
Harnessing Biomass for a Sustainable Future: The Role of Starch and Lignin




Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Origins and Classification
2.1. Starch
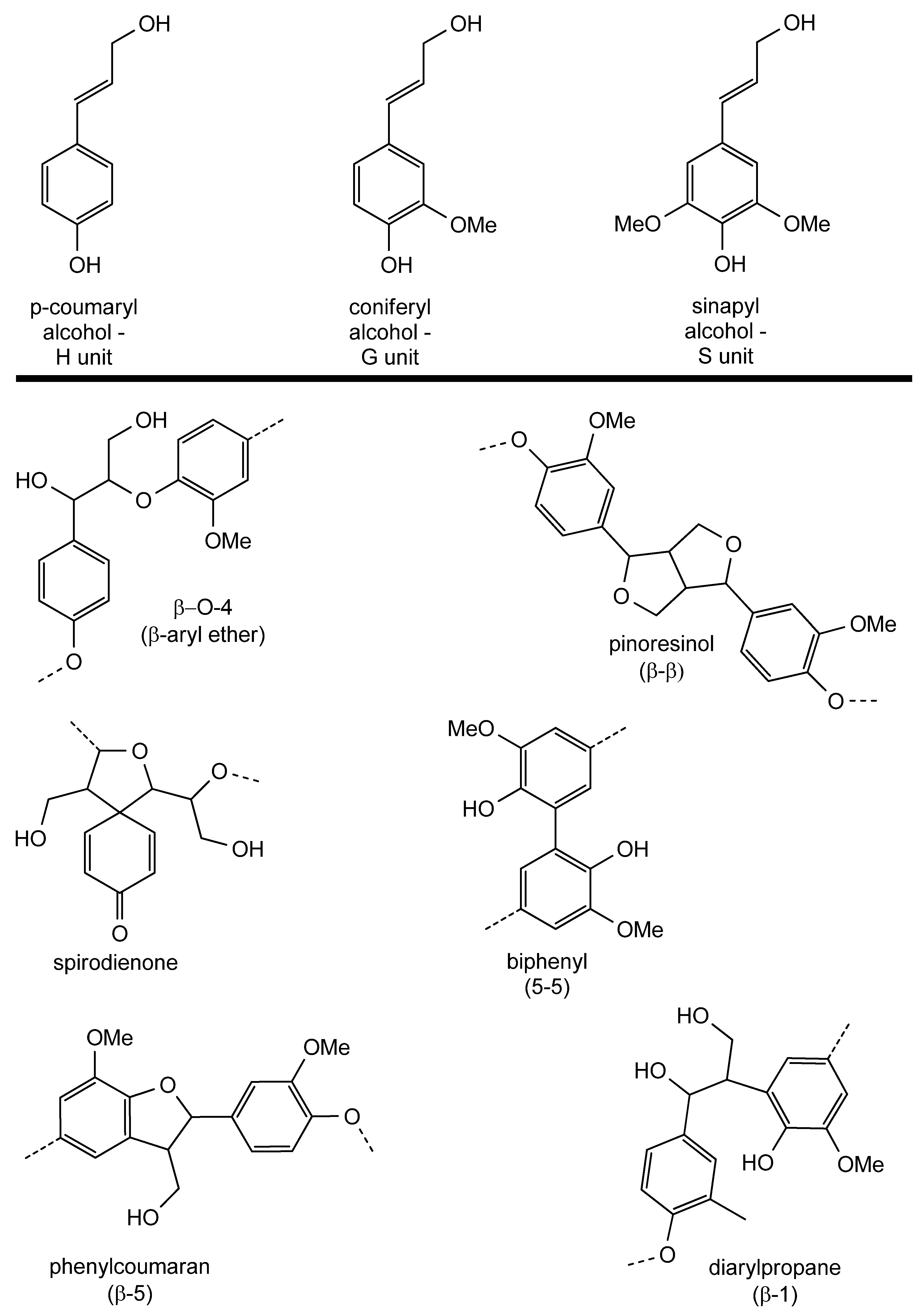
2.2. Lignin
3. Catalysts for Lignin and Starch Valorization
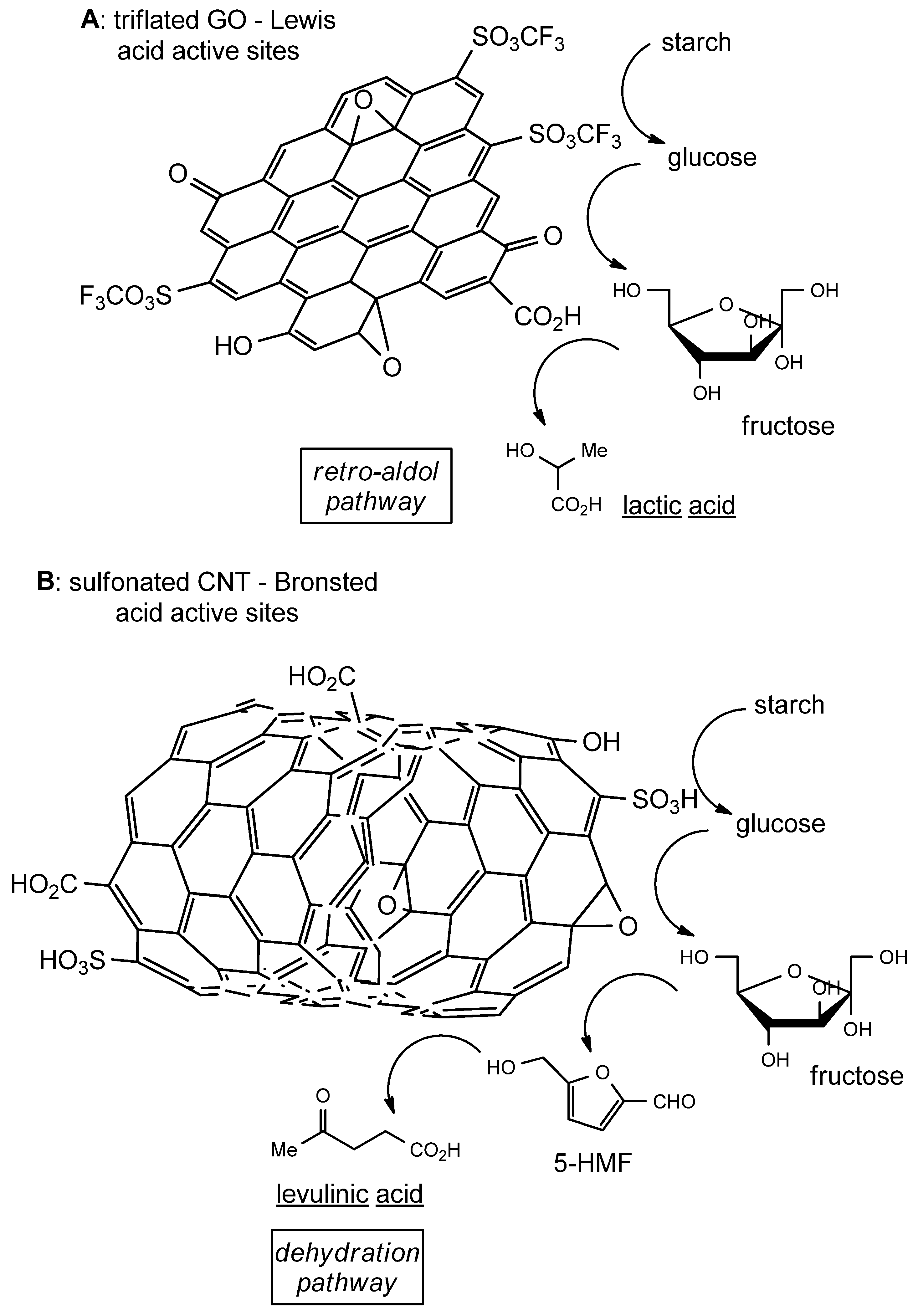
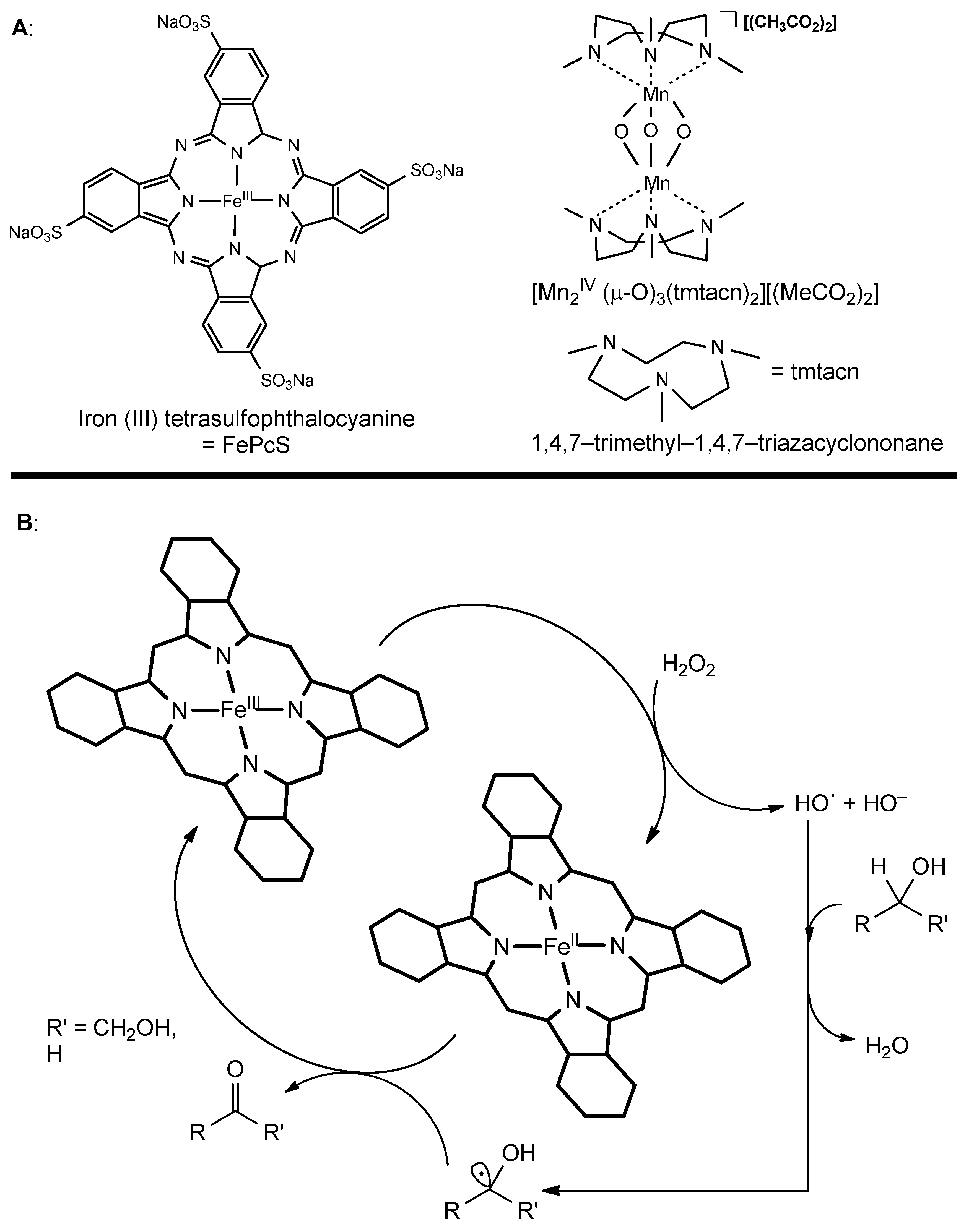
3.1. Catalytic Modifications of Starch
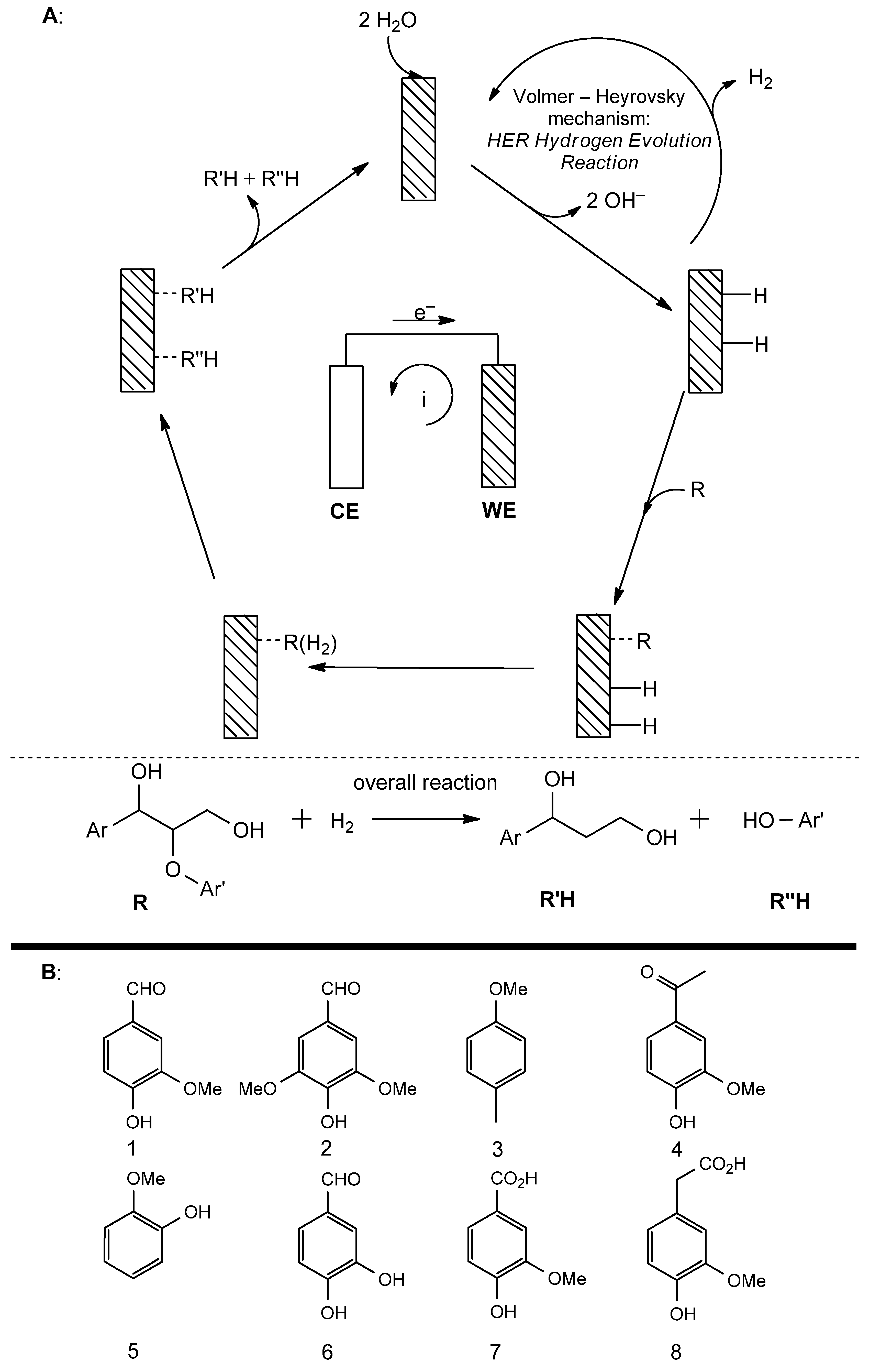
3.2. Catalytic Modifications of Lignin
4. Starch and Lignin-Derived Catalysts
4.1. Starch-Based Catalysts
4.2. Lignin-Based Catalysts
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rhodes, C.J. Plastic Pollution and Potential Solutions. Sci. Prog. 2018, 101, 207–260. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, S.; Aboobacker, V.M.; Seegobin, V.O.; Al Khayat, J.A.; Rangel-Buitrago, N.; Al-Kuwari, H.A.-S.; Sadooni, F.N.; Vethamony, P. History of a Disaster: A Baseline Assessment of the Wakashio Oil Spill on the Coast of Mauritius, Indian Ocean. Mar. Pollut. Bull. 2022, 175, 113330. [Google Scholar] [CrossRef] [PubMed]
- Cazacu, G.; Pascu, M.C.; Profire, L.; Kowarski, A.I.; Mihaes, M.; Vasile, C. Lignin Role in a Complex Polyolefin Blend. Ind. Crops Prod. 2004, 20, 261–273. [Google Scholar] [CrossRef]
- Wang, S.-K.; Yang, K.-X.; Zhu, Y.-R.; Zhu, X.-Y.; Nie, D.-F.; Jiao, N.; Angelidaki, I. One-Step Co-Cultivation and Flocculation of Microalgae with Filamentous Fungi to Valorize Starch Wastewater into High-Value Biomass. Bioresour. Technol. 2022, 361, 127625. [Google Scholar] [CrossRef]
- Beckham, G.T.; Johnson, C.W.; Karp, E.M.; Salvachúa, D.; Vardon, D.R. Opportunities and Challenges in Biological Lignin Valorization. Curr. Opin. Biotechnol. 2016, 42, 40–53. [Google Scholar] [CrossRef]
- Burritt, R.L.; Schaltegger, S. Measuring the (Un-)Sustainability of Industrial Biomass Production and Use. Sustain. Account. Manag. Policy J. 2012, 3, 109–133. [Google Scholar] [CrossRef]
- Cornejo-Ramírez, Y.I.; Martínez-Cruz, O.; Del Toro-Sánchez, C.L.; Wong-Corral, F.J.; Borboa-Flores, J.; Cinco-Moroyoqui, F.J. The Structural Characteristics of Starches and Their Functional Properties. CyTA J. Food 2018, 16, 1003–1017. [Google Scholar] [CrossRef]
- Avancini, S.R.P.; Faccin, G.L.; Vieira, M.A.; Rovaris, A.A.; Podestá, R.; Tramonte, R.; De Souza, N.M.A.; Amante, E.R. Cassava Starch Fermentation Wastewater: Characterization and Preliminary Toxicological Studies. Food Chem. Toxicol. 2007, 45, 2273–2278. [Google Scholar] [CrossRef] [PubMed]
- Goswami, K.P.; Pugazhenthi, G. Treatment of Starch-rich Wastewater Using Fly Ash-based Low-cost Tubular Ceramic Membrane. Environ. Prog. Sustain. Energy 2022, 41, e13906. [Google Scholar] [CrossRef]
- Kringel, D.H.; Dias, A.R.G.; Zavareze, E.D.R.; Gandra, E.A. Fruit Wastes as Promising Sources of Starch: Extraction, Properties, and Applications. Starch Stärke 2020, 72, 1900200. [Google Scholar] [CrossRef]
- Devereux, S.; Shuttleworth, P.S.; Macquarrie, D.J.; Paradisi, F. Isolation and Characterization of Recovered Starch from Industrial Wastewater. J. Polym. Environ. 2011, 19, 971–979. [Google Scholar] [CrossRef]
- Abe, M.M.; Martins, J.R.; Sanvezzo, P.B.; Macedo, J.V.; Branciforti, M.C.; Halley, P.; Botaro, V.R.; Brienzo, M. Advantages and Disadvantages of Bioplastics Production from Starch and Lignocellulosic Components. Polymers 2021, 13, 2484. [Google Scholar] [CrossRef] [PubMed]
- Gökçe, E. Rethinking Sustainability: A Research on Starch Based Bioplastic. J. Sustain. Constr. Mater. Technol. 2018, 3, 249–260. [Google Scholar] [CrossRef]
- Lu, Y.; Ding, Y.; Wu, Q. Simultaneous Saccharification of Cassava Starch and Fermentation of Algae for Biodiesel Production. J. Appl. Phycol. 2011, 23, 115–121. [Google Scholar] [CrossRef]
- Welker, C.; Balasubramanian, V.; Petti, C.; Rai, K.; DeBolt, S.; Mendu, V. Engineering Plant Biomass Lignin Content and Composition for Biofuels and Bioproducts. Energies 2015, 8, 7654–7676. [Google Scholar] [CrossRef]
- Santos, R.B.; Hart, P.; Jameel, H.; Chang, H. Wood Based Lignin Reactions Important to the Biorefinery and Pulp and Paper Industries. BioResources 2013, 8, 1456–1477. [Google Scholar] [CrossRef]
- Bajwa, D.S.; Pourhashem, G.; Ullah, A.H.; Bajwa, S.G. A Concise Review of Current Lignin Production, Applications, Products and Their Environmental Impact. Ind. Crops Prod. 2019, 139, 111526. [Google Scholar] [CrossRef]
- Sun, R. Lignin Source and Structural Characterization. ChemSusChem 2020, 13, 4385–4393. [Google Scholar] [CrossRef]
- Ariyanta, H.A.; Sari, F.P.; Sohail, A.; Restu, W.K.; Septiyanti, M.; Aryana, N.; Fatriasari, W.; Kumar, A. Current Roles of Lignin for the Agroindustry: Applications, Challenges, and Opportunities. Int. J. Biol. Macromol. 2023, 240, 124523. [Google Scholar] [CrossRef]
- Wang, Z.; Deuss, P.J. The Isolation of Lignin with Native-like Structure. Biotechnol. Adv. 2023, 68, 108230. [Google Scholar] [CrossRef]
- Melro, E.; Filipe, A.; Sousa, D.; Medronho, B.; Romano, A. Revisiting Lignin: A Tour through Its Structural Features, Characterization Methods and Applications. New J. Chem. 2021, 45, 6986–7013. [Google Scholar] [CrossRef]
- Chakar, F.S.; Ragauskas, A.J. Review of Current and Future Softwood Kraft Lignin Process Chemistry. Ind. Crops Prod. 2004, 20, 131–141. [Google Scholar] [CrossRef]
- Zhou, H.; Lou, H.; Yang, D.; Zhu, J.Y.; Qiu, X. Lignosulfonate To Enhance Enzymatic Saccharification of Lignocelluloses: Role of Molecular Weight and Substrate Lignin. Ind. Eng. Chem. Res. 2013, 52, 8464–8470. [Google Scholar] [CrossRef]
- Aro, T.; Fatehi, P. Production and Application of Lignosulfonates and Sulfonated Lignin. ChemSusChem 2017, 10, 1861–1877. [Google Scholar] [CrossRef]
- José Borges Gomes, F.; De Souza, R.E.; Brito, E.O.; Costa Lelis, R.C. A Review on Lignin Sources and Uses. J. Appl. Biotechnol. Bioeng. 2020, 7, 100–105. [Google Scholar] [CrossRef]
- De La Torre, M.J.; Moral, A.; Hernández, M.D.; Cabeza, E.; Tijero, A. Organosolv Lignin for Biofuel. Ind. Crops Prod. 2013, 45, 58–63. [Google Scholar] [CrossRef]
- Rabelo, S.C.; Nakasu, P.Y.S.; Scopel, E.; Araújo, M.F.; Cardoso, L.H.; Costa, A.C.D. Organosolv Pretreatment for Biorefineries: Current Status, Perspectives, and Challenges. Bioresour. Technol. 2023, 369, 128331. [Google Scholar] [CrossRef]
- Li, J.; Gellerstedt, G.; Toven, K. Steam Explosion Lignins; Their Extraction, Structure and Potential as Feedstock for Biodiesel and Chemicals. Bioresour. Technol. 2009, 100, 2556–2561. [Google Scholar] [CrossRef]
- Hu, Z.; Yeh, T.-F.; Chang, H.; Matsumoto, Y.; Kadla, J.F. Elucidation of the Structure of Cellulolytic Enzyme Lignin. Holzforschung 2006, 60, 389–397. [Google Scholar] [CrossRef]
- Vanier, N.L.; El Halal, S.L.M.; Dias, A.R.G.; Da Rosa Zavareze, E. Molecular Structure, Functionality and Applications of Oxidized Starches: A Review. Food Chem. 2017, 221, 1546–1559. [Google Scholar] [CrossRef]
- Podolean, I.; Anita, F.; García, H.; Parvulescu, V.I.; Coman, S.M. Efficient Magnetic Recoverable Acid-Functionalized-Carbon Catalysts for Starch Valorization to Multiple Bio-Chemicals. Catal. Today 2017, 279, 45–55. [Google Scholar] [CrossRef]
- Dookheh, M.; Najafi Chermahini, A. Starch Valorization: Direct Conversion of Starch to Hexyl Levulinate over SO4/ZrO2-KIT5 Composite. Int. J. Biol. Macromol. 2024, 262, 130093. [Google Scholar] [CrossRef] [PubMed]
- Lerf, A.; He, H.; Forster, M.; Klinowski, J. Structure of Graphite Oxide Revisited. J. Phys. Chem. B 1998, 102, 4477–4482. [Google Scholar] [CrossRef]
- Guo, H.; Bian, K.; Ding, S.; Cai, H.; Zhang, H.; Chen, X.; Wang, C.; Yao, S.; Chen, X. Efficient Utilization of Biomass Hydrolysis Residues in Preparing a Metal/Acid Bifunctional Catalyst for Butyl Levulinate Hydrogenation to γ-Valerolactone. Ind. Eng. Chem. Res. 2023, 62, 5502–5514. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, X.; Dong, M.; Wu, Y.; Zheng, G.; Huang, J.; Guan, X.; Zheng, X. MCM-41 Immobilized 12-Silicotungstic Acid Mesoporous Materials: Structural and Catalytic Properties for Esterification of Levulinic Acid and Oleic Acid. J. Taiwan Inst. Chem. Eng. 2016, 61, 147–155. [Google Scholar] [CrossRef]
- Appaturi, J.N.; Selvaraj, M.; Rajabathar, J.R.; Khoerunnisa, F.; Rigolet, S.; Daou, T.J.; Maireles-Torres, P.; El-Bahy, S.M.; El-Bahy, Z.M.; Ng, E.-P. Highly Efficient Non-Microwave Instant Heating Synthesis of Hexyl Levulinate Fuel Additive Enhanced by Sulfated Nanosilica Catalyst. Microporous Mesoporous Mater. 2022, 331, 111645. [Google Scholar] [CrossRef]
- Parovuori, P.; Hamunen, A.; Forssell, P.; Autio, K.; Poutanen, K. Oxidation of Potato Starch by Hydrogen Peroxide. Starch Stärke 1995, 47, 19–23. [Google Scholar] [CrossRef]
- Sangseethong, K.; Termvejsayanon, N.; Sriroth, K. Characterization of Physicochemical Properties of Hypochlorite- and Peroxide-Oxidized Cassava Starches. Carbohydr. Polym. 2010, 82, 446–453. [Google Scholar] [CrossRef]
- Maniglia, B.C.; Castanha, N.; Le-Bail, P.; Le-Bail, A.; Augusto, P.E.D. Starch Modification through Environmentally Friendly Alternatives: A Review. Crit. Rev. Food Sci. Nutr. 2021, 61, 2482–2505. [Google Scholar] [CrossRef]
- Zhang, Y.-R.; Wang, X.-L.; Zhao, G.-M.; Wang, Y.-Z. Preparation and Properties of Oxidized Starch with High Degree of Oxidation. Carbohydr. Polym. 2012, 87, 2554–2562. [Google Scholar] [CrossRef]
- Floor, M.; Schenk, K.M.; Kieboom, A.P.G.; Van Bekkum, H. Oxidation of Maltodextrins and Starch by the System Tungstate-Hydrogen Peroxide. Starch Stärke 1989, 41, 303–309. [Google Scholar] [CrossRef]
- Sorokin, A.B.; Kachkarova-Sorokina, S.L.; Donzé, C.; Pinel, C.; Gallezot, P. From Native Starch to Hydrophilic and Hydrophobic Products: A Catalytic Approach. Top. Catal. 2004, 27, 67–76. [Google Scholar] [CrossRef]
- Wang, H.; Poya, Y.; Chen, X.; Jia, T.; Wang, X.; Shi, J. Hydrogen Peroxide as an Oxidant in Starch Oxidation Using Molybdovanadophosphate for Producing a High Carboxylic Content. RSC Adv. 2015, 5, 45725–45730. [Google Scholar] [CrossRef]
- Broekman, J.O.P.; Genuino, H.C.; Heeres, H.J.; Brinksma, J.; Wielema, T.; Deuss, P.J. Benign Catalytic Oxidation of Potato Starch Using a Homogeneous Binuclear Manganese Catalyst and Hydrogen Peroxide. Catal. Sci. Technol. 2023, 13, 1233–1243. [Google Scholar] [CrossRef]
- Zhao, Y.; Deng, L.; Liao, B.; Fu, Y.; Guo, Q.-X. Aromatics Production via Catalytic Pyrolysis of Pyrolytic Lignins from Bio-Oil. Energy Fuels 2010, 24, 5735–5740. [Google Scholar] [CrossRef]
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The Catalytic Valorization of Lignin for the Production of Renewable Chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef]
- Rahman, M.M.; Liu, R.; Cai, J. Catalytic Fast Pyrolysis of Biomass over Zeolites for High Quality Bio-Oil—A Review. Fuel Process. Technol. 2018, 180, 32–46. [Google Scholar] [CrossRef]
- Carlson, T.R.; Jae, J.; Lin, Y.-C.; Tompsett, G.A.; Huber, G.W. Catalytic Fast Pyrolysis of Glucose with HZSM-5: The Combined Homogeneous and Heterogeneous Reactions. J. Catal. 2010, 270, 110–124. [Google Scholar] [CrossRef]
- Aho, A.; Kumar, N.; Eränen, K.; Salmi, T.; Hupa, M.; Murzin, D.Y. Catalytic Pyrolysis of Woody Biomass in a Fluidized Bed Reactor: Influence of the Zeolite Structure. Fuel 2008, 87, 2493–2501. [Google Scholar] [CrossRef]
- Park, H.J.; Heo, H.S.; Jeon, J.-K.; Kim, J.; Ryoo, R.; Jeong, K.-E.; Park, Y.-K. Highly Valuable Chemicals Production from Catalytic Upgrading of Radiata Pine Sawdust-Derived Pyrolytic Vapors over Mesoporous MFI Zeolites. Appl. Catal. B Environ. 2010, 95, 365–373. [Google Scholar] [CrossRef]
- Ben, H.; Ragauskas, A.J. Influence of Si/Al Ratio of ZSM-5 Zeolite on the Properties of Lignin Pyrolysis Products. ACS Sustain. Chem. Eng. 2013, 1, 316–324. [Google Scholar] [CrossRef]
- Widayatno, W.B.; Guan, G.; Rizkiana, J.; Du, X.; Hao, X.; Zhang, Z.; Abudula, A. Selective Catalytic Conversion of Bio-Oil over High-Silica Zeolites. Bioresour. Technol. 2015, 179, 518–523. [Google Scholar] [CrossRef]
- Ambursa, M.M.; Juan, J.C.; Yahaya, Y.; Taufiq-Yap, Y.H.; Lin, Y.-C.; Lee, H.V. A Review on Catalytic Hydrodeoxygenation of Lignin to Transportation Fuels by Using Nickel-Based Catalysts. Renew. Sustain. Energy Rev. 2021, 138, 110667. [Google Scholar] [CrossRef]
- Bjelić, A.; Likozar, B.; Grilc, M. Scaling of Lignin Monomer Hydrogenation, Hydrodeoxygenation and Hydrocracking Reaction Micro-Kinetics over Solid Metal/Acid Catalysts to Aromatic Oligomers. Chem. Eng. J. 2020, 399, 125712. [Google Scholar] [CrossRef]
- Mahdavi, B.; Lafrance, A.; Martel, A.; Lessard, J.; Me’Nard, H.; Brossard, L. Electrocatalytic hydrogenolysis of lignin model dimers at Raney nickel electrodes. J. Appl. Electrochem. 1997, 27, 605–611. [Google Scholar] [CrossRef]
- Robin, D.; Comtois, M.; Martel, A.; Lemieux, R.; Cheong, A.K.; Belot, G.; Lessard, J. The Electrocatalytic Hydrogenation of Fused PolyCyclic Aromatic Compounds at Raney Nickel Electrodes: The Influence of Catalyst Activation and Electrolysis Conditions. Can. J. Chem. 1990, 68, 1218–1227. [Google Scholar] [CrossRef]
- Mahdavi, B.; Chambrion, P.; Binette, J.; Martel, E.; Lessard, J. Electrocatalytic Hydrogenation of Conjugated Enones on Nickel Boride, Nickel, and Raney Nickel Electrodes. Can. J. Chem. 1995, 73, 846–852. [Google Scholar] [CrossRef]
- Mahdavi, B.; Chapuzet, J.M.; Lessard, J. The Electrocatalytic Hydrogenation of Phenanthrene at Raney Nickel Electrodes: The Effect of Periodic Current Control. Electrochim. Acta 1993, 38, 1377–1380. [Google Scholar] [CrossRef]
- Wijaya, Y.P.; Smith, K.J.; Kim, C.S.; Gyenge, E.L. Electrocatalytic Hydrogenation and Depolymerization Pathways for Lignin Valorization: Toward Mild Synthesis of Chemicals and Fuels from Biomass. Green Chem. 2020, 22, 7233–7264. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, L.; Chen, Y.; Li, G.; Li, H.; Tang, Y.; Wan, P. Electrochemical Depolymerization of Lignin into Renewable Aromatic Compounds in a Non-Diaphragm Electrolytic Cell. RSC Adv. 2014, 4, 29917. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, F.; Liu, Z.; Yuan, L.; Li, G. Electrocatalytic Degradation of Aspen Lignin over Pb/PbO2 Electrode in Alkali Solution. Catal. Commun. 2015, 67, 49–53. [Google Scholar] [CrossRef]
- Di Marino, D.; Stöckmann, D.; Kriescher, S.; Stiefel, S.; Wessling, M. Electrochemical Depolymerisation of Lignin in a Deep Eutectic Solvent. Green Chem. 2016, 18, 6021–6028. [Google Scholar] [CrossRef]
- Sun, Z.; Fridrich, B.; De Santi, A.; Elangovan, S.; Barta, K. Bright Side of Lignin Depolymerization: Toward New Platform Chemicals. Chem. Rev. 2018, 118, 614–678. [Google Scholar] [CrossRef]
- Sales, F.G.; Maranhão, L.C.A.; Filho, N.M.L.; Abreu, C.A.M. Experimental Evaluation and Continuous Catalytic Process for Fine Aldehyde Production from Lignin. Chem. Eng. Sci. 2007, 62, 5386–5391. [Google Scholar] [CrossRef]
- Pourjafar, S.; Kreft, J.; Bilek, H.; Kozliak, E.; Seames, W. Exploring Large Pore Size Alumina and Silica-Alumina Based Catalysts for Decomposition of Lignin. AIMS Energy 2018, 6, 993–1008. [Google Scholar] [CrossRef]
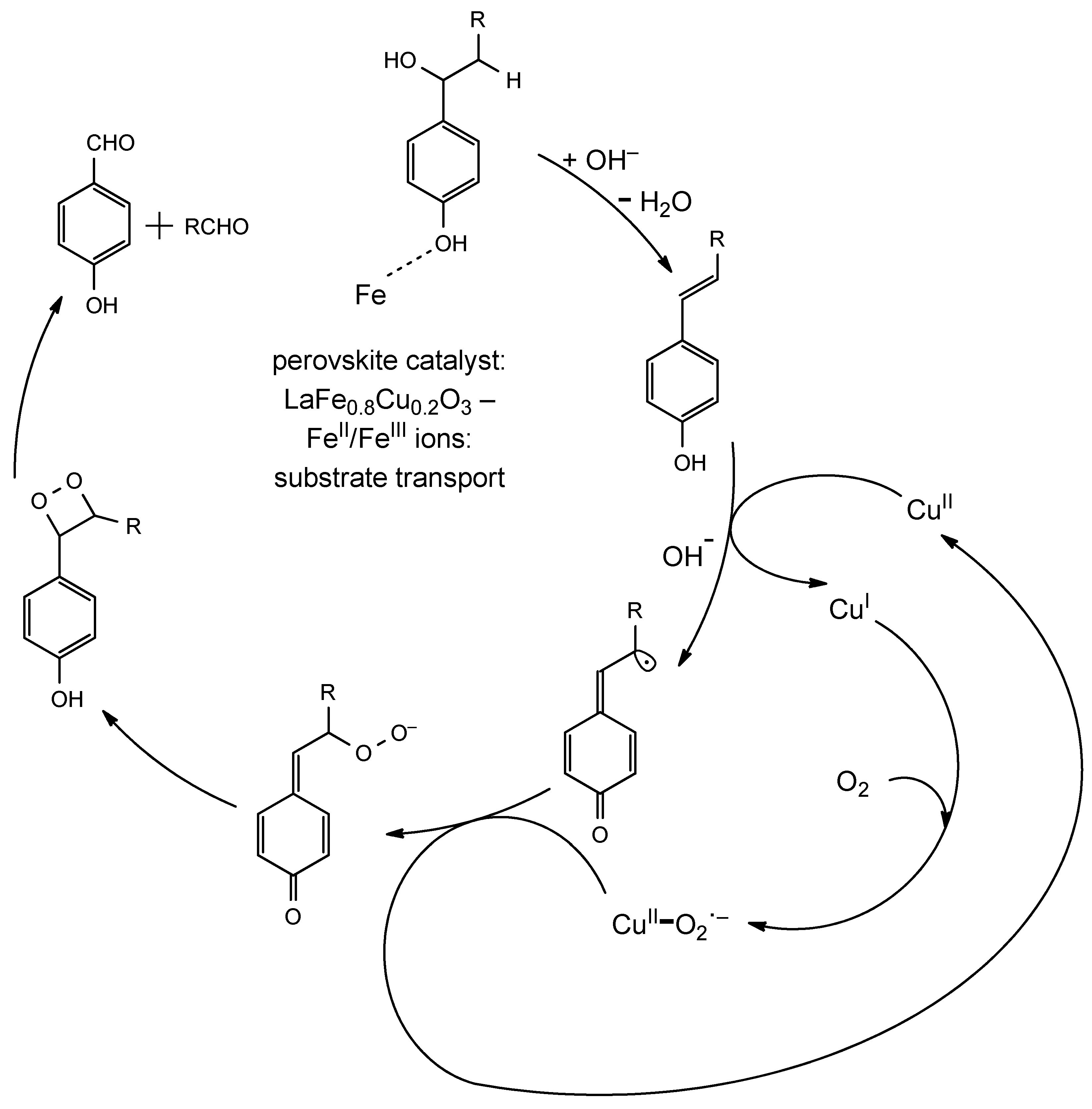
- Zhang, J.; Deng, H.; Lin, L. Wet Aerobic Oxidation of Lignin into Aromatic Aldehydes Catalysed by a Perovskite-Type Oxide: LaFe1−xCuxO3 (X = 0, 0.1, 0.2). Molecules 2009, 14, 2747–2757. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Xu, A.; Du, H.; Sun, C.; Li, C. Removal of Salicylic Acid on Perovskite-Type Oxide LaFeO3 Catalyst in Catalytic Wet Air Oxidation Process. J. Hazard. Mater. 2007, 139, 86–92. [Google Scholar] [CrossRef]
- Royer, S.; Levasseur, B.; Alamdari, H.; Barbier, J., Jr.; Duprez, D.; Kaliaguine, S. Mechanism of Stearic Acid Oxidation over Nanocrystalline La1−xA′xBO3La1−xA′xBO3 (A′ = Sr, Ce; B = Co, Mn): The Role of Oxygen Mobility. Appl. Catal. B Environ. 2008, 80, 51–61. [Google Scholar] [CrossRef]
- Fortuny, A. Bimetallic Catalysts for Continuous Catalytic Wet Air Oxidation of Phenol. J. Hazard. Mater. 1999, 64, 181–193. [Google Scholar] [CrossRef]
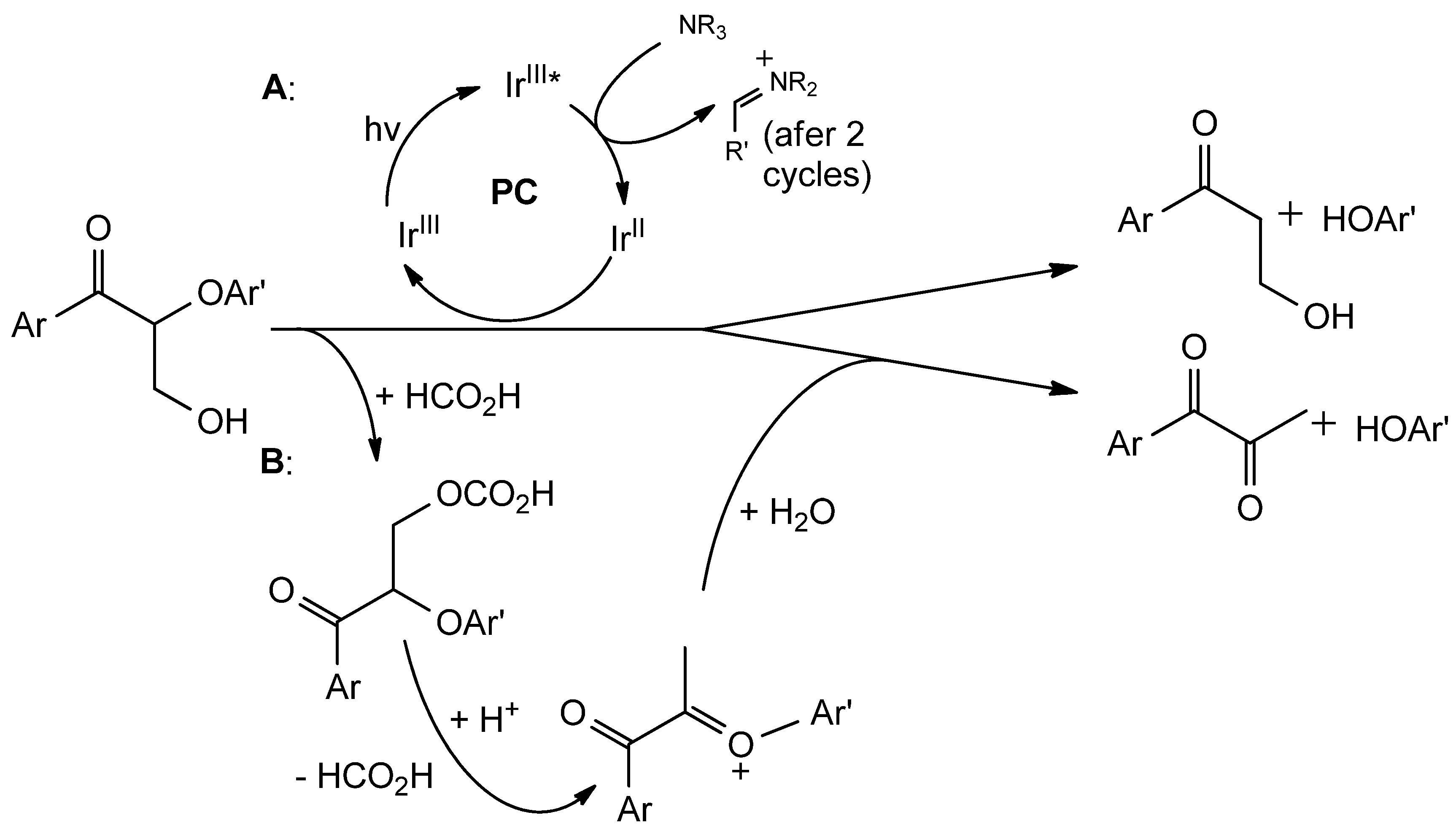
- Nguyen, J.D.; Matsuura, B.S.; Stephenson, C.R.J. A Photochemical Strategy for Lignin Degradation at Room Temperature. J. Am. Chem. Soc. 2014, 136, 1218–1221. [Google Scholar] [CrossRef]
- Lancefield, C.S.; Ojo, O.S.; Tran, F.; Westwood, N.J. Isolation of Functionalized Phenolic Monomers through Selective Oxidation and C-O Bond Cleavage of the β-O-4 Linkages in Lignin. Angew. Chem. 2015, 127, 260–264. [Google Scholar] [CrossRef]
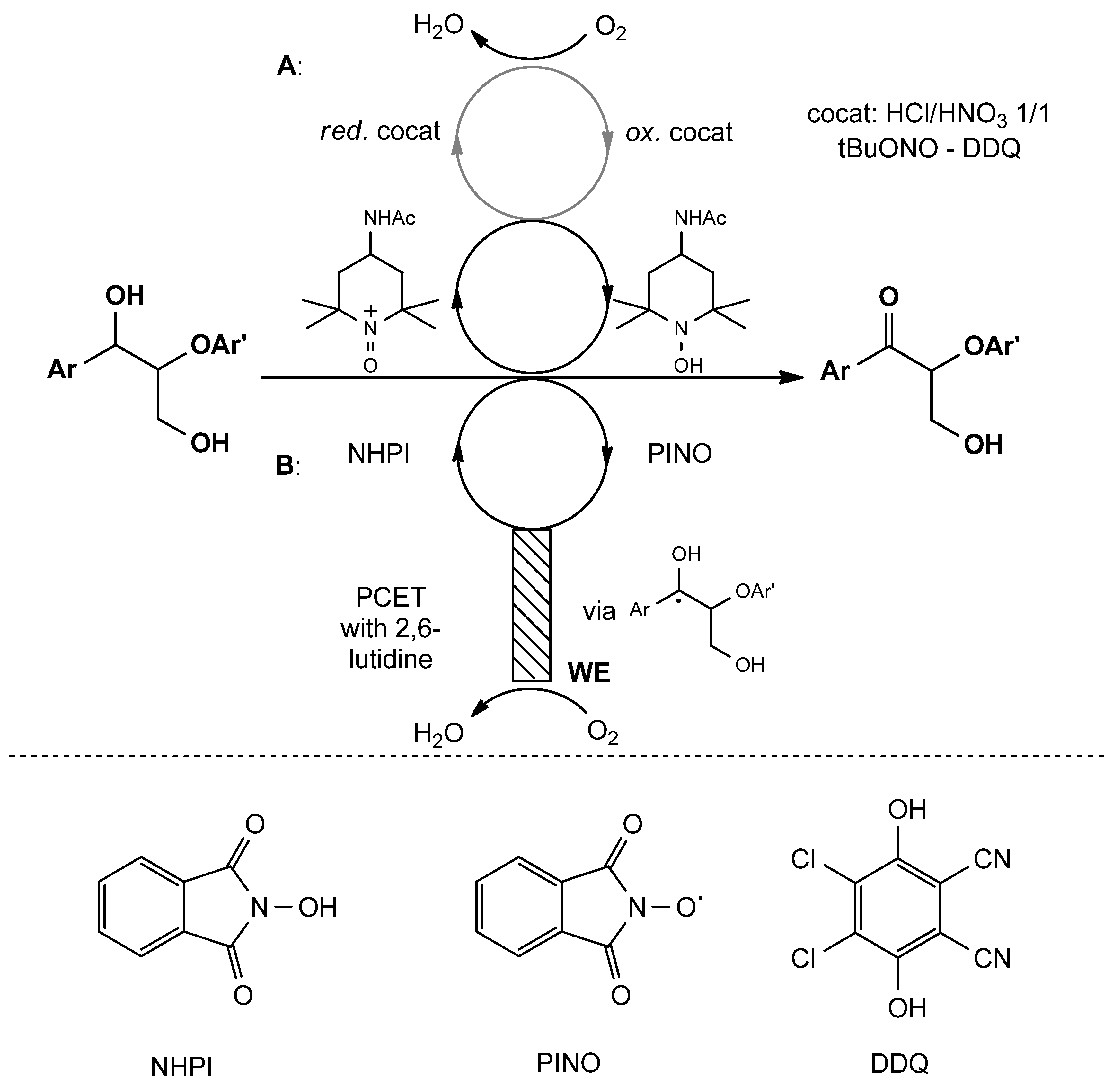
- Rahimi, A.; Azarpira, A.; Kim, H.; Ralph, J.; Stahl, S.S. Chemoselective Metal-Free Aerobic Alcohol Oxidation in Lignin. J. Am. Chem. Soc. 2013, 135, 6415–6418. [Google Scholar] [CrossRef] [PubMed]
- Bosque, I.; Magallanes, G.; Rigoulet, M.; Kärkäs, M.D.; Stephenson, C.R.J. Redox Catalysis Facilitates Lignin Depolymerization. ACS Cent. Sci. 2017, 3, 621–628. [Google Scholar] [CrossRef]
- Rahimi, A.; Ulbrich, A.; Coon, J.J.; Stahl, S.S. Formic-Acid-Induced Depolymerization of Oxidized Lignin to Aromatics. Nature 2014, 515, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.T.; Jusoh, M.; Zakaria, Z.Y. Starch-Derived Solid Acid Catalyst for Biodiesel Production: A Mini Review. Chem. Eng. Trans. 2021, 89, 487–492. [Google Scholar] [CrossRef]
- Lou, W.-Y.; Zong, M.-H.; Duan, Z.-Q. Efficient Production of Biodiesel from High Free Fatty Acid-Containing Waste Oils Using Various Carbohydrate-Derived Solid Acid Catalysts. Bioresour. Technol. 2008, 99, 8752–8758. [Google Scholar] [CrossRef]
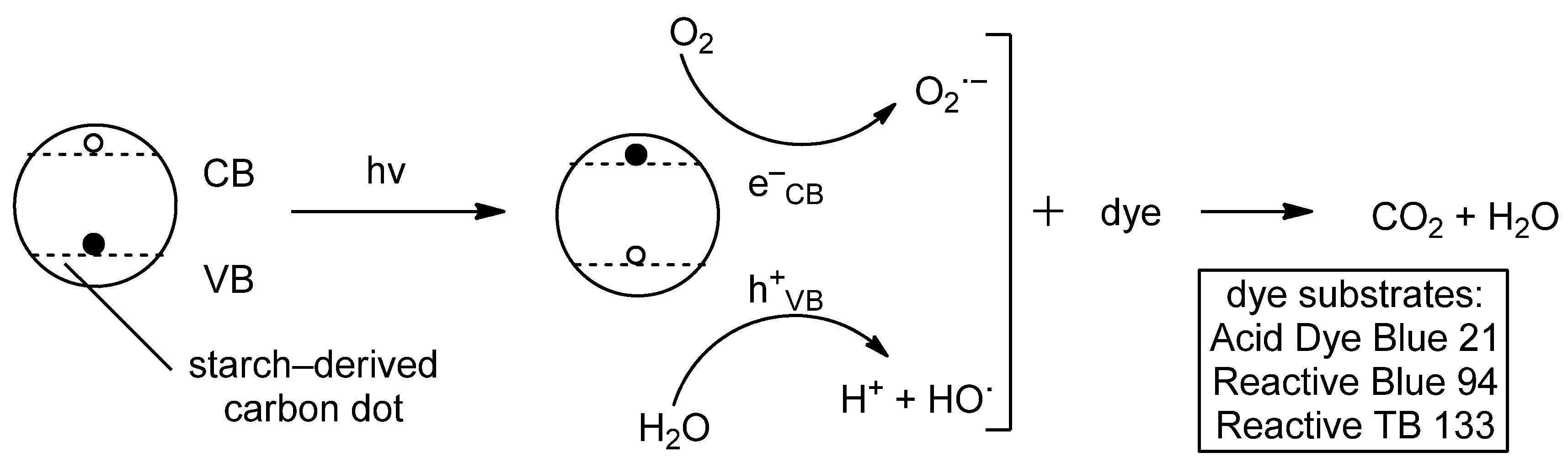
- Sheshmani, S.; Mardali, M.; Shokrollahzadeh, S.; Bide, Y. Starch-Derived Carbon Quantum Dots: Unveiling Structural Insights and Photocatalytic Potential as a Bio-Sourced Metal-Free Semiconductor. Int. J. Biol. Macromol. 2024, 271, 132535. [Google Scholar] [CrossRef] [PubMed]
- Safari, J.; Aftabi, P.; Ahmadzadeh, M.; Sadeghi, M.; Zarnegar, Z. Sulfonated Starch Nanoparticles: An Effective, Heterogeneous and Bio-Based Catalyst for Synthesis of 14-Aryl-14-H-Dibenzo[a,j]Xanthenes. J. Mol. Struct. 2017, 1142, 33–39. [Google Scholar] [CrossRef]
- Patra, D.; Panja, S.; Saha, A. C–C Cross-Coupling Reactions of Organosilanes with Terminal Alkenes and Allylic Acetates Using PdII Catalyst Supported on Starch Coated Magnetic Nanoparticles. Eur. J. Org. Chem. 2020, 2020, 878–883. [Google Scholar] [CrossRef]
- Dharmendra, D.; Chundawat, P.; Vyas, Y.; Ameta, C. Ultrasound-Assisted Efficient Synthesis and Antimicrobial Evaluation of Pyrazolopyranopyrimidine Derivatives Using Starch Functionalized Magnetite Nanoparticles as a Green Biocatalyst in Water. J. Chem. Sci. 2022, 134, 47. [Google Scholar] [CrossRef]
- Dohendou, M.; Pakzad, K.; Nezafat, Z.; Nasrollahzadeh, M.; Dekamin, M.G. Progresses in Chitin, Chitosan, Starch, Cellulose, Pectin, Alginate, Gelatin and Gum Based (Nano)Catalysts for the Heck Coupling Reactions: A Review. Int. J. Biol. Macromol. 2021, 192, 771–819. [Google Scholar] [CrossRef] [PubMed]
- Arghan, M.; Koukabi, N.; Kolvari, E. Magnetic Apple Seed Starch Functionalized with 2,2′-furil as a Green Host for Cobalt Nanoparticles: Highly Active and Reusable Catalyst for Mizoroki–Heck and the Suzuki–Miyaura Reactions. Appl. Organomet. Chem. 2019, 33, e5075. [Google Scholar] [CrossRef]
- Bahadorikhalili, S.; Ansari, S.; Hamedifar, H.; Mahdavi, M. The Use of Magnetic Starch as a Support for an Ionic Liquid-β-Cyclodextrin Based Catalyst for the Synthesis of Imidazothiadiazolamine Derivatives. Int. J. Biol. Macromol. 2019, 135, 453–461. [Google Scholar] [CrossRef]
- Pandit, N.; Shah, K.; Agrawal, N.; Upmanyu, N.; Shrivastava, S.K.; Mishra, P. Synthesis, Characterization and Biological Evaluation of Some Novel Fluoroquinolones. Med. Chem. Res. 2016, 25, 843–851. [Google Scholar] [CrossRef]
- Lee, A.-L. Enantioselective Oxidative Boron Heck Reactions. Org. Biomol. Chem. 2016, 14, 5357–5366. [Google Scholar] [CrossRef]
- Ma, L.; Zhan, X. Dye-Sensitized Solar Cells (DSSCs). In Organic Optoelectronics; Hu, W., Ed.; Wiley: Hoboken, NJ, USA, 2013; pp. 437–465. ISBN 978-3-527-32968-7. [Google Scholar]
- Mariotti, N.; Bonomo, M.; Fagiolari, L.; Barbero, N.; Gerbaldi, C.; Bella, F.; Barolo, C. Recent Advances in Eco-Friendly and Cost-Effective Materials towards Sustainable Dye-Sensitized Solar Cells. Green Chem. 2020, 22, 7168–7218. [Google Scholar] [CrossRef]
- Boschloo, G.; Hagfeldt, A. Characteristics of the Iodide/Triiodide Redox Mediator in Dye-Sensitized Solar Cells. Acc. Chem. Res. 2009, 42, 1819–1826. [Google Scholar] [CrossRef]
- De Haro, J.C.; Tatsi, E.; Fagiolari, L.; Bonomo, M.; Barolo, C.; Turri, S.; Bella, F.; Griffini, G. Lignin-Based Polymer Electrolyte Membranes for Sustainable Aqueous Dye-Sensitized Solar Cells. ACS Sustain. Chem. Eng. 2021, 9, 8550–8560. [Google Scholar] [CrossRef]
- Chen, W.-J.; Zhao, C.-X.; Li, B.-Q.; Yuan, T.-Q.; Zhang, Q. Lignin-Derived Materials and Their Applications in Rechargeable Batteries. Green Chem. 2022, 24, 565–584. [Google Scholar] [CrossRef]
- Zhang, W.; Yin, J.; Lin, Z.; Lin, H.; Lu, H.; Wang, Y.; Huang, W. Facile Preparation of 3D Hierarchical Porous Carbon from Lignin for the Anode Material in Lithium Ion Battery with High Rate Performance. Electrochim. Acta 2015, 176, 1136–1142. [Google Scholar] [CrossRef]
- Xi, Y.; Wang, Y.; Yang, D.; Zhang, Z.; Liu, W.; Li, Q.; Qiu, X. K2CO3 Activation Enhancing the Graphitization of Porous Lignin Carbon Derived from Enzymatic Hydrolysis Lignin for High Performance Lithium-Ion Storage. J. Alloys Compd. 2019, 785, 706–714. [Google Scholar] [CrossRef]
- Xi, Y.; Huang, S.; Yang, D.; Qiu, X.; Su, H.; Yi, C.; Li, Q. Hierarchical Porous Carbon Derived from the Gas-Exfoliation Activation of Lignin for High-Energy Lithium-Ion Batteries. Green Chem. 2020, 22, 4321–4330. [Google Scholar] [CrossRef]
- Wang, H.; Shao, Y.; Mei, S.; Lu, Y.; Zhang, M.; Sun, J.; Matyjaszewski, K.; Antonietti, M.; Yuan, J. Polymer-Derived Heteroatom-Doped Porous Carbon Materials. Chem. Rev. 2020, 120, 9363–9419. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Yu, L.; Lou, X.W. (David) Nanostructured Conversion-Type Anode Materials for Advanced Lithium-Ion Batteries. Chem 2018, 4, 972–996. [Google Scholar] [CrossRef]
- Chen, F.; Wu, L.; Zhou, Z.; Ju, J.; Zhao, Z.; Zhong, M.; Kuang, T. MoS2 Decorated Lignin-Derived Hierarchical Mesoporous Carbon Hybrid Nanospheres with Exceptional Li-Ion Battery Cycle Stability. Chin. Chem. Lett. 2019, 30, 197–202. [Google Scholar] [CrossRef]
- Zhou, Z.; Chen, F.; Kuang, T.; Chang, L.; Yang, J.; Fan, P.; Zhao, Z.; Zhong, M. Lignin-Derived Hierarchical Mesoporous Carbon and NiO Hybrid Nanospheres with Exceptional Li-Ion Battery and Pseudocapacitive Properties. Electrochim. Acta 2018, 274, 288–297. [Google Scholar] [CrossRef]
- Zhao, Y.; Wen, M.; He, C.; Liu, C.; Li, Z.; Liu, Y. Preparation of Graphene by Catalytic Pyrolysis of Lignin and Its Electrochemical Properties. Mater. Lett. 2020, 274, 128047. [Google Scholar] [CrossRef]
- Reddy, K.R.; Jyothi, M.S.; Raghu, A.V.; Sadhu, V.; Naveen, S.; Aminabhavi, T.M. Nanocarbons-Supported and Polymers-Supported Titanium Dioxide Nanostructures as Efficient Photocatalysts for Remediation of Contaminated Wastewater and Hydrogen Production. In Nanophotocatalysis and Environmental Applications; Inamuddin, Asiri, A.M., Lichtfouse, E., Eds.; Environmental Chemistry for a Sustainable World; Springer International Publishing: Cham, Switzerland, 2020; Volume 30, pp. 139–169. ISBN 978-3-030-12618-6. [Google Scholar]
- Ramesh Reddy, N.; Bhargav, U.; Mamatha Kumari, M.; Cheralathan, K.K.; Sakar, M. Review on the Interface Engineering in the Carbonaceous Titania for the Improved Photocatalytic Hydrogen Production. Int. J. Hydrogen Energy 2020, 45, 7584–7615. [Google Scholar] [CrossRef]
- Zhang, Q.; Bao, N.; Wang, X.; Hu, X.; Miao, X.; Chaker, M.; Ma, D. Advanced Fabrication of Chemically Bonded Graphene/TiO2 Continuous Fibers with Enhanced Broadband Photocatalytic Properties and Involved Mechanisms Exploration. Sci. Rep. 2016, 6, 38066. [Google Scholar] [CrossRef] [PubMed]
- Vadivel, D.; Branciforti, D.S.; Speltini, A.; Sturini, M.; Bellani, V.; Malaichamy, I.; Dondi, D. Pyrolytic Formation of TiO2/Carbon Nanocomposite from Kraft Lignin: Characterization and Photoactivities. Catalysts 2020, 10, 270. [Google Scholar] [CrossRef]
- Speltini, A.; Sturini, M.; Maraschi, F.; Mandelli, E.; Vadivel, D.; Dondi, D.; Profumo, A. Preparation of Silica-Supported Carbon by Kraft Lignin Pyrolysis, and Its Use in Solid-Phase Extraction of Fluoroquinolones from Environmental Waters. Microchim. Acta 2016, 183, 2241–2249. [Google Scholar] [CrossRef]
- Dondi, D.; Zeffiro, A.; Speltini, A.; Tomasi, C.; Vadivel, D.; Buttafava, A. The Role of Inorganic Sulfur Compounds in the Pyrolysis of Kraft Lignin. J. Anal. Appl. Pyrolysis 2014, 107, 53–58. [Google Scholar] [CrossRef]
- Vadivel, D.; Suryakumar, S.; Casella, C.; Speltini, A.; Dondi, D. Advancements in Materials Science and Photocatalysts for Sustainable Development. Catalysts 2024, 14, 378. [Google Scholar] [CrossRef]
- Dondi, D.; Vadivel, D. Preparation of Catalysts from Renewable and Waste Materials. Catalysts 2020, 10, 662. [Google Scholar] [CrossRef]
- Vadivel, D.; Malaichamy, I. Pyrolytic Formation and Photoactivity of Reactive Oxygen Species in a SiO2/Carbon Nanocomposite from Kraft Lignin. F1000Research 2018, 7, 1574. [Google Scholar] [CrossRef]
- Vadivel, D.; Speltini, A.; Zeffiro, A.; Bellani, V.; Pezzini, S.; Buttafava, A.; Dondi, D. Reactive Carbons from Kraft Lignin Pyrolysis: Stabilization of Peroxyl Radicals at Carbon/Silica Interface. J. Anal. Appl. Pyrolysis 2017, 128, 346–352. [Google Scholar] [CrossRef]
- Wang, H.; Qiu, X.; Zhong, R.; Fu, F.; Qian, Y.; Yang, D. One-Pot in-Situ Preparation of a Lignin-Based Carbon/ZnO Nanocomposite with Excellent Photocatalytic Performance. Mater. Chem. Phys. 2017, 199, 193–202. [Google Scholar] [CrossRef]
- Donar, Y.O.; Bilge, S.; Sinağ, A. Utilisation of Lignin as a Model Biomass Component for Preparing a Highly Active Photocatalyst under UV and Visible Light. Mater. Sci. Semicond. Process. 2020, 118, 105151. [Google Scholar] [CrossRef]
- Mabuti, L.A.; Manding, I.K.S.; Mercado, C.C. Photovoltaic and Photocatalytic Properties of Bismuth Oxyiodide–Graphene Nanocomposites. RSC Adv. 2018, 8, 42254–42261. [Google Scholar] [CrossRef]
- Matos, J.; García, A.; Zhao, L.; Titirici, M.M. Solvothermal Carbon-Doped TiO2 Photocatalyst for the Enhanced Methylene Blue Degradation under Visible Light. Appl. Catal. A Gen. 2010, 390, 175–182. [Google Scholar] [CrossRef]
- Mennani, M.; Kasbaji, M.; Benhamou, A.A.; Boussetta, A.; Ablouh, E.-H.; Bayousfi, O.; Grimi, N.; Moubarik, A. Effects of Direct Sulfonation on the Catalytic Activity and Recyclability of Novel Lignin-Based Solid Acid Catalysts from Agri-Food Waste. Int. J. Biol. Macromol. 2023, 230, 123242. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, Z.; Chen, J. Applications of Lignin-Derived Catalysts for Green Synthesis. Green Energy Environ. 2019, 4, 210–244. [Google Scholar] [CrossRef]
- Hu, S.; Jiang, F.; Hsieh, Y.-L. 1D Lignin-Based Solid Acid Catalysts for Cellulose Hydrolysis to Glucose and Nanocellulose. ACS Sustain. Chem. Eng. 2015, 3, 2566–2574. [Google Scholar] [CrossRef]
- Liang, F.; Song, Y.; Huang, C.; Zhang, J.; Chen, B. Preparation and Performance Evaluation of a Lignin-Based Solid Acid from Acid Hydrolysis Lignin. Catal. Commun. 2013, 40, 93–97. [Google Scholar] [CrossRef]
- Zhu, S.; Xu, J.; Cheng, Z.; Kuang, Y.; Wu, Q.; Wang, B.; Gao, W.; Zeng, J.; Li, J.; Chen, K. Catalytic Transformation of Cellulose into Short Rod-like Cellulose Nanofibers and Platform Chemicals over Lignin-Based Solid Acid. Appl. Catal. B Environ. 2020, 268, 118732. [Google Scholar] [CrossRef]
- Mennani, M.; Kasbaji, M.; Ait Benhamou, A.; Boussetta, A.; Mekkaoui, A.A.; Grimi, N.; Moubarik, A. Current Approaches, Emerging Developments and Functional Prospects for Lignin-Based Catalysts—A Review. Green Chem. 2023, 25, 2896–2929. [Google Scholar] [CrossRef]
- Hayashi, J.; Kazehaya, A.; Muroyama, K.; Watkinson, A.P. Preparation of Activated Carbon from Lignin by Chemical Activation. Carbon 2000, 38, 1873–1878. [Google Scholar] [CrossRef]
- Bergna, D.; Varila, T.; Romar, H.; Lassi, U. Activated Carbon from Hydrolysis Lignin: Effect of Activation Method on Carbon Properties. Biomass Bioenergy 2022, 159, 106387. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, H.; Yu, H.; Yang, H.; Chen, T. Synthesis of Lignin-Derived Nitrogen-Doped Carbon as a Novel Catalyst for 4-NP Reduction Evaluation. Sci. Rep. 2020, 10, 20075. [Google Scholar] [CrossRef]
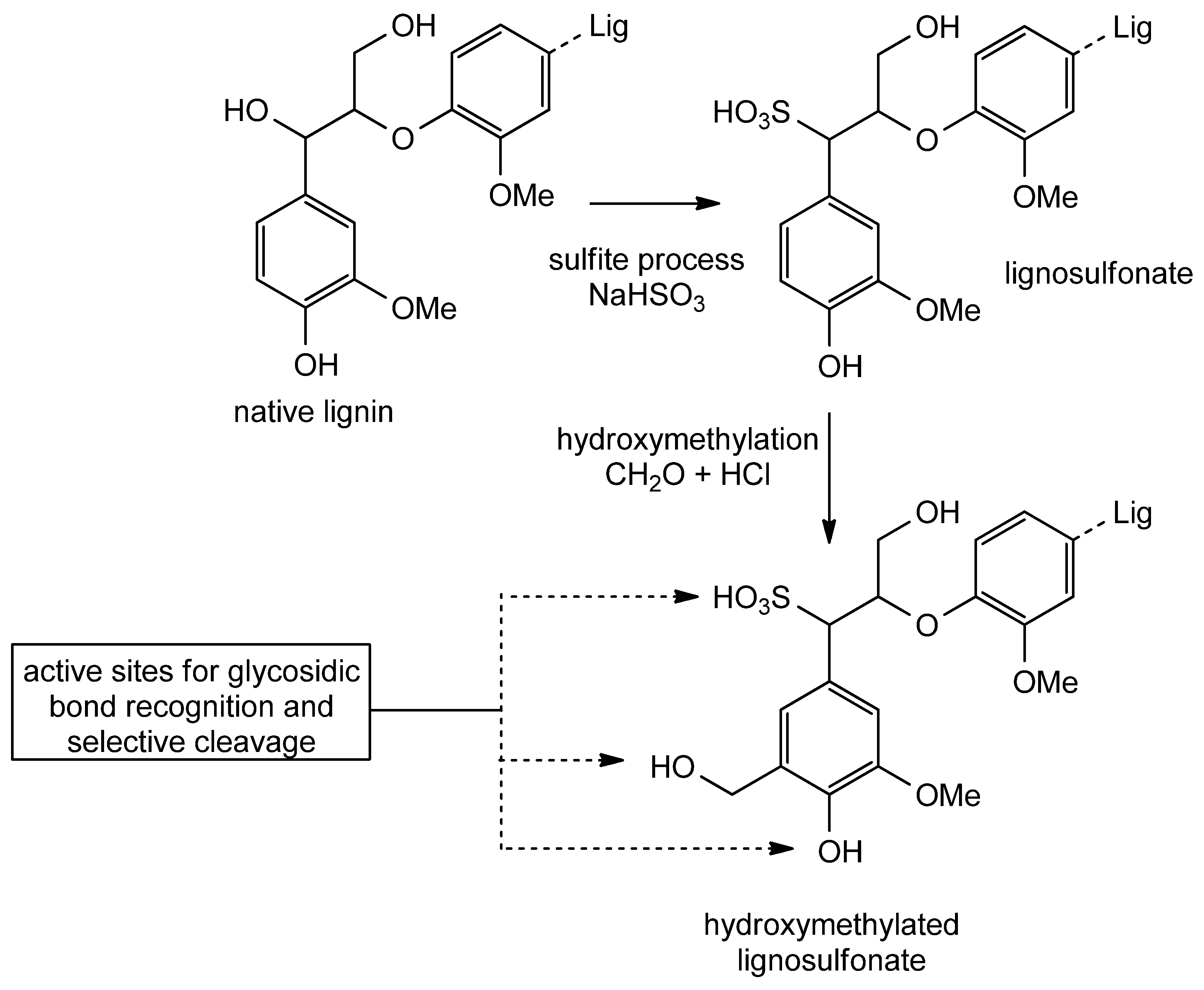
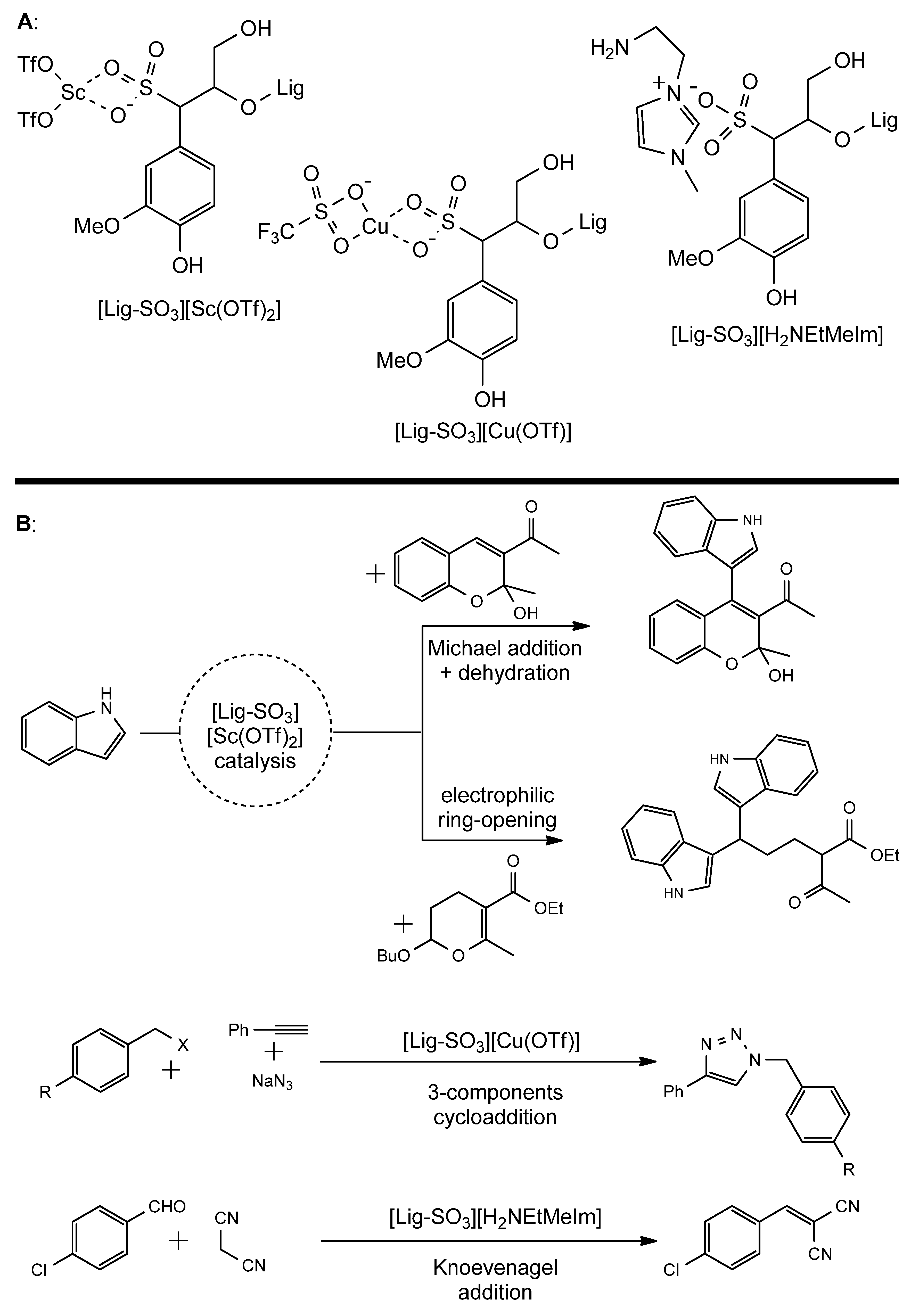
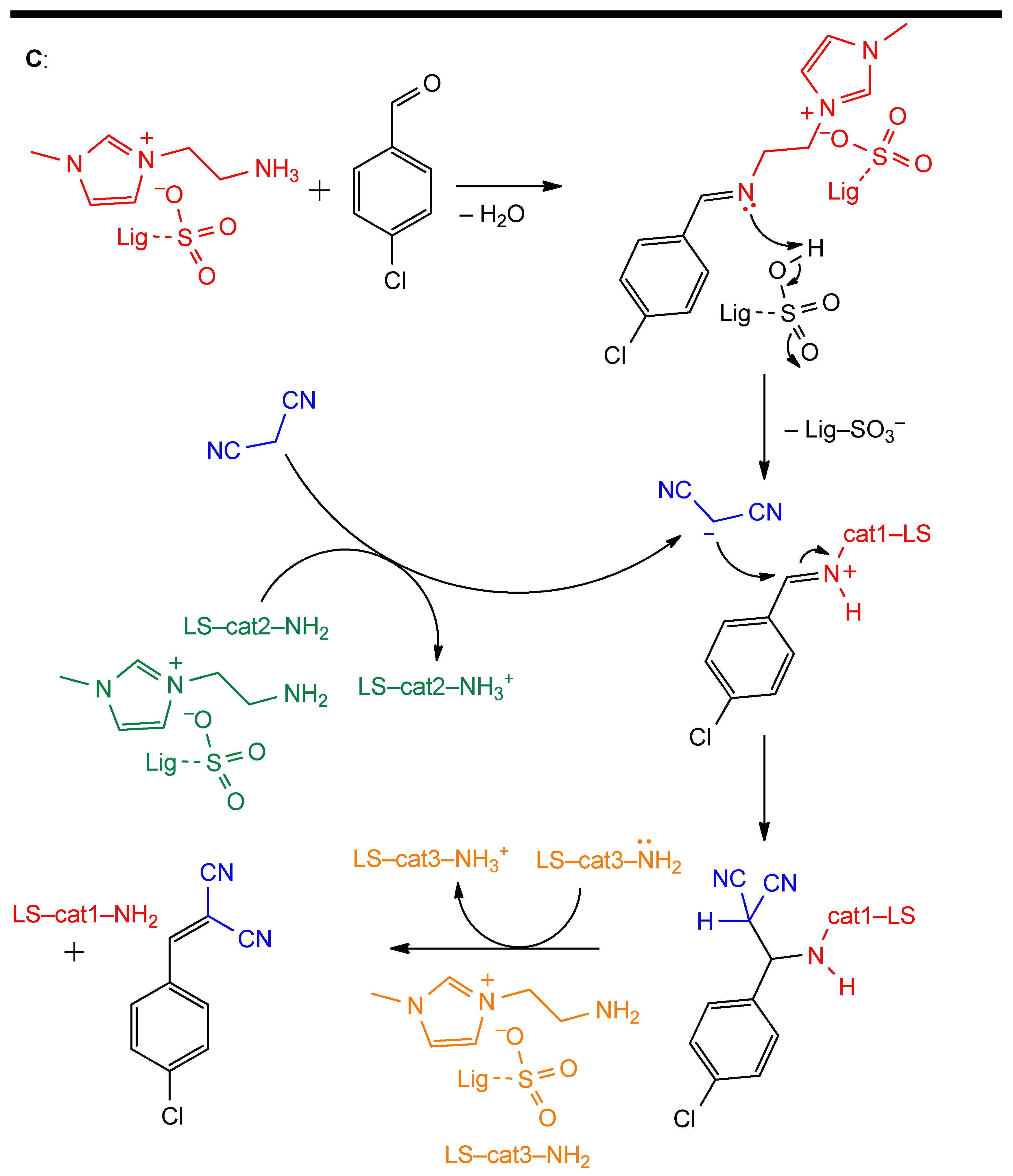
- Zhang, X.; Zhang, Z.; Wang, F.; Wang, Y.; Song, Q.; Xu, J. Lignosulfonate-Based Heterogeneous Sulfonic Acid Catalyst for Hydrolyzing Glycosidic Bonds of Polysaccharides. J. Mol. Catal. A Chem. 2013, 377, 102–107. [Google Scholar] [CrossRef]
- Sun, S.; Bai, R.; Gu, Y. From Waste Biomass to Solid Support: Lignosulfonate as a Cost-Effective and Renewable Supporting Material for Catalysis. Chem. A Eur. J 2014, 20, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gu, J.; Shan, R.; Yuan, H.; Chen, Y. Advances in Thermochemical Valorization of Biomass towards Carbon Neutrality. Resour. Conserv. Recycl. 2025, 212, 107905. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vadivel, D.; Ferraro, F.; Dondi, D. Harnessing Biomass for a Sustainable Future: The Role of Starch and Lignin. Catalysts 2024, 14, 747. https://doi.org/10.3390/catal14110747
Vadivel D, Ferraro F, Dondi D. Harnessing Biomass for a Sustainable Future: The Role of Starch and Lignin. Catalysts. 2024; 14(11):747. https://doi.org/10.3390/catal14110747
Chicago/Turabian StyleVadivel, Dhanalakshmi, Francesco Ferraro, and Daniele Dondi. 2024. "Harnessing Biomass for a Sustainable Future: The Role of Starch and Lignin" Catalysts 14, no. 11: 747. https://doi.org/10.3390/catal14110747
APA StyleVadivel, D., Ferraro, F., & Dondi, D. (2024). Harnessing Biomass for a Sustainable Future: The Role of Starch and Lignin. Catalysts, 14(11), 747. https://doi.org/10.3390/catal14110747