Mechanisms for the Production and Suppression of Hydrogen Peroxide at the Hydrogen Electrode in Proton Exchange Membrane Fuel Cells and Water Electrolyzers: Theoretical Considerations




Abstract

1. Introduction
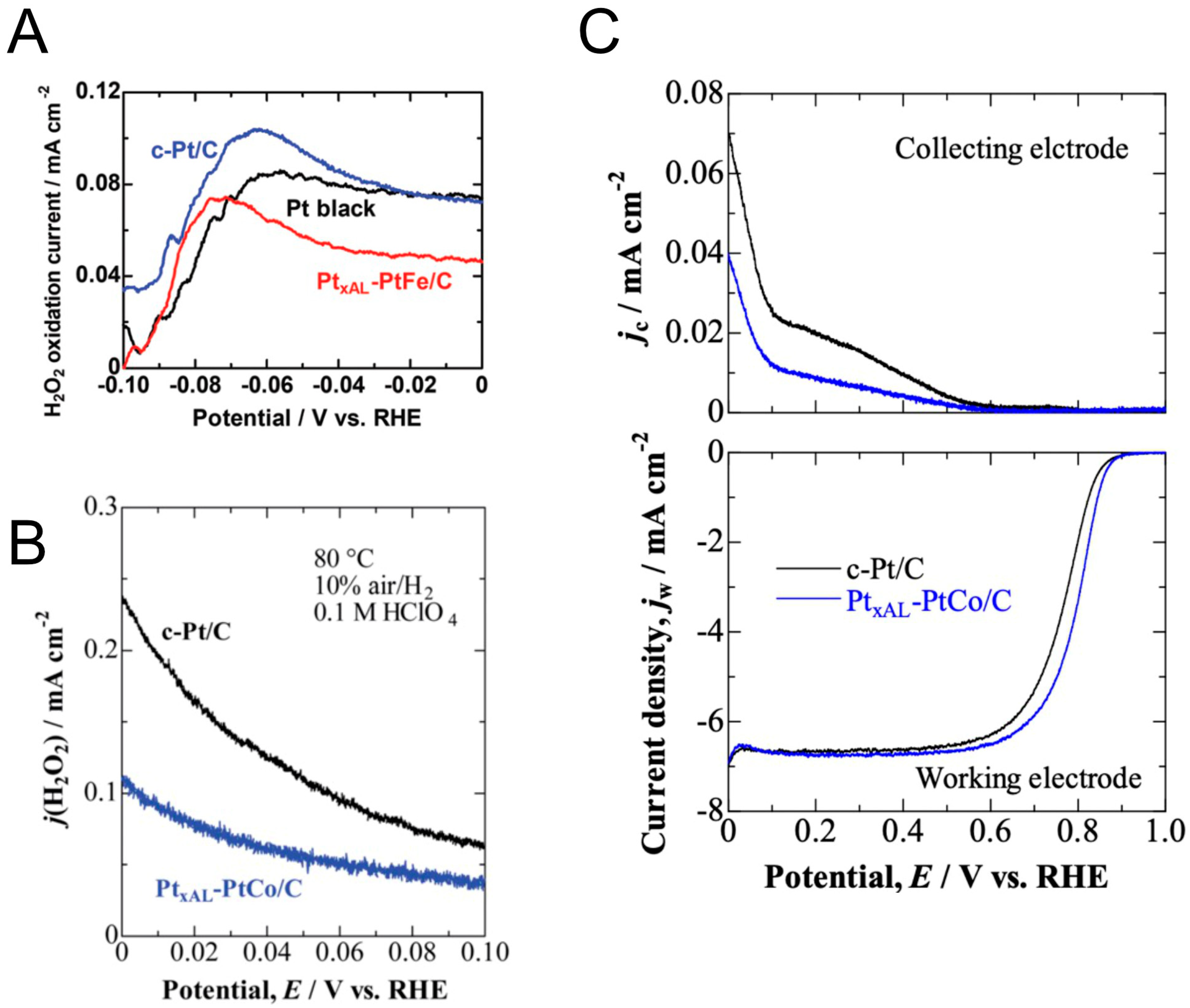
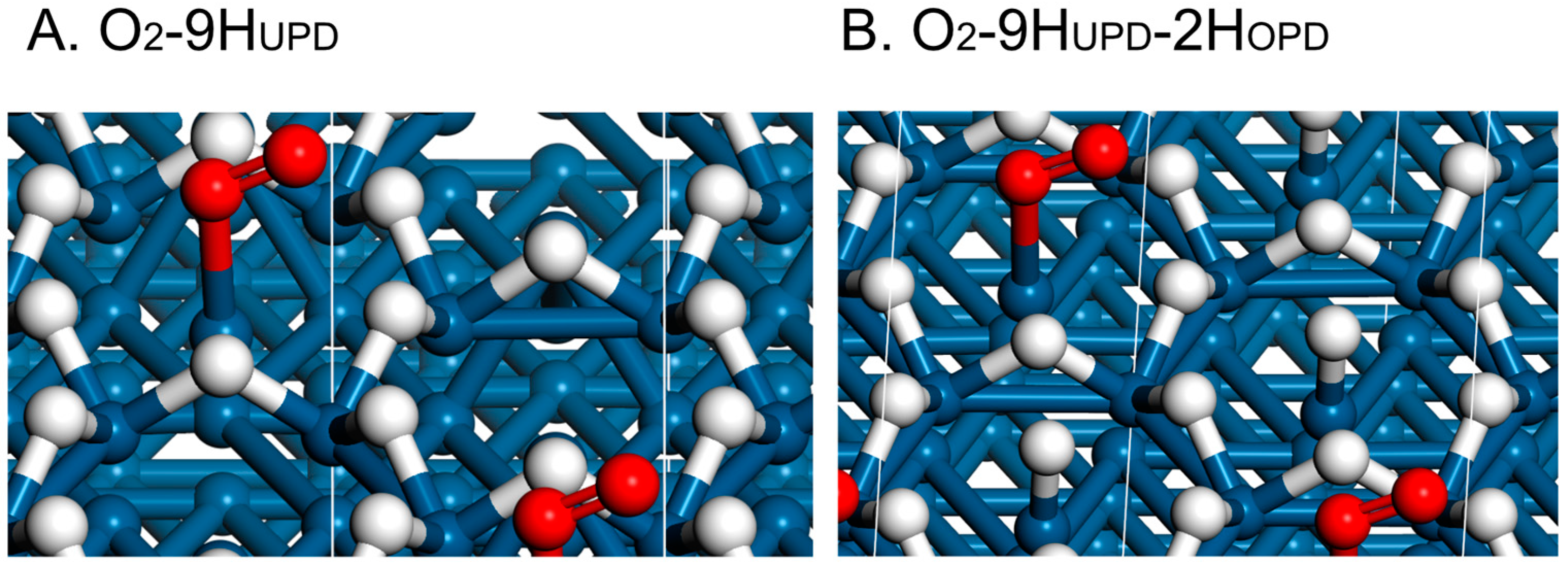
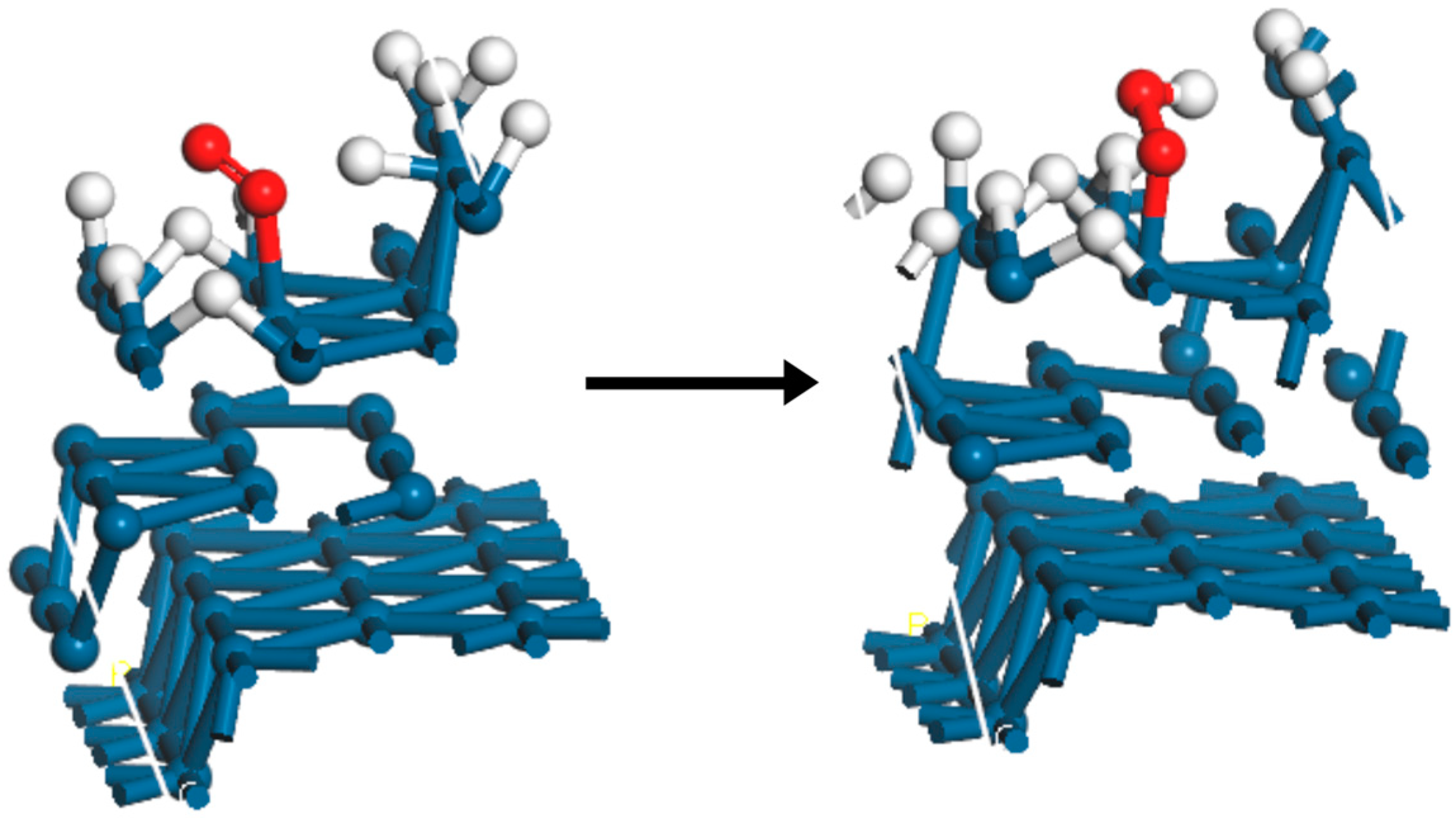
2. Results and Discussion
- The O2,br adsorption should be stronger than that of HUPD, so that the 4-electron ORR will be favored vs. the 2-electron process to produce H2O2 (Pathways 2 and 3);
- Even if #1 is not satisfied, θH can be decreased significantly, both by decreasing the adsorption energy Ead,H and the appearance potentials UMHUPD (Pathway 2) and UMH(110),edge (Pathway 3) via alloying;
- The coverage of HUPD is also important, because high coverage tends to stabilize the end-on adsorption of O2 on the (111) terraces. These coverages are dependent upon the UMHUPD values, which are very close to the UMH(110),edge values;
- The HOR and HER activities are favored by decreased H adsorption energies at both the (110) step and the (111) terrace, with the effect of the latter being particularly apparent for the HOR [8];
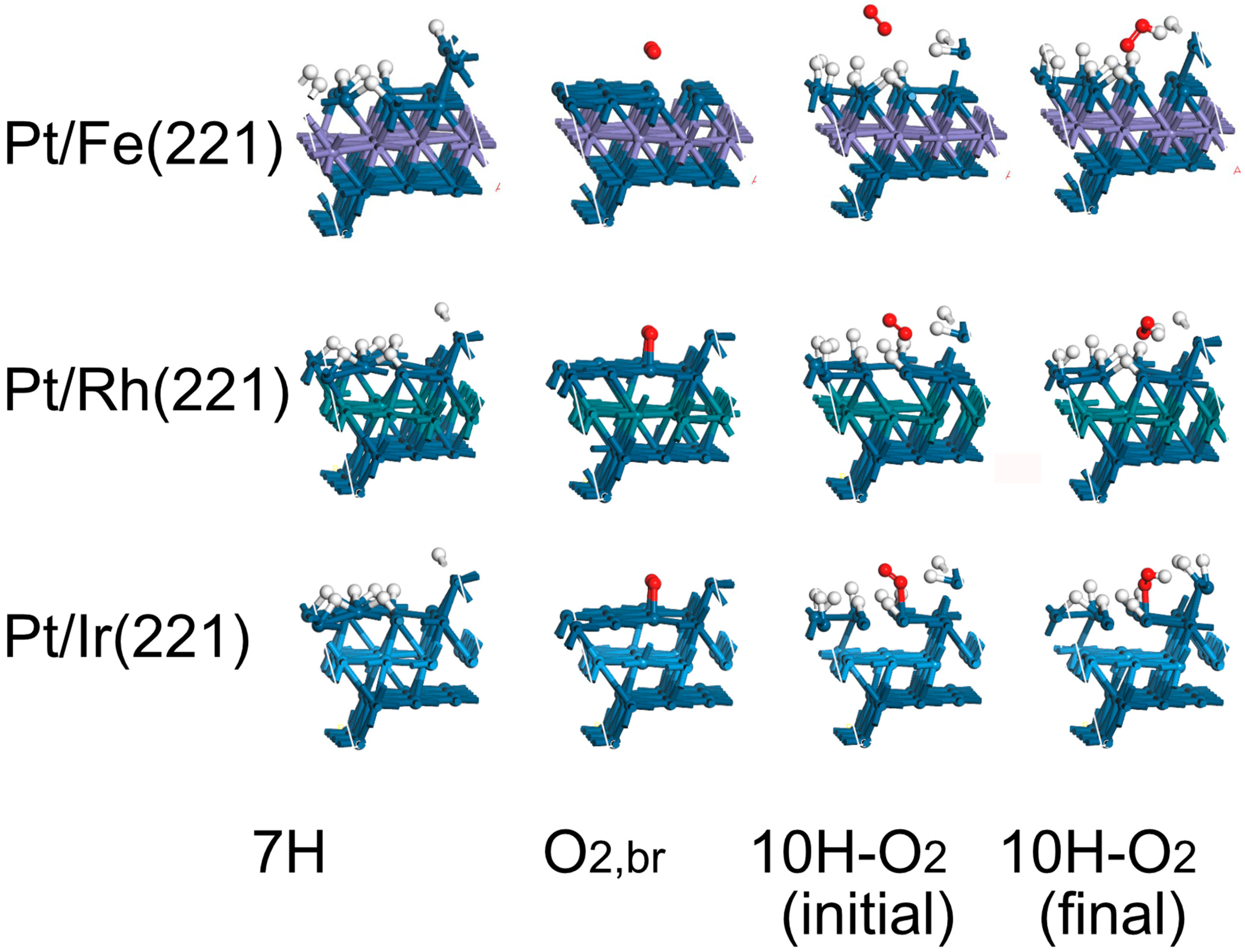
- Based on the above guidelines, PtFe, PtRh and PtIr all are predicted to have high activity for the HOR and HER with low activity for H2O2 production. The Pt monolayer on Ir(111) model is also attractive if it is possible to prepare in high area form;
- Of course, the final choice of catalyst also requires consideration of cost, durability and ability to prepare a suitable material. The durability of both Rh and Ir is expected to be superior to that of Fe as an alloying element.
3. DFT Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Watanabe, M.; Tryk, D.A. Fuel Cells: An Overview with Emphasis on Polymer Electrolyte Fuel Cells. In Electrochemical Science for a Sustainable Society: A Tribute to John O’M Bockris; Uosaki, K., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 51–94. [Google Scholar] [CrossRef]
- Yap, J.; McLellan, B. A Historical Analysis of Hydrogen Economy Research, Development, and Expectations, 1972 to 2020. Environments 2023, 10, 11. [Google Scholar] [CrossRef]
- Link, S.; Stephan, A.; Speth, D.; Plötz, P. Rapidly Declining Costs of Truck Batteries and Fuel Cells Enable Large-Scale Road Freight Electrification. Nat. Energy 2024, 9, 1032–1039. [Google Scholar] [CrossRef]
- Jerkiewicz, G. Hydrogen Sorption at/in Electrodes. Prog. Surf. Sci. 1998, 57, 137–186. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Mateo, J.J.; Tryk, D.A.; Cabrera, C.R. Direct Molecular Dynamics and Density-Functional Theoretical Study of the Electrochemical Hydrogen Oxidation Reaction and Underpotential Deposition of H on Pt(111). J. Electroanal. Chem. 2007, 607, 37–46. [Google Scholar] [CrossRef]
- Mateo, J.J.; Tryk, D.A.; Cabrera, C.R.; Ishikawa, Y. Underpotential Deposition of Hydrogen on Pt(111): A Combined Direct Molecular Dynamics/Density Functional Theory Study. Mol. Simul. 2008, 34, 1065–1072. [Google Scholar] [CrossRef]
- Shi, G.; Yano, H.; Tryk, D.A.; Nohara, S.; Uchida, H. High Hydrogen Evolution Activity and Suppressed H2O2 Production on Pt-Skin/PtFe Alloy Nanocatalysts for Proton Exchange Membrane Water Electrolysis. Phys. Chem. Chem. Phys. 2019, 21, 2861–2865. [Google Scholar] [CrossRef]
- Shi, G.; Yano, H.; Tryk, D.A.; Iiyama, A.; Uchida, H. Highly Active, Co-Tolerant, and Robust Hydrogen Anode Catalysts: Pt-M(M = Fe, Co, Ni) Alloys with Stabilized Pt-Skin Layers. ACS Catal. 2017, 7, 267–274. [Google Scholar] [CrossRef]
- Shi, G.; Tano, T.; Tryk, D.A.; Iiyama, A.; Uchida, M.; Kakinuma, K. Temperature Dependence of Oxygen Reduction Activity at Pt/Nb-Doped SnO2 Catalysts with Varied Pt Loading. ACS Catal. 2021, 11, 5222–5230. [Google Scholar] [CrossRef]
- Shi, G.; Hashimoto, T.; Tryk, D.A.; Tano, T.; Iiyama, A.; Uchida, M.; Kakinuma, K. Enhanced Oxygen Reduction Electrocatalysis on Ptcosn Alloy Nanocatalyst Mediated by Ta-Doped SnO2 Support for Polymer Electrolyte Fuel Cells. Electrochim. Acta 2021, 390, 138894. [Google Scholar] [CrossRef]
- Shi, G.; Tryk, D.A.; Iwataki, T.; Yano, H.; Uchida, M.; Iiyama, A.; Uchida, H. Unparalleled Mitigation of Membrane Degradation in Fuel Cells Via a Counter-Intuitive Approach: Suppression of H2O2 Production at the Hydrogen Anode Using a Ptskin–PtCo Catalyst. J. Mater. Chem. A 2020, 8, 1091–1094. [Google Scholar] [CrossRef]
- Uchida, H.; Shi, G.; Imran, M.; Tryk, D.A. Particle-Size Effect of Pt Anode Catalysts on H2O2 Production Rate and H2 Oxidation Activity at 20 to 80 °C. J. Electrochem. Soc. 2022, 169, 014516. [Google Scholar] [CrossRef]
- Conway, B.E.; Jerkiewicz, G. Nature of Electrosorbed H and Its Relation to Metal Dependence of Catalysis in Cathodic H2 Evolution. Solid State Ionics 2002, 150, 93–103. [Google Scholar] [CrossRef]
- Markovic, N.M.; Adzic, R.R.; Cahan, B.D.; Yeager, E.B. Structural Effects in Electrocatalysis: Oxygen Reduction on Platinum Low Index Single-Crystal Surfaces in Perchloric Acid Solutions. J. Electroanal. Chem. 1994, 377, 249–259. [Google Scholar] [CrossRef]
- MacNaughton, J.B.; Naslund, L.A.; Anniyev, T.; Ogasawara, H.; Nilsson, A. Peroxide-Like Intermediate Observed at Hydrogen Rich Condition on Pt(111) after Interaction with Oxygen. Phys. Chem. Chem. Phys. 2010, 12, 5712–5716. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.S.; Tryk, D.; Yeager, E. The Electrochemistry of Graphite and Modified Graphite Surfaces: The Reduction of O2. Electrochim. Acta 1989, 34, 1733–1737. [Google Scholar] [CrossRef]
- Conway, B.E.; Jerkiewicz, G. Relation of Energies and Coverages of Underpotential and Overpotential Deposited H at Pt and Other Metals to the ‘Volcano Curve’ for Cathodic H2 Evolution Kinetics. Electrochim. Acta 2000, 45, 4075–4083. [Google Scholar] [CrossRef]
- Climent, V.; Feliu, J. Single Crystal Electrochemistry as an in Situ Analytical Characterization Tool. Ann. Rev. Anal. Chem. 2020, 13, 201–222. [Google Scholar] [CrossRef]
- Santana, J.A.; Mateo, J.J.; Ishikawa, Y. Electrochemical Hydrogen Oxidation on Pt(110): A Combined Direct Molecular Dynamics/Density Functional Theory Study. J. Phys. Chem. C 2010, 114, 4995–5002. [Google Scholar] [CrossRef]
- Kunimatsu, K.; Senzaki, T.; Tsushima, M.; Osawa, M. A Combined Surface-Enhanced Infrared and Electrochemical Kinetics Study of Hydrogen Adsorption and Evolution on a Pt Electrode. Chem. Phys. Lett. 2005, 401, 451–454. [Google Scholar] [CrossRef]
- Kunimatsu, K.; Senzaki, T.; Samjeske, G.; Tsushima, M.; Osawa, M. Hydrogen Adsorption and Hydrogen Evolution Reaction on a Polycrystalline Pt Electrode Studied by Surface-Enhanced Infrared Absorption Spectroscopy. Electrochim. Acta 2007, 52, 5715–5724. [Google Scholar] [CrossRef]
- Ogasawara, H.; Ito, M. Hydrogen Adsorption on Pt(100), Pt(110), Pt(111) and Pt(1111) Electrode Surfaces Studied by in Situ Infrared Reflection Absorption Spectroscopy. Chem. Phys. Lett. 1994, 221, 213–218. [Google Scholar] [CrossRef]
- Nakamura, M.; Kobayashi, T.; Hoshi, N. Structural Dependence of Intermediate Species for the Hydrogen Evolution Reaction on Single Crystal Electrodes of Pt. Surf. Sci. 2011, 605, 1462–1465. [Google Scholar] [CrossRef]
- Nakamori, Y.; Suzuki, N.; Tanaka, K.; Aoki, T.; Nohira, T.; Hagiwara, R. Pt–Ru Anode Catalyst to Suppress H2O2 Formation Due to Oxygen Crossover. J. Electrochem. Soc. 2018, 165, F463. [Google Scholar] [CrossRef]
- Neergat, M.; Gunasekar, V.; Singh, R.K. Oxygen Reduction Reaction and Peroxide Generation on Ir, Rh, and Their Selenides–a Comparison with Pt and Ruse. J. Electrochem. Soc. 2011, 158, B1060. [Google Scholar] [CrossRef]
- Hayashi, K.; Tomimori, T.; Chida, Y.; Todoroki, N.; Wadayama, T. Hydrogen Peroxide Generation and Hydrogen Oxidation Reaction Properties of Ir(111)-, (100)-, and (110)-Low-Index Single-Crystal Surfaces. J. Phys. Chem. C 2021, 125, 21481–21487. [Google Scholar] [CrossRef]
- Liu, C.; Zhu, L.; Wen, X.; Yang, Y.; Li, Y.-W.; Jiao, H. Hydrogen Adsorption on Ir(111), Ir(100) and Ir(110)—Surface and Coverage Dependence. Surf. Sci. 2020, 692, 121514. [Google Scholar] [CrossRef]
- Hayashi, K.; Tomimori, T.; Sato, R.; Todoroki, N.; Wadayama, T. Enhanced Electrochemical Hydrogen Oxidation Reaction and Suppressed Hydrogen Peroxide Generation Properties on Pt/Ir(111) Bimetallic Surfaces. Phys. Chem. Chem. Phys. 2023, 25, 2770–2775. [Google Scholar] [CrossRef]
- Shi, G.; Tano, T.; Tryk, D.A.; Iiyama, A.; Uchida, M.; Terao, K.; Osada, H.; Yamaguchi, M.; Tamoto, K.; Kakinuma, K. Nanostructured Pt-NiFe Oxide Catalyst for Hydrogen Evolution Reaction in Alkaline Electrolyte Membrane Water Electrolyzers. ACS Catal. 2024, 14, 9460–9468. [Google Scholar] [CrossRef]
- Jerkiewicz, G.; Zolfaghari, A. Determination of the Energy of the Metal-Underpotential-Deposited Hydrogen Bond for Rhodium Electrodes. J. Phys. Chem. 1996, 100, 8454–8461. [Google Scholar] [CrossRef]
- Greeley, J.; Mavrikakis, M. Surface and Subsurface Hydrogen: Adsorption Properties on Transition Metals and near-Surface Alloys. J. Phys. Chem. B 2005, 109, 3460–3471. [Google Scholar] [CrossRef]
- Klyukin, K.; Zagalskaya, A.; Alexandrov, V. Ab Initio Thermodynamics of Iridium Surface Oxidation and Oxygen Evolution Reaction. J. Phys. Chem. C 2018, 122, 29350–29358. [Google Scholar] [CrossRef]
- Comelli, G.; Dhanak, V.R.; Kiskinova, M.; Prince, K.C.; Rosei, R. Oxygen and Nitrogen Interaction with Rhodium Single Crystal Surfaces. Surf. Sci. Rep. 1998, 32, 165–231. [Google Scholar] [CrossRef]
- Gee, A.T.; Hayden, B.E. The Dynamics of O2 Adsorption on Pt(533): Step Mediated Molecular Chemisorption and Dissociation. J. Chem. Phys. 2000, 113, 10333–10343. [Google Scholar] [CrossRef]
- Ueta, H.; Kurahashi, M. Dynamics of O2 Chemisorption on a Flat Platinum Surface Probed by an Alignment-Controlled O2 Beam. Angew. Chem. Int. Ed. 2017, 56, 4174–4177. [Google Scholar] [CrossRef] [PubMed]
- Ou, L.; Yang, F.; Liu, Y.; Chen, S. First-Principle Study of the Adsorption and Dissociation of O2 on Pt(111) in Acidic Media. J. Phys. Chem. C 2009, 113, 20657–20665. [Google Scholar] [CrossRef]
- Eichler, A.; Hafner, J. Molecular Precursors in the Dissociative Adsorption of O2 on Pt(111). Phys. Rev. Lett. 1997, 79, 4481–4484. [Google Scholar] [CrossRef]
- Watanabe, M.; Motoo, S. Electrocatalysis by Ad-Atoms Part II. Enhancement of the Oxidation of Methanol on Platinum by Ruthenium Ad-Atoms. J. Electroanal. Chem. 1975, 60, 267–273. [Google Scholar] [CrossRef]
- Watanabe, M.; Motoo, S. Electrocatalysis by Ad-Atoms: Part III. Enhancement of the Oxidation of Carbon Monoxide on Platinum by Ruthenium Ad-Atoms. J. Electroanal. Chem. 1975, 60, 275–283. [Google Scholar] [CrossRef]
- Perez, A.; Vilkas, M.J.; Cabrera, C.R.; Ishikawa, Y. Density Functional Theory Study of Water Activation and COads + OHads Reaction on Pure Platinum and Bimetallic Platinum/Ruthenium Nanoclusters. J. Phys. Chem. B 2005, 109, 23571–23578. [Google Scholar] [CrossRef]
- Siahrostami, S.; Verdaguer-Casadevall, A.; Karamad, M.; Deiana, D.; Malacrida, P.; Wickman, B.; Escudero-Escribano, M.; Paoli, E.A.; Frydendal, R.; Hansen, T.W.; et al. Enabling Direct H2O2 Production through Rational Electrocatalyst Design. Nat. Mater. 2013, 12, 1137–1143. [Google Scholar] [CrossRef]
- Chang, Y.; Yang, J.; Zhang, M.; Yue, M.; Wang, W.; Li, J.; Wang, J.; Song, K.; Liu, Y.; Zuo, Y.; et al. Pt-Based Electrocatalysts Design for Oxygen Reduction toward Hydrogen Peroxide. J. Alloys Compd. 2024, 1005, 176091. [Google Scholar] [CrossRef]
- Sheng, W.C.; Zhuang, Z.B.; Gao, M.R.; Zheng, J.; Chen, J.G.G.; Yan, Y.S. Correlating Hydrogen Oxidation and Evolution Activity on Platinum at Different pH with Measured Hydrogen Binding Energy. Nat. Commun. 2015, 6, 5848. [Google Scholar] [CrossRef] [PubMed]
- Delley, B. From Molecules to Solids with the DMol3 Approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Delley, B. Hardness Conserving Semilocal Pseudopotentials. Phys. Rev. B 2002, 66, 155125. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Basiuk, V.A.; Prezhdo, O.V.; Basiuk, E.V. Thermal Smearing in DFT Calculations: How Small Is Really Small? A Case of La and Lu Atoms Adsorbed on Graphene. Mater. Today Commun. 2020, 25, 101595. [Google Scholar] [CrossRef]
- Xu, S.; Feng, S.; Yu, Y.; Xue, D.; Liu, M.; Wang, C.; Zhao, K.; Xu, B.; Zhang, J.-N. Dual-Site Segmentally Synergistic Catalysis Mechanism: Boosting CoFeSx Nanocluster for Sustainable Water Oxidation. Nat. Commun. 2024, 15, 1720. [Google Scholar] [CrossRef]
- Zhou, M.; Kong, W.; Xue, M.; Li, H.; Khan, M.A.; Liu, B.; Lu, F.; Zeng, X. Recent Progress of Dual-Site Catalysts in Emerging Electrocatalysis: A Review. Catal. Sci. Technol. 2023, 13, 4615–4634. [Google Scholar] [CrossRef]
- Hyman, M.P.; Medlin, J.W. Effects of Electronic Structure Modifications on the Adsorption of Oxygen Reduction Reaction Intermediates on Model Pt(111)-Alloy Surfaces. J. Phys. Chem. C 2007, 111, 17052–17060. [Google Scholar] [CrossRef]
- Li, R.; Li, H.; Liu, J. First Principles Study of O2 Dissociation on Pt(111) Surface: Stepwise Mechanism. Int. J. Quantum Chem. 2016, 116, 908–914. [Google Scholar] [CrossRef]
- De Darwent, B.B. NSRDS-NBS 31; Bond Dissociation Energies in Simple Molecules. National Bureau of Standards: Gaithersburg, MD, USA, 1970.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Surface | Hbr (HUPD) | O2,br | 10H–O2, Initial | 10H–O2, Final |
|---|---|---|---|---|
| Pt(221) | −0.54 | −0.45 | 0.16 | −0.45 |
| Rh(221) | −0.45 | −1.46 | −0.99 | −1.41 |
| Ir(221) | −0.38 | −1.26 | −1.20 | −0.76 |
| Pt/Fe(221) | −0.23 | −0.20 | 0.03 | 0.31 |
| Pt/Rh(221) | −0.42 | −0.53 | −0.09 | −0.89 |
| Pt/Ir(221) | −0.44 | −0.65 | 0.36 | −0.14 |
| M-nH | Pt(221) | Rh(221) | Ir(221) | Pt/Fe(221) | Pt/Rh(221) | Pt/Ir(221) | 10H–O2, Final |
|---|---|---|---|---|---|---|---|
| M-7H | 0.118 | −0.294 | 0.058 | −0.032 | 0.086 | 0.083 | −0.45 |
| M-10H | 0.100 | −0.295 | 0.045 | −0.014 | 0.034 | 0.044 | −1.41 |
| Pathway | H | O2 | Potential a | Onset | Remedy b |
|---|---|---|---|---|---|
| 1 | OPD | (110) edge | <0.0 V | sudden | Decr. θH |
| 2 | UPD | End-on | <0.5 V | gradual | Decr. θH, incr. Ead,O2 |
| 3 | (110) edge | End-on | <0.1 V | sudden | Decr. θH, incr. Ead,O2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tryk, D.A.; Shi, G.; Kakinuma, K.; Uchida, M.; Iiyama, A. Mechanisms for the Production and Suppression of Hydrogen Peroxide at the Hydrogen Electrode in Proton Exchange Membrane Fuel Cells and Water Electrolyzers: Theoretical Considerations. Catalysts 2024, 14, 890. https://doi.org/10.3390/catal14120890
Tryk DA, Shi G, Kakinuma K, Uchida M, Iiyama A. Mechanisms for the Production and Suppression of Hydrogen Peroxide at the Hydrogen Electrode in Proton Exchange Membrane Fuel Cells and Water Electrolyzers: Theoretical Considerations. Catalysts. 2024; 14(12):890. https://doi.org/10.3390/catal14120890
Chicago/Turabian StyleTryk, Donald A., Guoyu Shi, Katsuyoshi Kakinuma, Makoto Uchida, and Akihiro Iiyama. 2024. "Mechanisms for the Production and Suppression of Hydrogen Peroxide at the Hydrogen Electrode in Proton Exchange Membrane Fuel Cells and Water Electrolyzers: Theoretical Considerations" Catalysts 14, no. 12: 890. https://doi.org/10.3390/catal14120890
APA StyleTryk, D. A., Shi, G., Kakinuma, K., Uchida, M., & Iiyama, A. (2024). Mechanisms for the Production and Suppression of Hydrogen Peroxide at the Hydrogen Electrode in Proton Exchange Membrane Fuel Cells and Water Electrolyzers: Theoretical Considerations. Catalysts, 14(12), 890. https://doi.org/10.3390/catal14120890