
Effect of Calcination Temperature in Large-Aperture Medium-Entropy Oxide (FeCoCuZnNa)O on CO2 Hydrogenation for Light Olefins


Abstract

1. Introduction
2. Results and Discussion
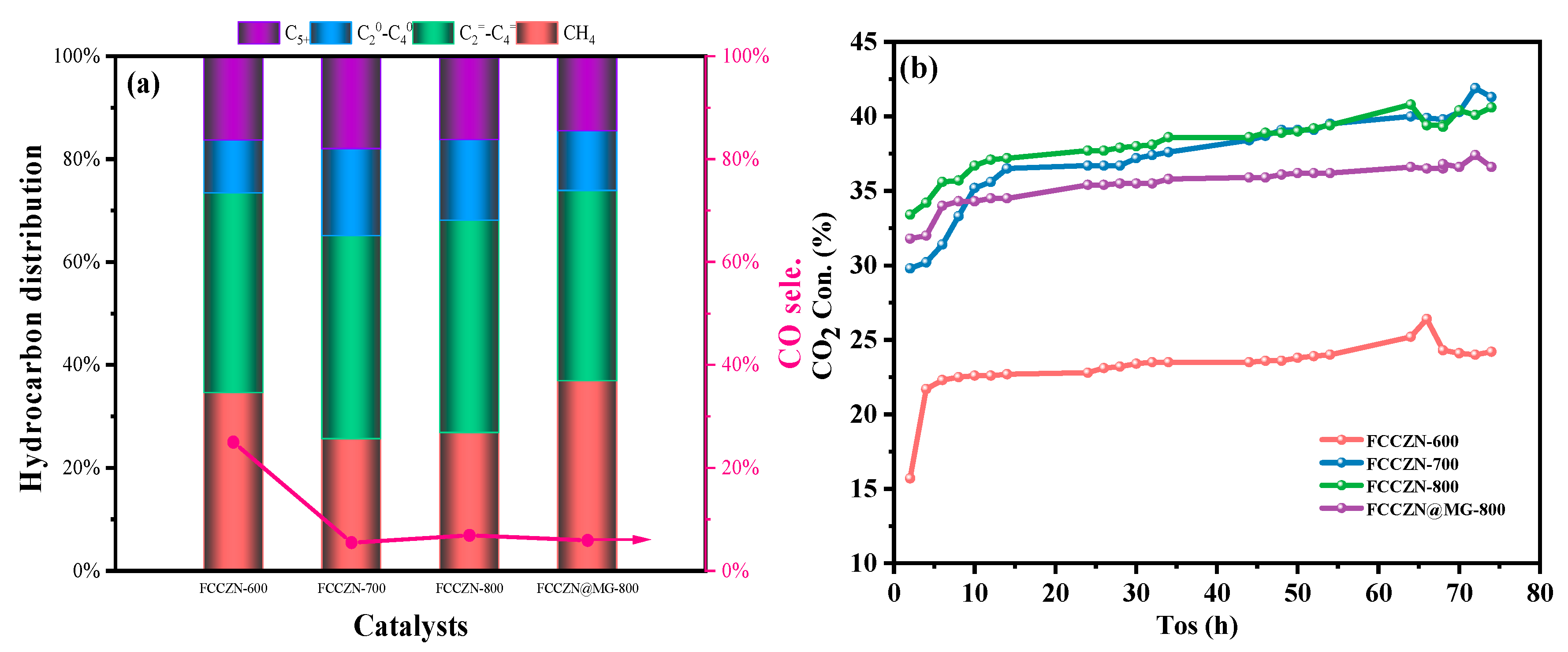
2.1. Catalytic Performances of Catalysts
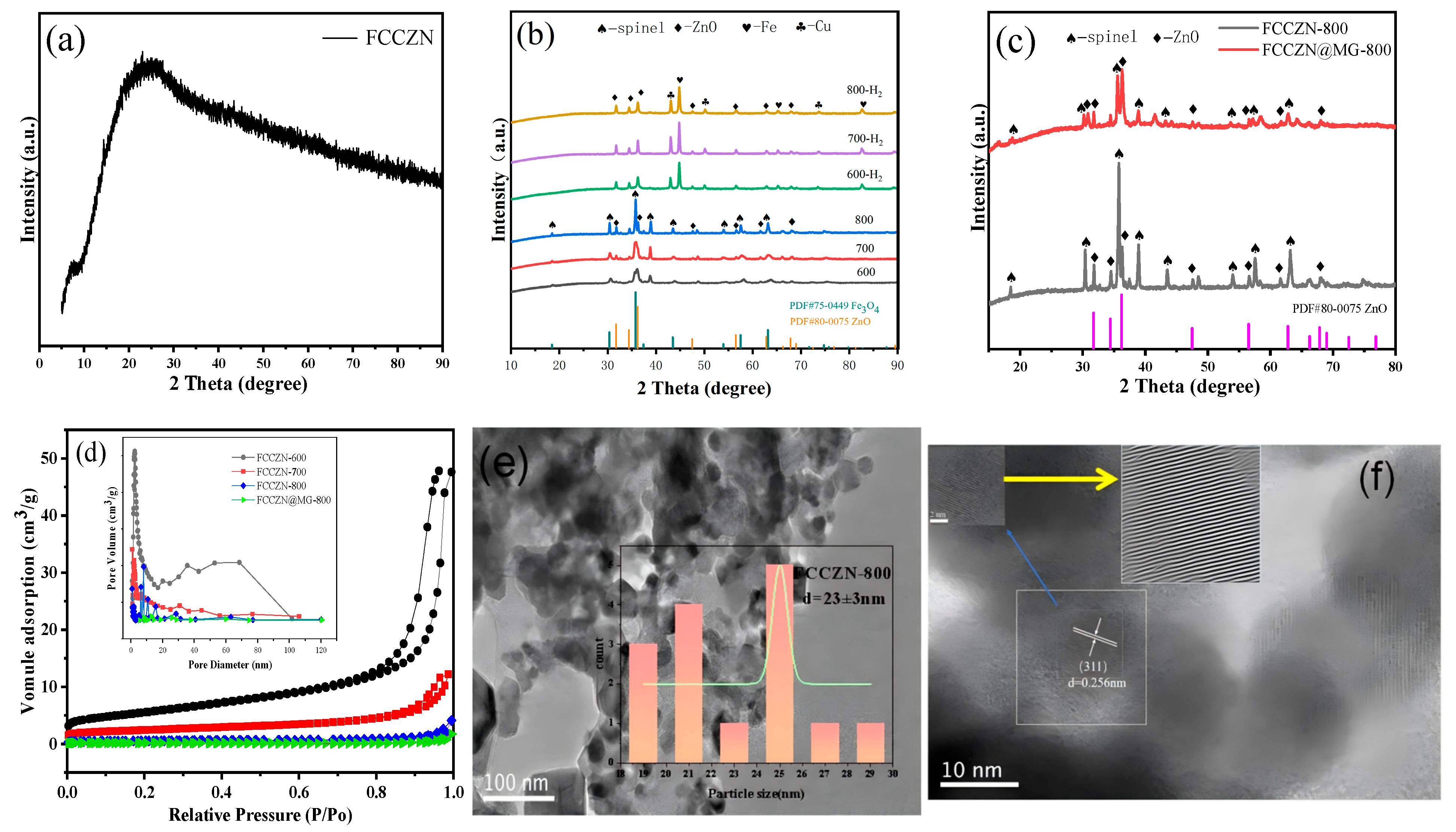
2.2. Textural and Morphological Characteristics of FCCZN Catalysts
2.3. Chemical Adsorption Properties of FCCZN Catalysts
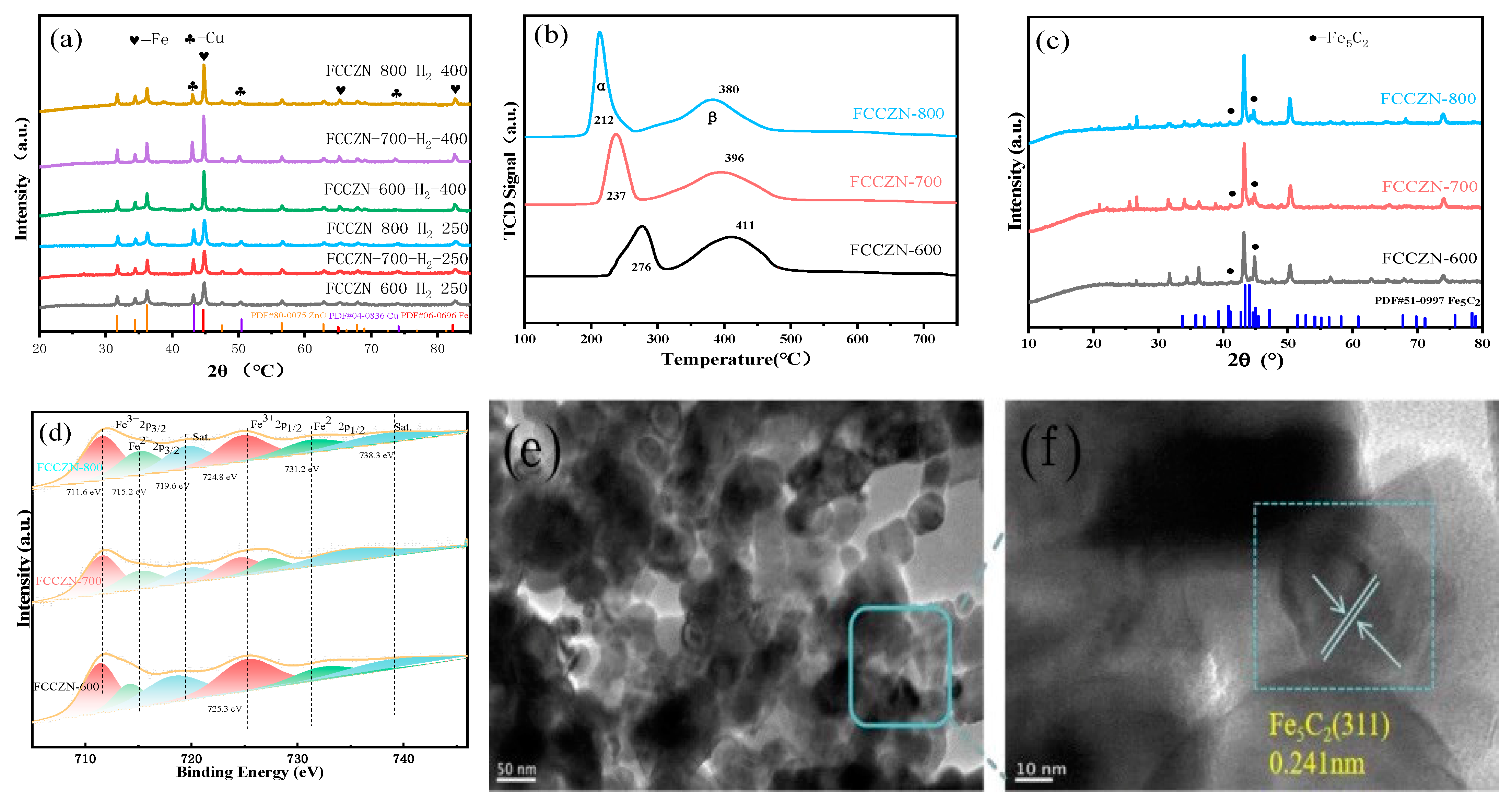
2.4. Electronic Characteristics of FCCZN Catalysts
2.5. Effect of Different Oxygen Vacancy Content on Catalysts
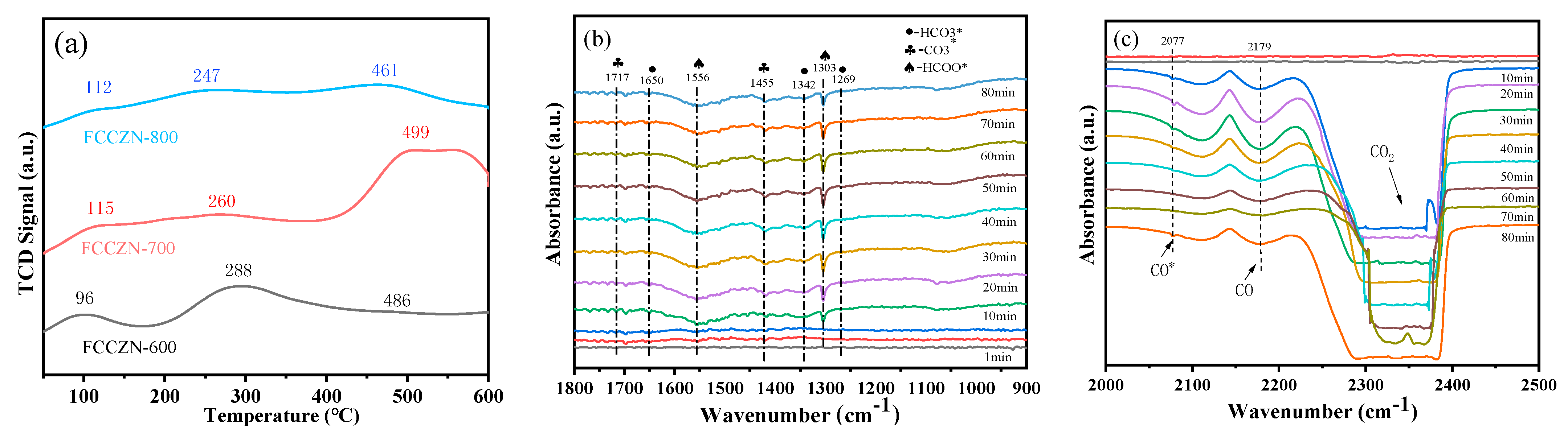
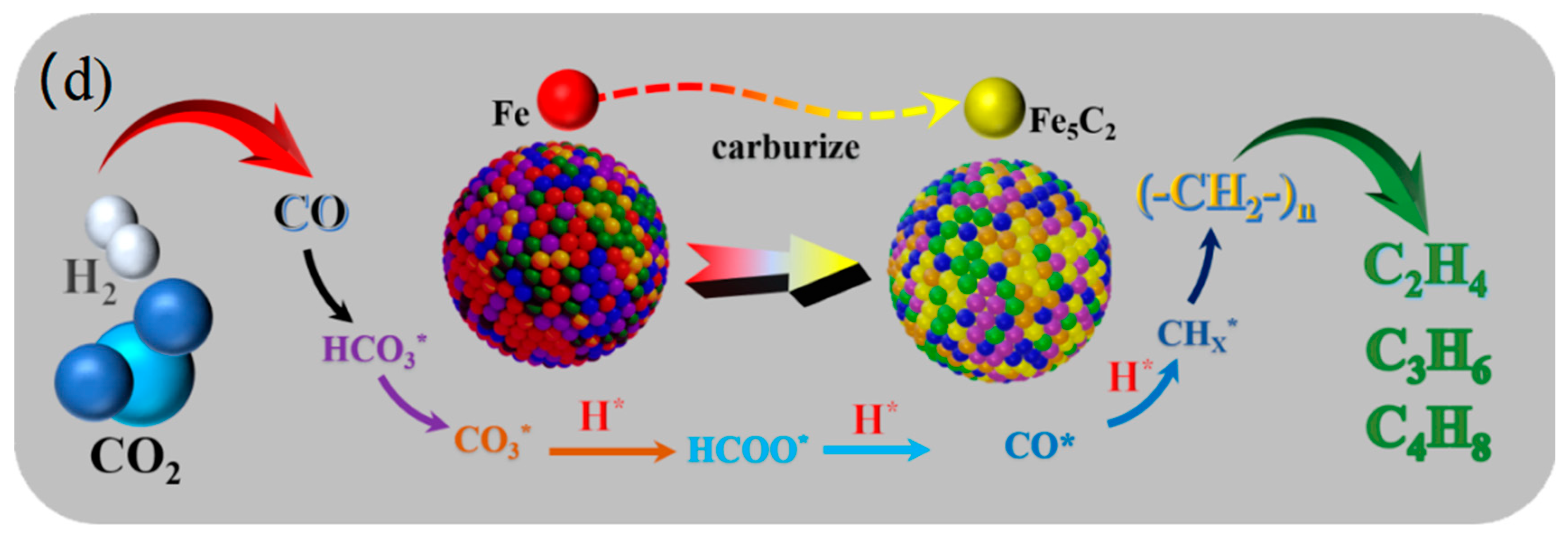
2.6. Reaction Mechanism Study of CO2 Hydrogenation to Light Olefins
2.7. Regulation of Calcination Temperature on Catalyst Structure and Activity
3. Experimental Section
3.1. Synthesis of Catalysts
3.2. Sample Characterization
3.3. Catalyst Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kondratenko, E.V.; Mul, G.; Baltrusaitis, J.; Larrazábal, G.O.; Pérez-Ramírez, J. Status and Perspectives of CO2 Conversion into Fuels and Chemicals by Catalytic, Photocatalytic and Electrocatalytic Processes. Energy Environ. Sci. 2013, 6, 3112. [Google Scholar] [CrossRef]
- Pan, Y.; Ding, X.; Zhang, C.; Zhu, M.; Yang, Z.; Han, Y.-F. Effects of Different Reductive Agents on Zn-Promoted Iron Oxide Phases in the CO2–Fischer–Tropsch to Linear α-Olefins. Catalysts 2023, 13, 594. [Google Scholar] [CrossRef]
- Sun, C.; Jiang, Z.; Li, W.; Hou, Q.; Li, L. Changes in Extreme Temperature over China When Global Warming Stabilized at 1.5 °C and 2.0 °C. Sci. Rep. 2019, 9, 14982. [Google Scholar] [CrossRef] [PubMed]
- Bootpakdeetam, P.; Igboenyesi, O.V.; Dennis, B.H.; MacDonnell, F.M. Superhydrophobic Surface Modification of a Co-Ru/SiO2 Catalyst for Enhanced Fischer-Tropsch Synthesis. Catalysts 2024, 14, 638. [Google Scholar] [CrossRef]
- Guo, L.; Sun, J.; Ge, Q.; Tsubaki, N. Recent Advances in Direct Catalytic Hydrogenation of Carbon Dioxide to Valuable C2+ Hydrocarbons. J. Mater. Chem. A 2018, 6, 23244–23262. [Google Scholar] [CrossRef]
- Davis, B.H. Fischer−Tropsch Synthesis: Comparison of Performances of Iron and Cobalt Catalysts. Ind. Eng. Chem. Res. 2007, 46, 8938–8945. [Google Scholar] [CrossRef]
- Hu, B.; Frueh, S.; Garces, H.F.; Zhang, L.; Aindow, M.; Brooks, C.; Kreidler, E.; Suib, S.L. Selective Hydrogenation of CO2 and CO to Useful Light Olefins over Octahedral Molecular Sieve Manganese Oxide Supported Iron Catalysts. Appl. Catal. B Environ. 2013, 132–133, 54–61. [Google Scholar] [CrossRef]
- Satthawong, R.; Koizumi, N.; Song, C.; Prasassarakich, P. Bimetallic Fe–Co Catalysts for CO2 Hydrogenation to Higher Hydrocarbons. J. CO2 Util. 2013, 3–4, 102–106. [Google Scholar] [CrossRef]
- Orege, J.I.; Liu, N.; Amoo, C.C.; Wei, J.; Ge, Q.; Sun, J. Boosting CO2 Hydrogenation to High-Value Olefins with Highly Stable Performance over Ba and Na Co-Modified Fe Catalyst. J. Energy Chem. 2023, 80, 614–624. [Google Scholar] [CrossRef]
- Xu, Y.; Zhai, P.; Deng, Y.; Xie, J.; Liu, X.; Wang, S.; Ma, D. Highly Selective Olefin Production from CO2 Hydrogenation on Iron Catalysts: A Subtle Synergy between Manganese and Sodium Additives. Angew. Chem. Int. Ed. 2020, 59, 21736–21744. [Google Scholar] [CrossRef]
- Zhang, Z.; Huang, G.; Tang, X.; Yin, H.; Kang, J.; Zhang, Q.; Wang, Y. Zn and Na Promoted Fe Catalysts for Sustainable Production of High-Valued Olefins by CO2 Hydrogenation. Fuel 2022, 309, 122105. [Google Scholar] [CrossRef]
- Chirik, P.J. Iron- and Cobalt-Catalyzed Alkene Hydrogenation: Catalysis with Both Redox-Active and Strong Field Ligands. Acc. Chem. Res. 2015, 48, 1687–1695. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Duan, H.; Sun, T.; Ma, J.; Liu, X.; Xu, J.; Su, X.; Huang, Y.; Zhang, T. Effect of Na Promoter on Fe-Based Catalyst for CO2 Hydrogenation to Alkenes. ACS Sustain. Chem. Eng. 2019, 7, 925–932. [Google Scholar] [CrossRef]
- Chernavskii, P.A.; Kazak, V.O.; Pankina, G.V.; Perfiliev, Y.D.; Li, T.; Virginie, M.; Khodakov, A.Y. Influence of Copper and Potassium on the Structure and Carbidisation of Supported Iron Catalysts for Fischer–Tropsch Synthesis. Catal. Sci. Technol. 2017, 7, 2325–2334. [Google Scholar] [CrossRef]
- Wang, W.; Jiang, X.; Wang, X.; Song, C. Fe–Cu Bimetallic Catalysts for Selective CO2 Hydrogenation to Olefin-Rich C2 + Hydrocarbons. Ind. Eng. Chem. Res. 2018, 57, 4535–4542. [Google Scholar] [CrossRef]
- Zhang, C.; Cao, C.; Zhang, Y.; Liu, X.; Xu, J.; Zhu, M.; Tu, W.; Han, Y.-F. Unraveling the Role of Zinc on Bimetallic Fe5C2 –ZnO Catalysts for Highly Selective Carbon Dioxide Hydrogenation to High Carbon α-Olefins. ACS Catal. 2021, 11, 2121–2133. [Google Scholar] [CrossRef]
- Shi, Z.; Wang, L.; Huang, Y.; Kong, X.Y.; Ye, L. High-Entropy Catalysts: New Opportunities toward Excellent Catalytic Activities. Mater. Chem. Front. 2024, 8, 179–191. [Google Scholar] [CrossRef]
- Gild, J.; Samiee, M.; Braun, J.L.; Harrington, T.; Vega, H.; Hopkins, P.E.; Vecchio, K.; Luo, J. High-Entropy Fluorite Oxides. J. Eur. Ceram. Soc. 2018, 38, 3578–3584. [Google Scholar] [CrossRef]
- Sarkar, A.; Wang, Q.; Schiele, A.; Chellali, M.R.; Bhattacharya, S.S.; Wang, D.; Brezesinski, T.; Hahn, H.; Velasco, L.; Breitung, B. High-Entropy Oxides: Fundamental Aspects and Electrochemical Properties. Adv. Mater. 2019, 31, 1806236. [Google Scholar] [CrossRef]
- Anand, G.; Wynn, A.P.; Handley, C.M.; Freeman, C.L. Phase Stability and Distortion in High-Entropy Oxides. Acta Mater. 2018, 146, 119–125. [Google Scholar] [CrossRef]
- Zhang, R.-Z.; Reece, M.J. Review of High Entropy Ceramics: Design, Synthesis, Structure and Properties. J. Mater. Chem. A 2019, 7, 22148–22162. [Google Scholar] [CrossRef]
- Mi, J.; Chen, X.; Ding, Y.; Zhang, L.; Ma, J.; Kang, H.; Wu, X.; Liu, Y.; Chen, J.; Wu, Z.-S. Activation of Partial Metal Sites in High-Entropy Oxides for Enhancing Thermal and Electrochemical Catalysis. Chin. J. Catal. 2023, 48, 235–246. [Google Scholar] [CrossRef]
- Meng, Z.; Gong, X.; Xu, J.; Sun, X.; Zeng, F.; Du, Z.; Hao, Z.; Shi, W.; Yu, S.; Hu, X.; et al. A General Strategy for Preparing Hollow Spherical Multilayer Structures of Oxygen-Rich Vacancy Transition Metal Oxides, Especially High Entropy Perovskite Oxides. Chem. Eng. J. 2023, 457, 141242. [Google Scholar] [CrossRef]
- Wang, G.; Qin, J.; Feng, Y.; Feng, B.; Yang, S.; Wang, Z.; Zhao, Y.; Wei, J. Sol–Gel Synthesis of Spherical Mesoporous High-Entropy Oxides. ACS Appl. Mater. Interfaces 2020, 12, 45155–45164. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Sun, Z.; Luo, W.; Li, Y.; Elzatahry, A.A.; Al-Enizi, A.M.; Deng, Y.; Zhao, D. New Insight into the Synthesis of Large-Pore Ordered Mesoporous Materials. J. Am. Chem. Soc. 2017, 139, 1706–1713. [Google Scholar] [CrossRef]
- Qiu, P.; Ma, B.; Hung, C.-T.; Li, W.; Zhao, D. Spherical Mesoporous Materials from Single to Multilevel Architectures. Acc. Chem. Res. 2019, 52, 2928–2938. [Google Scholar] [CrossRef]
- Cai, W.; Han, H.; Hu, C.; Ye, C.; Cao, Y.; Wang, Y.; Fu, J.; Zhao, Y.; Bu, Y. Fabrication of Transition Metal (Mn, Co, Ni, Cu)-Embedded Faveolate ZnFe2O4 Spinel Structure with Robust CO2 Hydrogenation into Value-Added C2+ Hydrocarbons. ChemCatChem 2023, 15, e202201403. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, J.; Gao, X.; Ma, Q.; Fan, S.; Zhao, T.-S. CO2 Hydrogenation to Linear α-Olefins on FeCx/ZnO Catalysts: Effects of Surface Oxygen Vacancies. Appl. Surf. Sci. 2023, 641, 158543. [Google Scholar] [CrossRef]
- Nolen, M.A.; Tacey, S.A.; Kwon, S.; Farberow, C.A. Theoretical Assessments of CO2 Activation and Hydrogenation Pathways on Transition-Metal Surfaces. Appl. Surf. Sci. 2023, 637, 157873. [Google Scholar] [CrossRef]
- Ra, E.C.; Kim, K.H.; Lee, J.H.; Jang, S.; Kim, H.E.; Lee, J.H.; Kim, E.H.; Kim, H.; Kwak, J.H.; Lee, J.S. Selective Light Hydrocarbon Production from CO2 Hydrogenation over Na/ZnFe2O4 and CHA-Zeolite Hybrid Catalysts. ACS Catal. 2024, 14, 3492–3503. [Google Scholar] [CrossRef]
- Zhao, K.; Wang, L.; Calizzi, M.; Moioli, E.; Züttel, A. In Situ Control of the Adsorption Species in CO2 Hydrogenation: Determination of Intermediates and Byproducts. J. Phys. Chem. C 2018, 122, 20888–20893. [Google Scholar] [CrossRef]
- Yanagisawa, Y.; Takaoka, K.; Yamabe, S.; Ito, T. Interaction of CO2 with Magnesium Oxide Surfaces: A TPD, FTIR, and Cluster-Model Calculation Study. J. Phys. Chem. 1995, 99, 3704–3710. [Google Scholar] [CrossRef]
- Yang, R.; Zhang, Y.; Iwama, Y.; Tsubaki, N. Mechanistic Study of a New Low-Temperature Methanol Synthesis on Cu/MgO Catalysts. Appl. Catal. Gen. 2005, 288, 126–133. [Google Scholar] [CrossRef]
- Borchert, H.; Jurgens, B.; Zielasek, V.; Rupprechter, G.; Giorgio, S.; Henry, C.; Baumer, M. Pd Nanoparticles with Highly Defined Structure on MgO as Model Catalysts: An FTIR Study of the Interaction with CO, O2, and H2 under Ambient Conditions. J. Catal. 2007, 247, 145–154. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | CO2 Con (%) | Selectivity a (%) | O/P | STY (mmol g−1·h−1) | ||||
|---|---|---|---|---|---|---|---|---|
| CH4 | C5+ | CO | ||||||
| FCCZN-600 | 23.3 | 34.6 | 38.8 | 10.3 | 16.3 | 25 | 3.1 | 0.0278 |
| FCCZN-700 | 38.6 | 29.1 | 39.5 | 13.5 | 17.9 | 5.5 | 2.2 | 0.0455 |
| FCCZN-800 | 39.0 | 26.8 | 41.3 | 15.7 | 16.2 | 6.9 | 2.6 | 0.0495 |
| FCCZN@MG-800 | 36.1 | 36.9 | 37.0 | 11.6 | 14.4 | 5.9 | 3.2 | 0.0324 |
| Catalyst | Fe Loading (wt%) a | Co Loading (wt%) a | Cu Loading (wt%) a | Zn Loading (wt%) a | SSA (m2/g) b | APS (nm) b |
|---|---|---|---|---|---|---|
| FCCZN-600 | 24.98 | 24.32 | 25.11 | 24.53 | 19.6 | 17.6 |
| FCCZN-700 | 25.05 | 24.63 | 24.99 | 24.30 | 8.6 | 14.8 |
| FCCZN-800 | 24.88 | 24.43 | 24.96 | 24.68 | 1.8 | 33.2 |
| FCCZN@MG-800 | 25.14 | 24.50 | 24.54 | 24.81 | 0.6 | 50.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ni, Z.; Chen, X.; Su, L.; Shen, H.; Yin, C. Effect of Calcination Temperature in Large-Aperture Medium-Entropy Oxide (FeCoCuZnNa)O on CO2 Hydrogenation for Light Olefins. Catalysts 2024, 14, 818. https://doi.org/10.3390/catal14110818
Ni Z, Chen X, Su L, Shen H, Yin C. Effect of Calcination Temperature in Large-Aperture Medium-Entropy Oxide (FeCoCuZnNa)O on CO2 Hydrogenation for Light Olefins. Catalysts. 2024; 14(11):818. https://doi.org/10.3390/catal14110818
Chicago/Turabian StyleNi, Zhijiang, Xiaoyu Chen, Lin Su, Hanyu Shen, and Chaochuang Yin. 2024. "Effect of Calcination Temperature in Large-Aperture Medium-Entropy Oxide (FeCoCuZnNa)O on CO2 Hydrogenation for Light Olefins" Catalysts 14, no. 11: 818. https://doi.org/10.3390/catal14110818
APA StyleNi, Z., Chen, X., Su, L., Shen, H., & Yin, C. (2024). Effect of Calcination Temperature in Large-Aperture Medium-Entropy Oxide (FeCoCuZnNa)O on CO2 Hydrogenation for Light Olefins. Catalysts, 14(11), 818. https://doi.org/10.3390/catal14110818