

Copper-Catalyzed Benzothiazolyldifluoroalkylation of Arylidenemalonitriles or para-Quinone Methides


Abstract

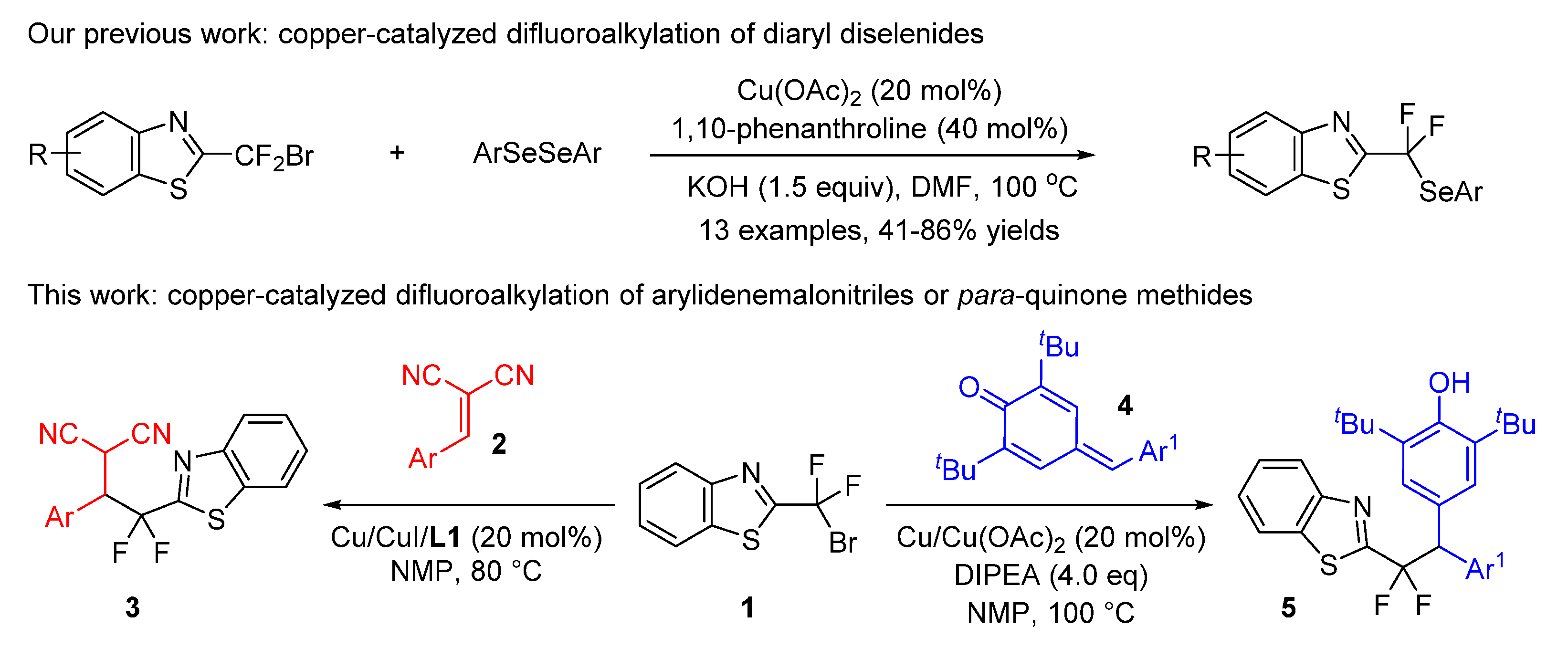
1. Introduction
2. Results
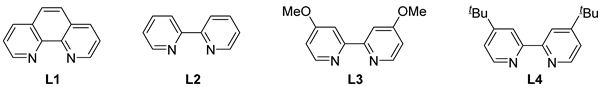
2.1. The Reaction Conditions Optimization of 1 and 2
2.2. Reaction Conditions Optimization of 1 and 4
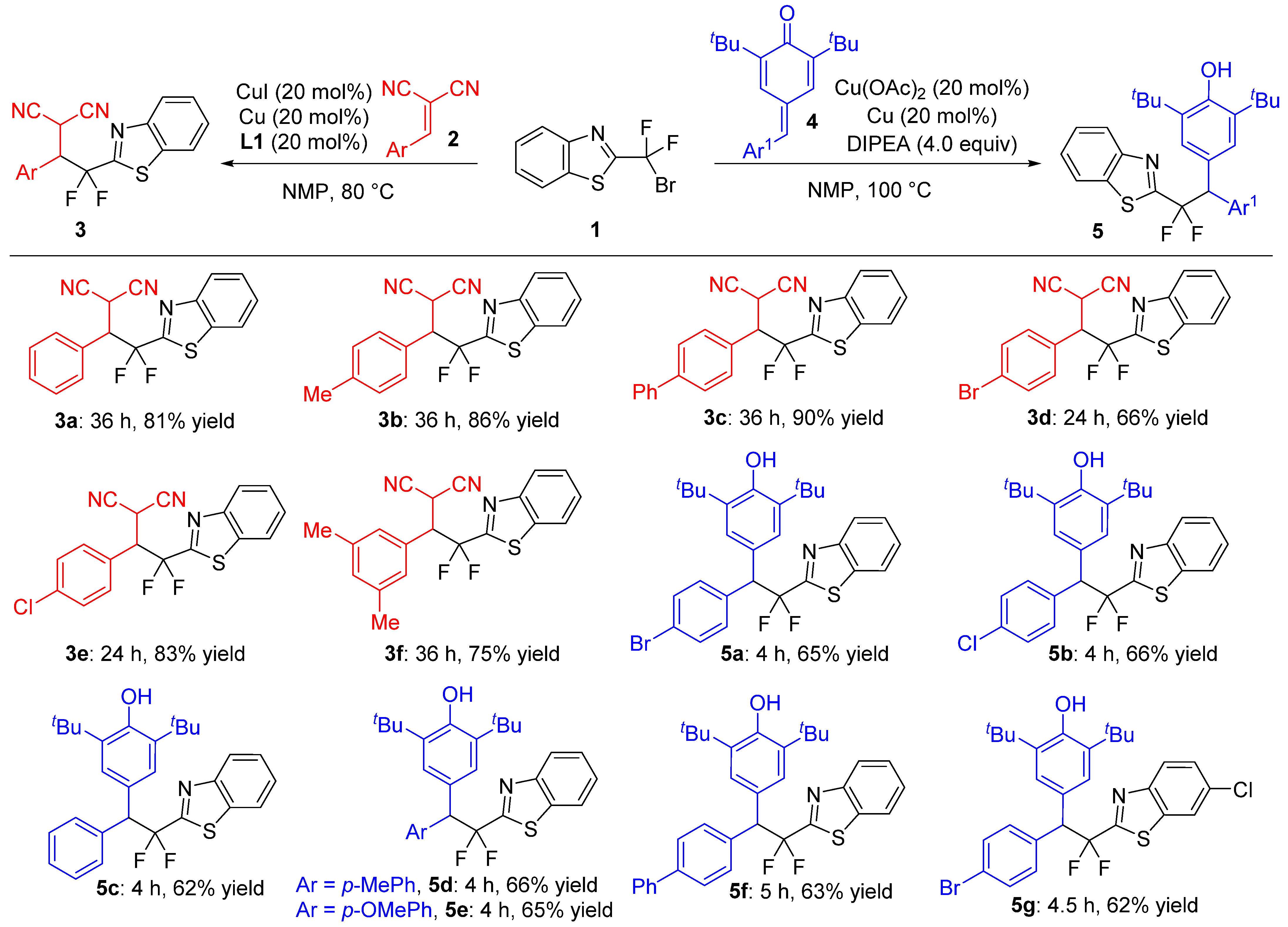
2.3. Substrate Scope
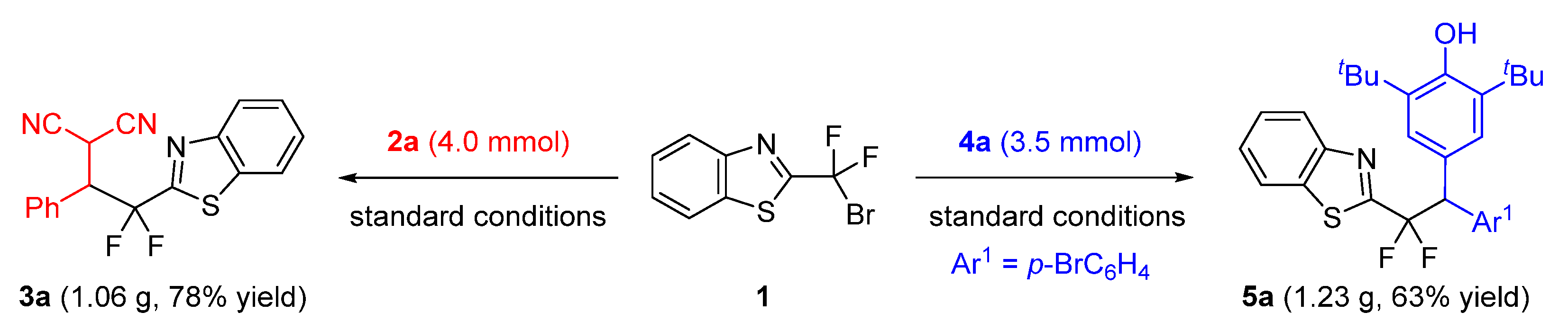
2.4. Gram-Scale Synthesis
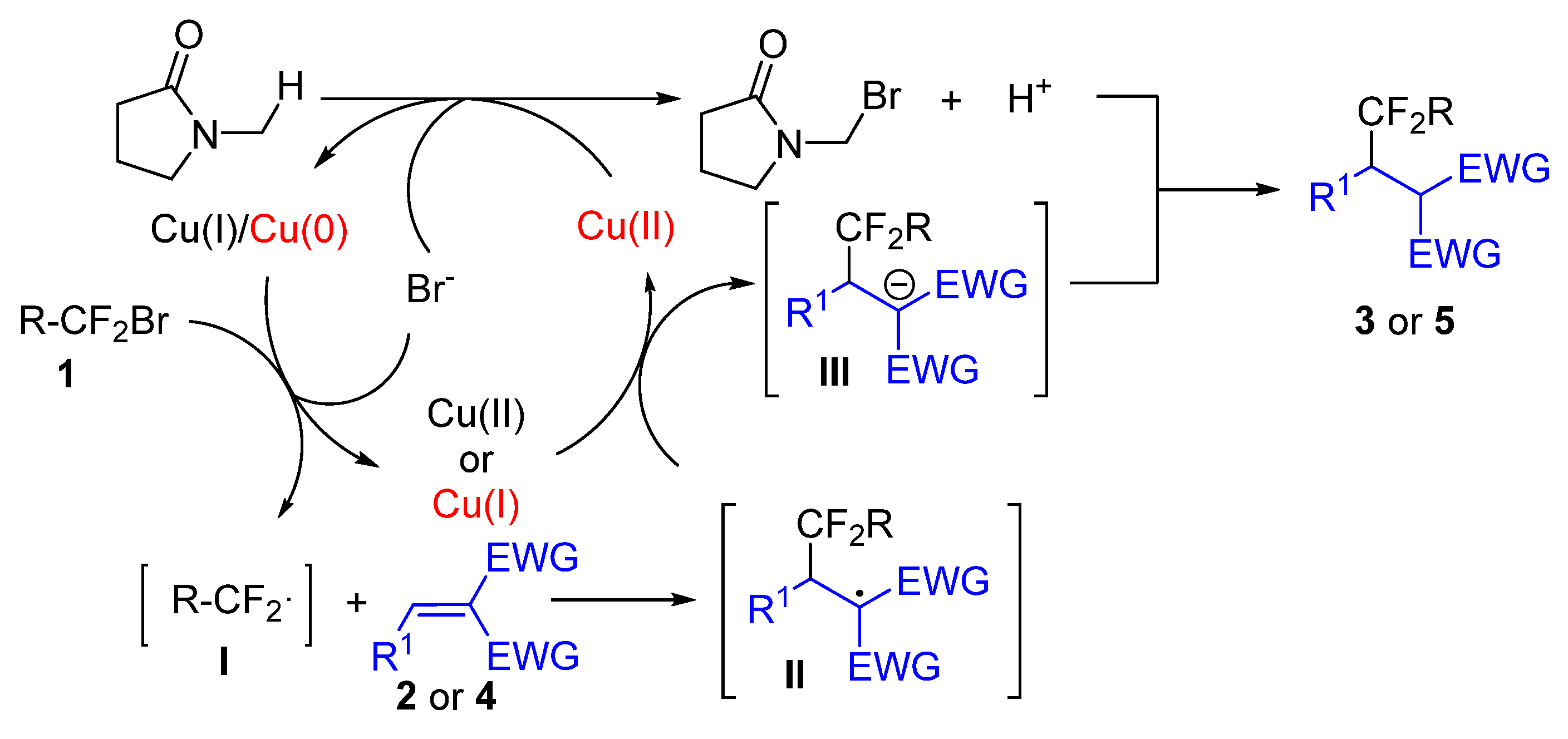
2.5. The Proposed Mechanism
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ma, J.-A.; Cahard, D. Asymmetric fluorination, trifluoromethylation, and perfluoroalkylation reactions. Chem. Rev. 2004, 104, 6119–6146. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D. Understanding organofluorine chemistry. An introduction to the C-F bond. Chem. Soc. Rev. 2008, 37, 308–319. [Google Scholar] [CrossRef]
- Zhang, C.-P.; Chen, Q.-Y.; Guo, Y.; Xiao, J.-C.; Gu, Y.-C. Progress in fluoroalkylation of organic compounds via sulfinatodehalogenation initiation system. Chem. Soc. Rev. 2012, 41, 4536–4559. [Google Scholar] [CrossRef]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in pharmaceutical industry: Fluorine-containing drugs introduced to the market in the last decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef]
- Yang, X.; Wu, T.; Phipp, R.J.; Toste, F.D. Advances in catalytic enantioselective fluorination, mono-, di-, and trifluoromethylation, and trifluoromethylthiolation reactions. Chem. Rev. 2015, 115, 826–870. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.-Q.; Yang, H.; Shi, J.-L.; Si, W.-J.; Wang, Z.-L.; Xu, X.-M. Promising reagents for difluoroalkylation. Org. Chem. Front. 2020, 7, 2538–2575. [Google Scholar] [CrossRef]
- Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley: Berlin, Germany, 2009. [Google Scholar]
- Bey, P.; Gerhart, F.; Dorsselaer, V.V.; Danzin, C. a-(Fluorornet hy1)dehydroornit hine and a-(Fluorornet hy1)dehydroputre an alogues as irreversible inhibitors of ornithine decarboxylase. J. Med. Chem. 1983, 26, 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- Pourquier, P.; Gioffre, C.; Kohlhagen, G.; Urasaki, Y.; Goldwasser, F.; Hertel, L.W.; Yu, S.; Pon, R.T.; Gmeiner, W.H.; Pommier, Y. Gemcitabine (2′,2′-difluoro-2′-deoxycytidine), an antimetabolite that poisons topoisomerase I. Clin. Cancer Res. 2002, 8, 2499–2504. [Google Scholar]
- Ando, K.; Koike, F.; Kondo, F.; Takayama, H. An improved synthesis of 24,24-difluoro-1α,25-dihydroxyvitamin D3 from readily available vitamin D2. Chem. Pharm. Bull. 1995, 43, 189–192. [Google Scholar] [CrossRef]
- Uoto, K.; Ohsuki, S.; Takenoshita, H.; Ishiyama, T.; Iimura, S.; Hirota, Y.; Mitsui, I.; Terasawa, H.; Soga, T. Synthesis and structure-activity relationships of novel 2′,2′-difluoro analogues of docetaxel. Chem. Pharm. Bull. 1997, 45, 1793–1804. [Google Scholar] [CrossRef]
- Nie, J.; Guo, H.-C.; Cahard, D.; Ma, J.-A. Asymmetric construction of stereogenic carbon centers featuring a trifluoromethyl group from prochiral trifluoromethylated substrates. Chem. Rev. 2011, 111, 455–529. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, W.; Wang, F. Selective difluoromethylation and monofluoromethylation reactions. Chem. Commun. 2009, 7465–7478. [Google Scholar] [CrossRef]
- Liu, Y.-L.; Yu, J.-S.; Zhou, J. Catalytic asymmetric construction of stereogenic carbon centers that feature a gem-difluoroalkyl group. Asian J. Org. Chem. 2013, 2, 194–206. [Google Scholar] [CrossRef]
- Lin, J.-H.; Xiao, J.-C. Recent advances in asymmetric fluorination and fluoroalkylation reactions via organocatalysis. Tetrahedron Lett. 2014, 55, 6147–6155. [Google Scholar] [CrossRef]
- Ni, C.; Zhu, L.; Hu, J. Advances in transition-metal-mediated di- and monofluoroalkylations. Acta Chim. Sin. 2015, 73, 90–115. [Google Scholar] [CrossRef]
- Qing, F.-L.; Liu, X.-Y.; Ma, J.-A.; Shen, Q.; Song, Q.; Tang, P. A fruitful decade of organofluorine chemistry: New reagents and reactions. CCS Chem. 2022, 4, 2518–2549. [Google Scholar] [CrossRef]
- Burkholder, C.R.; Dolbier, W.R., Jr.; Médebielle, M. The syntheses of nonnucleoside, HIV-1 reverse transcriptase inhibitors containing a CF2 group The SRN1 reactions of 2-(bromodifluoromethyl)benzoxazole with the anions derived from heterocyclic thiols and phenolic compounds. J. Fluor. Chem. 2000, 102, 369–376. [Google Scholar] [CrossRef]
- Jiang, H.; Yuan, S.; Cai, Y.; Wan, W.; Zhu, S.; Hao, J. A facile preparation of 2-bromodifluoromethyl benzo-1,3-diazoles and its application in the synthesis of gem-difluoromethylene linked aryl ether compounds. J. Fluor. Chem. 2012, 133, 167–170. [Google Scholar] [CrossRef]
- Burkholder, C.R.; Dolbier Jr, W.R.; Médebielle, M. Synthesis and reactivity of halogeno-difluoromethyl aromatics heterocycles: Application to synthesis of gem-difluorinated bioactive compounds. J. Fluor. Chem. 2001, 109, 39–48. [Google Scholar] [CrossRef]
- Jiang, H.; Lu, W.; Yang, K.; Ma, G.; Xu, M.; Li, J.; Yao, J.; Wan, W.; Deng, H.; Wu, S.; et al. Enhancement of neighbouring group participation in Cu0 promoted cross-coupling gem-difluoromethylenation of aryl/alkenyl halides with 1,3-azolic difluoromethyl bromides. Chem.-Eur. J. 2014, 20, 10084–10092. [Google Scholar] [CrossRef]
- Ma, G.; Wan, W.; Hu, Q.; Jiang, H.; Wang, J.; Zhu, S.; Hao, J. Highly effective copper-mediated gem-difluoromethylenation of arylboronic acids. Chem. Commun. 2014, 50, 7527–7530. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Xu, M.; Lu, W.; Tian, W.; Wan, W.; Chen, Y.; Deng, H.; Wu, S.; Hao, J. Direct gem-difluoromethylenation of sp3-hybridized carbon center through copper-mediated radical/radical cross-coupling for the construction of a CH2-CF2 linkage. Chem. Commun. 2015, 51, 15756–15759. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.-L.; Zhang, B.; Feng, Z.; Zhang, X. Heteroaryldifluoromethylation of organoborons catalyzed by palladium: Facile access to aryl(heteroaryl)difluoromethanes. Org. Lett. 2014, 16, 4822–4825. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.-W.; Zhang, X. Palladium-catalyzed difluoroalkylation of isocyanides: Access to difluoroalkylated phenanthridine derivatives. Org. Lett. 2015, 17, 5384–5387. [Google Scholar] [CrossRef]
- Xu, C.; Yang, Z.-F.; An, L.; Zhang, X. Nickel-catalyzed difluoroalkylation-alkylation of enamides. ACS Catal. 2019, 9, 8224–8229. [Google Scholar] [CrossRef]
- Xu, H.; Wang, D.; Chen, Y.; Wan, W.; Deng, H.; Ma, K.; Wu, S.; Hao, J.; Jiang, H. Copper-catalyzed gem-difluoromethylenation of C(sp2)-H bonds of alkenes. Org. Chem. Front. 2017, 4, 1239–1243. [Google Scholar] [CrossRef]
- Zhang, T.; Chen, B.; Wang, W.; Zhang, Q.; Wang, P.; Wan, W.; Deng, H.; Hao, J.; Jiang, H. Copper-promoted aryldifluoromethylenation of N-arylacrylamides to 3-Benzodiazolyldifluoromethylene substituted 2-oxindoles. Asian J. Org. Chem. 2019, 8, 671–674. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, W.; Wang, P.; Deng, H.; Jiang, H. Copper-catalyzed aryldifluoromethylenation of N-arylacrylamides to synthesis of the diheterocyclic compounds linked by gem-difluoromethylene moiety. Tetrahedron 2019, 75, 130778. [Google Scholar] [CrossRef]
- Da, Y.; Han, S.; Du, X.; Liu, S.; Liu, L.; Li, J. Copper(I)-catalyzed oxydifluoroalkylation of alkenes: A route to functionalization of lactones. Org. Lett. 2018, 20, 5149–5152. [Google Scholar] [CrossRef]
- Li, X.; Hu, K.; Zhang, J.; Jiang, H. Cu0-promoted Truce-Smiles rearrangement for aryl-difluoromethylenation of C=C bonds via a reductive radical-polar crossover process. J. Org. Chem. 2024, 89, 13947–13952. [Google Scholar] [CrossRef]
- Malapit, C.A.; Reeves, J.T.; Busacca, C.A.; Howell, A.R.; Senanayake, C.H. Rhodium-catalyzed transnitrilation of aryl boronic acids with dimethylmalononitrile. Angew. Chem. Int. Ed. 2016, 55, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Malapit, C.A.; Caldwell, D.R.; Luvaga, I.K.; Reeves, J.T.; Volchkov, I.; Gonnella, N.C.; Han, Z.S.; Busacca, C.A.; Howell, A.R.; Senanayake, C.H. Rhodium-catalyzed addition of aryl boronic acids to 2,2-disubstituted malononitriles. Angew. Chem. Int. Ed. 2017, 56, 6999–7002. [Google Scholar] [CrossRef] [PubMed]
- Peter, M.G. Chemical modifications of biopolymers by quinones and quinone methides. Angew. Chem. Int. Ed. Engl. 1989, 28, 555–570. [Google Scholar] [CrossRef]
- Abrams, P.; Freeman, R.; Anderström, C.; Mattiasson, A. Tolterodine, a new antimuscarinic agent: As effective but better tolerated than oxybutynin in patients with an overactive bladder. Br. J. Urol. 1998, 81, 801–810. [Google Scholar] [CrossRef] [PubMed]
- An, R.-B.; Jeong, G.-S.; Kim, Y.-C. Flavonoids from the heartwood of dalbergia odorifera and their protective effect on glutamate-induced oxidative injury in HT22 cells. Chem. Pharm. Bull. 2008, 56, 1722–1724. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.J.; Barker, D. Synthesis of various lignans via the rearrangements of 1,4-diarylbutane-1,4-diols. Tetrahedron Lett. 2015, 56, 4549–4553. [Google Scholar] [CrossRef]
- Yu, J.-S.; Liao, F.-M.; Gao, W.-M.; Liao, K.; Zuo, R.-L.; Zhou, J. Michael addition catalyzed by chiral secondary amine phosphoramide using fluorinated silyl enol ethers: Formation of quaternary carbon stereocenters. Angew. Chem. Int. Ed. 2015, 54, 7381–7385. [Google Scholar] [CrossRef]
- Hao, Y.-J.; Hu, X.-S.; Yu, J.-S.; Zhou, F.; Zhou, Y.; Zhou, J. An efficient Fe(III)-catalyzed 1,6-conjugate addition of para-quinone methides with fluorinated silyl enol ethers toward b,b-diaryl afluorinated ketones. Tetrahedron 2018, 74, 7395–7398. [Google Scholar] [CrossRef]
- Ke, M.; Song, Q. Copper-catalyzed 1,6-hydrodifluoroacetylation of para-quinone methides at ambient temperature with bis(pinacolato)diboron as reductant. Adv. Synth. Catal. 2017, 359, 384–389. [Google Scholar] [CrossRef]
- Zhao, Y.-N.; Luo, Y.-C.; Wang, Z.-Y.; Xu, P.-F. A new approach to access difluoroalkylated diarylmethanes via visible-light photocatalytic cross-coupling reactions. Chem. Commun. 2018, 54, 3993–3996. [Google Scholar] [CrossRef]
- Qu, C.-H.; Song, G.-T.; Tang, D.-Y.; Shao, J.-W.; Li, H.-Y.; Xu, Z.-G.; Chen, Z.-Z. Microwave-assisted copper catalysis of α-difluorinated gem-diol toward difluoroalkyl radical for hydrodifluoroalkylation of para-quinone methides. J. Org. Chem. 2020, 85, 12785–12796. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Liao, F.; Cai, Y.; Liu, J. A convenient synthesis of 2-bromodifluoromethyl benzo-1,3-thiazoles. Chin. J. Syn. Chem. 2020, 30, 588–592. [Google Scholar]
- Xu, R.; Cai, Y.; Liao, F.; Liu, J. 2-[Difluoro(phenylselenyl)methyl]benzo-1,3-thiazole. Molbank 2022, 2022, 1450. [Google Scholar] [CrossRef]
- Xu, R.; Liao, F.; Cai, Y.; Liu, J. Copper-catalyzed aryl selenylation of 2-bromodifluoromethylbenzo-1,3-thi(x)azoles with diaryl diselenides. Tetrahedron 2022, 128, 133118. [Google Scholar] [CrossRef]
- Petrier, C.; Dupuy, C.; Luche, J.L. Conjugate additions to α,β-unsaturated carbonyl compounds in aqueous media. Tetrahedron Lett. 1986, 27, 3149–3152. [Google Scholar] [CrossRef]
- Suárez, R.M.; Sestelo, J.P.; Sarandeses, L.A. Diastereoselective conjugate addition to chiral α,β-unsaturated carbonyl systems in aqueous media: An enantioselective entry to α- and γ-hydroxy acids and α-amino acids. Chem.-Eur. J. 2003, 9, 4179–4187. [Google Scholar] [CrossRef]
- Shin, J.-A.; Kim, J.; Lee, H.; Ha, S.; Lee, H.-Y. Cu(OTf)2-promoted 1,4-addition of alkyl bromides to dehydroalanine. J. Org. Chem. 2019, 84, 4558–4565. [Google Scholar] [CrossRef]
- Gugkaeva, Z.T.; Smol’yakov, A.F.; Maleev, V.I.; Larionov, V.A. A general asymmetric synthesis of artificial aliphatic and perfluoroalkylated α-amino acids by Luche’s cross-electrophile coupling reaction. Org. Biomol. Chem. 2021, 19, 5327–5332. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Catalyst | [Ligands] | Solvent | Time (h) | Yield (%) b |
| 1 | CuI | - | DMF | 72 | 18 |
| 2 | CuI | L1 | DMF | 72 | 39 |
| 3 | CuI | L2 | DMF | 48 | 12 |
| 4 | CuI | L3 | DMF | 48 | 10 |
| 5 | CuI | L4 | DMF | 48 | 10 |
| 6 | CuI | L1 | DMAc | 48 | trace |
| 7 | CuI | L1 | DMSO | 48 | 22 |
| 8 | CuI | L1 | NMP | 48 | 57 |
| 9 c | CuI | L1 | NMP | 36 | 41 |
| 10 d | CuI | L1 | NMP | 48 | 25 |
| 11 | CuBr | L1 | NMP | 46 | 43 |
| 12 | CuCl | L1 | NMP | 46 | 45 |
| 13 | Cu(OAc)2 | L1 | NMP | 46 | 45 |
| 14 | CuI + Cu | L1 | NMP | 36 | 81 |
| 15 e | CuI + Cu | L1 | NMP | 24 | 79 |
| 16 f | CuI + Cu | L1 | NMP | 42 | 71 |
 | |||||
 | ||||
|---|---|---|---|---|
| Entry | Catalyst (20 mol%) | Base | Time (h) | Yield (%) b |
| 1 | Cu/CuI/L1 | - | 48 | 43 |
| 2 | Cu (1.0 eq)/CuI/L1 | - | 9 | 53 |
| 3 | Cu (1.0 eq)/Cu2O/L1 | - | 9 | 57 |
| 4 | Cu (1.0 eq)/Cu(OAc)2/L1 | - | 9 | 60 |
| 5 | Cu (1.0 eq)/Cu(OAc)2/L2 | - | 12 | 46 |
| 6 | Cu (1.0 eq)/Cu(OAc)2/L3 | - | 12 | 49 |
| 7 | Cu (1.0 eq)/Cu(OAc)2/L4 | - | 12 | 45 |
| 8 | Cu (1.0 eq)/Cu(OAc)2 | - | 9 | 61 |
| 9 | Cu (1.0 eq)/Cu(OAc)2 | NaHCO3 (2.0 eq) | 18 | 61 |
| 10 | Cu (1.0 eq)/Cu(OAc)2 | Et3N (2.0 eq) | 18 | 59 |
| 11 | Cu (1.0 eq)/Cu(OAc)2 | Pyridine (2.0 eq) | 18 | 38 |
| 12 | Cu (1.0 eq)/Cu(OAc)2 | DIPEA (2.0 eq) | 6 | 62 |
| 13 | Cu (1.0 eq)/Cu(OAc)2 | DIPEA (3.0 eq) | 4 | 63 |
| 14 | Cu (1.0 eq)/Cu(OAc)2 | DIPEA (4.0 eq) | 4 | 66 |
| 15 | Cu (1.0 eq)/Cu(OAc)2 | DIPEA (5.0 eq) | 4 | 45 |
| 16 c | Cu (1.0 eq)/Cu(OAc)2 | DIPEA (4.0 eq) | 4 | 48 |
| 17 d | Cu (1.0 eq)/Cu(OAc)2 | DIPEA (4.0 eq) | 24 | 52 |
| 18 e | -/Cu(OAc)2 | DIPEA (4.0 eq) | 24 | 39 |
| 19 f | Cu (1.0 eq)/- | DIPEA (4.0 eq) | 14 | 53 |
| 20 | Cu (0.2 eq)/Cu(OAc)2 | DIPEA (4.0 eq) | 12 | 57 |
| 21 g | Cu (0.2 eq)/Cu(OAc)2 | DIPEA (4.0 eq) | 4 | 65 |
 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, J.; Cai, Y.; Xu, R.; Liao, F.; Liu, J. Copper-Catalyzed Benzothiazolyldifluoroalkylation of Arylidenemalonitriles or para-Quinone Methides. Catalysts 2024, 14, 777. https://doi.org/10.3390/catal14110777
Xiao J, Cai Y, Xu R, Liao F, Liu J. Copper-Catalyzed Benzothiazolyldifluoroalkylation of Arylidenemalonitriles or para-Quinone Methides. Catalysts. 2024; 14(11):777. https://doi.org/10.3390/catal14110777
Chicago/Turabian StyleXiao, Jilin, Ying Cai, Rongfu Xu, Fumin Liao, and Jinbiao Liu. 2024. "Copper-Catalyzed Benzothiazolyldifluoroalkylation of Arylidenemalonitriles or para-Quinone Methides" Catalysts 14, no. 11: 777. https://doi.org/10.3390/catal14110777
APA StyleXiao, J., Cai, Y., Xu, R., Liao, F., & Liu, J. (2024). Copper-Catalyzed Benzothiazolyldifluoroalkylation of Arylidenemalonitriles or para-Quinone Methides. Catalysts, 14(11), 777. https://doi.org/10.3390/catal14110777