
Hydrogenation of Simulated Bio-Syngas in the Presence of GdBO3 (B = Fe, Co, Mn) Perovskite-Type Oxides

 ,
,  and
and
Abstract
1. Introduction
2. Results
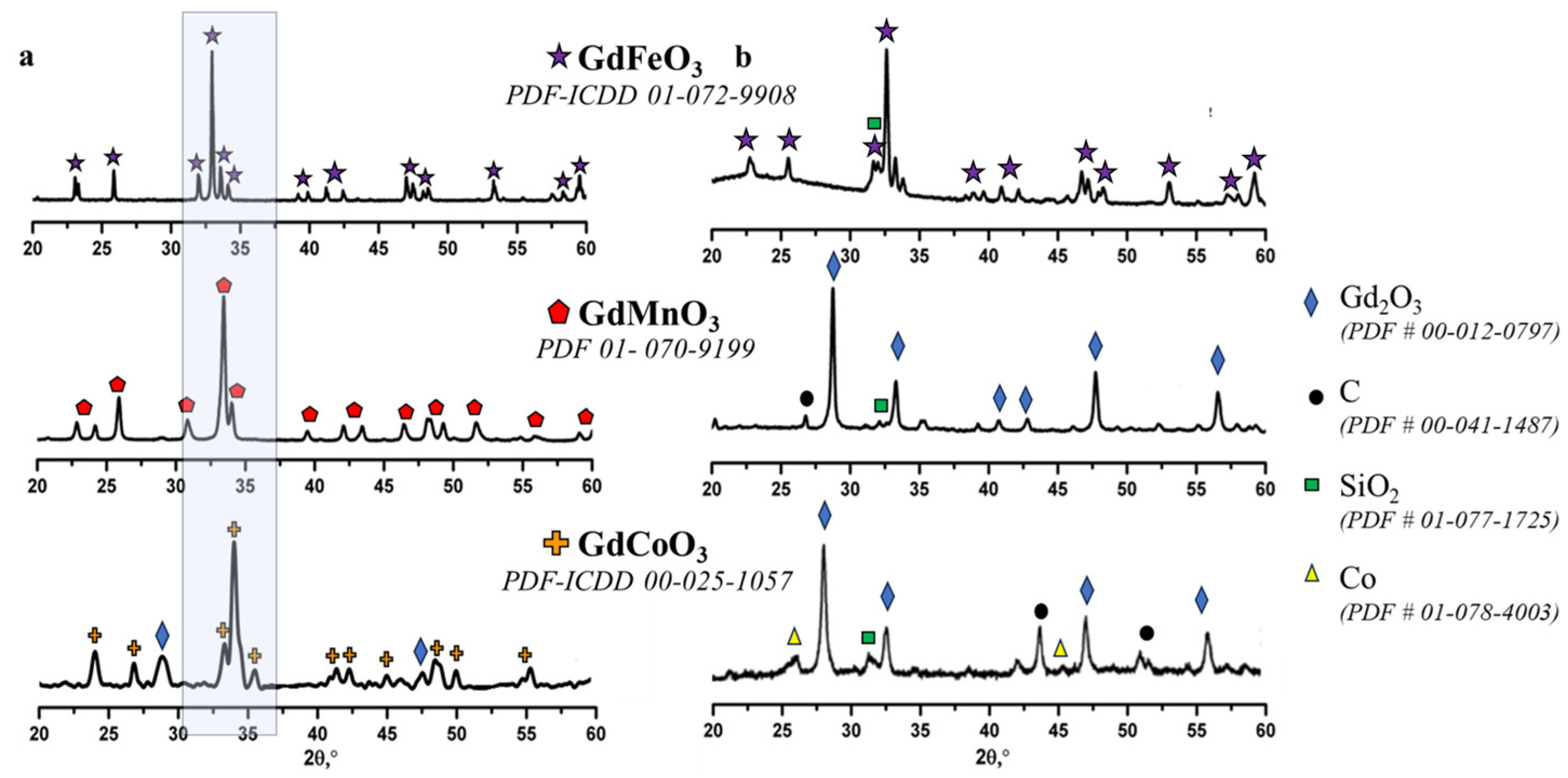
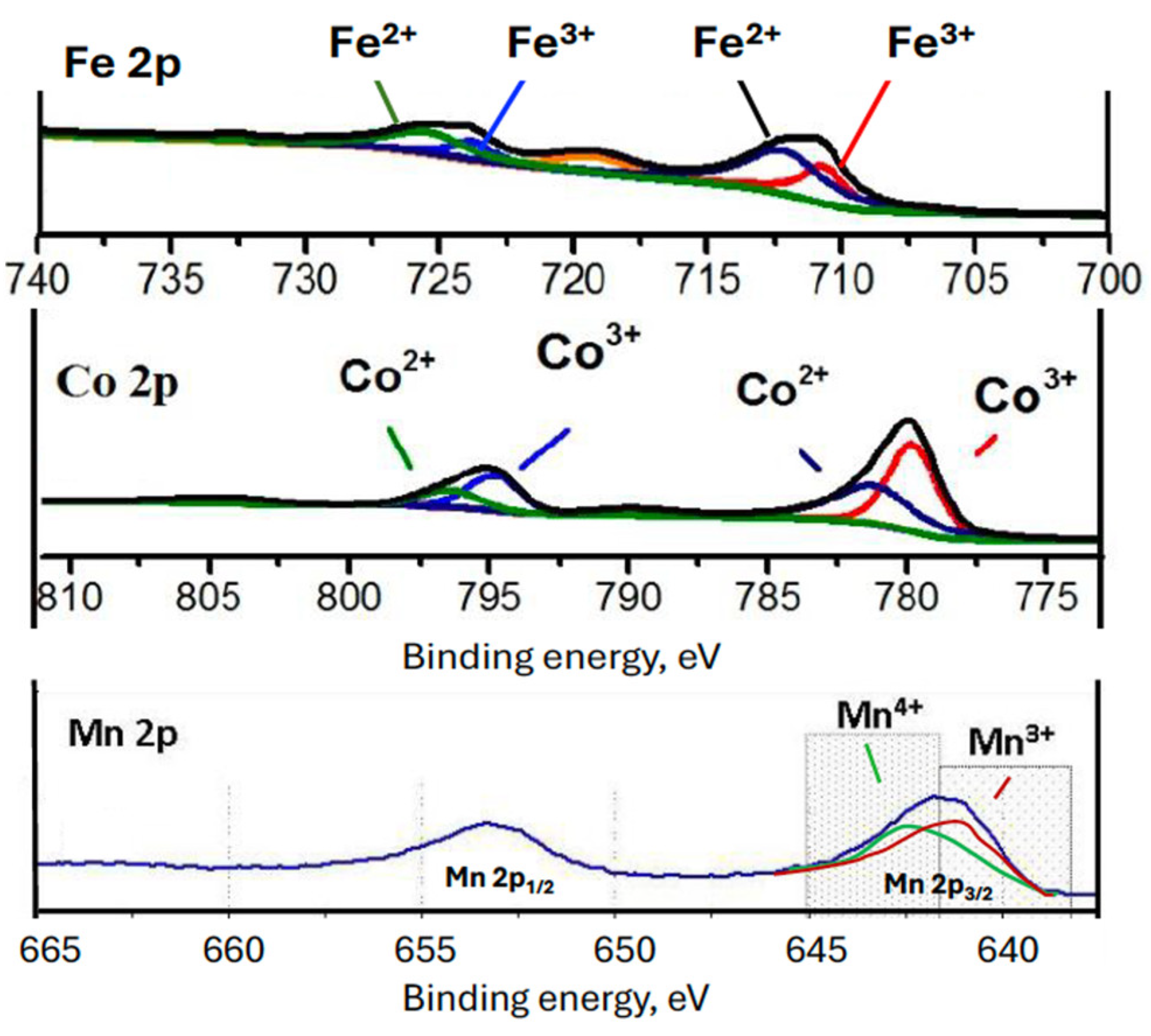
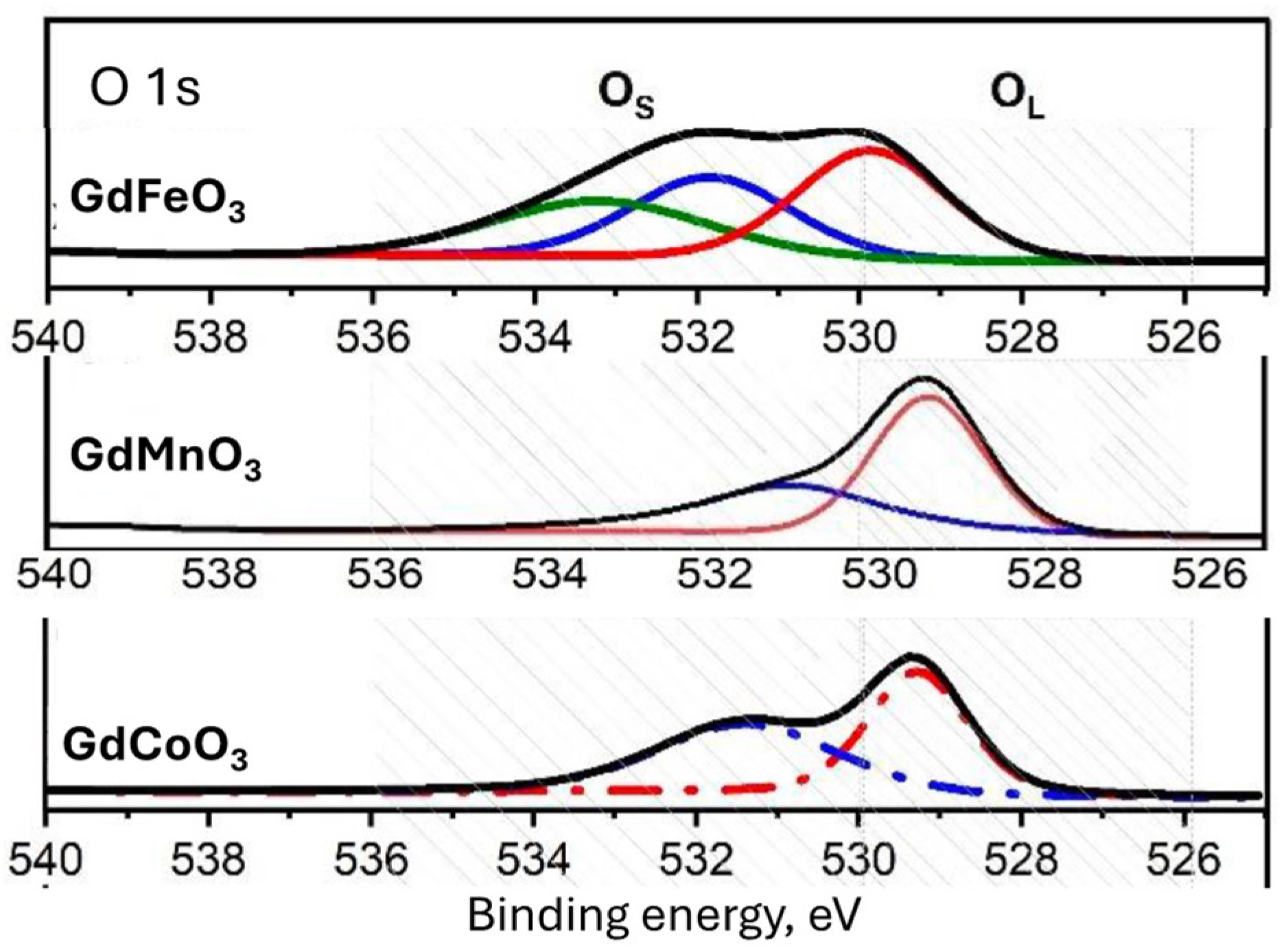
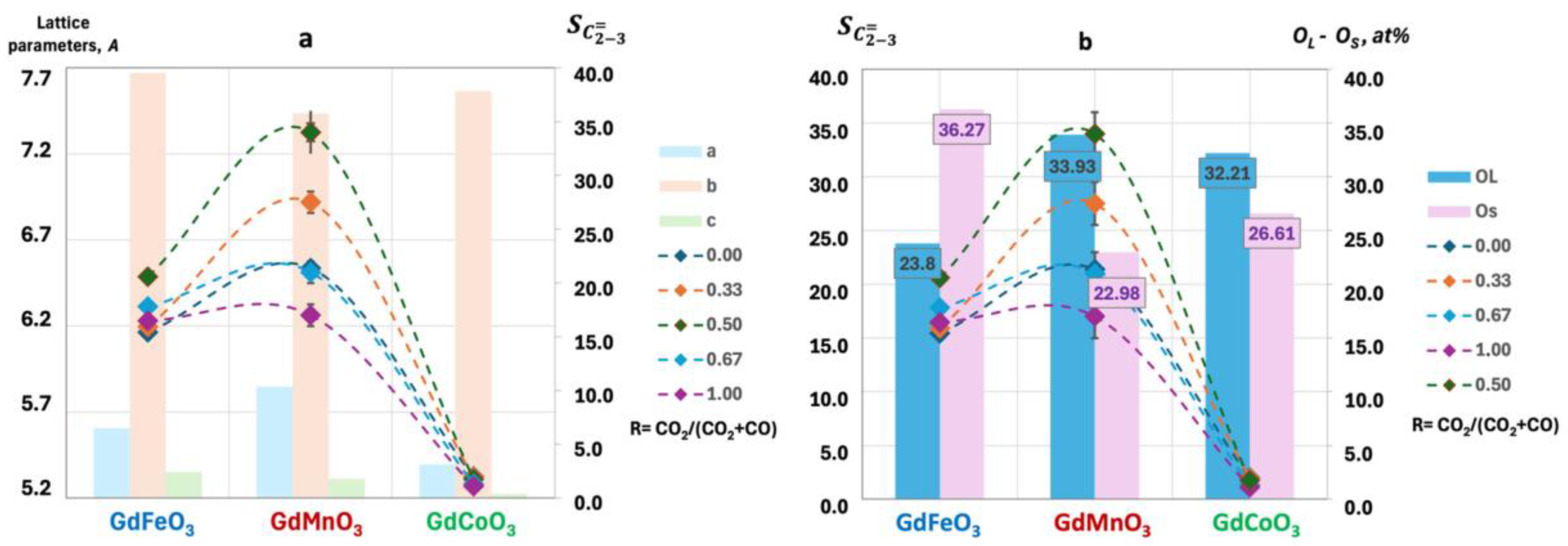
2.1. Characterization of the Catalysts
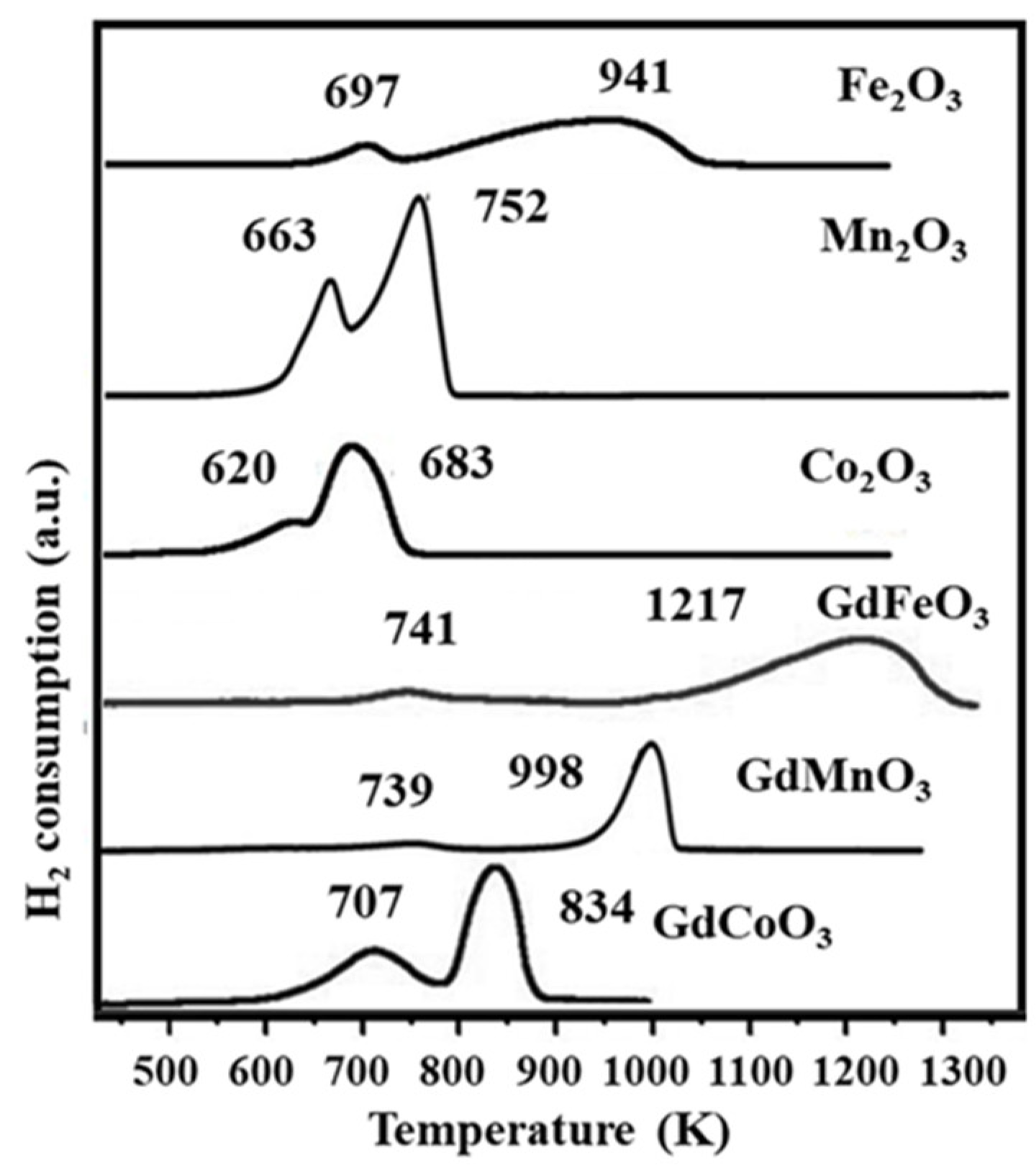
2.2. Temperature-Programmed Reduction by Hydrogen
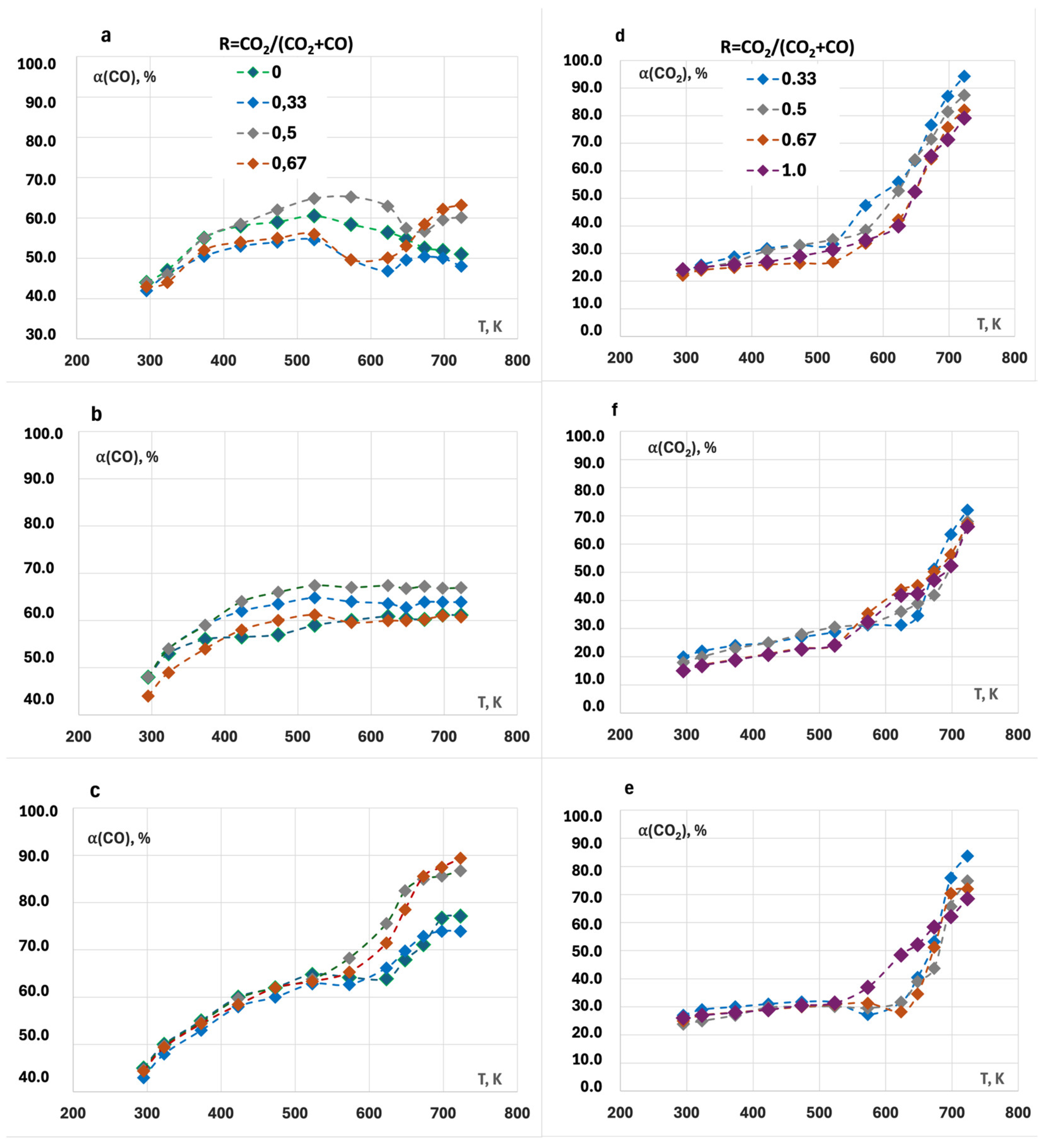
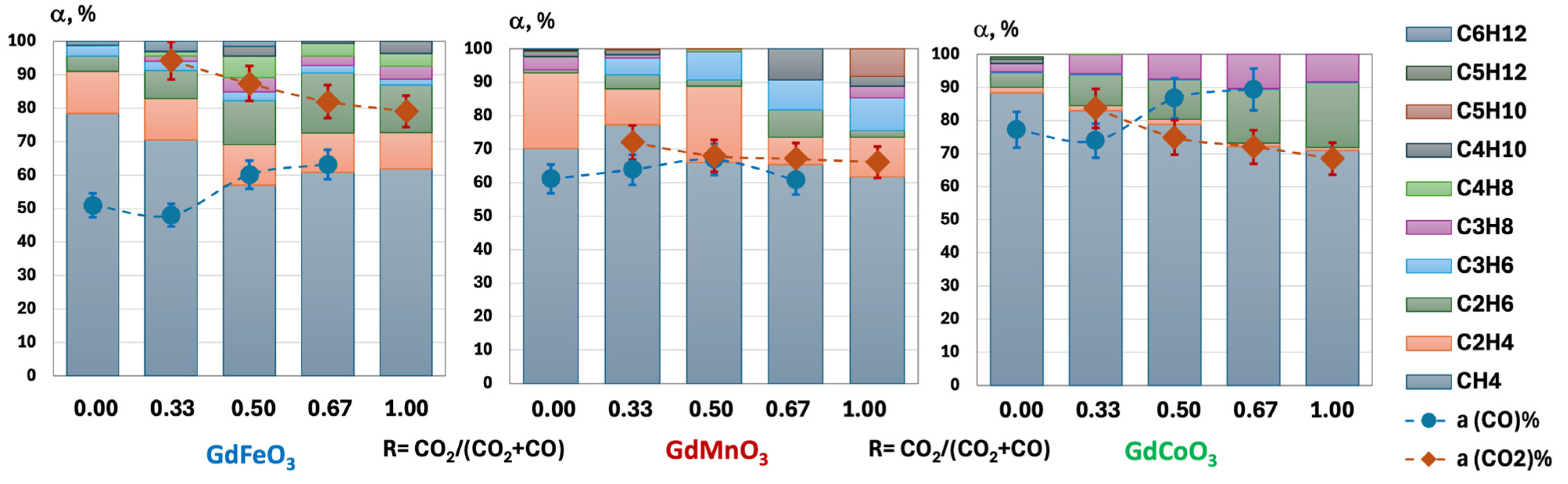
2.3. Catalytic Activity
3. Materials and Methods
3.1. Materials
3.2. Catalyst Preparation
3.3. Characterization
3.4. Catalytic Activity Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Olah, G.A.; Goeppert, A.; Prakash, G.K.S. Chemical Recycling of Carbon Dioxide to Methanol and Dimethyl Ether: From Greenhouse Gas to Renewable, Environmentally Carbon Neutral Fuels and Synthetic Hydrocarbons. J. Org. Chem. 2008, 74, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Riduan, S.N.; Zhang, Y. Recent Developments in Carbon Dioxide Utilization under Mild Conditions. Dalton Trans. 2010, 39, 3347. [Google Scholar] [CrossRef] [PubMed]
- Nomoto, K.; Okazaki, T.; Beppu, K.; Shishido, T.; Amano, F. Highly Selective Formate Formation via Bicarbonate Conversions. EES. Catal. 2024, 2, 1277–1284. [Google Scholar] [CrossRef]
- Hirunsit, P.; Senocrate, A.; Gómez-Camacho, C.E.; Kiefer, F. From CO2 to Sustainable Aviation Fuel: Navigating the Technology Landscape. ACS Sustain. Chem. Eng. 2024, 12, 12143–12160. [Google Scholar] [CrossRef]
- Song, C. Global Challenges and Strategies for Control, Conversion and Utilization of CO2 for Sustainable Development Involving Energy, Catalysis, Adsorption and Chemical Processing. Catal. Today 2006, 115, 2–32. [Google Scholar] [CrossRef]
- Muradov, N. Low to near-zero CO2 production of hydrogen from fossil fuels: Status and perspectives. Int. J. Hydrogen Energy 2017, 42, 14058–14088. [Google Scholar] [CrossRef]
- dos Santos, R.G.; Alencar, A.C. Biomass-derived syngas production via gasification process and its catalytic conversion into fuels by Fischer Tropsch synthesis: A review. Int. J. Hydrogen Energy 2020, 45, 18114–18132. [Google Scholar] [CrossRef]
- Gutierrez-Martı´n, F.; Rodrı´guez-Anton, L.M. Power-to-SNG technologies by hydrogenation of CO2 and biomass resources: A comparative chemical engineering process analysis. Int. J. Hydrogen Energy 2019, 44, 12544–12553. [Google Scholar] [CrossRef]
- Haryanto, A.; Fernando, S.D.; Pordesimo, L.O.; Adhikari, S. Upgrading of syngas derived from biomass gasification: A thermodynamic analysis. Biomass Bioenergy 2009, 33, 882–889. [Google Scholar] [CrossRef]
- Alauddin, Z.A.B.Z.; Lahijani, P.; Mohammadi, M.; Mohamed, A.R. Gasification of lignocellulosic biomass in fluidized beds for renewable energy development: A review. Renew. Sustain. Energy Rev. 2010, 14, 2852–2862. [Google Scholar] [CrossRef]
- Dinca, C.; Slavu, N.; Cormos‚, C.-C.; Badea, A. CO2 capture from syngas generated by a biomass gasification power plant with chemical absorption process. Energy 2018, 149, 925–936. [Google Scholar] [CrossRef]
- Zhao, B.; Zhang, X.; Xu, A.; Ding, W.; Sun, L.; Chen, L.; Guan, H.; Yang, S.; Zhou, W. A study of the in-situ CO2 removal pyrolysis of Chinese herb residue for syngas production. Sci. Total Environ. 2018, 626, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Tijmensen, M.J.A.; Faaij, A.P.C.; Hamelinck, C.N.; Van Hardeveld, M.R.M. Exploration of the possibilities for production of Fischer Tropsch liquids and power via biomass gasification. Biomass Bioenergy 2002, 23, 129–152. [Google Scholar] [CrossRef]
- Ma, J.; Sun, N.; Zhang, X.; Zhao, N.; Xiao, F.; Wei, W.; Sun, Y. A Short Review of Catalysis for CO2 Conversion. Catal. Today 2009, 148, 221–231. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent Advances in Catalytic Hydrogenation of Carbon Dioxide. Chem. Soc. Rev. 2011, 40, 3703. [Google Scholar] [CrossRef]
- Zhou, Z.; Gao, P. Direct Carbon Dioxide Hydrogenation to Produce Bulk Chemicals and Liquid Fuels via Heterogeneous Catalysis. Chin. J. Catal. 2022, 43, 2045–2056. [Google Scholar] [CrossRef]
- Choi, Y.H.; Jang, Y.J.; Park, H.; Kim, W.Y.; Lee, Y.H.; Choi, S.H.; Lee, J.S. Carbon Dioxide Fischer-Tropsch Synthesis: A New Path to Carbon-Neutral Fuels. Appl. Catal. B Environ. 2017, 202, 605–610. [Google Scholar] [CrossRef]
- Guo, L.; Sun, J.; Ji, X.; Wei, J.; Wen, Z.; Yao, R.; Xu, H.; Ge, Q. Directly converting carbon dioxide to linear α-olefins on bio-promoted catalysts. Commun. Chem. 2018, 1, 11. [Google Scholar] [CrossRef]
- Zhang, K.; Guo, D.; Wang, X.; Qin, Y.; Hu, L.; Zhang, Y.; Zou, R.; Gao, S. Sustainable CO2 management through integrated CO2 capture and conversion. J. CO2 Util. 2023, 72, 102493. [Google Scholar] [CrossRef]
- Zhang, R.; Xie, Z.; Ge, Q.; Zhu, X. Recent advancements in integrating CO2 capture from flue gas and ambient air with thermal catalytic conversion for efficient CO2 utilization. J. CO2 Util. 2024, 89, 102973. [Google Scholar] [CrossRef]
- Latsiou, A.I.; Charisiou, N.D.; Frontistis, Z.; Goula, M.A. From CO2 to value added chemicals: The promise of single atom catalysts. Int. J. Hydrogen Energy 2024, 92, 465–481. [Google Scholar] [CrossRef]
- Zhang, J.; Ma, L.; Fan, S.; Zhao, T.-S.; Sun, Y. Synthesis of Light Olefins from CO Hydrogenation over Fe–Mn Catalysts: Effect of Carburization Pretreatment. Fuel 2013, 109, 116–123. [Google Scholar] [CrossRef]
- Kuddusi, Y.; Piveteau, L.; Mensi, M.; Cano-Blanco, D.C.; Züttel, A. Selective light olefin synthesis with high ethylene abundance via CO2 hydrogenation over (Ga-In)2O3/SSZ-13 catalysts. J. CO2 Util. 2024, 91, 103001. [Google Scholar] [CrossRef]
- Riedel, T.; Schaub, G.; Jun, K.-W.; Lee, K.-W. Kinetics of CO2 Hydrogenation on a K-Promoted Fe Catalyst. Ind. Eng. Chem. Res. 2001, 40, 1355–1363. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Yan, B.; Chen, J.G. Catalytic Reduction of CO2 by H2 for Synthesis of CO, Methanol and Hydrocarbons: Challenges and Opportunities. Energy Environ. Sci. 2016, 9, 62–73. [Google Scholar] [CrossRef]
- Green Carbon Dioxide: Advances in CO2 Utilization; Centi, G., Perathoner, S., Eds.; Wiley: Hoboken, NJ, USA, 2014. [Google Scholar] [CrossRef]
- Dorner, R.W.; Hardy, D.R.; Williams, F.W.; Davis, B.H.; Willauer, H.D. Influence of Gas Feed Composition and Pressure on the Catalytic Conversion of CO2 to Hydrocarbons Using a Traditional Cobalt-Based Fischer−Tropsch Catalyst. Energy Fuels 2009, 23, 4190–4195. [Google Scholar] [CrossRef]
- Dorner, R.W.; Hardy, D.R.; Williams, F.W.; Willauer, H.D. K and Mn Doped Iron-Based CO2 Hydrogenation Catalysts: Detection of KAlH4 as Part of the Catalyst’s Active Phase. Appl. Catal. A Gen. 2010, 373, 112–121. [Google Scholar] [CrossRef]
- Matveyeva, A.N.; Omarov, S.O. Comparison of Perovskite Systems Based on AFeO3 (A = Ce, La, Y) in CO2 Hydrogenation to CO. Trans. Tianjin Univ. 2024, 30, 337–358. [Google Scholar] [CrossRef]
- Atta, N.F.; Galal, A.; El-Ads, E.H. Perovskite Nanomaterials—Synthesis, Characterization, and Applications. Perovskite Mater.–Synth. Characterisation Prop. Appl. 2016, 107–151. [Google Scholar] [CrossRef]
- Silva, C.L.S.; Marchetti, S.G.; Faro Júnior, A. da C.; de Silva, T.F.; Assaf, J.M.; do Rangel, M.C. Effect of gadolinium on the catalytic properties of iron oxides for WGSR. Catal. Today 2013, 213, 127–134. [Google Scholar] [CrossRef]
- Kost, V.V.; Kryuchkova, T.A.; Zimina, V.D.; Sheshko, T.F.; Cherednichenko, A.G.; Kurilkin, V.V.; Safronenko, M.G.; Yafarova, L.V. Catalytic performance of LnFeO3 complex oxides for dry reforming of methane to synthesis gas. Bulg. Chem. Commun. 2019, 51, 138–142. Available online: http://bcc.bas.bg/BCC_Contens_Volume_51-D_Special.html (accessed on 8 January 2025).
- Zhang, C.; Jun, K.-W.; Ha, K.-S.; Lee, Y.-J.; Kang, S.C. Efficient Utilization of Greenhouse Gases in a Gas-to-Liquids Process Combined with CO2 /Steam-Mixed Reforming and Fe-Based Fischer–Tropsch Synthesis. Environ. Sci. Technol. 2014, 48, 8251–8257. [Google Scholar] [CrossRef] [PubMed]
- Rafati, M.; Wang, L.; Shahbazi, A. Effect of Silica and Alumina Promoters on Co-Precipitated Fe–Cu–K Based Catalysts for the Enhancement of CO2 Utilization during Fischer–Tropsch Synthesis. J. CO2 Util. 2015, 12, 34–42. [Google Scholar] [CrossRef]
- Sheshko, T.F.; Borodina, E.M.; Yafarova, L.V.; Markova, E.B.; Kryuchkova, T.A.; Cherednichenko, A.G.; Zvereva, I.A.; Terent’ev, A.O. Insights into the Reactivity of Gd2−xSrxFe2O7 (x = 0 ÷ 0.4) in CO Radical Hydrogenation. Catalysts 2023, 13, 1256. [Google Scholar] [CrossRef]
- Sheshko, T.F.; Markova, E.B.; Sharaeva, A.A.; Kryuchkova, T.A.; Zvereva, I.A.; Chislova, I.V.; Yafarova, L.V. Carbon Monoxide Hydrogenation over Gd(Fe/Mn)O3 Perovskite-Type Catalysts. Pet. Chem. 2019, 59, 1307–1313. [Google Scholar] [CrossRef]
- Borodina, E.M.; Yafarova, L.V.; Kryuchkova, T.A.; Sheshko, T.F.; Cherednichenko, A.G.; Zvereva, I.A. Influences of Co-Content on the Physico-Chemical and Catalytic Properties of Perovskite GdCoxFe1−xO3 in CO Hydrogenation. Catalysts 2022, 13, 8. [Google Scholar] [CrossRef]
- Marezio, M.; Remeika, J.; Dernier, P. The crystal chemistry of the rare earth orthoferrites. Acta Crystallogr. 1970, 26, 2008–2022. [Google Scholar] [CrossRef]
- Vasquez, J.A.C.; Téllez, D.A.L.; A Collazos, C.; Rojas, J.R. Structural and magnetic characterization of the new GdMn1-xFexO3 perovskite material. J. Phys. Conf. Ser. 2016, 687, 012087. [Google Scholar] [CrossRef]
- Khan, H.; Ahmed, J.; Lofland, S.E.; Ramanujachary, K.V.; Ahmad, T. Synergistic Effect of Multiferroicity in GdFeO3 Nanoparticles for Significant Hydrogen Production through Photo/Electrocatalysis. Mater. Today Chem. 2023, 33, 101713. [Google Scholar] [CrossRef]
- Graat, P.C.J.; Somers, M.A.J. Simultaneous Determination of Composition and Thickness of Thin Iron-Oxide Films from XPS Fe 2p Spectra. Appl. Surf. Sci. 1996, 100–101, 36–40. [Google Scholar] [CrossRef]
- Tholkappiyan, R.; Vishista, K. Tuning the Composition and Magnetostructure of Dysprosium Iron Garnets by Co-Substitution: An XRD, FT-IR, XPS and VSM Study. Appl. Surf. Sci. 2015, 351, 1016–1024. [Google Scholar] [CrossRef]
- Zhao, K.; Shen, Y.; Huang, Z.; He, F.; Wei, G.; Zheng, A.; Li, H.; Zhao, Z. Different Oxidation Routes for Lattice Oxygen Recovery of Double-Perovskite Type Oxides LaSrFeCoO6 as Oxygen Carriers for Chemical Looping Steam Methane Reforming. J. Energy Chem. 2017, 26, 501–509. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Grosvenor, A.P.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving Surface Chemical States in XPS Analysis of First Row Transition Metals, Oxides and Hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar] [CrossRef]
- Uppara, H.P.; Pasuparthy, J.S.; Pradhan, S.; Singh, S.K.; Labhsetwar, N.K.; Dasari, H. The Comparative Experimental Investigations of SrMn(Co3+/Co2+)O3±δ and SrMn(Cu2+)O3±δ Perovskites towards Soot Oxidation Activity. Mol. Catal. 2020, 482, 110665. [Google Scholar] [CrossRef]
- Merino, N.A.; Barbero, B.P.; Eloy, P.; Cadús, L.E. La1−xCaxCoO3 Perovskite-Type Oxides: Identification of the Surface Oxygen Species by XPS. Appl. Surf. Sci. 2006, 253, 1489–1493. [Google Scholar] [CrossRef]
- Wang, Y.; Ren, J.; Wang, Y.; Zhang, F.; Liu, X.; Guo, Y.; Lu, G. Nanocasted Synthesis of Mesoporous LaCoO3 Perovskite with Extremely High Surface Area and Excellent Activity in Methane Combustion. J. Phys. Chem. C 2008, 112, 15293–15298. [Google Scholar] [CrossRef]
- Sun, J.; Zhao, Z.; Li, Y.; Yu, X.; Zhao, L.; Li, J.; Wei, Y.; Liu, J. Synthesis and Catalytic Performance of Macroporous La1-xCexCoO3 Perovskite Oxide Catalysts with High Oxygen Mobility for Catalytic Combustion of Soot. J. Rare Earths 2020, 38, 584–593. [Google Scholar] [CrossRef]
- Fuwa, Y.; Wakeshima, M.; Hinatsu, Y. Crystal Structure and Magnetic Properties of a New Layered Cobalt Oxyselenide. Solid. State Commun. 2010, 150, 1698–1701. [Google Scholar] [CrossRef]
- Yafarova, L.V.; Silyukov, O.I.; Kryuchkova, T.A.; Sheshko, T.F.; Zvereva, I.A. The Influence of Fe Substitution in GdFeO3 on Redox and Catalytic Properties. Russ. J. Phys. Chem. 2020, 94, 2679–2684. [Google Scholar] [CrossRef]
- Kang, S.C.; Jun, K.-W.; Lee, Y.-J. Effects of the CO/CO2 Ratio in Synthesis Gas on the Catalytic Behavior in Fischer–Tropsch Synthesis Using K/Fe–Cu–Al Catalysts. Energy Fuels 2013, 27, 6377–6387. [Google Scholar] [CrossRef]
- Sonal Ahmad, E.; Upadhyayula, S.; Pant, K.K. Biomass-Derived CO2 Rich Syngas Conversion to Higher Hydrocarbon via Fischer-Tropsch Process over Fe–Co Bimetallic Catalyst. Int. J. Hydrogen Energy 2019, 44, 27741–27748. [Google Scholar] [CrossRef]
- Wang, Z.X.; Dong, T.; Yuan, L.X.; Kan, T.; Zhu, X.F.; Torimoto, Y.; Sadakata, M.; Li, Q.X. Characteristics of Bio-Oil-Syngas and Its Utilization in Fischer−Tropsch Synthesis. Energy Fuels 2007, 21, 2421–2432. [Google Scholar] [CrossRef]
- Yao, Y.; Liu, X.; Hildebrandt, D.; Glasser, D. The Effect of CO2 on a Cobalt-Based Catalyst for Low Temperature Fischer–Tropsch Synthesis. Chem. Eng. J. 2012, 193–194, 318–327. [Google Scholar] [CrossRef]
- Daza, Y.A.; Kuhn, J.N. CO2 Conversion by Reverse Water Gas Shift Catalysis: Comparison of Catalysts, Mechanisms and Their Consequences for CO2 Conversion to Liquid Fuels. RSC Adv. 2016, 6, 49675–49691. [Google Scholar] [CrossRef]
- Álvarez, A.; Bansode, A.; Urakawa, A.; Bavykina, A.V.; Wezendonk, T.A.; Makkee, M.; Gascon, J.; Kapteijn, F. Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes. Chem. Rev. 2017, 117, 9804–9838. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Porosoff, M.D. Development of Tandem Catalysts for CO2 Hydrogenation to Olefins. ACS Catal. 2019, 9, 2639–2656. [Google Scholar] [CrossRef]
- Ronda-Lloret, M.; Rothenberg, G.; Shiju, N.R. A Critical Look at Direct Catalytic Hydrogenation of Carbon Dioxide to Olefins. ChemSusChem 2019, 12, 3896–3914. [Google Scholar] [CrossRef]
- Visconti, C.G.; Lietti, L.; Tronconi, E.; Forzatti, P.; Zennaro, R.; Finocchio, E. Fischer–Tropsch Synthesis on a Co/Al2O3 Catalyst with CO2 Containing Syngas. Appl. Catal. A Gen. 2009, 355, 61–68. [Google Scholar] [CrossRef]
- Liang, B.; Sun, T.; Ma, J.; Duan, H.; Li, L.; Yang, X.; Zhang, Y.; Su, X.; Huang, Y.; Zhang, T. Mn Decorated Na/Fe Catalysts for CO2 Hydrogenation to Light Olefins. Catal. Sci. Technol. 2019, 9, 456–464. [Google Scholar] [CrossRef]
- Kulikova, M.V.; Chudakova, M.V.; Ivantsov, M.I.; Dementyva, O.S.; Maksimov, A.L. Hydrocarbon Synthesis from CO2 and H2 Using the Ultrafine Iron-Containing Catalytic Systems Based on Carbonized Cellulose. Eurasian Chem. Tech. J. 2022, 24, 149. [Google Scholar] [CrossRef]
- Al-Dossary, M.; Ismail, A.A.; Fierro, J.L.G.; Bouzid, H.; Al-Sayari, S.A. Effect of Mn Loading onto MnFeO Nanocomposites for the CO2 Hydrogenation Reaction. Appl. Catal. B Environ. 2015, 165, 651–660. [Google Scholar] [CrossRef]
- Bligaard, T.; Honkala, K.; Logadottir, A.; Nørskov, J.K.; Dahl, S.; Jacobsen, C.J.H. On the Compensation Effect in Heterogeneous Catalysis. J. Phys. Chem. B 2003, 107, 9325–9331. [Google Scholar] [CrossRef]
- Mota, N.; Millán Ordoñez, E.; Pawelec, B.; Fierro, J.L.G.; Navarro, R.M. Direct Synthesis of Dimethyl Ether from CO2: Recent Advances in Bifunctional/Hybrid Catalytic Systems. Catalysts 2021, 11, 411. [Google Scholar] [CrossRef]
- Kryuchkova, T.A.; Kost’, V.V.; Sheshko, T.F.; Chislova, I.V.; Yafarova, L.V.; Zvereva, I.A. Effect of Cobalt in GdFeO3 Catalyst Systems on Their Activity in the Dry Reforming of Methane to Synthesis Gas. Pet. Chem. 2020, 60, 609–615. [Google Scholar] [CrossRef]
- Malhi, H.S.; Sun, C.; Zhang, Z.; Liu, Y.; Liu, W.; Ren, P.; Tu, W.; Han, Y.-F. Catalytic Consequences of the Decoration of Sodium and Zinc Atoms during CO2 Hydrogenation to Olefins over Iron-Based Catalyst. Catal. Today 2022, 387, 28–37. [Google Scholar] [CrossRef]
- Tu, W.; Ghoussoub, M.; Singh, C.V.; Chin, Y.-H.C. Consequences of Surface Oxophilicity of Ni, Ni-Co, and Co Clusters on Methane Activation. J. Am. Chem. Soc. 2017, 139, 6928–6945. [Google Scholar] [CrossRef] [PubMed]
- Sadykov, V.A.; Bulgakov, N.N.; Muzykantov, V.S.; Kuznetsova, T.G.; Alikina, G.M.; Lukashevich, A.I.; Neophytides, S. Mobility and reactivity of the surface and lattice oxygen of some complex oxides with perovskite structure. In Mixed Ionic Electronic Conducting Perovskites for Advanced Energy Systems; Springer: Dordrecht, The Netherlands, 2004; Volume 183, pp. 53–74. [Google Scholar] [CrossRef]
- Goguet, A.; Meunier, F.C.; Tibiletti, D.; Breen, J.P.; Burch, R. Spectrokinetic Investigation of Reverse Water-Gas-Shift Reaction Intermediates over a Pt/CeO2 Catalyst. J. Phys. Chem. B 2004, 108, 20240–20246. [Google Scholar] [CrossRef]
- Mebel, A.M.; Hwang, D.-Y. Theoretical Study on the Reaction Mechanism of Nickel Atoms with Carbon Dioxide. J. Phys. Chem. A 2000, 104, 11622–11627. [Google Scholar] [CrossRef]
- Popova, N.M.; Babenkova, L.V.; Savel’eva, G.A. Adsorption and Interaction of the Simplest Gases with VIII Group Metals; Nauka: Alma-Ata, Kazakhstan, 1979; pp. 83–97. (In Russian) [Google Scholar]
- Weber, D.; He, T.; Wong, M.; Moon, C.; Zhang, A.; Foley, N.; Ramer, N.J.; Zhang, C. Recent Advances in the Mitigation of the Catalyst Deactivation of CO2 Hydrogenation to Light Olefins. Catalysts 2021, 11, 1447. [Google Scholar] [CrossRef]
- Li, W.H.; Wang, H.Z.; Jiang, X.; Zhu, J.; Liu, Z.M.; Guo, X.W.; Song, C.S. A short review of recent advances in CO2 hydrogenation to hydrocarbons over heterogeneous catalysts. RCS Adv. 2018, 8, 7651–7669. [Google Scholar] [CrossRef]
- Chislova, I.V.; Matveeva, A.A.; Volkova, A.V.; Zvereva, I.A. Sol-Gel Synthesis of Nanostructured Perovskite-like Gadolinium Ferrites. Glass Phys. Chem. 2011, 37, 653–660. [Google Scholar] [CrossRef]
- Yafarova, L.V.; Chislova, I.V.; Zvereva, I.A.; Kryuchkova, T.A.; Kost, V.V.; Sheshko, T.F. Sol–gel synthesis and investigation of catalysts on the basis of perovskite-type oxides GdMO3 (M = Fe, Co). J. Sol-Gel Sci. Technol. 2019, 92, 264–272. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Lattice Parameters (Å) | Crystallite Size (nm) | Space | Content (at%) | SBET (m2/g) | ||
|---|---|---|---|---|---|---|---|
| Group | Gd | B | O | ||||
| GdFeO3 | a = 5.606 b = 7.671 c = 5.352 | 53.4 | Pnma (62) | 18.53 | 19.15 | 62.32 | 4.6 |
| GdMnO3 | a = 5.847 b = 7.435 c = 5.312 | 55.9 | Pnma (62) | 14.60 | 14.04 | 65.17 | 8.5 |
| Compound | Content (at%) | |||||
|---|---|---|---|---|---|---|
| Gd | B | B2+/(B2++B3+) | OS | OL | OS/OL | |
| GdFeO3 | 23.60 | 16.32 | 0.64 | 36.27 | 23.80 | 1.52 |
| GdMnO3 | 18.20 | 24.90 | - | 22.98 | 33.93 | 0.67 |
| GdCoO3 | 18.19 | 21.99 | 0.47 | 26.61 | 32.21 | 0.83 |
| Oxides | Temperature (K) | H2 Consumption, mmol/g | |||
|---|---|---|---|---|---|
| Peak 1 | Peak 1 | Peak 1 | Peak 1 | Total | |
| GdFeO3 | 741 K (Fe3+→Fe+2) | 1217 K (Fe2+→Fe0) | 141 | 752 | 893 |
| GdMnO3 | 739 K (Mn4+→Mn3+) | 998 K (Mn3+→Mn2+) | 286 | 528 | 814 |
| GdCoO3 | 707 K (Co3+→Co+2) | 834 K (Co2+→Co0) | 381 | 592 | 973 |
| R = CO2/(CO2 + CO) | CH4 | C2H4 | C3H6 | |||
|---|---|---|---|---|---|---|
| Ea, kJ/mol | lnK0 | Ea, kJ/mol | lnK0 | Ea, kJ/mol | lnK0 | |
| GdFeO3 | ||||||
| 0 | 30 | 4.74 | 34 | 6.37 | 44 | 5.94 |
| 0.33 | 22 | 6.28 | 29 | 7.07 | 39 | 6.70 |
| 0.50 | 22 | 6.45 | 30 | 7.01 | 38 | 7.20 |
| 0.67 | 21 | 6.57 | 28 | 7.32 | 38 | 7.26 |
| 1.00 | 20 | 6.52 | 28 | 7.60 | 42 | 6.79 |
| GdMnO3 | ||||||
| 0 | 67 | 1.57 | 89 | 3.47 | 87 | 11.7 |
| 0.33 | 74 | 6.47 | 93 | 15.01 | 74 | 15.9 |
| 0.50 | 73 | 10.7 | 73 | 9.60 | 74 | 9.1 |
| 0.67 | 73 | 16.6 | 81 | 18.90 | 91 | 12.5 |
| 1.00 | 54 | 6.51 | 77 | 15.17 | 73 | 12.9 |
| GdCoO3 | ||||||
| 0 | 33 | 2.15 | 36 | 5.74 | 35 | 7.13 |
| 0.33 | 38 | 1.48 | 46 | 4.19 | 43 | 6.27 |
| 0.50 | 39 | 1.08 | 46 | 3.80 | 45 | 5.43 |
| 0.67 | 38 | 1.79 | 43 | 5.23 | 41 | 6.98 |
| 1.00 | 31 | 2.94 | 35 | 6.89 | 40 | 7.35 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheshko, T.F.; Akhmina, P.V.; Skvortsova, L.G.; Borodina, E.M.; Kryuchkova, T.A.; Zvereva, I.A.; Cherednichenko, A.G. Hydrogenation of Simulated Bio-Syngas in the Presence of GdBO3 (B = Fe, Co, Mn) Perovskite-Type Oxides. Catalysts 2025, 15, 67. https://doi.org/10.3390/catal15010067
Sheshko TF, Akhmina PV, Skvortsova LG, Borodina EM, Kryuchkova TA, Zvereva IA, Cherednichenko AG. Hydrogenation of Simulated Bio-Syngas in the Presence of GdBO3 (B = Fe, Co, Mn) Perovskite-Type Oxides. Catalysts. 2025; 15(1):67. https://doi.org/10.3390/catal15010067
Chicago/Turabian StyleSheshko, Tatiana F., Polina V. Akhmina, Liliya G. Skvortsova, Elizaveta M. Borodina, Tatiana A. Kryuchkova, Irina A. Zvereva, and Alexander G. Cherednichenko. 2025. "Hydrogenation of Simulated Bio-Syngas in the Presence of GdBO3 (B = Fe, Co, Mn) Perovskite-Type Oxides" Catalysts 15, no. 1: 67. https://doi.org/10.3390/catal15010067
APA StyleSheshko, T. F., Akhmina, P. V., Skvortsova, L. G., Borodina, E. M., Kryuchkova, T. A., Zvereva, I. A., & Cherednichenko, A. G. (2025). Hydrogenation of Simulated Bio-Syngas in the Presence of GdBO3 (B = Fe, Co, Mn) Perovskite-Type Oxides. Catalysts, 15(1), 67. https://doi.org/10.3390/catal15010067