, 17, 2304–2324. [Google Scholar] [CrossRef]
- Joghee, P.; Malik, J.N.; Pylypenko, S.; O’Hayre, R. A review on direct methanol fuel cells–In the perspective of energy and sustainability. MRS Energy Sustain. 2015, 2, E3. [Google Scholar] [CrossRef]
- Tabibian, S.S.; Sharifzadeh, M. Statistical and analytical investigation of methanol applications, production technologies, value-chain and economy with a special focus on renewable methanol. Renew. Sustain. Energy Rev. 2023, 179, 113281. [Google Scholar] [CrossRef]
- Panafidin, M.A.; Bukhtiyarov, A.V.; Fedorov, A.Y.; Bukhtiyarova, M.V.; Prosvirin, I.P.; Bukhtiyarov, V.I. In situ XPS study of methanol oxidation over a copper catalyst derived from layered double hydroxides. Catal. Sci. Technol. 2024, 14, 4986–4996. [Google Scholar] [CrossRef]
- Marcandalli, G.; Monteiro, M.C.O.; Goyal, A.; Koper, M.T.M. Electrolyte Effects on CO2 Electrochemical Reduction to CO. Acc Chem. Res. 2022, 55, 1900–1911. [Google Scholar] [CrossRef]
- Kumar, A.; Aeshala, L.M.; Palai, T. Electrochemical reduction of CO2 to useful fuel: Recent advances and prospects. J. Appl. Electrochem. 2023, 53, 1295–1319. [Google Scholar] [CrossRef]
- Razzaq, A.; In, S.-I. TiO2 based nanostructures for photocatalytic CO2 conversion to valuable chemicals. Micromachines 2019, 10, 326. [Google Scholar] [CrossRef] [PubMed]
- Kiendl, B. Application of Diamond Nanomaterials in Catalysis. Doctoral Dissertation, Universität Würzburg, Würzburg, Germany, 2020. [Google Scholar]
- Siwek, H.; Łukaszewski, M.; Czerwiński, A. Electrochemical study on the adsorption of carbon oxides and oxidation of their adsorption products on platinum group metals and alloys. Phys. Chem. Chem. Phys. 2008, 10, 3752–3765. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-Y.; Park, K.T.; Lee, W.; Lim, H.; Kwon, Y.; Kang, S. Current achievements and the future direction of electrochemical CO2 reduction: A short review. Crit. Rev. Environ. Sci. Technol. 2020, 50, 769–815. [Google Scholar] [CrossRef]
- Hussain, I.; Alasiri, H.; Ullah Khan, W.; Alhooshani, K. Advanced electrocatalytic technologies for conversion of carbon dioxide into methanol by electrochemical reduction: Recent progress and future perspectives. Coord. Chem. Rev. 2023, 482, 215081. [Google Scholar] [CrossRef]
- Ait Ahsaine, H.; BaQais, A. Metal and metal oxide electrocatalysts for the electrochemical reduction of CO2-to-C1 chemicals: Are we there yet? Green Chem. Lett. Rev. 2023, 16, 2160215. [Google Scholar] [CrossRef]
- An, X.; Wang, P.; Ma, X.; Du, X.; Hao, X.; Yang, Z.; Guan, G. Application of ionic liquids in CO2 capture and electrochemical reduction: A review. Carbon Resour. Convers. 2023, 6, 85–97. [Google Scholar] [CrossRef]
- Alsunni, Y.A.; Alherz, A.W.; Musgrave, C.B. Electrocatalytic reduction of CO2 to CO over Ag (110) and Cu (211) modeled by grand-canonical density functional theory. J. Phys. Chem. C 2021, 125, 23773–23783. [Google Scholar] [CrossRef]
- Guzmán, H.; Russo, N.; Hernández, S. CO2 valorisation towards alcohols by Cu-based electrocatalysts: Challenges and perspectives. Green Chem. 2021, 23, 1896–1920. [Google Scholar] [CrossRef]
- Alamro, F.S.; Medany, S.S.; Al-Kadhi, N.S.; Mostafa, A.M.; Zaher, W.F.; Ahmed, H.A.; Hefnawy, M.A. Controllable Synthesis of Fe2O3/Nickel Cobaltite Electrocatalyst to Enhance Oxidation of Small Molecules. Catalysts 2024, 14, 329. [Google Scholar] [CrossRef]
- Shi, H.; Wu, L.; Zhang, Q.; Zhang, Y.; Sun, W.; Liu, C.; Zhang, R. Preparation and Electrocatalytic Properties of One-Dimensional Nanorod-Shaped N, S Co-Doped Bimetallic Catalysts of FeCuS-N-C. Catalysts 2024, 14, 849. [Google Scholar] [CrossRef]
- Wang, Y.; Zheng, M.; Wang, X.; Zhou, X. Electrocatalytic Reduction of CO2 to C1 Compounds by Zn-Based Monatomic Alloys: A DFT Calculation. Catalysts 2022, 12, 1617. [Google Scholar] [CrossRef]
- Yun, G.; Young Hwang, S.; Young Kim, S.; Gwon, Y.; Bae, S.; Kyun Rhee, C.; Sohn, Y. Exploring Pd-Ag/Cu electrodes in electrochemical CO2 reduction: Insights into C1, C2, and C3+ chemistry. Appl. Surf. Sci. 2024, 665, 160279. [Google Scholar] [CrossRef]
- Jia, S.; Wu, L.; Xu, L.; Sun, X.; Han, B. Multicomponent catalyst design for CO2/N2/NOx electroreduction. Ind. Chem. Mater. 2023, 1, 93–105. [Google Scholar] [CrossRef]
- Roy, A.; Jadhav, H.S.; Gil Seo, J. Cu2O/CuO Electrocatalyst for Electrochemical Reduction of Carbon Dioxide to Methanol. Electroanalysis 2020, 33, 705–712. [Google Scholar] [CrossRef]
- Wang, P.; Yang, H.; Tang, C.; Wu, Y.; Zheng, Y.; Cheng, T.; Davey, K.; Huang, X.; Qiao, S.Z. Boosting electrocatalytic CO2-to-ethanol production via asymmetric C-C coupling. Nat Commun. 2022, 13, 3754. [Google Scholar] [CrossRef]
- Ouyang, Y.; Shi, L.; Bai, X.; Li, Q.; Wang, J. Breaking scaling relations for efficient CO2 electrochemical reduction through dual-atom catalysts. Chem. Sci. 2020, 11, 1807–1813. [Google Scholar] [CrossRef]
- Yun, G.; Hwang, S.Y.; Maeng, J.Y.; Kim, Y.J.; Rhee, C.K.; Sohn, Y. Electrochemical CO2/CO reduction on Ag/Cu electrodes and exploring minor Fischer–Tropsch reaction pathways. Appl. Surf. Sci. 2024, 649, 159179. [Google Scholar] [CrossRef]
- Liu, Y.; Li, F.; Zhang, X.; Ji, X. Recent progress on electrochemical reduction of CO2 to methanol. Curr. Opin. Green Sustain. Chem. 2020, 23, 10–17. [Google Scholar] [CrossRef]
- Darban, M.A.; Lock, S.S.M.; Ilyas, S.U.; Kang, D.Y.; Othman, M.H.D.; Yiin, C.L.; Waqas, S.; Bashir, Z. Molecular simulation of [P8883][Tf(2)N] ionic liquid decorated silica in 6FDA-ODA based mixed matrix membrane for enhanced CO2/CH4 separation. RSC Adv. 2024, 14, 22894–22915. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Xu, X.; Li, Y.; Zhang, Y.; Song, Y. CO2 reduction mechanism on the Nb2CO2 MXene surface: Effect of nonmetal and metal modification. Comput. Mater. Sci. 2022, 202, 110971. [Google Scholar] [CrossRef]
- Mohammed, S.A.S.; Yahya, W.Z.N.; Bustam, M.A. Computational studies of ionic liquids as co-catalyst for CO2 electrochemical reduction to produce syngas using COSMO-RS. E3S Web Conf. 2021, 287, 02016. [Google Scholar] [CrossRef]
- Almajed, H. A Theoretical Study of the Electrochemical CO2 Reduction over Single Atom Catalysts Using Grand-Canonical DFT; University of Colorado at Boulder: Boulder, CO, USA, 2021. [Google Scholar]
- Kuo, T.-C.; Chou, J.-W.; Shen, M.-H.; Hong, Z.-S.; Chao, T.-H.; Lu, Q.; Cheng, M.-J. First-principles study of C–C coupling pathways for CO2 electrochemical reduction catalyzed by Cu (110). J. Phys. Chem. C 2021, 125, 2464–2476. [Google Scholar] [CrossRef]
- Bi, X.; Yan, Y.; Wang, H.; Zhao, Y.; Zhang, J.; Wu, M. Electroreduction of CO2 to C2H4 Regulated by Spacing Effect: Mechanistic Insights from DFT Studies. Energy Mater. Adv. 2023, 4, 0037. [Google Scholar] [CrossRef]
- Li, C.W.; Kanan, M.W. CO2 reduction at low overpotential on Cu electrodes resulting from the reduction of thick Cu2O films. J. Am. Chem. Soc. 2012, 134, 7231–7234. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Suo, W.; Zhang, Z.; Wang, Y.; Sun, S.; Liu, G. Insights on the catalytic active site for CO2 reduction on copper-based catalyst: A DFT study. Mol. Catal. 2021, 511, 111725. [Google Scholar] [CrossRef]
- Xiao, Y.; Tang, L.; Zhang, W.; Shen, C. Theoretical insights into the selective and activity of CuAu catalyst for O2 and CO2 electroreduction. Comput. Mater. Sci. 2021, 192, 110402. [Google Scholar] [CrossRef]
- Chu, S.; Kang, C.; Park, W.; Han, Y.; Hong, S.; Hao, L.; Zhang, H.; Lo, T.W.B.; Robertson, A.W.; Jung, Y. Single atom and defect engineering of CuO for efficient electrochemical reduction of CO2 to C2H4. SmartMat 2022, 3, 194–205. [Google Scholar] [CrossRef]
- Liu, T.; Song, G.; Liu, X.; Chen, Z.; Shen, Y.; Wang, Q.; Peng, Z.; Wang, G. Insights into the mechanism in electrochemical CO2 reduction over single-atom copper alloy catalysts: A DFT study. Iscience 2023, 26, 107953. [Google Scholar] [CrossRef]
- Masood, Z.; Ge, Q. Electrochemical reduction of CO2 at the earth-abundant transition metal-oxides/copper interfaces. Catal. Today 2023, 409, 53–62. [Google Scholar] [CrossRef]
- Gao, Y.; Wu, Q.; Liang, X.; Wang, Z.; Zheng, Z.; Wang, P.; Liu, Y.; Dai, Y.; Whangbo, M.H.; Huang, B. Cu2O Nanoparticles with Both 100 and 111 Facets for Enhancing the Selectivity and Activity of CO2 Electroreduction to Ethylene. Adv. Sci. 2020, 7, 1902820. [Google Scholar] [CrossRef]
- Haouas, H.; El Atouani, L.; Sbiaai, K.; Hasnaoui, A. Size and temperature effects on surface energy of Au and Fe nanoparticles from atomistic simulations. Comput. Mater. Sci. 2022, 214, 111695. [Google Scholar] [CrossRef]
- Gholizadeh, R.; Pavlin, M.; Huš, M.; Likozar, B. Multiscale Modeling of CO2 Electrochemical Reduction on Copper Electrocatalysts: A Review of Advancements, Challenges, and Future Directions. ChemSusChem 2024, 18, e202400898. [Google Scholar] [CrossRef]
- Chang, K.; Zhang, H.; Chen, J.G.; Lu, Q.; Cheng, M.-J. Constant Electrode Potential Quantum Mechanical Study of CO2 Electrochemical Reduction Catalyzed by N-Doped Graphene. ACS Catal. 2019, 9, 8197–8207. [Google Scholar] [CrossRef]
- Masood, Z.; Ge, Q. Comparative Study of Computational Hydrogen Electrodes and Constant Electrode Potential Models Applied to Electrochemical Reduction of CO2 and Oxygen Evolution Reaction on Metal Oxides/Copper Catalysts. J. Phys. Chem. C 2023, 127, 23170–23179. [Google Scholar] [CrossRef]
- Garza, A.J.; Bell, A.T.; Head-Gordon, M. Mechanism of CO2 Reduction at Copper Surfaces: Pathways to C2 Products. ACS Catal. 2018, 8, 1490–1499. [Google Scholar] [CrossRef]
- Xue, Y.; Guo, Y.; Cui, H.; Zhou, Z. Catalyst Design for Electrochemical Reduction of CO2 to Multicarbon Products. Small Methods 2021, 5, e2100736. [Google Scholar] [CrossRef]
- Li, S.; Tong, L.; Peng, Z.; Zhang, B.; Fu, X. Efficient CO2 reduction for CO production using triatomic catalyst screening: A DFT study. Appl. Surf. Sci. 2024, 644, 158804. [Google Scholar] [CrossRef]
- Wu, R.; Liu, D.; Geng, J.; Bai, H.; Li, F.; Zhou, P.; Pan, H. Electrochemical reduction of CO2 on single-atom catalysts anchored on N-terminated TiN (1 1 1): Low overpotential and high selectivity. Appl. Surf. Sci. 2022, 602, 154239. [Google Scholar] [CrossRef]
- Kundappaden, I.; Chatanathodi, R. A DFT study of CO adsorption on Pt (111) using van der Waals functionals. Surf. Sci. 2019, 681, 143–148. [Google Scholar] [CrossRef]
- Yang, Y.B.; Hao, Q.; Muller-Plathe, F.; Bohm, M.C. Monte Carlo simulations of SO2, H2S, and CO2 adsorption in charged single-walled carbon nanotube arrays. J. Phys. Chem. C 2020, 124, 5838–5852. [Google Scholar] [CrossRef]
- Jain, A.; Ong, S.; Hautier, G. Commentary: The materials project: A materials genome approach to accelerating materials. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef]
- Gražulis, S.; Chateigner, D.; Downs, R.T.; Yokochi, A.; Quirós, M.; Lutterotti, L.; Manakova, E.; Butkus, J.; Moeck, P.; Le Bail, A. Crystallography Open Database–an open-access collection of crystal structures. J. Appl. Crystallogr. 2009, 42, 726–729. [Google Scholar] [CrossRef]
- Coordinators, N.R. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2018, 46, D8. [Google Scholar] [CrossRef]
- Bae, G.-T. Computational Studies of the Properties of Copper Oxide Clusters and the Reactions of Phenol and Chlorinated Phenols with Copper Oxide Clusters; Louisiana State University and Agricultural & Mechanical College: Baton Rouge, LA, USA, 2009. [Google Scholar]
- Yadav, T.P.; Kaphle, G.C.; Srivastava, A. Computational Study of the Structural, Electronic and Magnetic Properties of Nanoclusters of Cu2O and CuO: Ab-Initio Approach. J. Nepal Phys. Soc. 2020, 6, 68–72. [Google Scholar] [CrossRef]
- Wang, C.; Tissot, H.; Escudero, C.; Pérez-Dieste, V.; Stacchiola, D.; Weissenrieder, J. Redox properties of Cu2O (100) and (111) surfaces. J. Phys. Chem. C 2018, 122, 28684–28691. [Google Scholar] [CrossRef]
- Gloystein, A.; Nilius, N.; Goniakowski, J.; Noguera, C. Nanopyramidal reconstruction of Cu2O (111): A long-standing surface puzzle solved by STM and DFT. J. Phys. Chem. C 2020, 124, 26937–26943. [Google Scholar] [CrossRef]
- Li, G.; Yu, X.; Weststrate, C.; Ren, P.; Xu, J.; Xu, Q.; Yang, Y.; Li, Y.; Niemantsverdriet, J.; Wen, X. Role of Interfaces in the Thermal Reduction Process of the FeO/Cu2O/Cu (100) Surface. J. Phys. Chem. C 2021, 125, 20863–20869. [Google Scholar] [CrossRef]
- Cheng, T.-K.; Jeromiyas, N.; Lin, Y.-K.; Yang, C.-C.; Kao, C.-L.; Chen, P.-Y.; Lee, C.-L. Spherical and porous Cu2O nanocages with Cu2O/Cu(OH)2 Surface: Synthesis and their promising selectivity for catalysing CO2 electroreduction to C2H4. Appl. Surf. Sci. 2024, 660, 159978. [Google Scholar] [CrossRef]
- Sieberer, M.; Redinger, J.; Mohn, P. Electronic and magnetic structure of cuprous oxide Cu2O doped with Mn, Fe, Co, and Ni: A density-functional theory study. Phys. Rev. B 2007, 75, 035203. [Google Scholar] [CrossRef]
- Li, X.; Chen, Y.; Zhan, X.; Xu, Y.; Hao, L.; Xu, L.; Li, X.; Umer, M.; Tan, X.; Han, B. Strategies for enhancing electrochemical CO2 reduction to multi-carbon fuels on copper. Innov. Mater. 2023, 1, 100014. [Google Scholar] [CrossRef]
- Zhang, R.; Li, L.; Frazer, L.; Chang, K.B.; Poeppelmeier, K.R.; Chan, M.K.; Guest, J.R. Atomistic determination of the surface structure of Cu2O (111): Experiment and theory. Phys. Chem. Chem. Phys. 2018, 20, 27456–27463. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.K.; Roldan, A.; de Leeuw, N.H. A density functional theory study of the adsorption behaviour of CO2 on Cu2O surfaces. J. Chem. Phys. 2016, 145, 044709. [Google Scholar] [CrossRef] [PubMed]
- Xing, W.; Meng, F.; Yu, R. Atomic structure of the Cu2O (111) surface: A transmission electron microscopy and DFT+ U study. Phys. Status Solidi 2021, 258, 2100185. [Google Scholar] [CrossRef]
- Goos, E.; Burcat, A. Overview of thermochemistry and its application to reaction kinetics. In Rate Constant Calculation for Thermal Reactions: Methods and Applications; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 1–32. [Google Scholar] [CrossRef]
- Ohya, S.; Kaneco, S.; Katsumata, H.; Suzuki, T.; Ohta, K. Electrochemical reduction of CO2 in methanol with aid of CuO and Cu2O. Catal. Today 2009, 148, 329–334. [Google Scholar] [CrossRef]
- Shen, Y.; Qian, L.; Xu, Q.; Wang, S.; Chen, Y.; Lu, H.; Zhou, Y.; Ye, J.; Zhao, J.; Gao, X. N-doped Cu2O with the tunable CuO and Cu+ sites for selective CO2 electrochemical reduction to ethylene. J. Environ. Sci. 2025, 150, 246–253. [Google Scholar] [CrossRef]
- Yan, C.; Luo, W.; Yuan, H.; Liu, G.; Hao, R.; Qin, N.; Wang, Z.; Liu, K.; Wang, Z.; Cui, D. Stabilizing intermediates and optimizing reaction processes with N doping in Cu2O for enhanced CO2 electroreduction. Appl. Catal. B Environ. 2022, 308, 121191. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, J.; Wei, W.; Gao, M.; Zhang, Y.-W.; Yu, Z.G.; Liang, Y.C.; Zhu, C. Pt single-atom electrocatalysts at Cu2O nanowires for boosting electrochemical sensing toward glucose. Chem. Eng. J. 2024, 495, 153564. [Google Scholar] [CrossRef]
- Wang, C.; Kong, Y.; Soldemo, M.; Wu, Z.; Tissot, H.; Karagoz, B.; Marks, K.; Stenlid, J.H.; Shavorskiy, A.; Kokkonen, E. Stabilization of Cu2O through site-selective formation of a Co1Cu hybrid single-atom catalyst. Chem. Mater. 2022, 34, 2313–2320. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, S.; Zhu, L.J.; Sun, M.J.; Zhang, T.; Cao, R. A Graphene-Supported Copper Complex as Site-Isolated Catalyst for Electrochemical CO2 Reduction. ChemElectroChem 2022, 9, e202200023. [Google Scholar] [CrossRef]
- Li, J.; Meng, C.; Gu, J.; Wang, H.; Dai, R.; Sha, H.; Zhu, H. High faradic efficiency of CO2 conversion to formic acid catalyzed by Cu2O hollow-dices. Carbon Neutrality 2022, 1, 36. [Google Scholar] [CrossRef]
- Fedorov, A.; Lund, H.; Kondratenko, V.A.; Kondratenko, E.V.; Linke, D. Elucidating reaction pathways occurring in CO2 hydrogenation over Fe-based catalysts. Appl. Catal. B Environ. 2023, 328, 122505. [Google Scholar] [CrossRef]
- Zhao, B.; Sun, M.; Chen, F.; Wang, W.; Lu, S.; Zhang, B. Photoinduced reaction pathway change for boosting CO2 hydrogenation over a MnO-Co catalyst. ACS Catal. 2021, 11, 10316–10323. [Google Scholar] [CrossRef]
- Huang, W. Oxide Nanocrystal Model Catalysts. Acc. Chem. Res. 2016, 49, 520–527. [Google Scholar] [CrossRef]
- Zhang, D.-F.; Zhang, H.; Guo, L.; Zheng, K.; Han, X.-D.; Zhang, Z. Delicate control of crystallographic facet-oriented Cu 2 O nanocrystals and the correlated adsorption ability. J. Mater. Chem. 2009, 19, 5220–5225. [Google Scholar] [CrossRef]
- Sun, Y.-F.; Liu, Z.; Liu, X.; Gan, L.-H.; Zhang, W.; Zhao, X.; Zhao, L.-B. Selective methanol production via CO2 reduction on Cu2O revealed by micro-kinetic study combined with constant potential model. Appl. Surf. Sci. 2025, 681, 161469. [Google Scholar] [CrossRef]
- Liu, H.; Liu, D.; Yu, Z.; Bai, H.; Pan, H. Electrochemical reduction of CO2 on pure and doped Cu2O (1 1 1). J. Colloid Interface Sci. 2025, 683, 170–177. [Google Scholar] [CrossRef]
- Mandal, L.; Yang, K.R.; Motapothula, M.R.; Ren, D.; Lobaccaro, P.; Patra, A.; Sherburne, M.; Batista, V.S.; Yeo, B.S.; Ager, J.W. Investigating the role of copper oxide in electrochemical CO2 reduction in real time. ACS Appl. Mater. Interfaces 2018, 10, 8574–8584. [Google Scholar] [CrossRef]
- Chang, X.; Wang, T.; Zhao, Z.J.; Yang, P.; Greeley, J.; Mu, R.; Zhang, G.; Gong, Z.; Luo, Z.; Chen, J. Tuning Cu/Cu2O interfaces for the reduction of carbon dioxide to methanol in aqueous solutions. Angew. Chem. Int. Ed. 2018, 57, 15415–15419. [Google Scholar] [CrossRef]
- Liu, B.; Yao, X.; Zhang, Z.; Li, C.; Zhang, J.; Wang, P.; Zhao, J.; Guo, Y.; Sun, J.; Zhao, C. Synthesis of Cu2O Nanostructures with Tunable Crystal Facets for Electrochemical CO2 Reduction to Alcohols. ACS Appl. Mater. Interfaces 2021, 13, 39165–39177. [Google Scholar] [CrossRef] [PubMed]
- Castro-Castillo, C.; Nanda, K.K.; Mardones-Herrera, E.; Gazzano, V.; Ruiz-León, D.; Aguirre, M.J.; García, G.; Armijo, F.; Isaacs, M. Growth direction and exposed facets of Cu/Cu2O nanostructures affect product selectivity in CO2 electroreduction. Mater. Chem. Phys. 2022, 278, 125650. [Google Scholar] [CrossRef]
- Luo, H.; Li, B.; Ma, J.G.; Cheng, P. Surface Modification of Nano-Cu2O for Controlling CO2 Electrochemical Reduction to Ethylene and Syngas. Angew. Chem. Int. Ed. 2022, 61, e202116736. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Wang, X.; Gu, C.; Sun, J.; Ding, W.; Wei, J.; Zuo, X.; Hao, C. Photoelectrocatalytic reduction of carbon dioxide to methanol at cuprous oxide foam cathode. RSC Adv. 2017, 7, 24933–24939. [Google Scholar] [CrossRef]
- Yang, Z.X.; Wen, X.; Gao, L.J.; Zhang, J.; Wei, R.P.; Pan, X.M.; Xiao, G.M. Facilitating CO2 electroreduction to C2H4 through facile regulating {100} & {111} grain boundary of Cu2O. Catal. Commun. 2023, 174, 106595. [Google Scholar]
- Hazarika, J.; Manna, M.S. Electrochemical reduction of CO2 to methanol with synthesized Cu2O nanocatalyst: Study of the selectivity. Electrochim. Acta 2019, 328, 135053. [Google Scholar] [CrossRef]
- Wu, J.C.; Lin, H.-M.; Lai, C.-L. Photo reduction of CO2 to methanol using optical-fiber photoreactor. Appl. Catal. A Gen. 2005, 296, 194–200. [Google Scholar] [CrossRef]
- Schmitt, K.G. Investigating Electrochemical Reduction Processes via In Situ Enhanced Raman Spectroscopy for Carbon Dioxide Conversion and Copper Deposition. Doctoral Dissertation, University of Illinois at Urbana-Champaign, Champaign, IL, USA, 2016. [Google Scholar]
- Besharat, Z.; Halldin Stenlid, J.; Soldemo, M.; Marks, K.; Önsten, A.; Johnson, M.; Öström, H.; Weissenrieder, J.; Brinck, T.; Göthelid, M. Dehydrogenation of methanol on Cu2O (100) and (111). J. Chem. Phys. 2017, 146, 244702. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fu, H.; Xiao, C.; Du, X.; Song, Z. Efficient electrochemical CO2 reduction to C2+ hydrocarbons on Zn-doped Cu films. Appl. Surf. Sci. 2024, 646, 158866. [Google Scholar] [CrossRef]
- Xue, Q.; Qi, X.; Li, K.; Zeng, Y.; Xu, F.; Zhang, K.; Qi, X.; Li, L.; Cabot, A. Axial heteroatom (P, S and Cl)-decorated Fe single-atom catalyst for the oxygen reduction reaction: A DFT study. RSC Adv. 2024, 14, 16379–16388. [Google Scholar] [CrossRef]
- Liu, Z.; Krösschell, R.D.; Filot, I.A.; Hensen, E.J. A combined density functional theory and microkinetics simulations study of electrochemical CO2 reduction on Cu8/SnO2 (110): The crucial role of hydrogen coverage. Electrochim. Acta 2024, 493, 144409. [Google Scholar] [CrossRef]
- Ramadhany, P.; Trần-Phú, T.; Yuwono, J.A.; Ma, Z.; Han, C.; Nguyen, T.K.A.; Leverett, J.; Kumar, P.; Hocking, R.K.; Tricoli, A. Triggering C–N Coupling on Metal Oxide Nanocomposite for the Electrochemical Reduction of CO2 and NOx− to Formamide. Adv. Energy Mater. 2024, 14, 2401786. [Google Scholar] [CrossRef]
- Tripkovic, V.; Björketun, M.E.; Skúlason, E.; Rossmeisl, J. Standard hydrogen electrode and potential of zero charge in density functional calculations. Phys. Rev. B 2011, 84, 115452. [Google Scholar] [CrossRef]
- Gong, L.; Wang, X.; Zheng, T.; Liu, J.; Wang, J.; Yang, Y.-C.; Zhang, J.; Han, X.; Zhang, L.; Xia, Z. Catalytic mechanism and design principle of coordinately unsaturated single metal atom-doped covalent triazine frameworks with high activity and selectivity for CO2 electroreduction. J. Mater. Chem. A 2021, 9, 3555–3566. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Compound Structure | After Activation on Cu2O (100) | After Activation on Cu2O (111) |
|---|---|---|---|
| Carbon dioxide (CO2) |  | ∠O–Cu–O = 177.645° | ∠O–Cu–O = 179.895° |
| Formate (HCOO) | 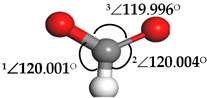 | 1∠H–C–O = 112.549° 2∠H–C–O = 67.422° 3∠O–C–O = 179.807° | 1∠H–C–O = 101.402° 2∠H–C–O = 101.549° 3∠O–C–O = 157.048° |
| Carboxyl (COOH) | 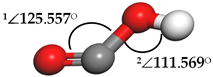 | 1∠O–C–O = 146.253° 2∠C–O-H = 106.324° | 1∠O–C–O = 175.551° 2∠C–O–H = 86.745° |
| Carbon Monoxide (CO) | 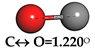 | C↔ O = 1.140° | C↔ O = 1.153° |
| Formic Acid (HCOOH) | 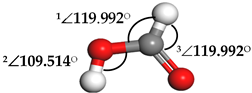 | 1∠H−O−C = 113.255° 2∠O−C−H = 104.742° 3∠H−C−O = 123.340° | 1∠H−O−C = 106.827° 2∠O−C−H = 107.428° 3∠H−C−O = 125.390° |
| Hydroxymethylene (HCOH) | 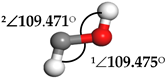 | 1∠H−O−C = 113.278° 2∠H−C−O = 96.812° | 1∠H−O−C = 107.966° 2∠H−C−O = 101.392° |
| Methoxy (H3CO) |  | 1∠H−C−O = 108.861° 2∠H−C−O = 119.843° 3∠H−C−O = 123.132° | 1∠H−O−C = 108.156° 2∠H−C−O = 110.627° 3∠H−C−O = 113.182° |
| Methanol (CH3OH) | 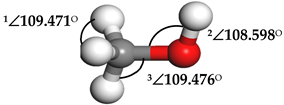 | 1∠H−C−H = 107.87° 2∠H−O−C = 108.644° 3∠O−C−H = 142.048° | 1∠H−C−H = 106.152° 2∠H−O−C = 103.948° 3∠O−C−H = 106.581° |
| Reaction | ΔG (eV) for Cu2O (100) | ΔG (eV) for Cu2O (111) |
|---|---|---|
| * + CO2(g) * | −0.37 | −0.11 |
| * | 0.22 | −0.12 |
| * | 0.30 | 0.08 |
| * | −0.10 | 0.16 |
| * | −0.43 | 0.43 |
| * | 0.27 | 0.50 |
| * | −0.14 | 0.23 |
| * | 0.14 | 0.41 |
| * | 0.003 | 0.29 |
| * | 0.05 | 0.46 |
| * | 0.86 | 0.63 |
| * | 0.37 | −0.05 |
| * | 0.60 | 0.45 |
| * | 0.07 | 0.08 |
| Adsorbate | Adsorbent | Free Energy (eV) | Reference |
|---|---|---|---|
| * | Cu2O (100) | −0.37 | This work |
| Cu2O (111) | −0.11 | ||
| *HCOO | Cu2O (100) | 0.22 | This work |
| Cu2O (111) | −0.12 | ||
| *COOH | Cu2O (100) | 0.30 | This work |
| Cu2O (111) | 0.08 | ||
| *CO | Cu2O (111) | −0.10 | This work |
| Cu2O (100) | 0.16 | ||
| *HCOOH | Cu2O (111) | 0.27 | This work |
| Cu2O (100) | 0.50 | ||
| *CO2 | Cu2O (111) | −0.09 | [82] |
| −0.14 | |||
| 0.35 | |||
| * | −0.50 | ||
| −0.40 | |||
| CO2 | Cu2O (111) | 0.00 | [83] |
| −0.50 | |||
| −0.01 | |||
| *CO2 | Cu2O (111) | −0.13 | [84] |
| −0.12 | |||
| −0.65 | |||
| CO2 | Cu2O | 0.00 | [85] |
| 0.10 | |||
| −1.00 | |||
| −0.50 |
| Material | Product | % Faradic Efficiency | References |
|---|---|---|---|
| Cu2O (100) | CH3OH | 26.20 | [86] |
| Cu2O (111) | CH3OH | 35.7 | |
| Cu2O (100) | CO | 20 | |
| Cu2O (111) | CO | 40 | |
| Cu2O (100) | C2H4 | 38 | [45] |
| Cu2O (111) | C2H4 | 45 | |
| Cu2O (111) | CH3OH | 29.1 | [89] |
| Cu2O (100) | C2H4 | 30.1 | [90] |
| Cu2O (111) | C2H4 | 44.9 | |
| Cu2O (111)/Cu2O (100) | C2H4 | 57.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laghari, Z.A.; Yahya, W.Z.N.; Mohammed, S.A.S.; Bustam, M.A. Electrocatalytic Pathways and Efficiency of Cuprous Oxide (Cu2O) Surfaces in CO2 Electrochemical Reduction (CO2ER) to Methanol: A Computational Approach. Catalysts 2025, 15, 130. https://doi.org/10.3390/catal15020130
Laghari ZA, Yahya WZN, Mohammed SAS, Bustam MA. Electrocatalytic Pathways and Efficiency of Cuprous Oxide (Cu2O) Surfaces in CO2 Electrochemical Reduction (CO2ER) to Methanol: A Computational Approach. Catalysts. 2025; 15(2):130. https://doi.org/10.3390/catal15020130
Chicago/Turabian StyleLaghari, Zubair Ahmed, Wan Zaireen Nisa Yahya, Sulafa Abdalmageed Saadaldeen Mohammed, and Mohamad Azmi Bustam. 2025. "Electrocatalytic Pathways and Efficiency of Cuprous Oxide (Cu2O) Surfaces in CO2 Electrochemical Reduction (CO2ER) to Methanol: A Computational Approach" Catalysts 15, no. 2: 130. https://doi.org/10.3390/catal15020130
APA StyleLaghari, Z. A., Yahya, W. Z. N., Mohammed, S. A. S., & Bustam, M. A. (2025). Electrocatalytic Pathways and Efficiency of Cuprous Oxide (Cu2O) Surfaces in CO2 Electrochemical Reduction (CO2ER) to Methanol: A Computational Approach. Catalysts, 15(2), 130. https://doi.org/10.3390/catal15020130