

Surface Redox Reaction for the Synthesis of NiPt Catalysts for the Upgrading of Renewable Ethanol/Methanol Mixtures


Abstract
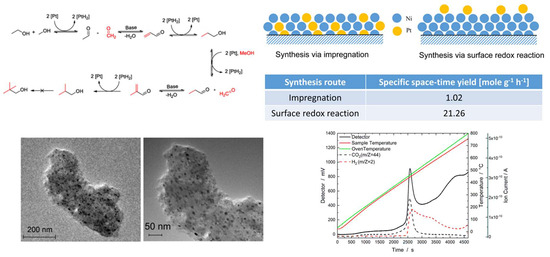
1. Introduction
2. Results and Discussion
2.1. NiPt Catalysts Prepared via Conventional Wet Impregnation
2.1.1. XRD Analysis
2.1.2. ICP–OES
2.1.3. H2–TPD
2.1.4. TEM/SEM Combined with EDX
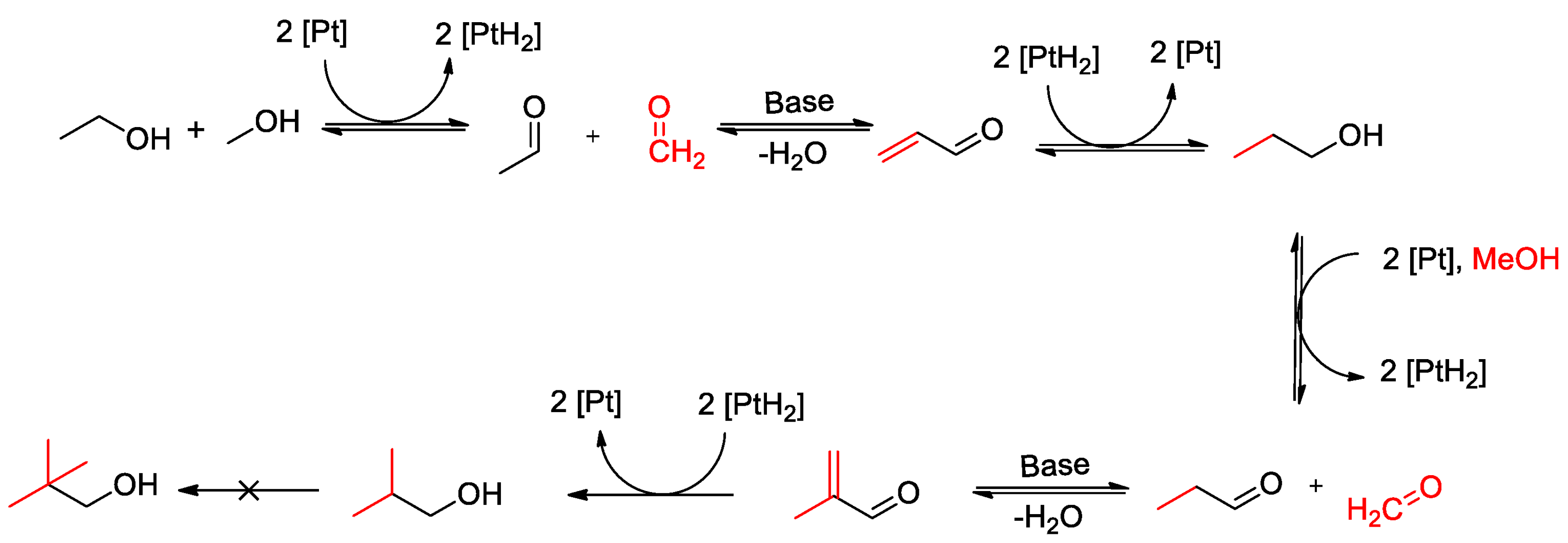
2.1.5. Catalytic Activity
2.2. NiPt Catalysts Prepared via Surface Redox Reactions
- NiPt/C synthesized in an HCl/KCl buffer
- ○
- Stirring for 15 min → SRR 15 buffer
- ○
- Stirring for 30 min → SRR 30 buffer
- ○
- Stirring for 60 min → SRR 60 buffer
- NiPt/C synthesized in a 0.1 M HCl solution
- ○
- Stirring for 15 min → SRR 15 0.1 HCl
- ○
- Stirring for 30 min → SRR 30 0.1 HCl
- NiPt/C synthesized in a 0.01 M HCl solution
- ○
- Stirring for 5 min → SRR 5 0.01 HCl
2.2.1. VIS Spectroscopy/ICP–OES
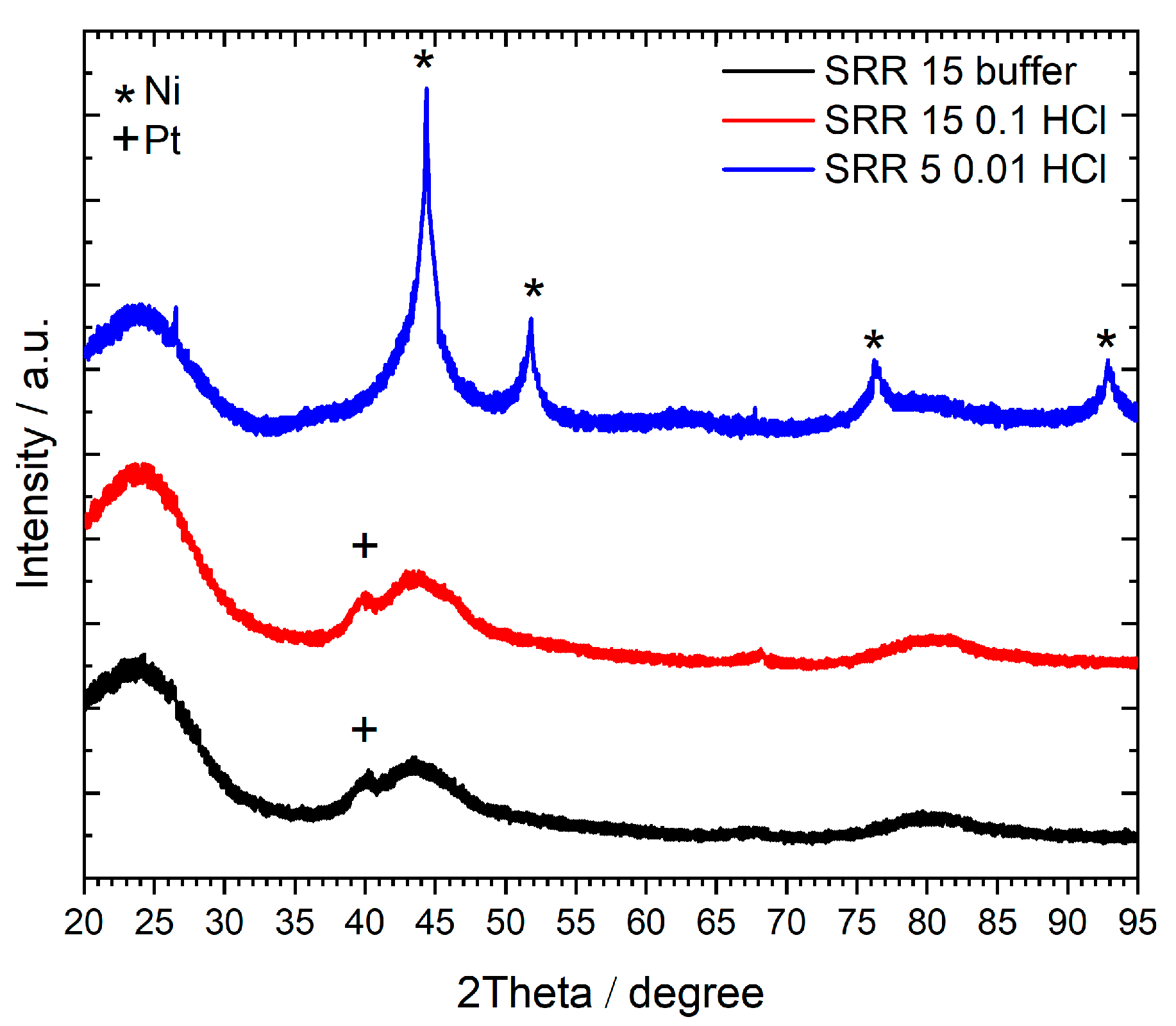
2.2.2. XRD Analysis
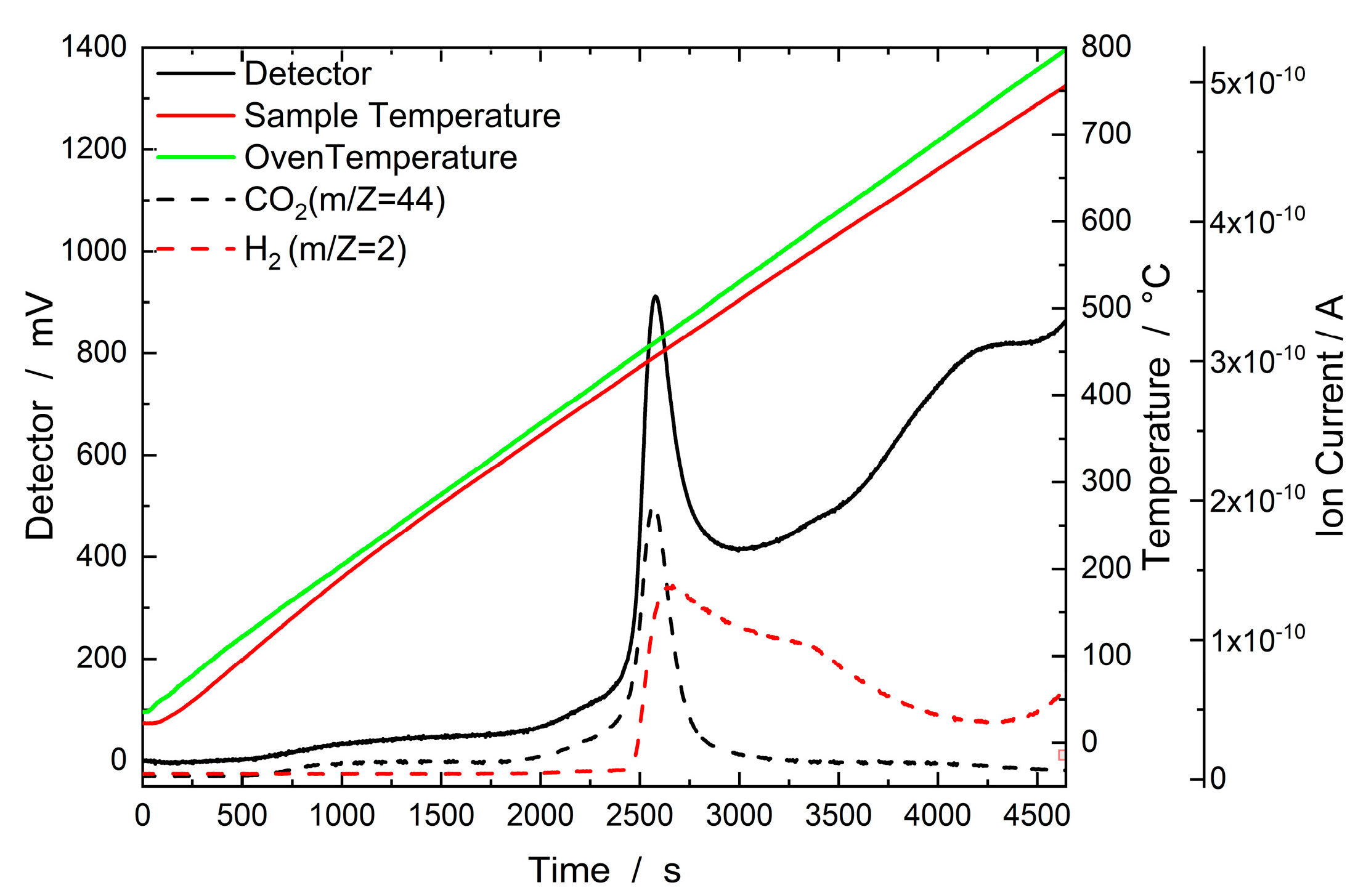
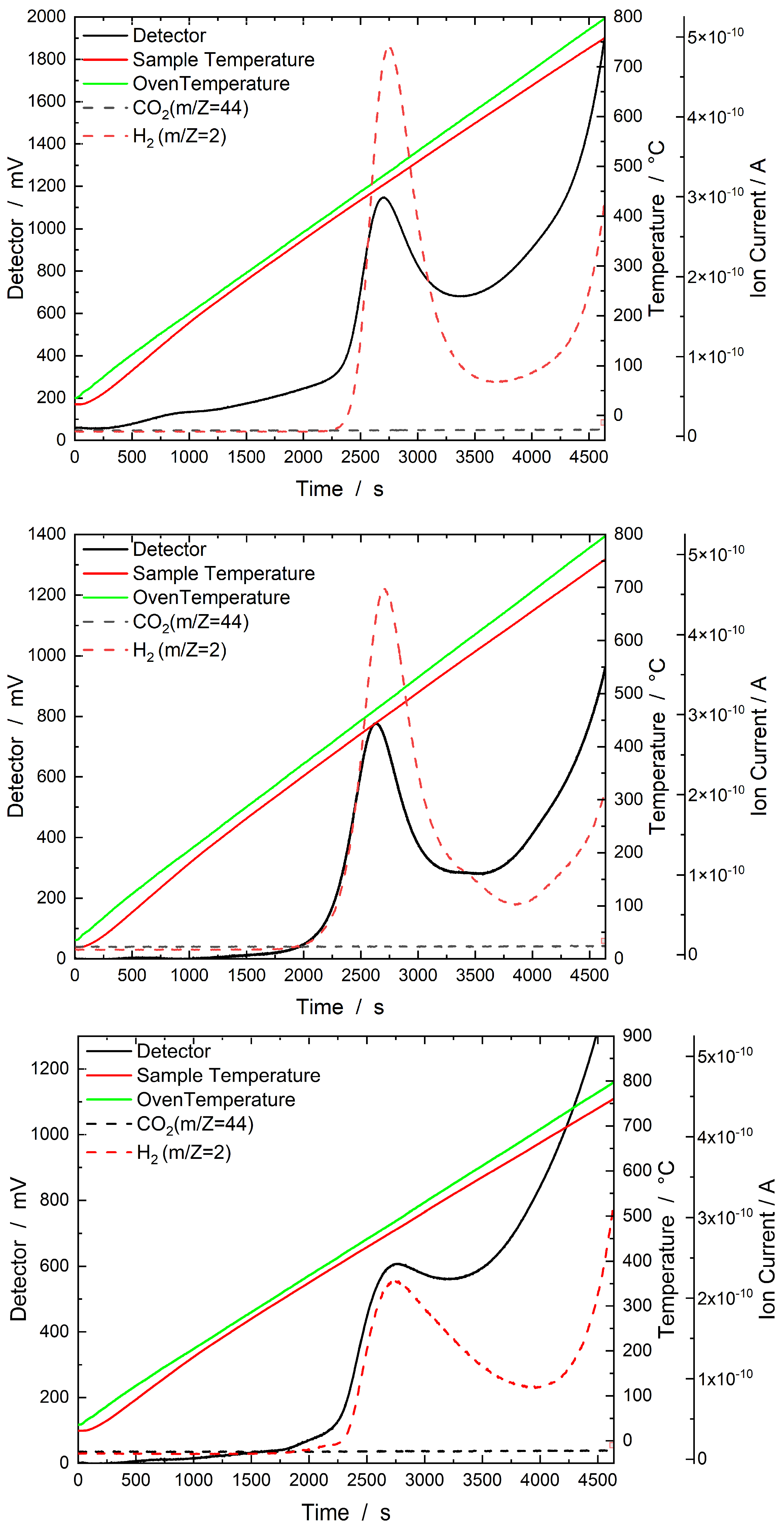
2.2.3. TGA
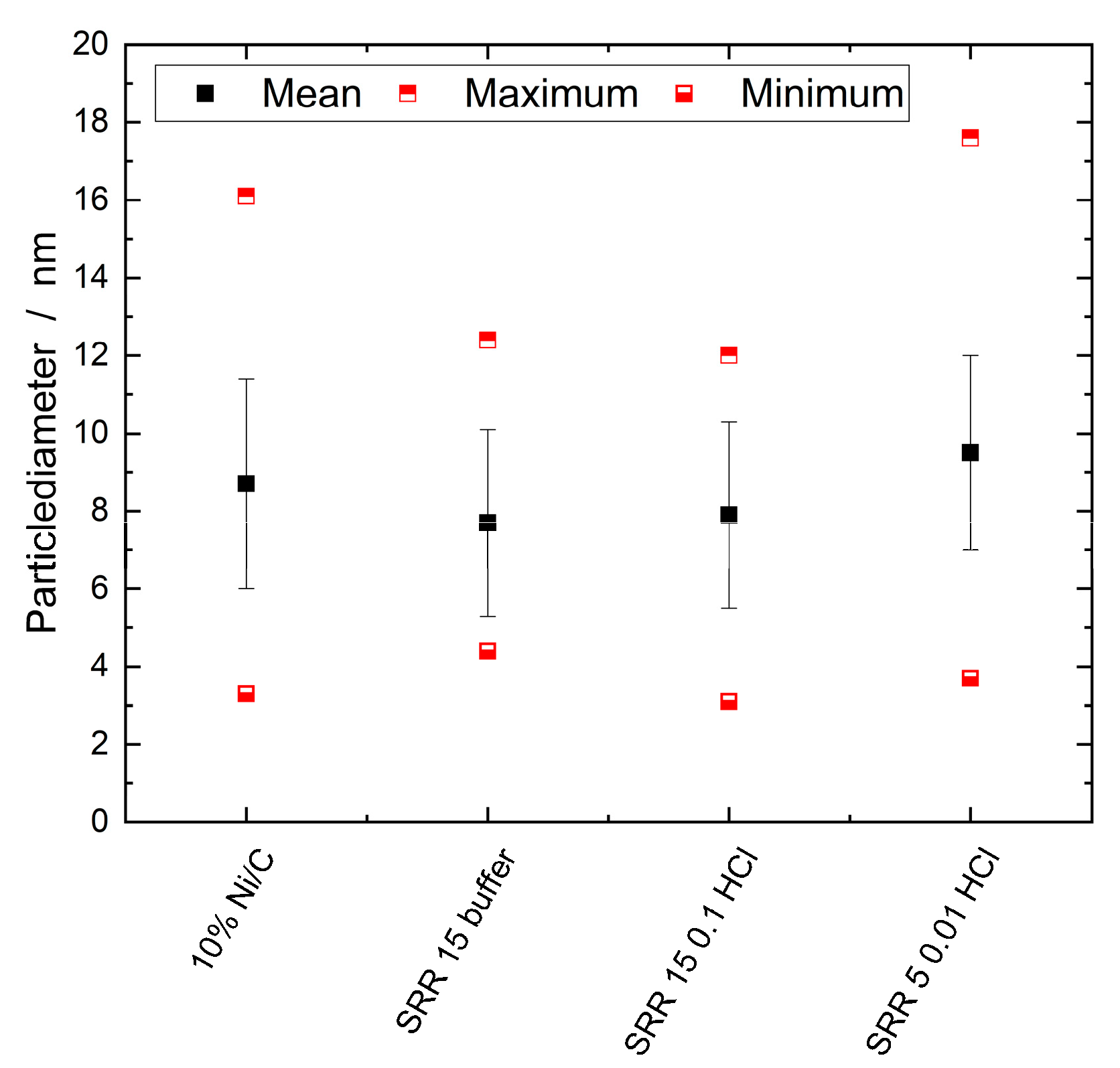
2.2.4. TEM/SEM Combined with EDX
2.2.5. H2–TPD
2.2.6. Catalytic Activity
3. Materials and Methods
3.1. Synthesis of the Impregnated Cataylsts
3.2. Synthesis of the Catalysts via the Surface Redox Reaction
3.2.1. KCl/HCl Buffer as a Polar Solution
3.2.2. 0.1 M HCl as a Polar Solution
3.2.3. 0.01 M HCl as a Polar Solution
3.3. Catalytic Experiments
3.4. Vis Spectroscopy
3.5. TGA
3.6. H2–TPD
3.7. ICP–OES
3.8. XRD
3.9. N2 Sorption
3.10. TEM
3.11. SEM/EDX
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Allen, M.R.; Dube, O.P.; Solecki, W.; Aragón-Durand, F.; Cramer, W.; Humphreys, S.; Kainuma, M.; Kala, J.; Mahowald, N.; Mulugetta, Y.; et al. 2018: Framing and Context Supplementary Material. In: Global Warming of 1.5 °C. Available online: https://www.ipcc.ch/sr15 (accessed on 18 September 2023).
- Rohde, R. Global Temperature Report for 2020. Available online: https://berkeleyearth.org/global-temperature-report-for-2020/ (accessed on 18 September 2023).
- Navarro-Jaén, S.; Virginie, M.; Bonin, J.; Robert, M.; Wojcieszak, R.; Khodakov, A.Y. Highlights and Challenges in the Selective Reduction of Carbon Dioxide to Methanol. Nat. Rev. Chem. 2021, 5, 564–579. [Google Scholar] [CrossRef] [PubMed]
- Pérez, J.J.; Escalante, D.J.; González, L.A.; Pérez, J.A. Catalytic Activity of Polymetallic and Hydrotalcite-Like Catalysts in the Synthesis of Methanol by Carbon Dioxide Hydrogenation. Renew. Energy Power Qual. J. 2022, 20, 569–573. [Google Scholar] [CrossRef]
- Santiago, R.G.; Coelho, J.A.; Lucena, S.M.P.; Musse, A.P.S.; Portilho, M.D.F.; Rodríguez-Castellón, E.; de Azevedo, D.C.S.; Bastos-Neto, M. Synthesis of MeOH and DME from CO2 Hydrogenation over Commercial and Modified Catalysts. Front. Chem. 2022, 10, 903053. [Google Scholar] [CrossRef]
- Navarro-Jaén, S.; Virginie, M.; Morin, J.-C.; Thuriot, J.; Wojcieszak, R.; Khodakov, A.Y. Hybrid Monometallic and Bimetallic Copper–palladium Zeolite Catalysts for Direct Synthesis of Dimethyl Ether From CO2. New J. Chem. 2022, 46, 3889–3900. [Google Scholar] [CrossRef]
- Navarro-Jaén, S.; Virginie, M.; Thuriot-Roukos, J.; Wojcieszak, R.; Khodakov, A.Y. Structure–performance Correlations in the Hybrid Oxide-Supported Copper–zinc SAPO-34 Catalysts for Direct Synthesis of Dimethyl Ether From CO2. J. Mater. Sci. 2022, 57, 3268–3279. [Google Scholar] [CrossRef]
- Mota, N.; Ordóñez, E.M.; Pawelec, B.; Fierro, J.L.G.; Navarro, R.M. Direct Synthesis of Dimethyl Ether From CO2: Recent Advances in Bifunctional/Hybrid Catalytic Systems. Catalysts 2021, 11, 411. [Google Scholar] [CrossRef]
- Tripathi, K.; Gupta, V.; Awasthi, V.; Pant, K.K.; Upadhyayula, S. Design Framework for Dimethyl Ether (DME) Production From Coal and Biomass-derived Syngas via Simulation Approach. Can. J. Chem. Eng. 2023, 101, 3213–3225. [Google Scholar] [CrossRef]
- Breitkreuz, C.F.; Hevert, N.; Schmitz, N.; Burger, J.; Hasse, H. Synthesis of Methylal and Poly(oxymethylene) Dimethyl Ethers From Dimethyl Ether and Trioxane. Ind. Eng. Chem. Res. 2022, 61, 7810–7822. [Google Scholar] [CrossRef]
- Tönges, Y.; Dieterich, V.; Fendt, S.; Spliethoff, H.; Burger, J. Techno-Economic Analysis of Large Scale Production of Poly(oxymethylene) Dimethyl Ether Fuels from Methanol in Water-Tolerant Processes. Fuels 2023, 4, 1–18. [Google Scholar] [CrossRef]
- Zhang, S.; Ding, X.; Shang, H.; Song, Y.; Wang, Y. Co-Production of Dimethyl Carbonate, Dimethoxymethane and Dimethyl Ether From Methanol: Process Design and Exergy Analysis. Entropy 2022, 24, 1438. [Google Scholar] [CrossRef]
- Li, Y.-L.; Yu, Y.-C.; Wang, Z.-W.; Wang, J.-F. Physical and chemical properties of isobutanol-gasoline blends. Environ. Prog. Sustain. Energy 2015, 34, 908–914. [Google Scholar] [CrossRef]
- Häusler, J. Synthese von Höheren Alkoholen als Zukünftige Kraftstoffe für den Verkehrssektor. Ph.D. Thesis, Fakultät für Maschinenwesen, RWTH Aachen, Jülich, Germany. in preparation.
- Pasel, J.; Häusler, J.; Schmitt, D.; Valencia, H.; Meledina, M.; Mayer, J.; Peters, R. Ethanol Dehydrogenation: A Reaction Path Study by Means of Temporal Analysis of Products. Catalysts 2020, 10, 1151. [Google Scholar] [CrossRef]
- Häusler, J.; Pasel, J.; Peters, R.; Woltmann, F. Catalyst System and Method for Producing Higher Alcohols from Methanol and Ethanol. International Patent WO2023213568A1, 9 November 2023. [Google Scholar]
- Brankovic, S.R.; Wang, J.X.; Adžić, R.R. Metal monolayer deposition by replacement of metal adlayers on electrode surfaces. Surf. Sci. 2001, 474, L173–L179. [Google Scholar] [CrossRef]
- Brankovic, S.R.; Wang, J.X.; Zhu, Y.; Sabatini, R.; McBreen, J.; Adžić, R.R. Electrosorption and catalytic properties of bare and Pt modified single crystal and nanostructured Ru surfaces. J. Electroanal. Chem. 2002, 524–525, 231–241. [Google Scholar] [CrossRef]
- Sasaki, K.; Mo, Y.; Wang, J.X.; Balasubramanian, M.; Uribe, F.; McBreen, J.; Adzic, R.R. Pt submonolayers on metal nanoparticles—Novel electrocatalysts for H2 oxidation and O2 reduction. Electrochim. Acta 2003, 48, 3841–3849. [Google Scholar] [CrossRef]
- Van Brussel, M.; Kokkinidis, G.; Hubin, A.; Buess-Herman, C. Oxygen reduction at platinum modified gold electrodes. Electrochim. Acta 2003, 48, 3909–3919. [Google Scholar] [CrossRef]
- Van Brussel, M.; Kokkinidis, G.; Vandendael, I.; Buess-Herman, C. High performance gold-supported platinum electrocatalyst for oxygen reduction. Electrochem. Commun. 2002, 4, 808–813. [Google Scholar] [CrossRef]
- Mintsouli, I.; Georgieva, J.; Valova, E.; Armyanov, S.; Kakaroglou, A.; Hubin, A.; Steenhaut, O.; Dille, J.; Papaderakis, A.; Kokkinidis, G.; et al. Pt–Ni carbon-supported catalysts for methanol oxidation prepared by Ni electroless deposition and its galvanic replacement by Pt. J. Solid State Electrochem. 2013, 17, 435–443. [Google Scholar] [CrossRef]
- Hu, Y.; Shao, Q.; Wu, P.; Zhang, H.; Cai, C. Synthesis of hollow mesoporous Pt–Ni nanosphere for highly active electrocatalysis toward the methanol oxidation reaction. Electrochem. Commun. 2012, 18, 96–99. [Google Scholar] [CrossRef]
- Tamašauskaitė-Tamašiūnaitė, L.; Balčiūnaitė, A.; Vaiciukevičienė, A.; Selskis, A.; Pakštas, V. Investigation of nanostructured platinum–nickel supported on the titanium surface as electrocatalysts for alkaline fuel cells. J. Power Sources 2012, 208, 242–247. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.; Wang, R.; Wang, Q.; Lei, Z. Carbon-supported platinum-decorated nickel nanoparticles for enhanced methanol oxidation in acid media. J. Solid State Electrochem. 2012, 16, 1049–1054. [Google Scholar] [CrossRef]
- Burger, T.; Augenstein, H.M.S.; Hnyk, F.; Döblinger, M.; Köhler, K.; Hinrichsen, O. Targeted Fe-Doping of Ni−Al Catalysts via the Surface Redox Reaction Technique for Unravelling its Promoter Effect in the CO2 Methanation Reaction. ChemCatChem 2020, 12, 649–662. [Google Scholar] [CrossRef]
- Badenes, P.; Daza, L.; Rodriguez-Ramos, I.; Guerrero-Ruiz, A. Mechanism of hydrogen spillover over carbon supported metal catalysts. In Studies in Surface Science and Catalysis; Li, C., Xin, Q., Eds.; Elsevier: Amsterdam, The Netherlands, 1997; pp. 241–250. [Google Scholar]
- Miller, J.T.; Meyers, B.L.; Barr, M.K.; Modica, F.S.; Koningsberger, D.C. Hydrogen Temperature-Programmed Desorptions in PlatinumCatalysts: Decomposition and Isotopic Exchange by SpilloverHydrogen of Chemisorbed Ammonia. J. Catal. 1996, 159, 41–49. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Percentage Values | IMP Ni99Pt1/C | IMP Ni95Pt5/C | ||
|---|---|---|---|---|
| ICP-OES | Calculated | ICP-OES | Calculated | |
| wt% Ni | 8.4 | 11.2 | 7.7 | 10.0 |
| wt% Pt | 0.1 | 0.3 | 0.4 | 1.5 |
| at% Ni | 99.6 | 99.2 | 98.5 | 95.7 |
| at% Pt | 0.4 | 0.8 | 1.5 | 4.3 |
| Reaction Products | IMP Ni99Pt1/C Y (%)] | IMP Ni99Pt1/C S (%) | IMP Ni95Pt5/C Y (%) | IMP Ni95Pt5/C S (%) |
|---|---|---|---|---|
| Iso-butanol | 2.45 | 61.03 | 3.01 | 69.49 |
| 2-methylpropanal | 0.34 | 8.55 | 0.39 | 8.94 |
| 1-propanol | 0.01 | 0.20 | 0.02 | 0.36 |
| 2-methylbutane-1-ol | 0.03 | 1.28 | 0.05 | 2.19 |
| 2-ethylbutane-1-ol | 0.02 | 1.20 | 0.03 | 2.20 |
| 1-hexanol | 0.21 | 15.32 | 0.22 | 15.47 |
| 2-ethylhexane-1-ol | 0.12 | 15.32 | 0 | 0 |
| Catalyst | βVIS (mg L−1) | βICP-OES (mg L−1) |
|---|---|---|
| SRR 15 buffer | 942 | 930 |
| SRR 30 buffer | 931 | 930 |
| SRR 60 buffer | 919 | 910 |
| Percentage Values | SRR 15 Buffer ICP–OES | SRR 15 Buffer Calculated | SRR 15 0.1 HCl ICP–OES | SRR 15 0.1 HCl Calculated | SRR 5 0.01 HCl ICP–OES | SRR 5 0.01 HCl Calculated |
|---|---|---|---|---|---|---|
| wt% Ni | 0.32 | 8.59 | 0.10 | 8.59 | 7.33 | 8.56 |
| wt% Pt | 0.25 | 0.34 | 0.19 | 0.33 | 0.24 | 0.38 |
| Catalyst | Specific STY (mole g−1 h−1) | Y (Iso-Butanol) (%) | S to Iso-Butanol (%) | Specific Surface Areas (m2 g−1) |
|---|---|---|---|---|
| IMP Ni99Pt1/C | 1.02 | 2.45 | 61.03 | 706 |
| IMP Ni95Pt5/C | 1.35 | 3.01 | 69.49 | 703 |
| SRR 15 buffer | 21.26 | 2.40 | 68.68 | 780 |
| SRR 30 buffer | 15.16 | 3.01 | 67.11 | - |
| SRR 60 buffer | 17.52 | 2.04 | 70.48 | - |
| SRR 15 0.1 HCl | 79.87 | 3.49 | 74.49 | 758 |
| SRR 30 0.1 HCl | 42.07 | 6.02 | 80.09 | - |
| SRR 5 0.01 HCl | 3.30 | 6.92 | 76.19 | 671 |
| Reaction Products | SRR 15 Buffer Y (%) | SRR 15 Buffer S (%) | SRR 15 0.1 HCl Y (%) | SRR 15 0.1 HCl S (%) | SRR 5 0.01 HCl Y (%) | SRR 5 0.01 HCl S (%) |
|---|---|---|---|---|---|---|
| Iso-butanol | 2.40 | 68.67 | 3.49 | 74.49 | 6.92 | 76.19 |
| 2-methylpropanal | 0.49 | 14.01 | 0.60 | 12.75 | 1.23 | 13.56 |
| 1-propanol | 0.00 | 0.04 | 0.00 | 0.05 | 0.02 | 0.23 |
| 2-methylbutane-1-ol | 0.01 | 0.83 | 0.01 | 0.39 | 0.06 | 1.35 |
| 2-ethylbutane-1-ol | 0.00 | 0.00 | 0.00 | 0.00 | 0.03 | 1.01 |
| 1-hexanol | 0.19 | 16.36 | 0.19 | 8.68 | 0.23 | 7.65 |
| 2-ethylhexane-1-ol | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasel, J.; Woltmann, F.; Häusler, J.; Peters, R. Surface Redox Reaction for the Synthesis of NiPt Catalysts for the Upgrading of Renewable Ethanol/Methanol Mixtures. Catalysts 2024, 14, 77. https://doi.org/10.3390/catal14010077
Pasel J, Woltmann F, Häusler J, Peters R. Surface Redox Reaction for the Synthesis of NiPt Catalysts for the Upgrading of Renewable Ethanol/Methanol Mixtures. Catalysts. 2024; 14(1):77. https://doi.org/10.3390/catal14010077
Chicago/Turabian StylePasel, Joachim, Friederike Woltmann, Johannes Häusler, and Ralf Peters. 2024. "Surface Redox Reaction for the Synthesis of NiPt Catalysts for the Upgrading of Renewable Ethanol/Methanol Mixtures" Catalysts 14, no. 1: 77. https://doi.org/10.3390/catal14010077
APA StylePasel, J., Woltmann, F., Häusler, J., & Peters, R. (2024). Surface Redox Reaction for the Synthesis of NiPt Catalysts for the Upgrading of Renewable Ethanol/Methanol Mixtures. Catalysts, 14(1), 77. https://doi.org/10.3390/catal14010077