
Density Functional Theory Study of CuAg Bimetal Electrocatalyst for CO2RR to Produce CH3OH


 ,
, 
Abstract
:
1. Introduction
2. Results and Discussion
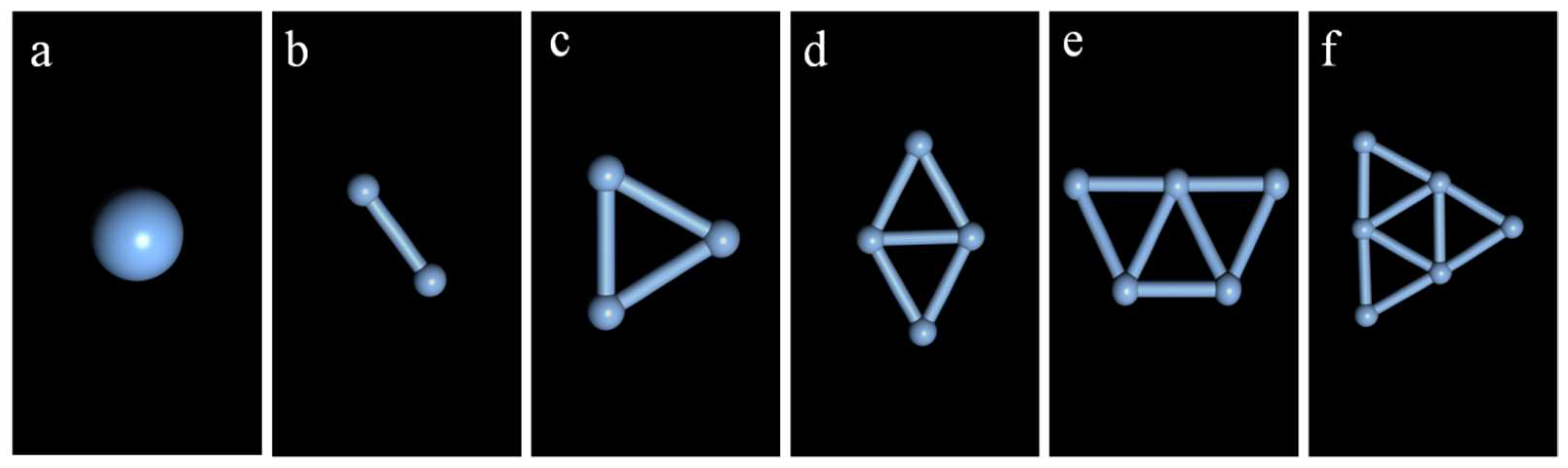
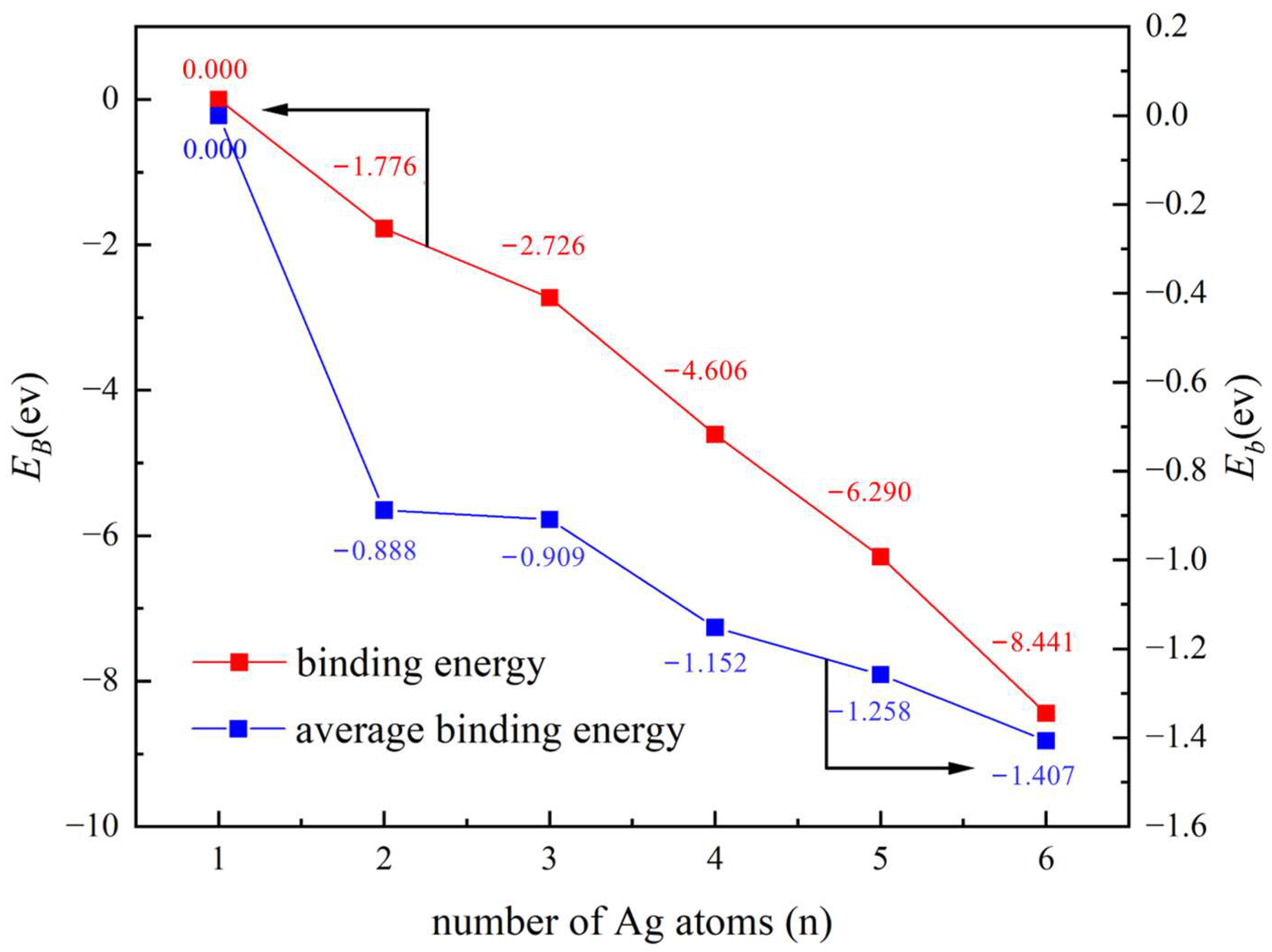
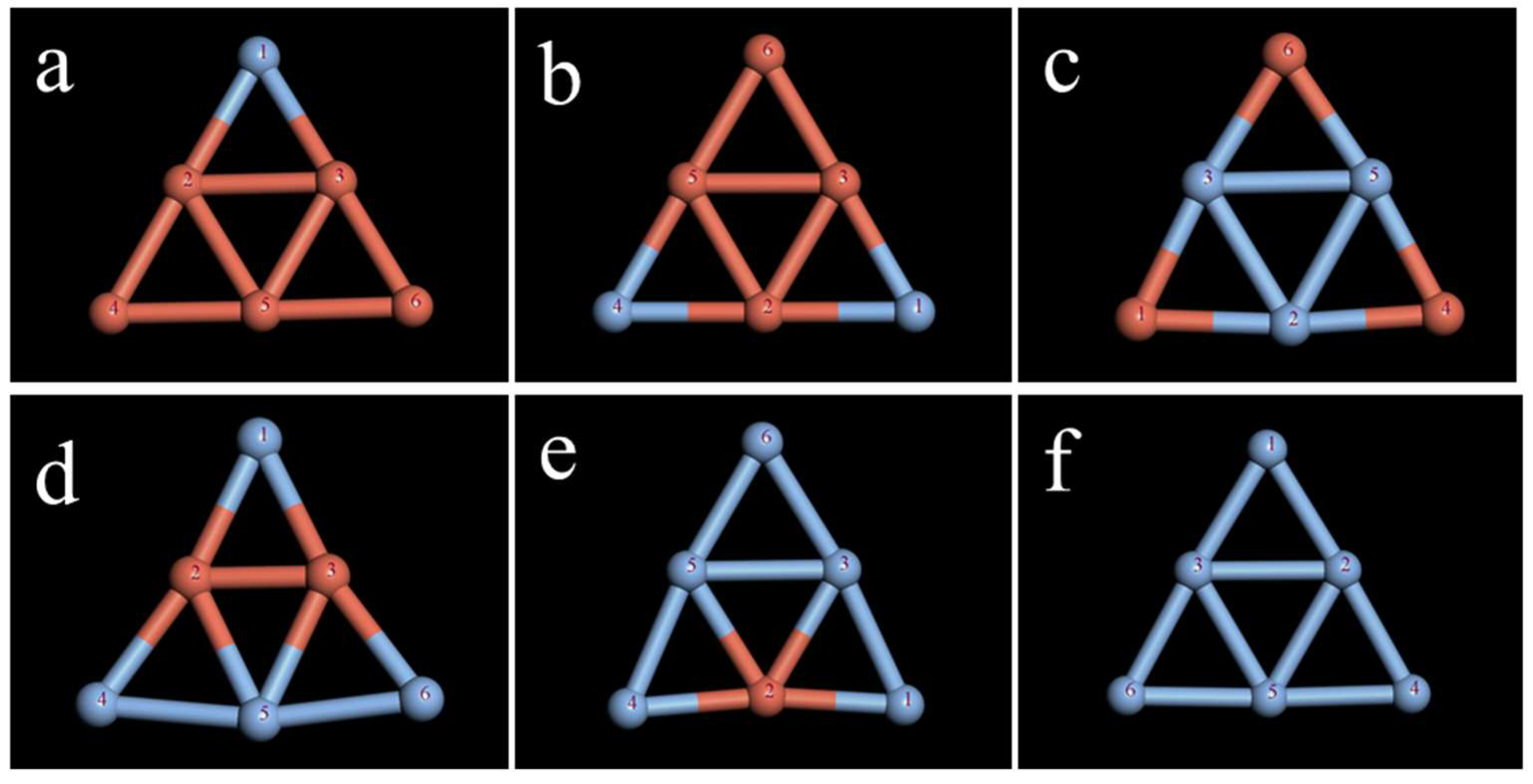
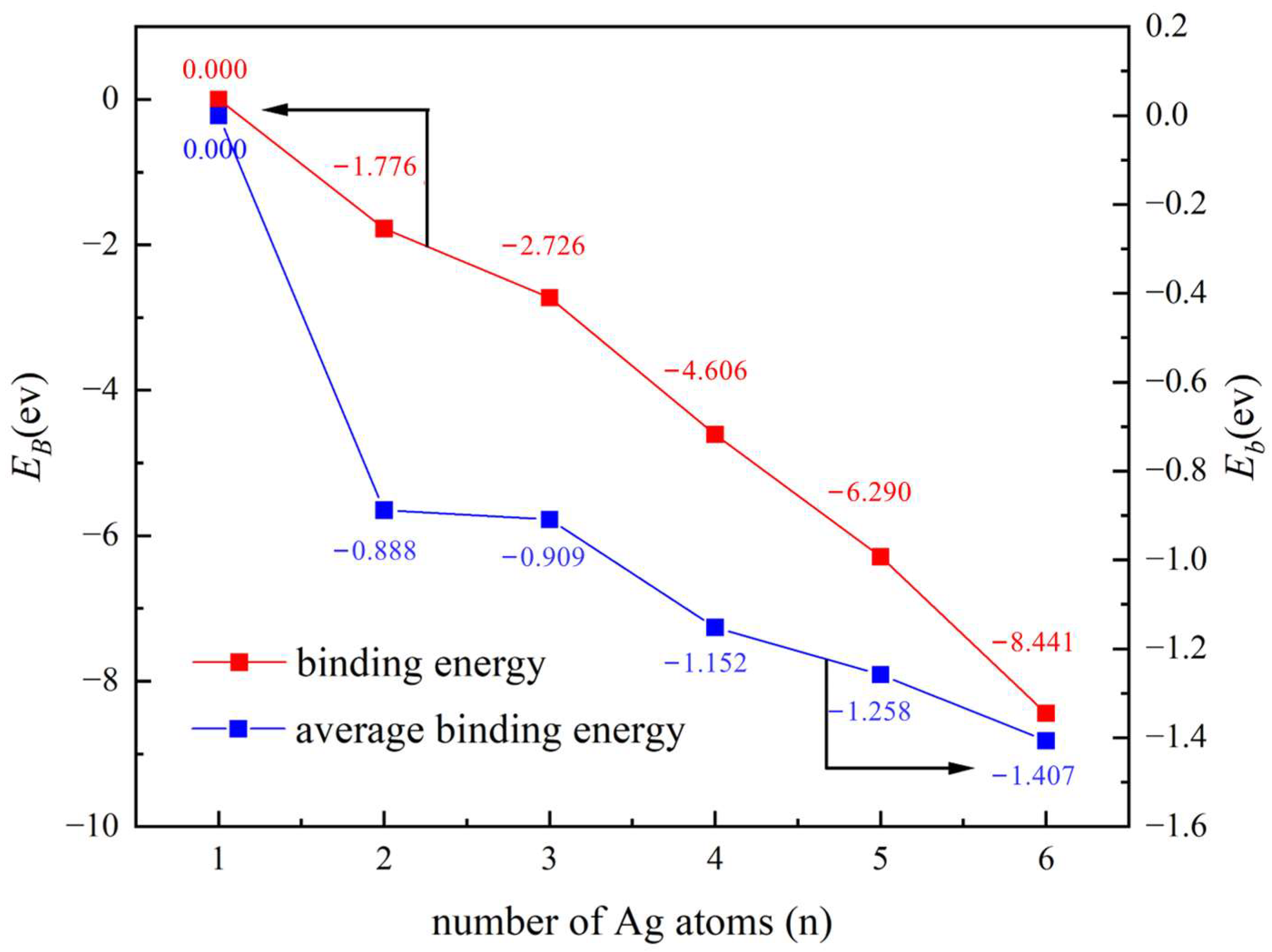
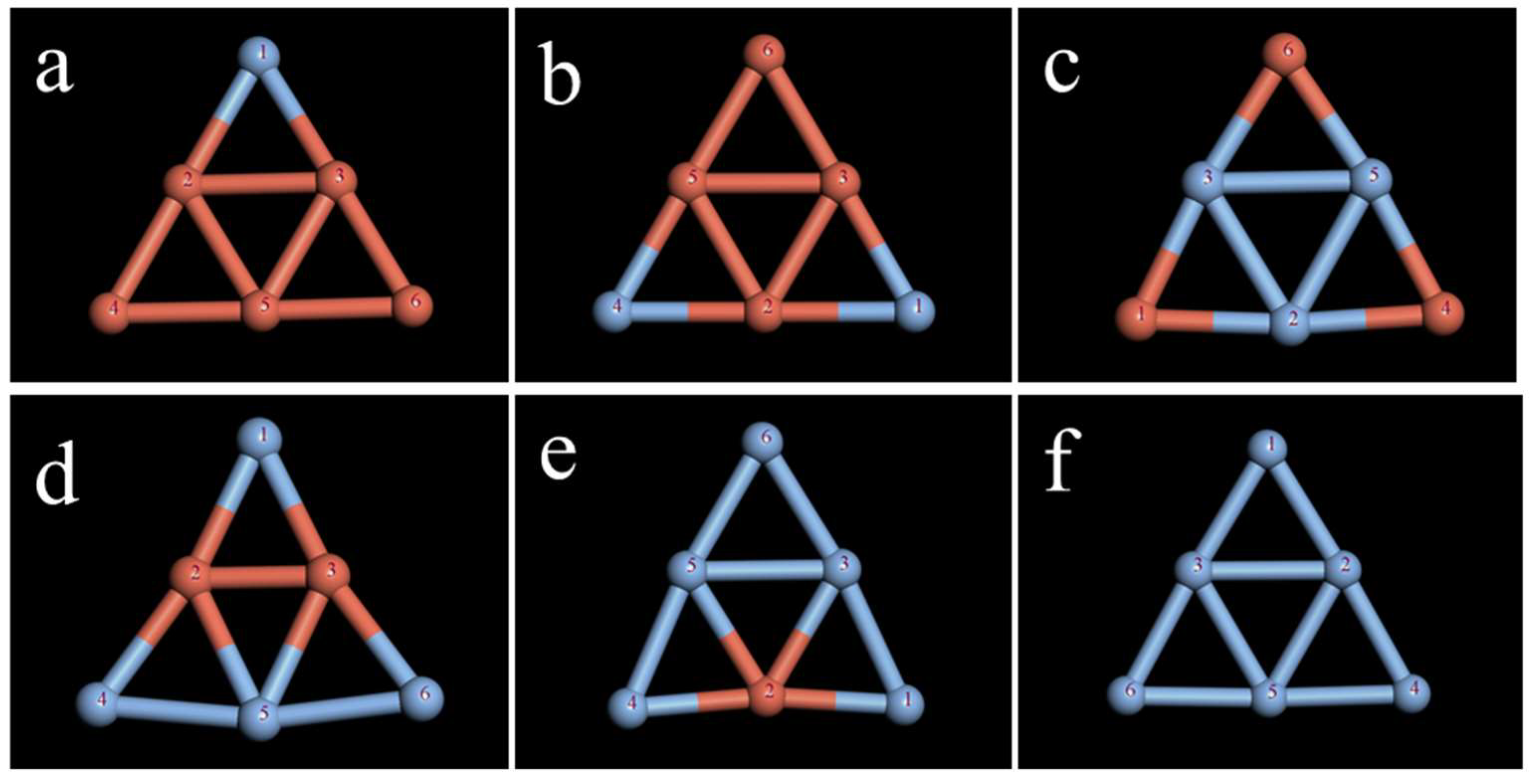
2.1. Study on the Stability of the Ag Clusters
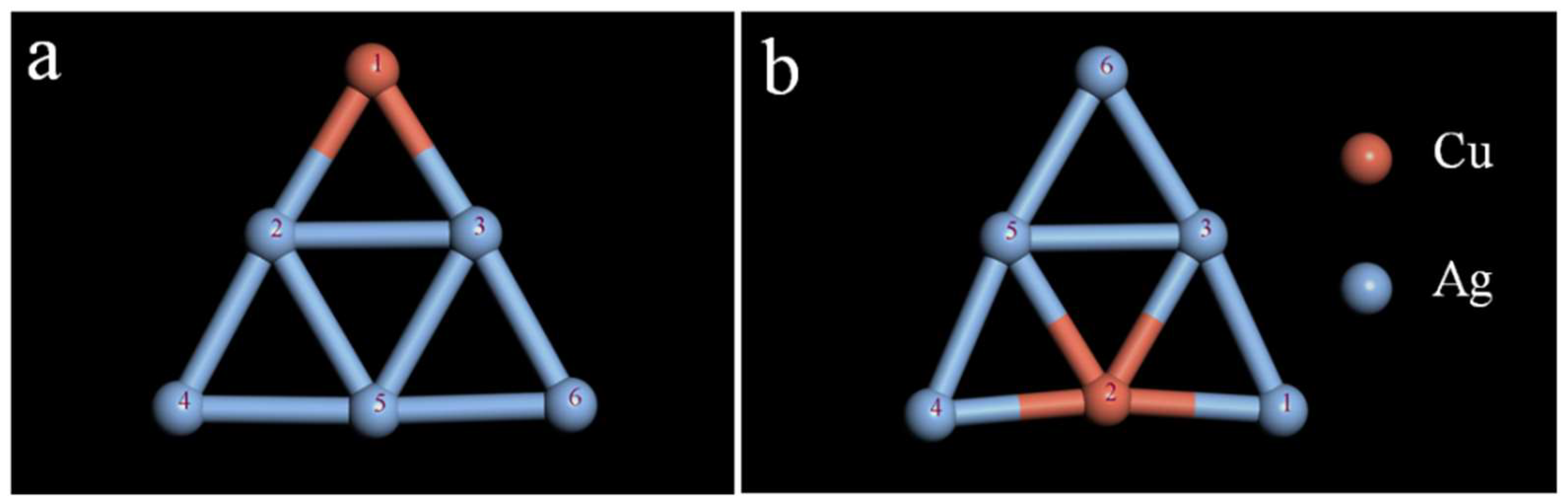
2.2. Study on the Stability of the CuAg Clusters
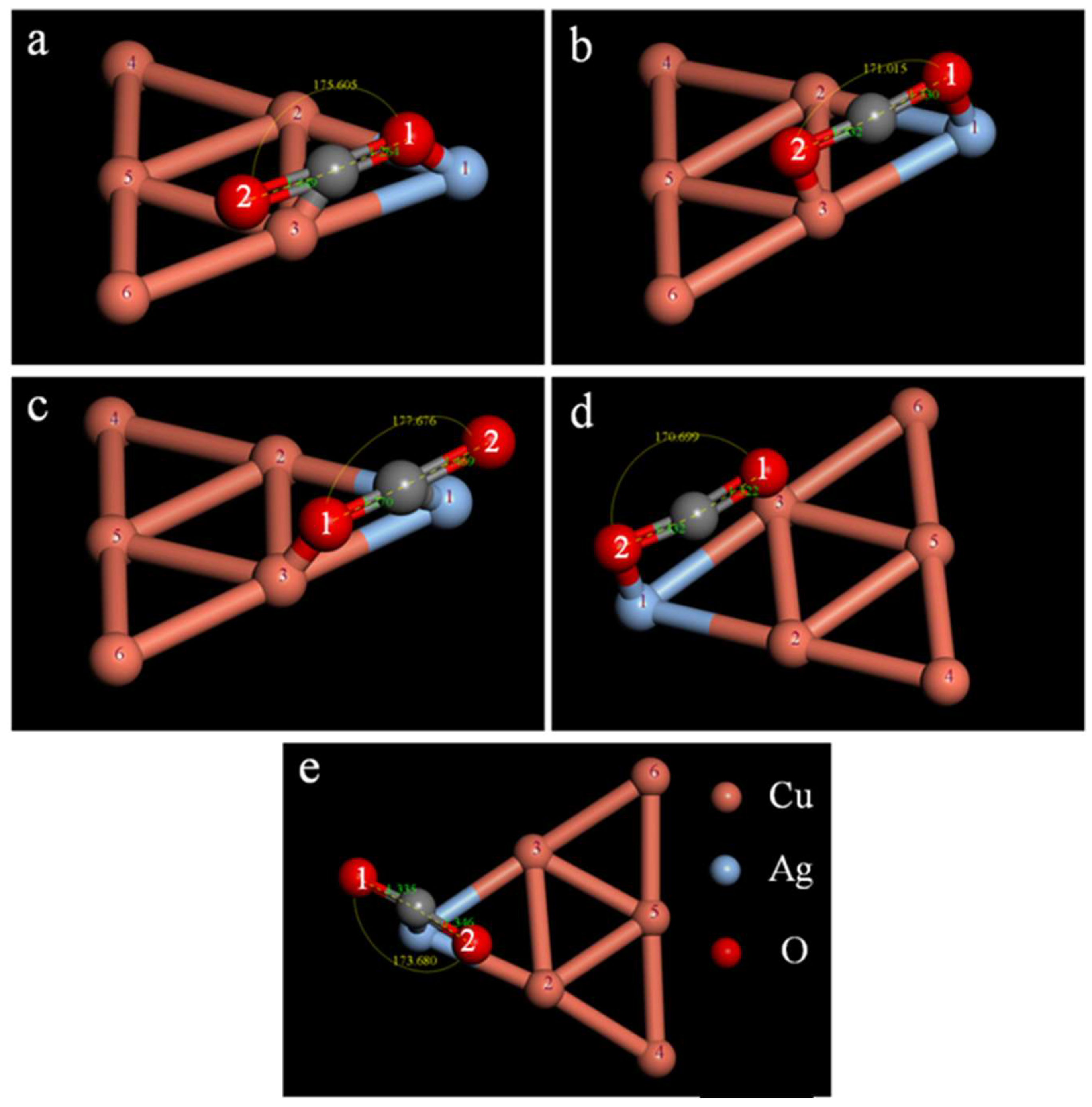
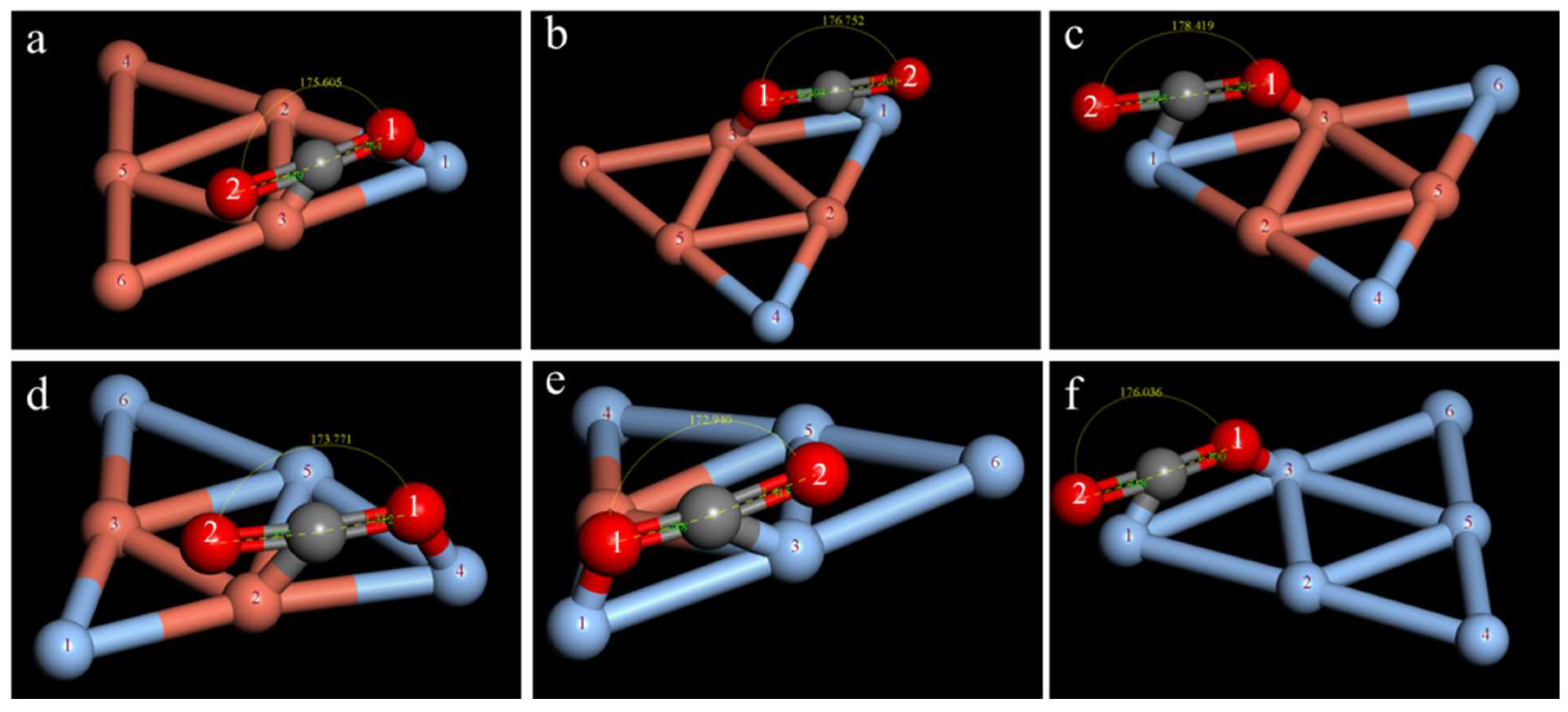
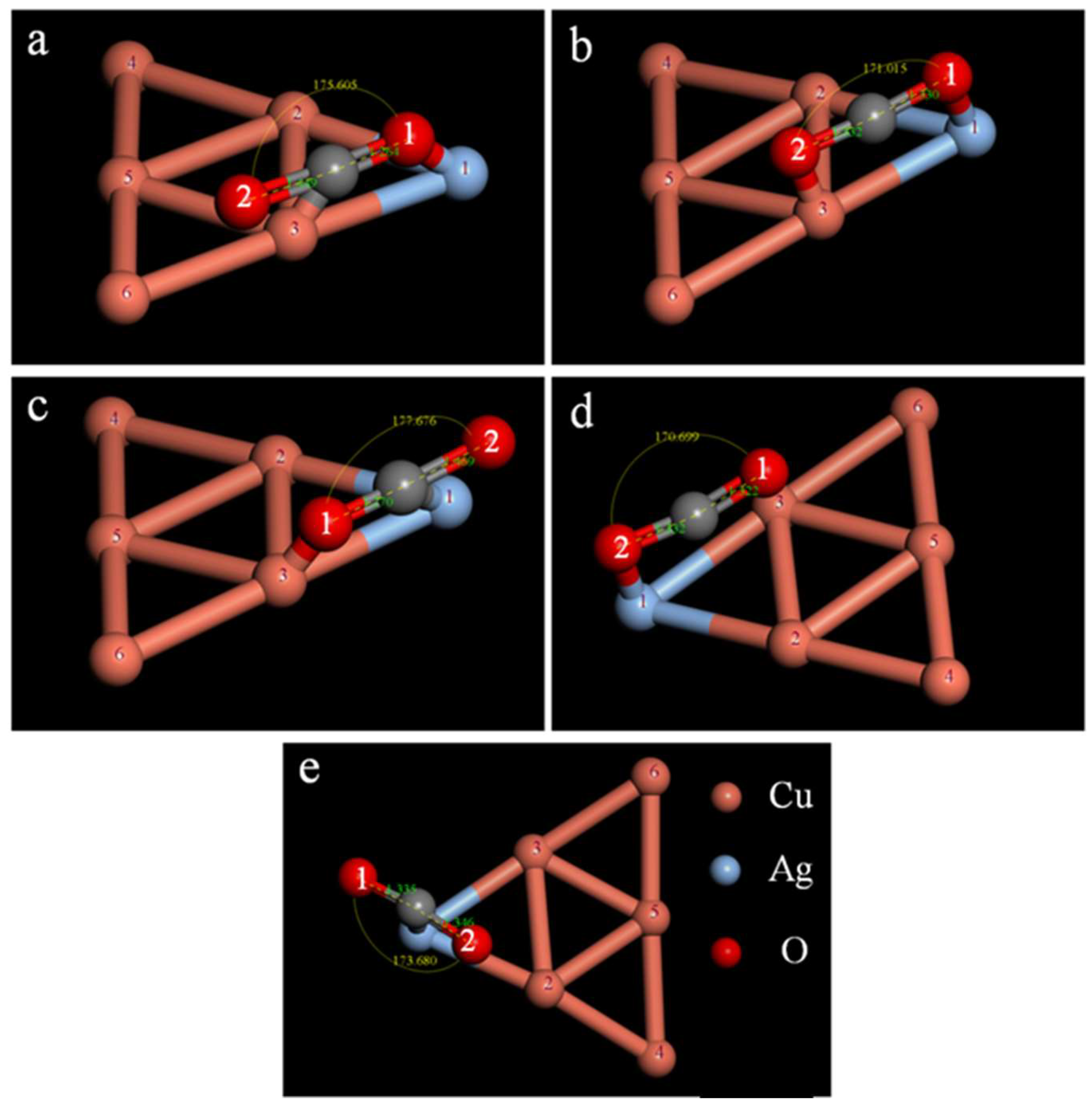
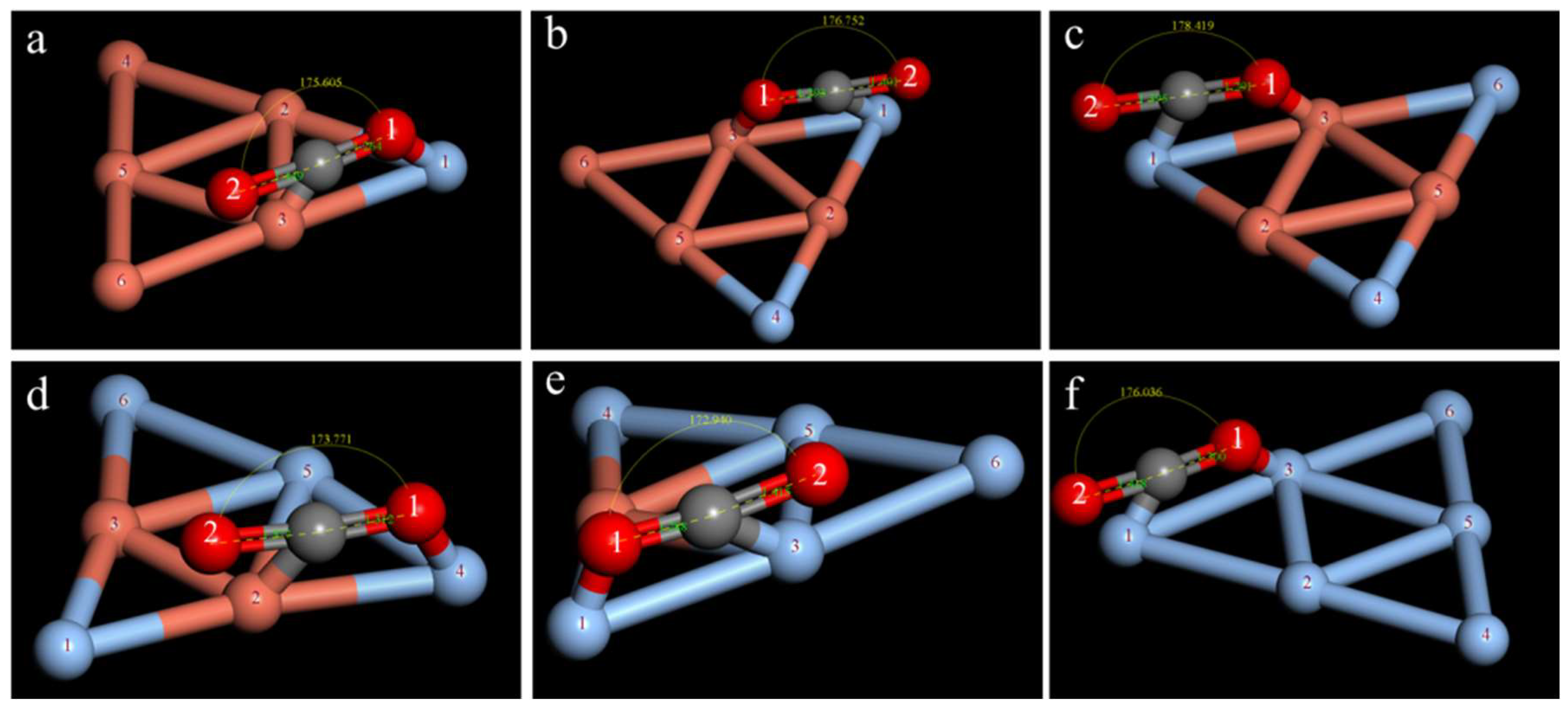
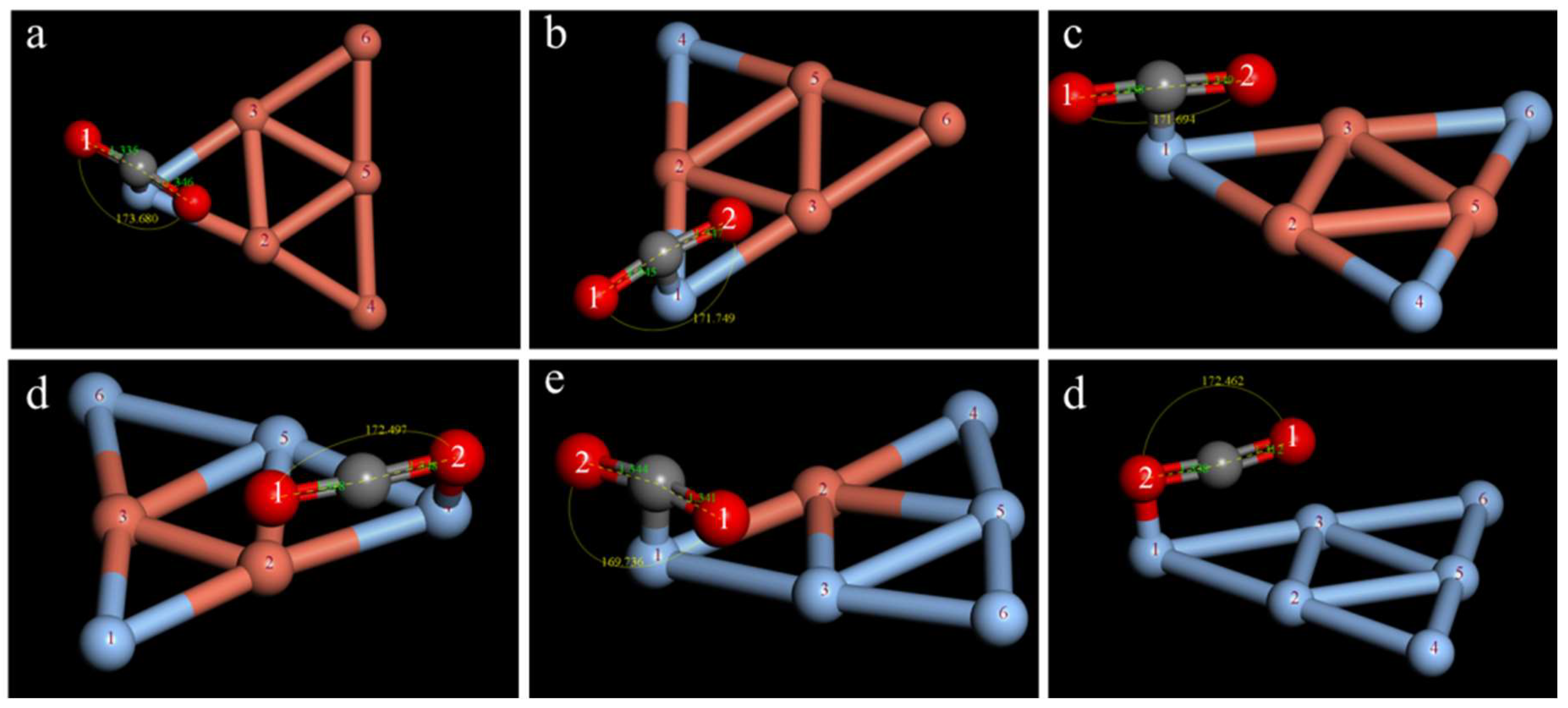
2.3. Study on the CO2 Adsorption Stability of the CuAg Clusters
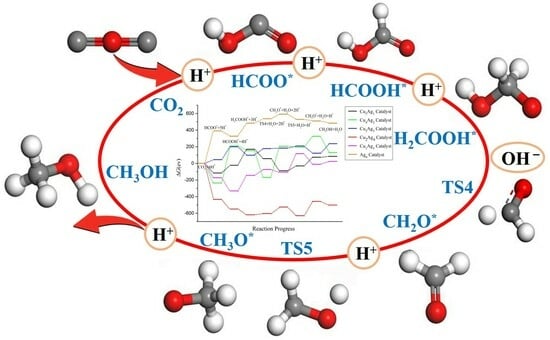
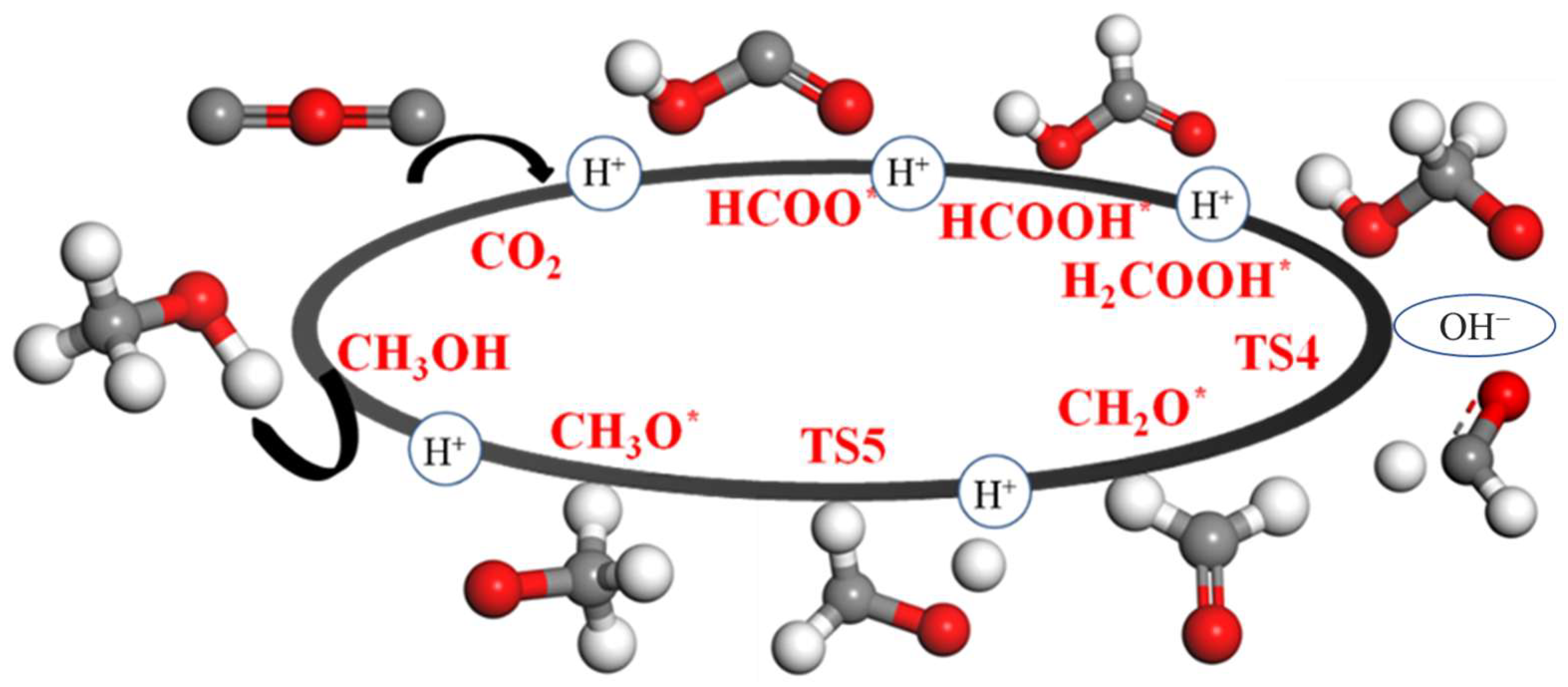
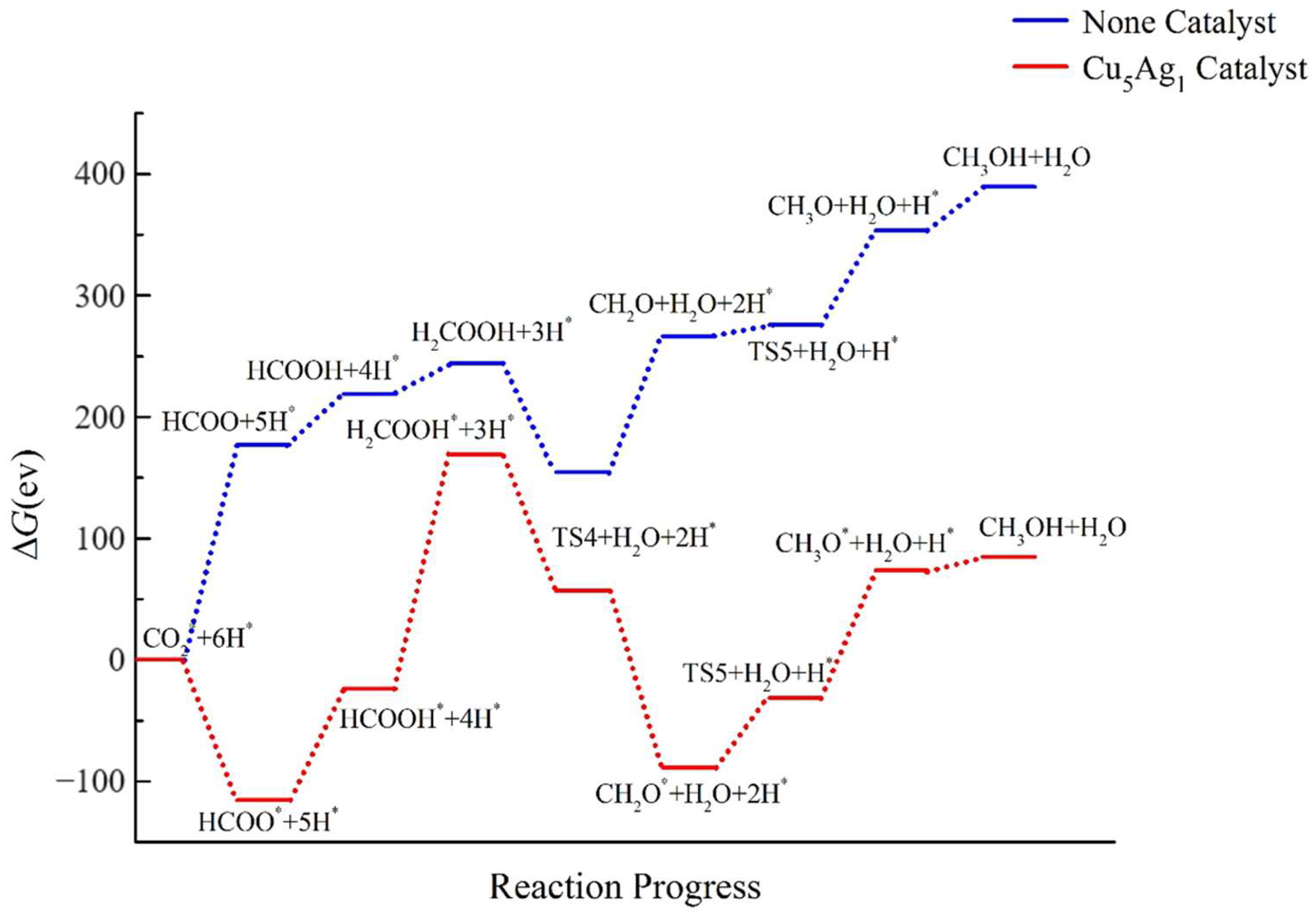
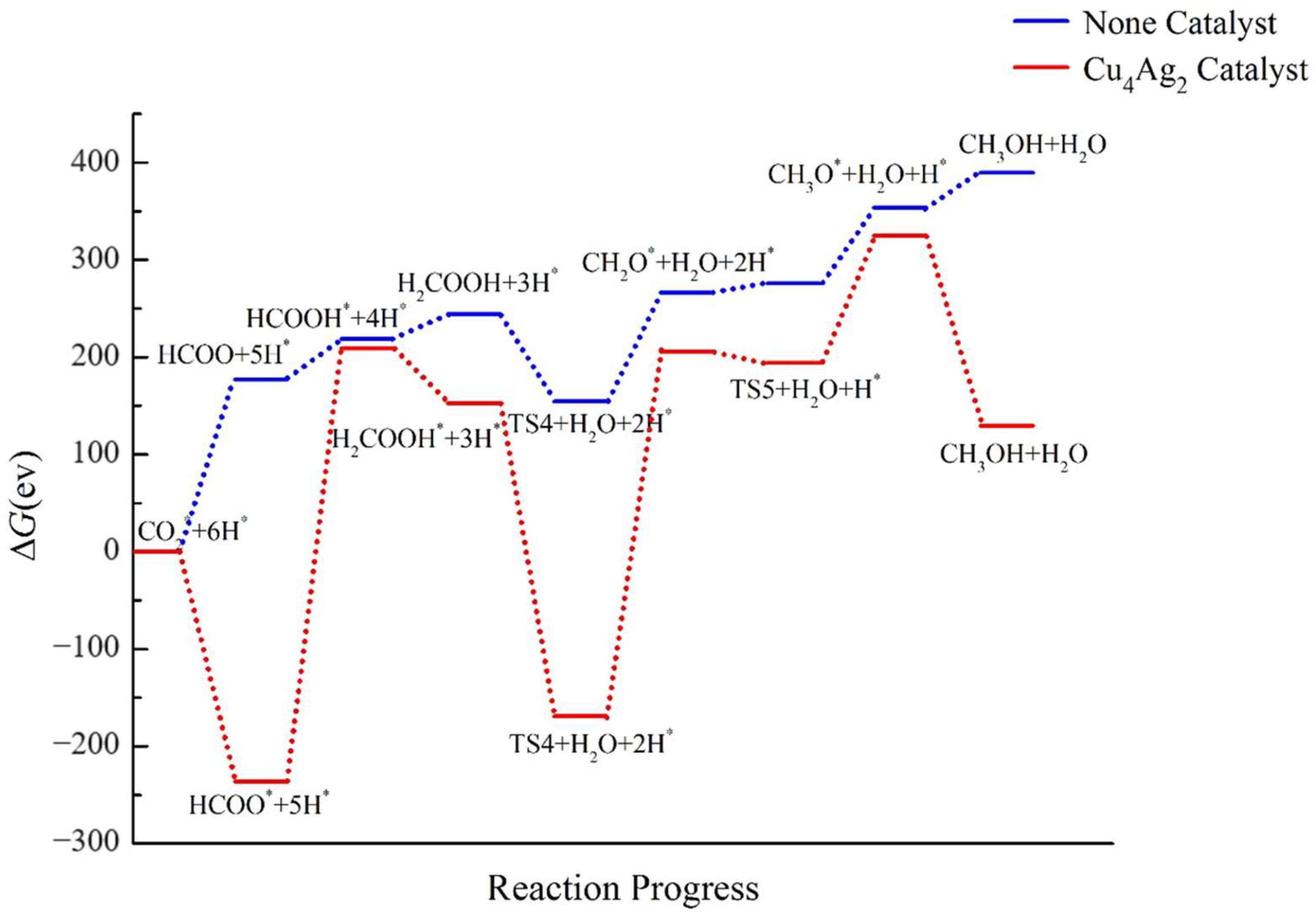
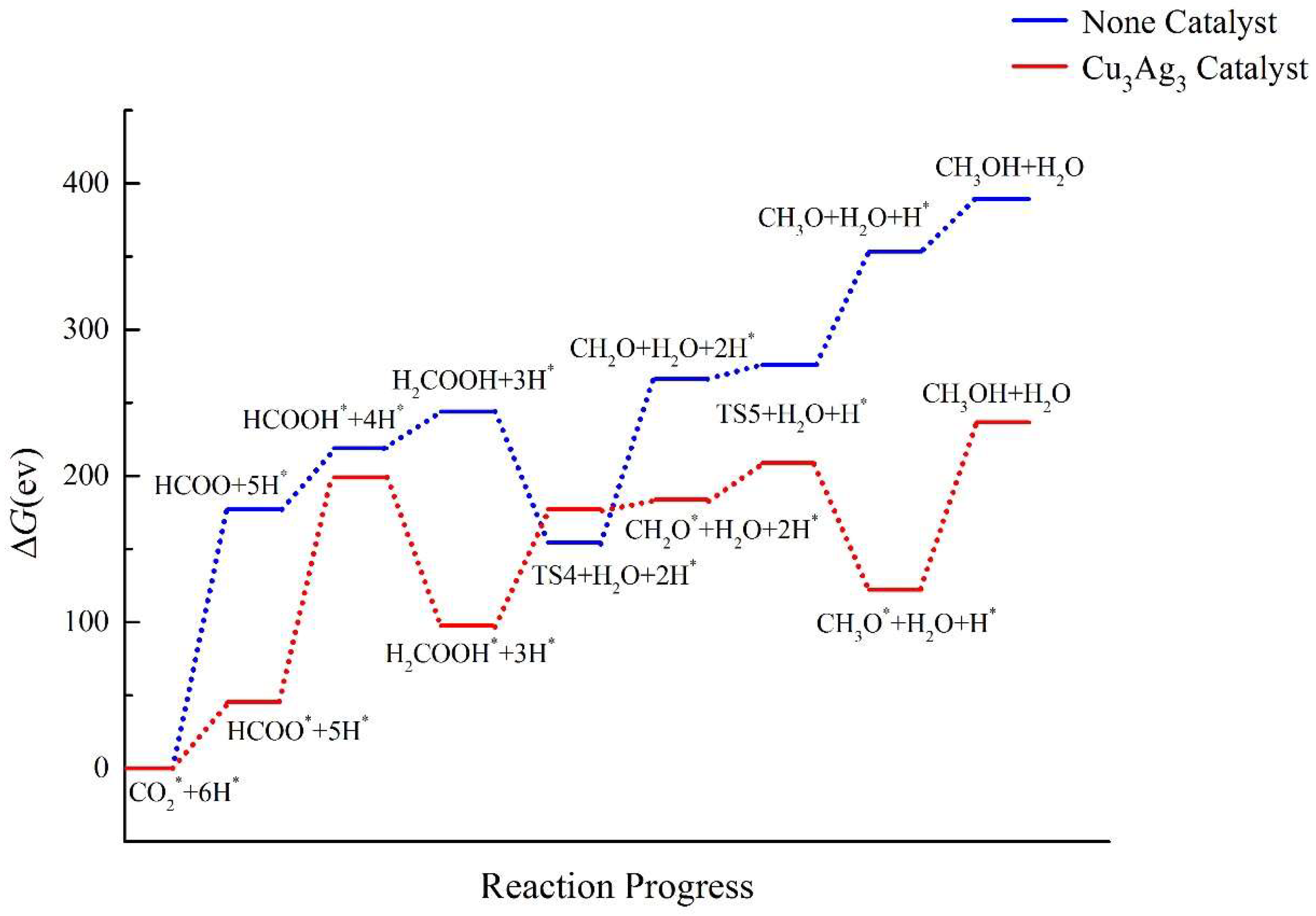
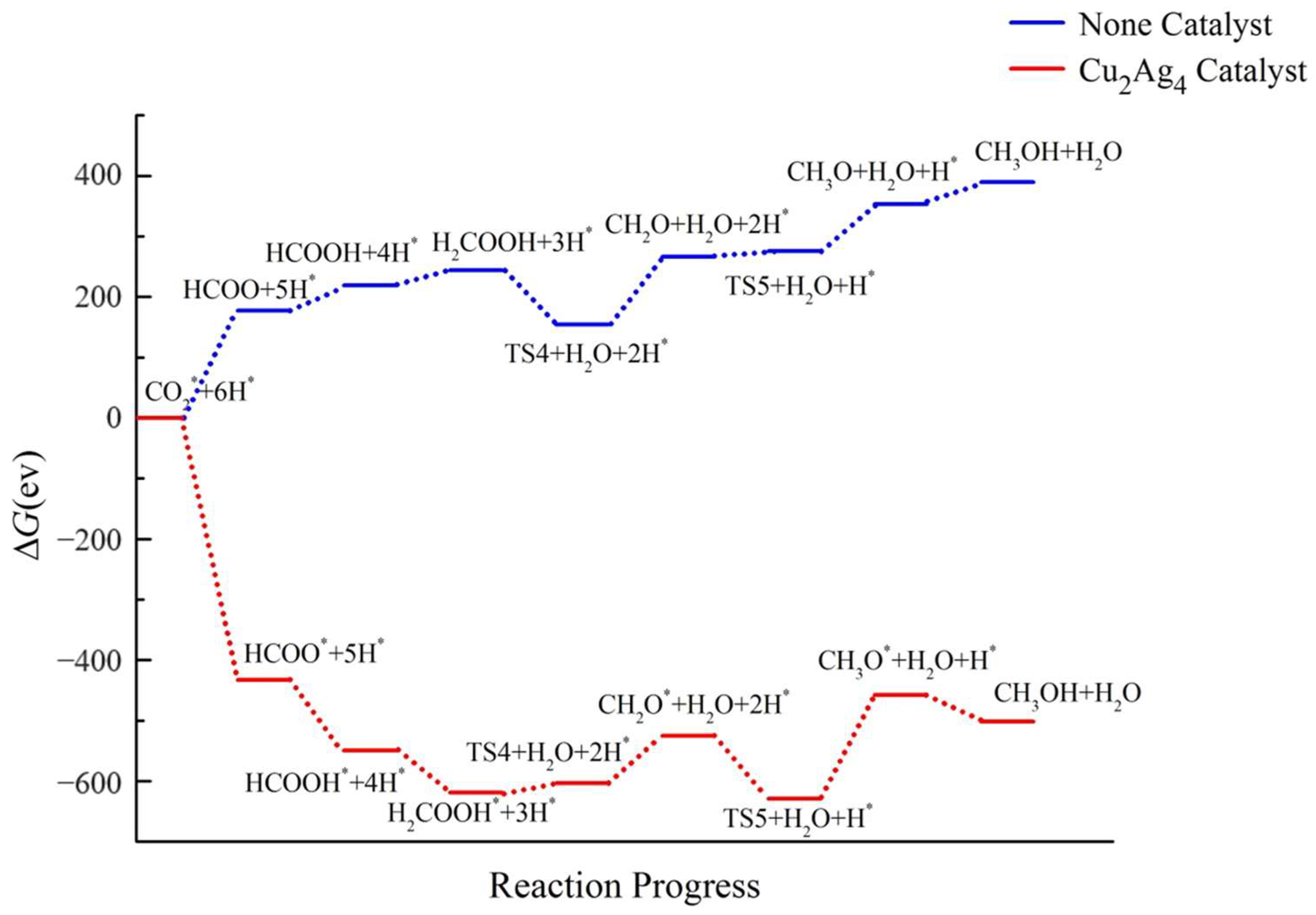
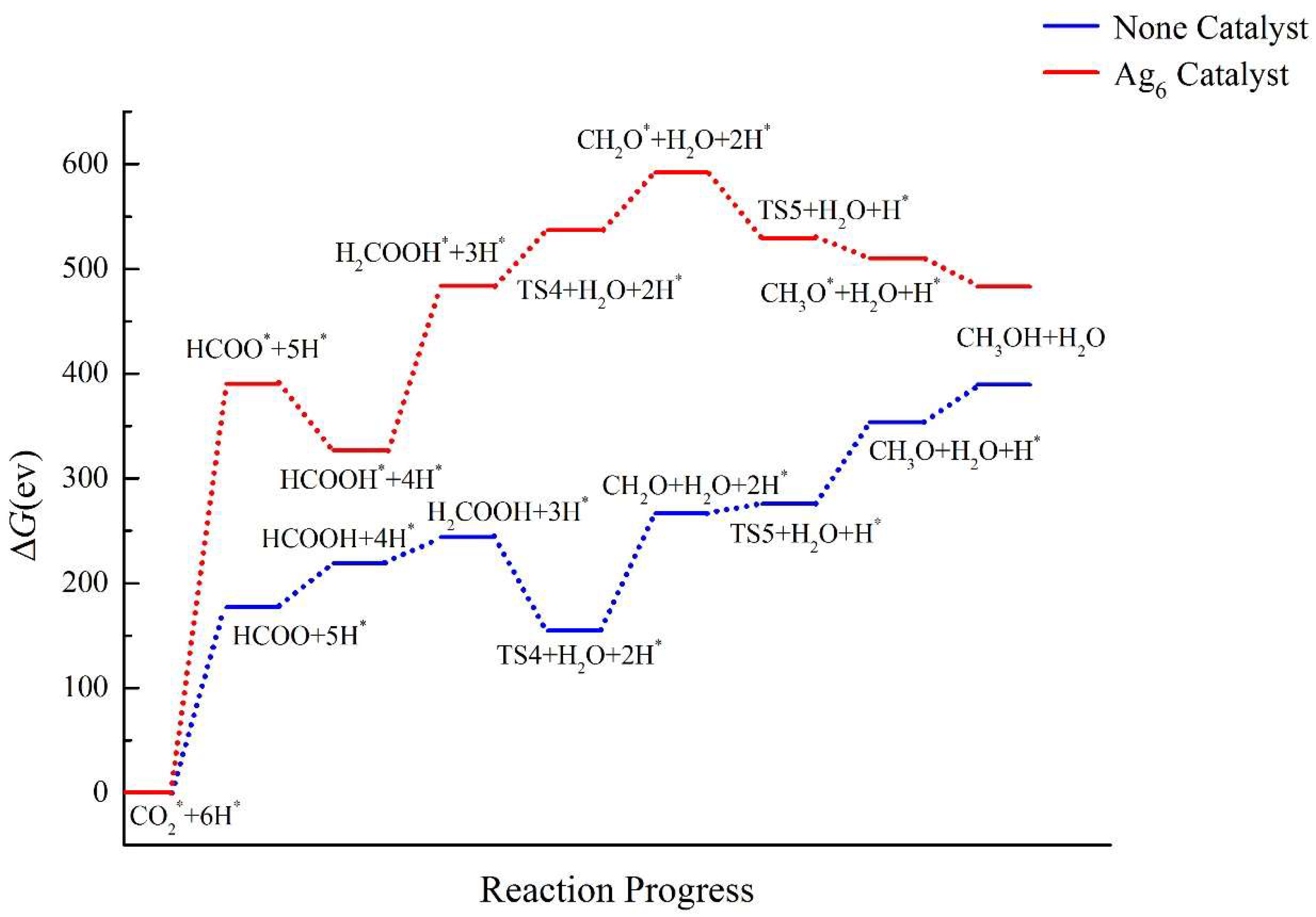
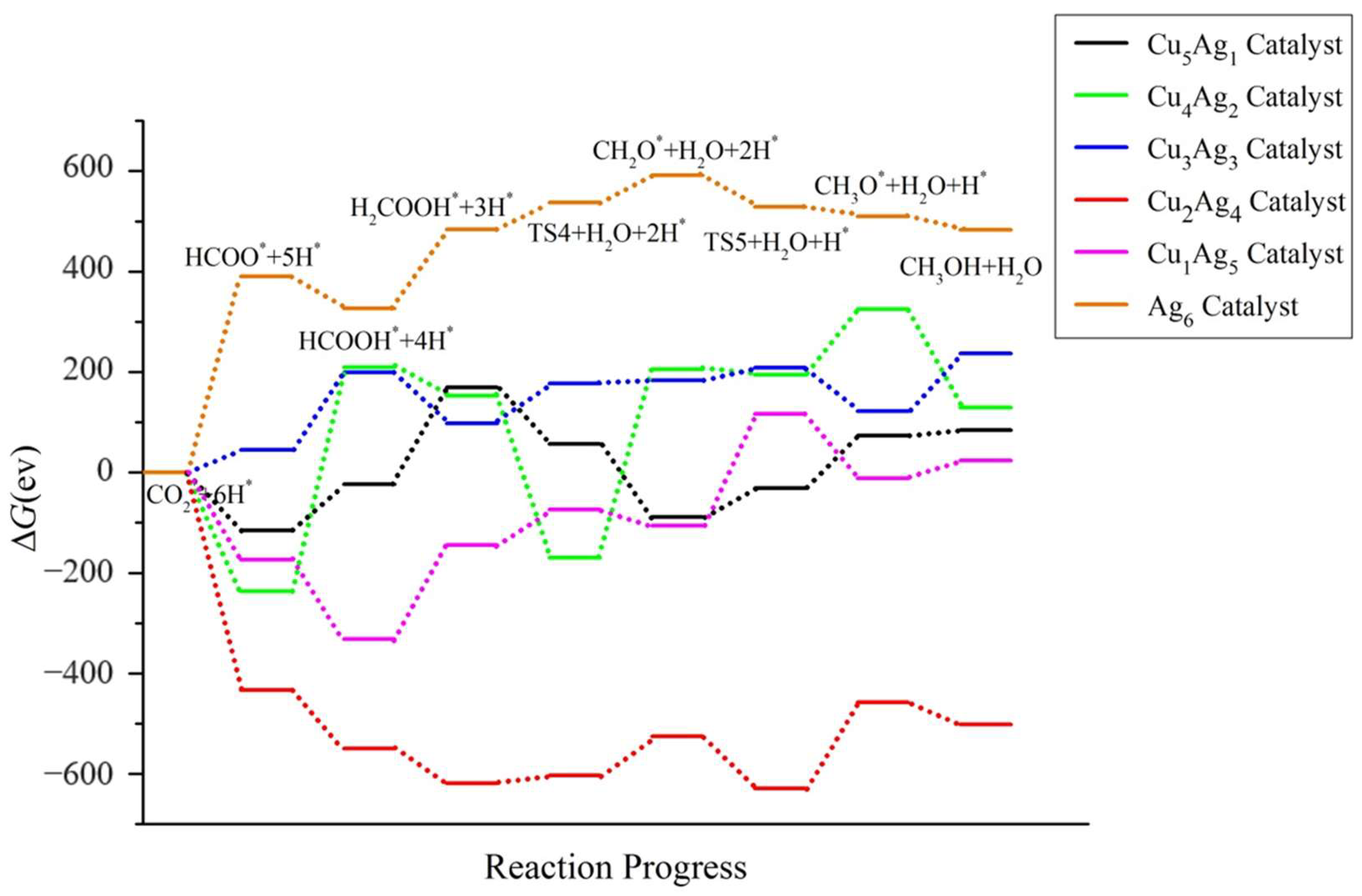
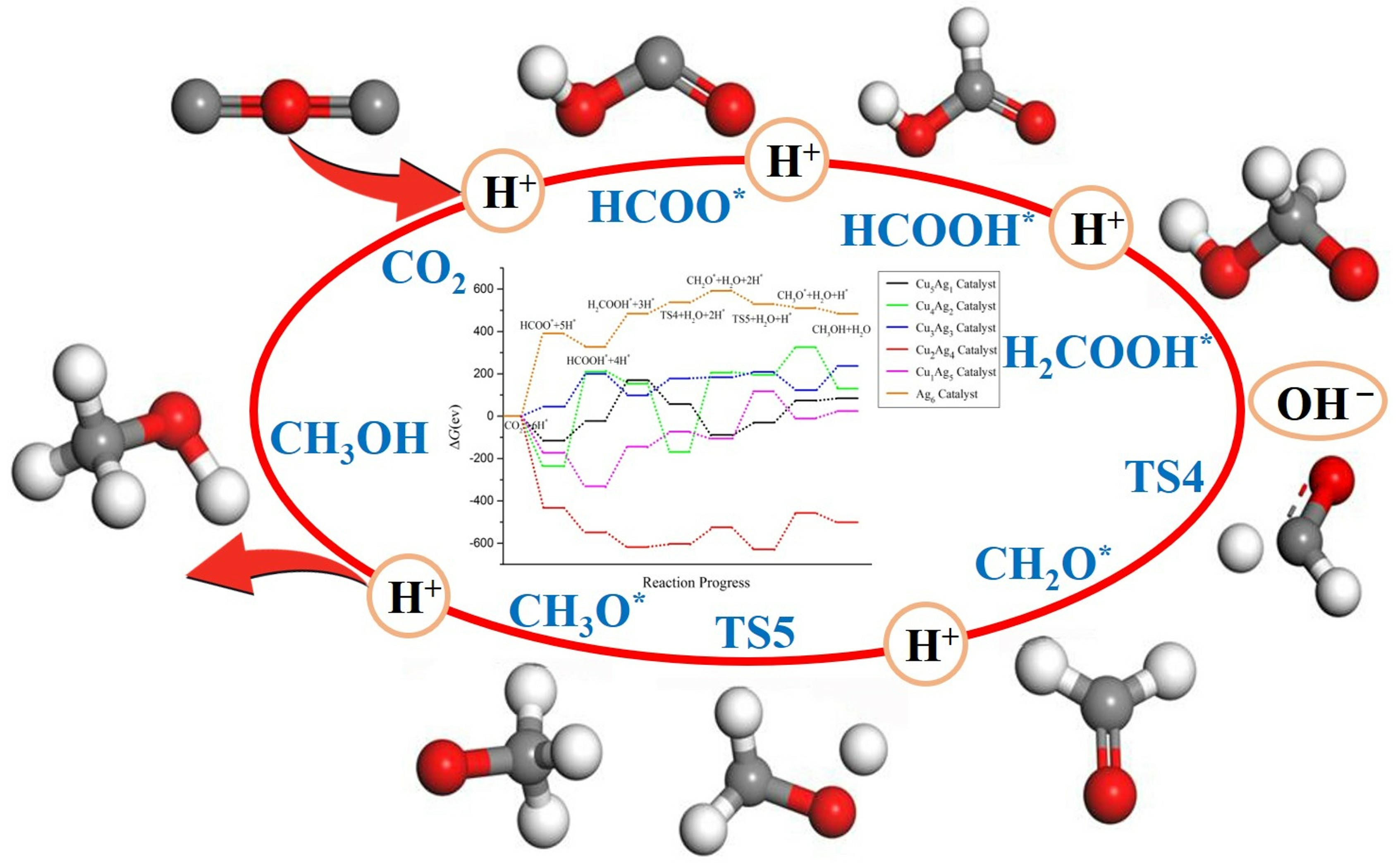
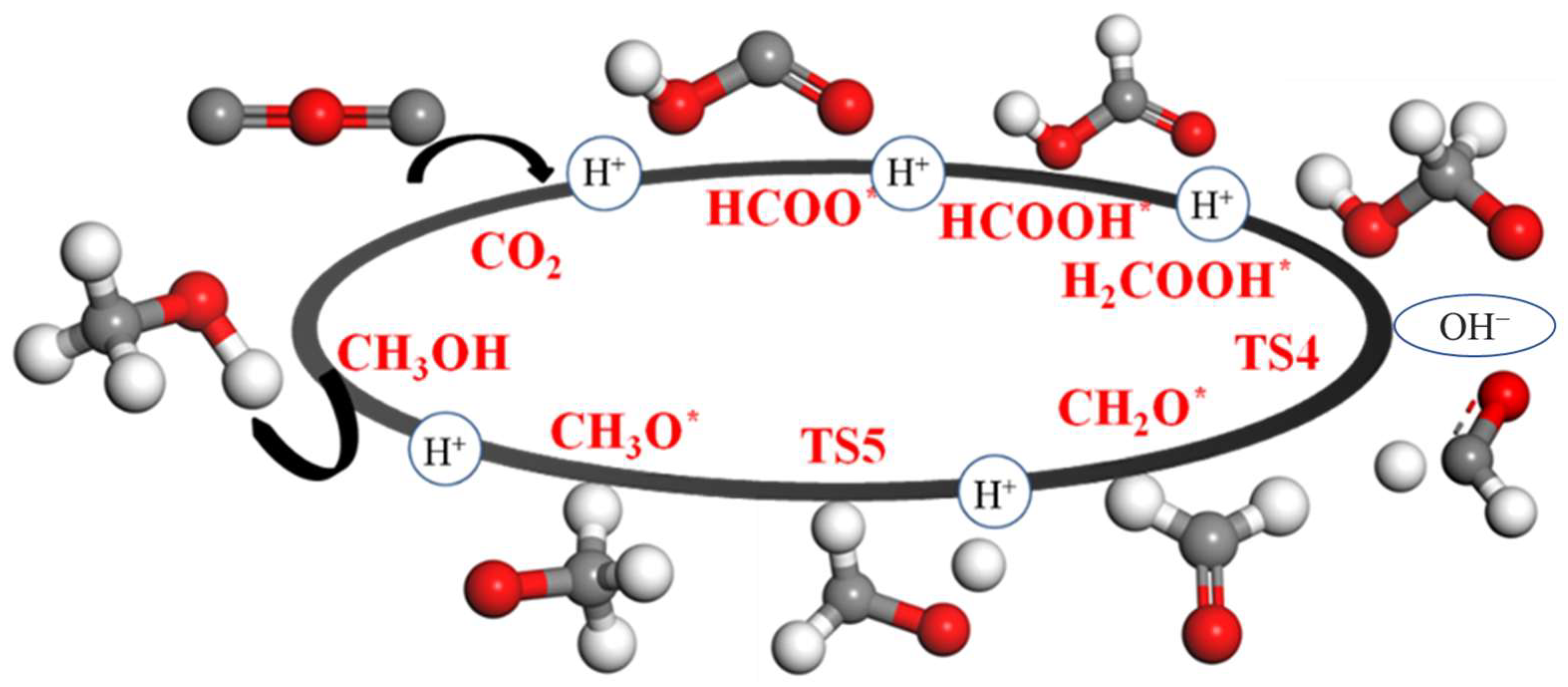
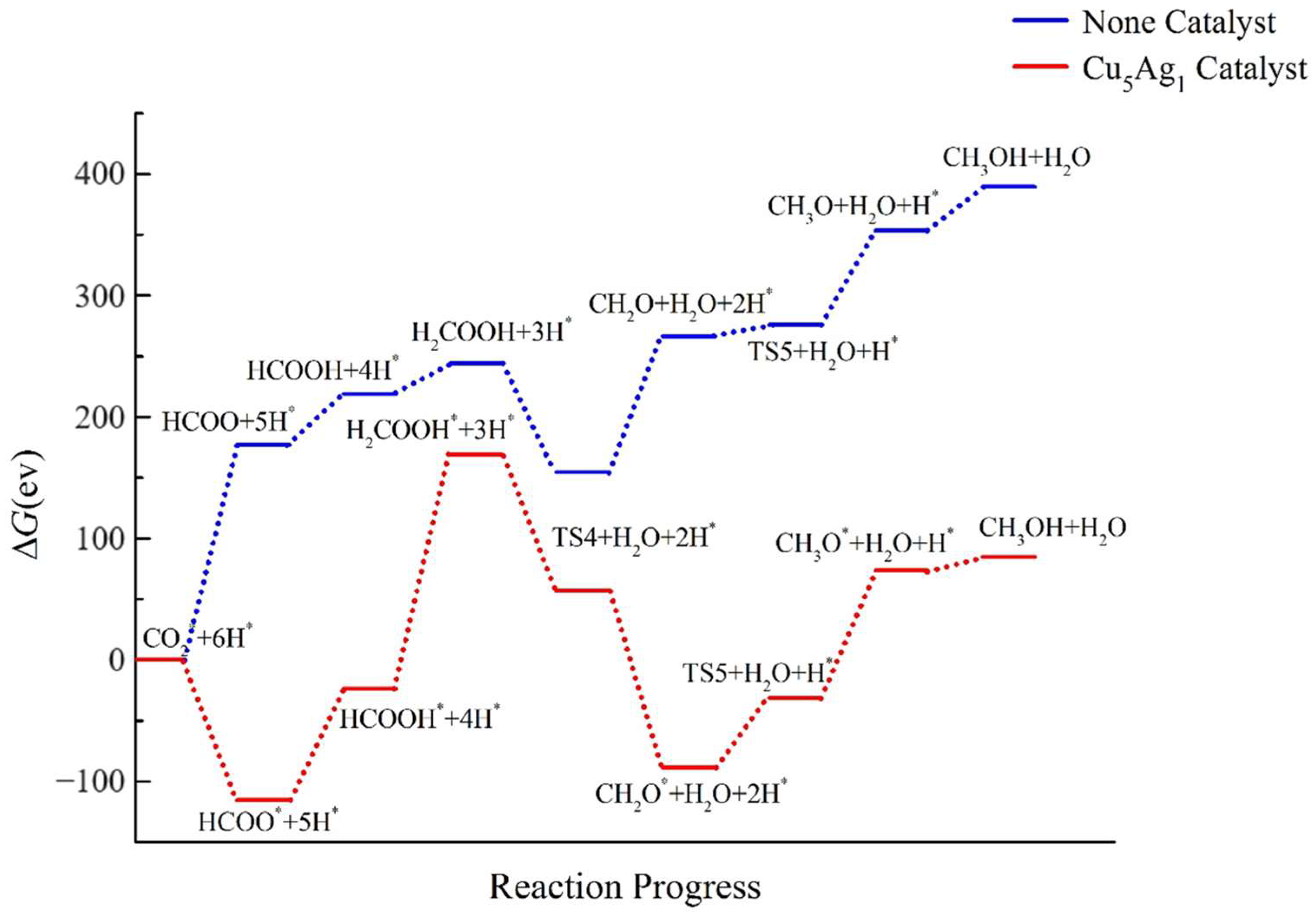
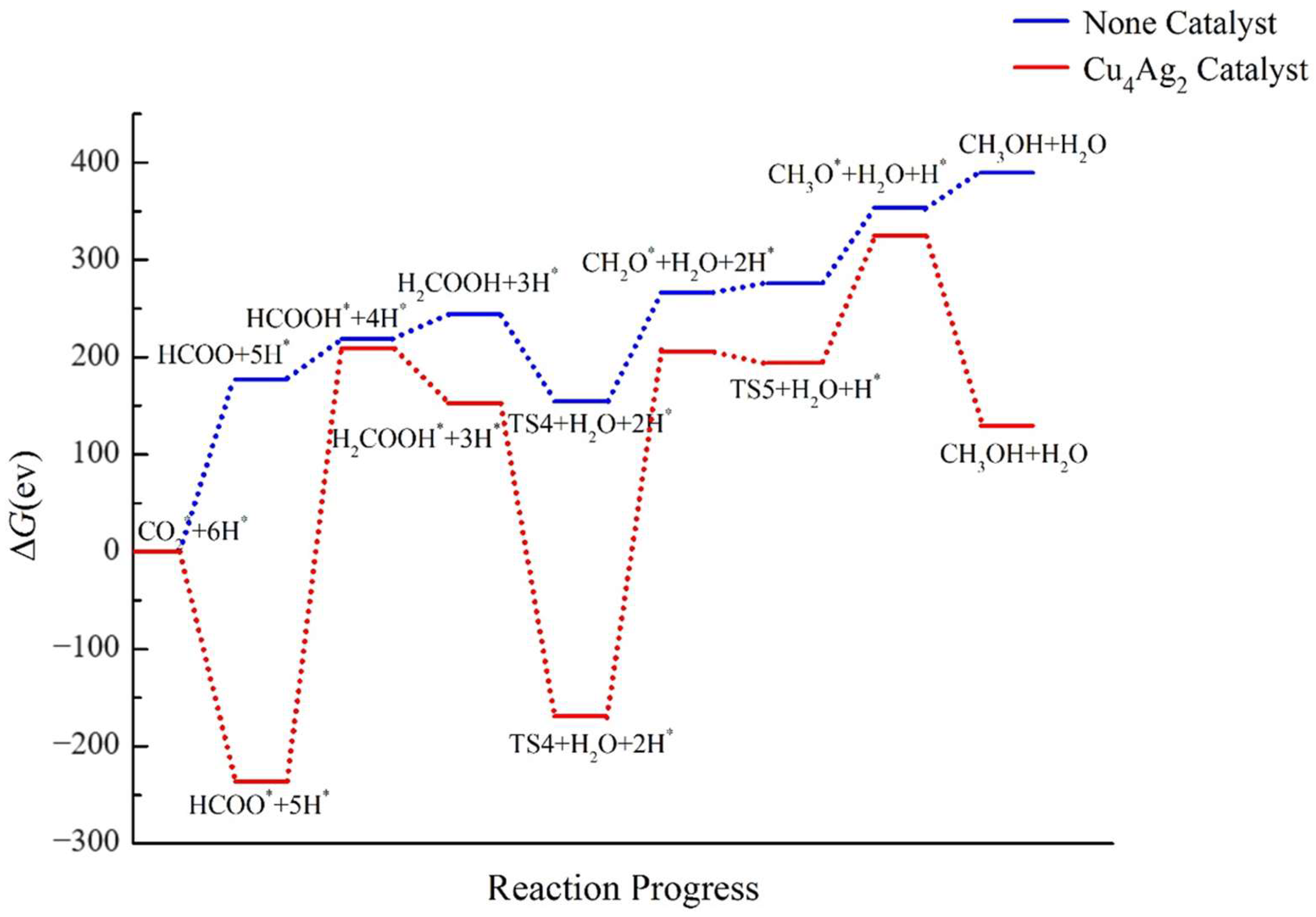
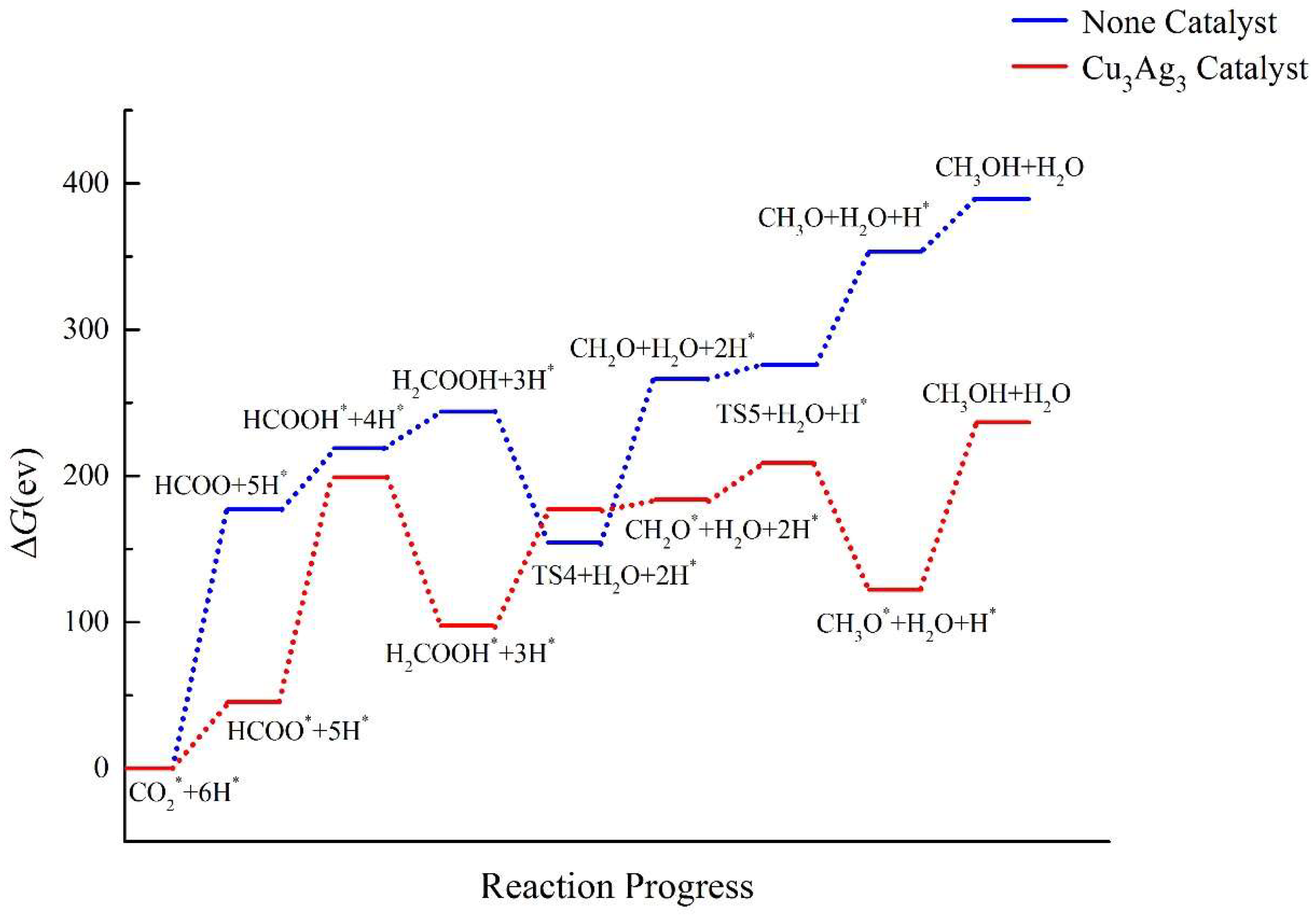
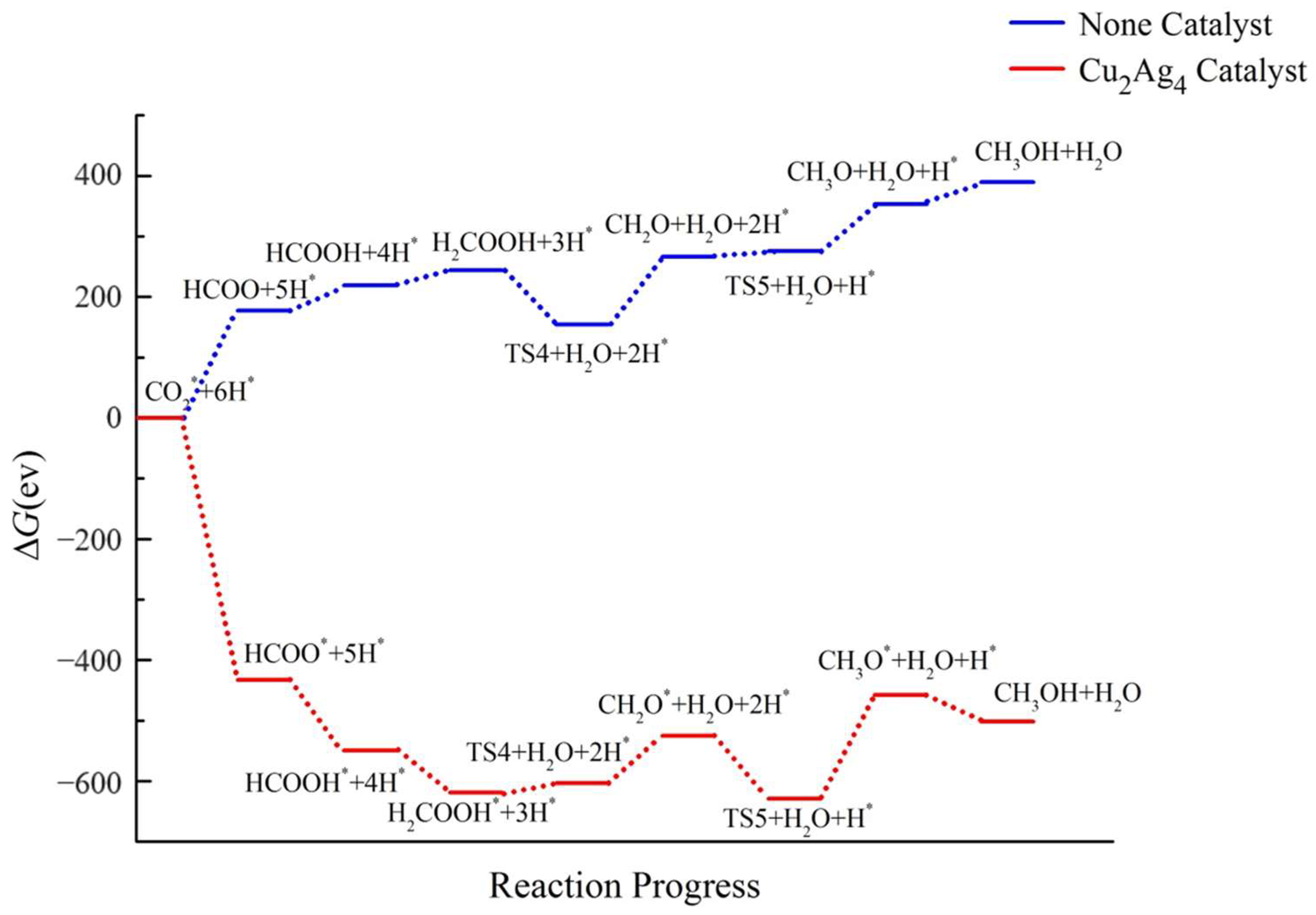
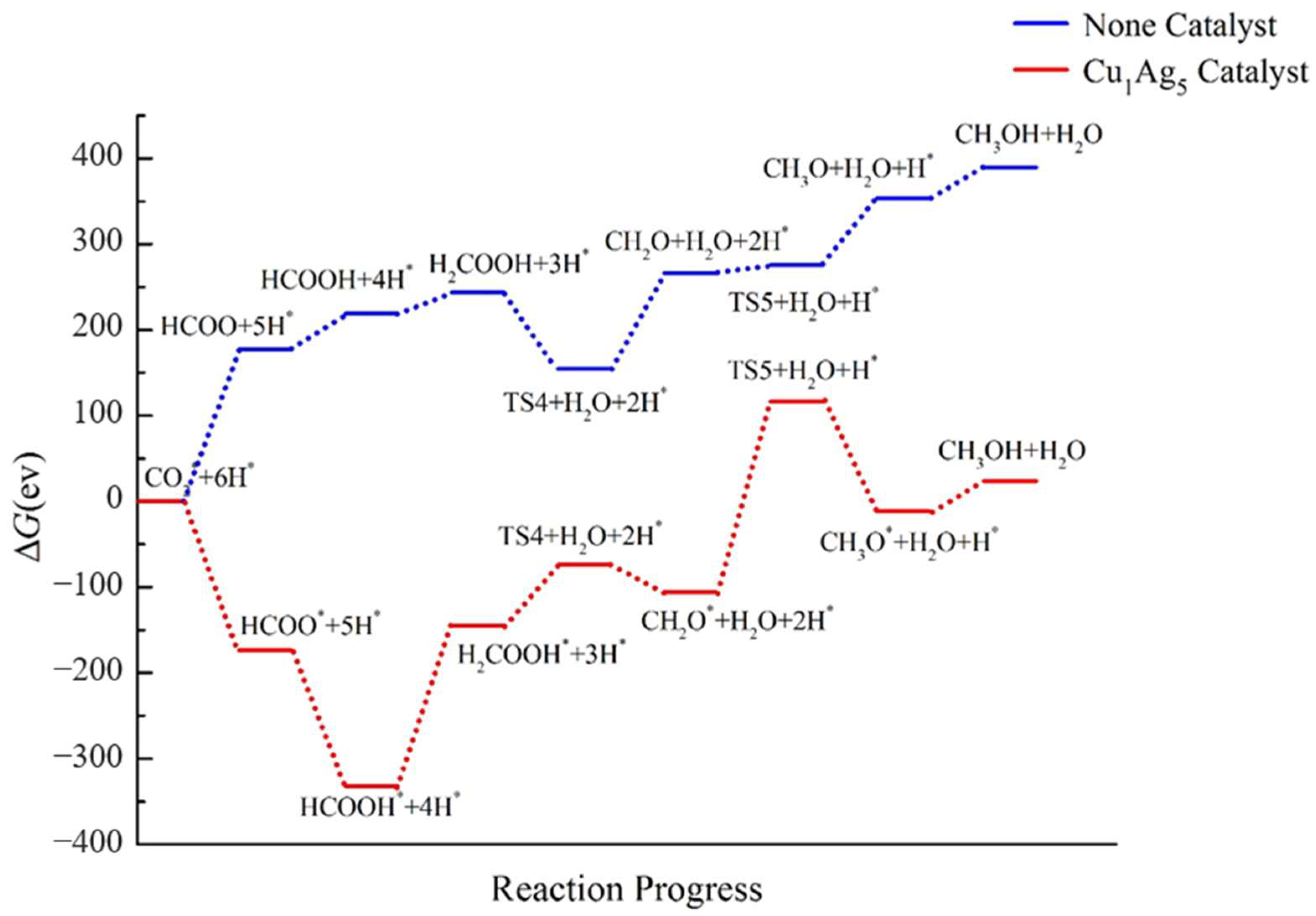
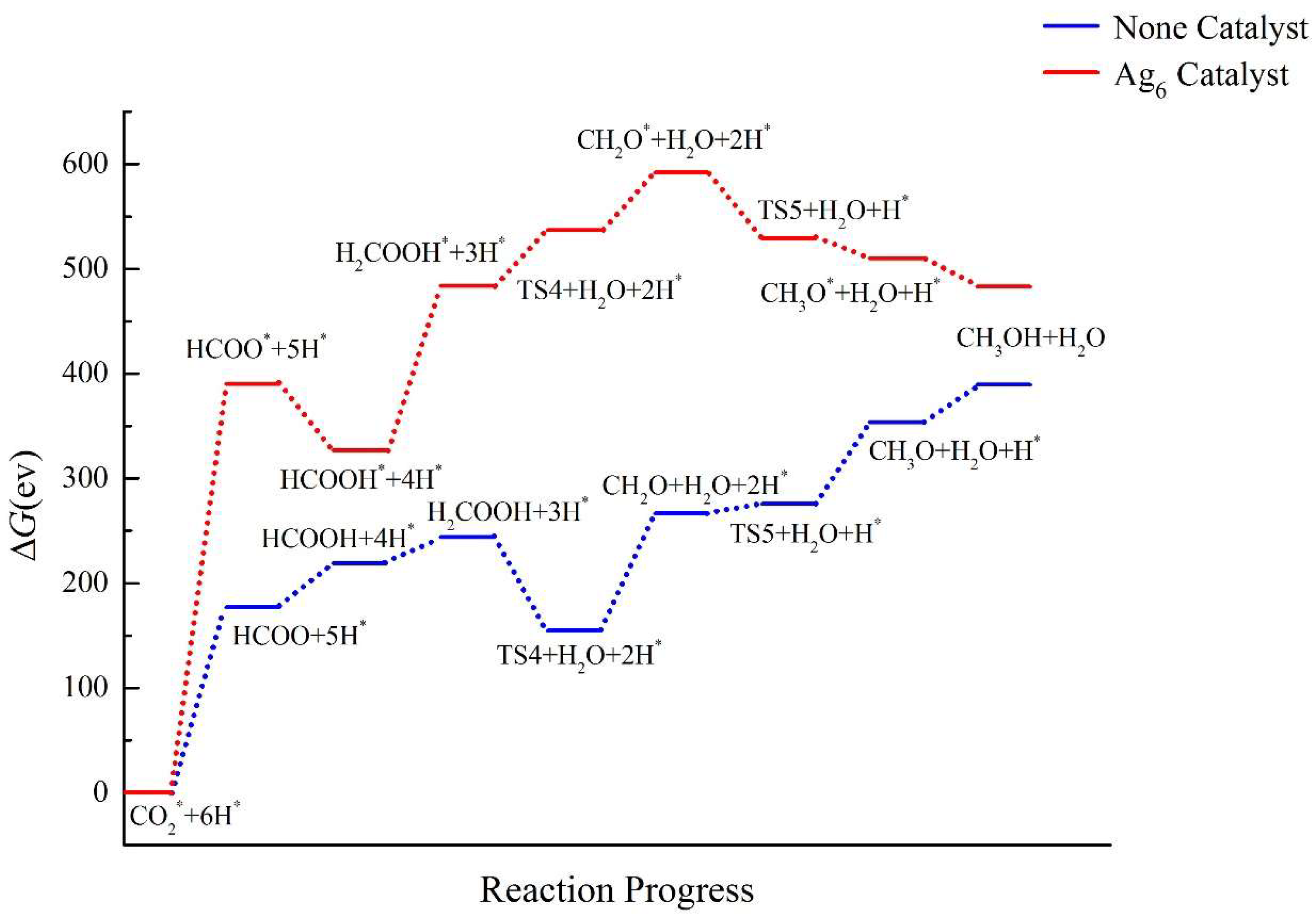
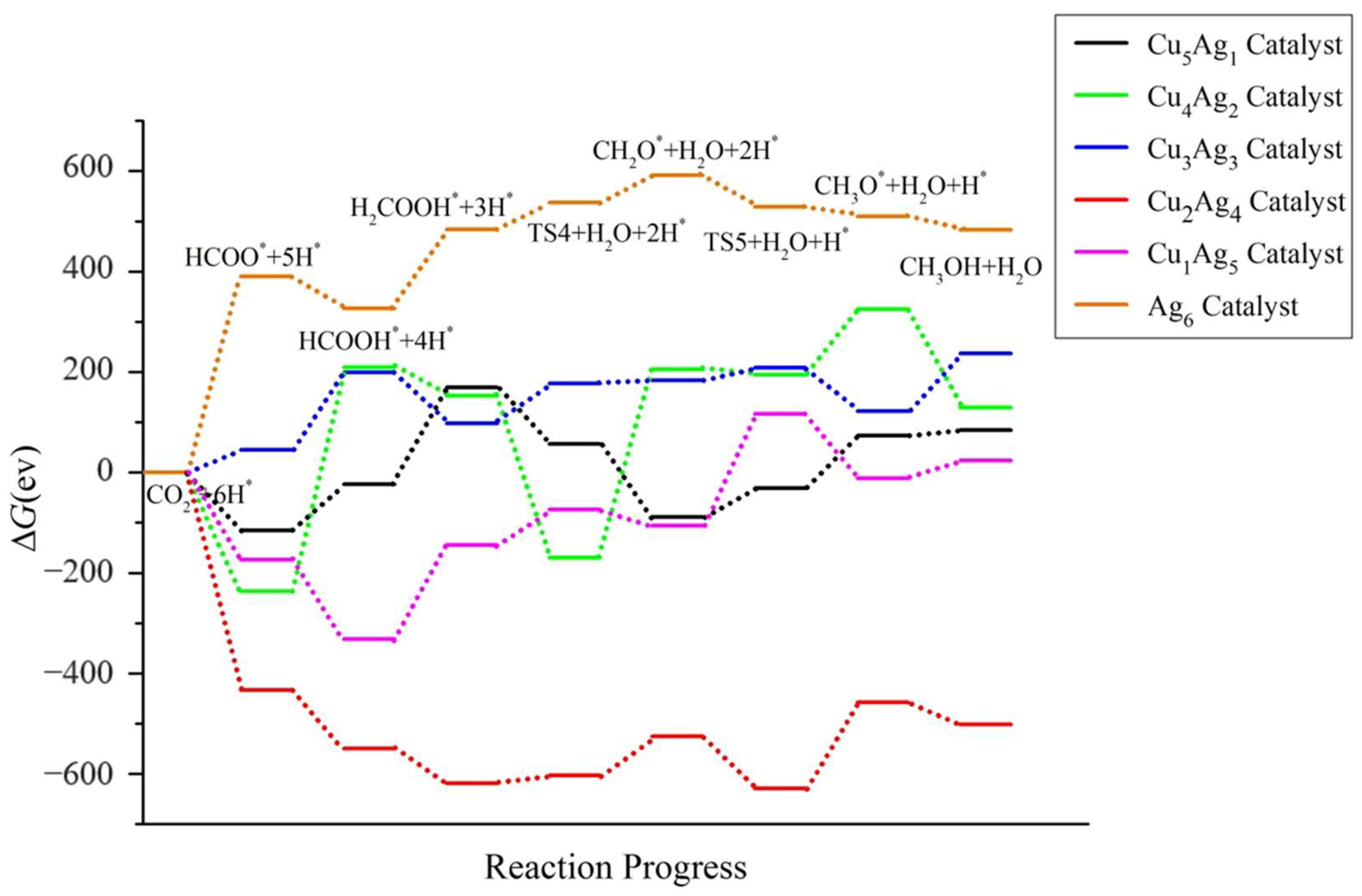
2.4. Pathway Study on CO2RR to CH3OH
3. Models and Methods
3.1. The Cluster Stability and CO2 Adsorption Structure Data Analysis
3.2. Data Analysis of CO2 Hydrogenation Reduction Path
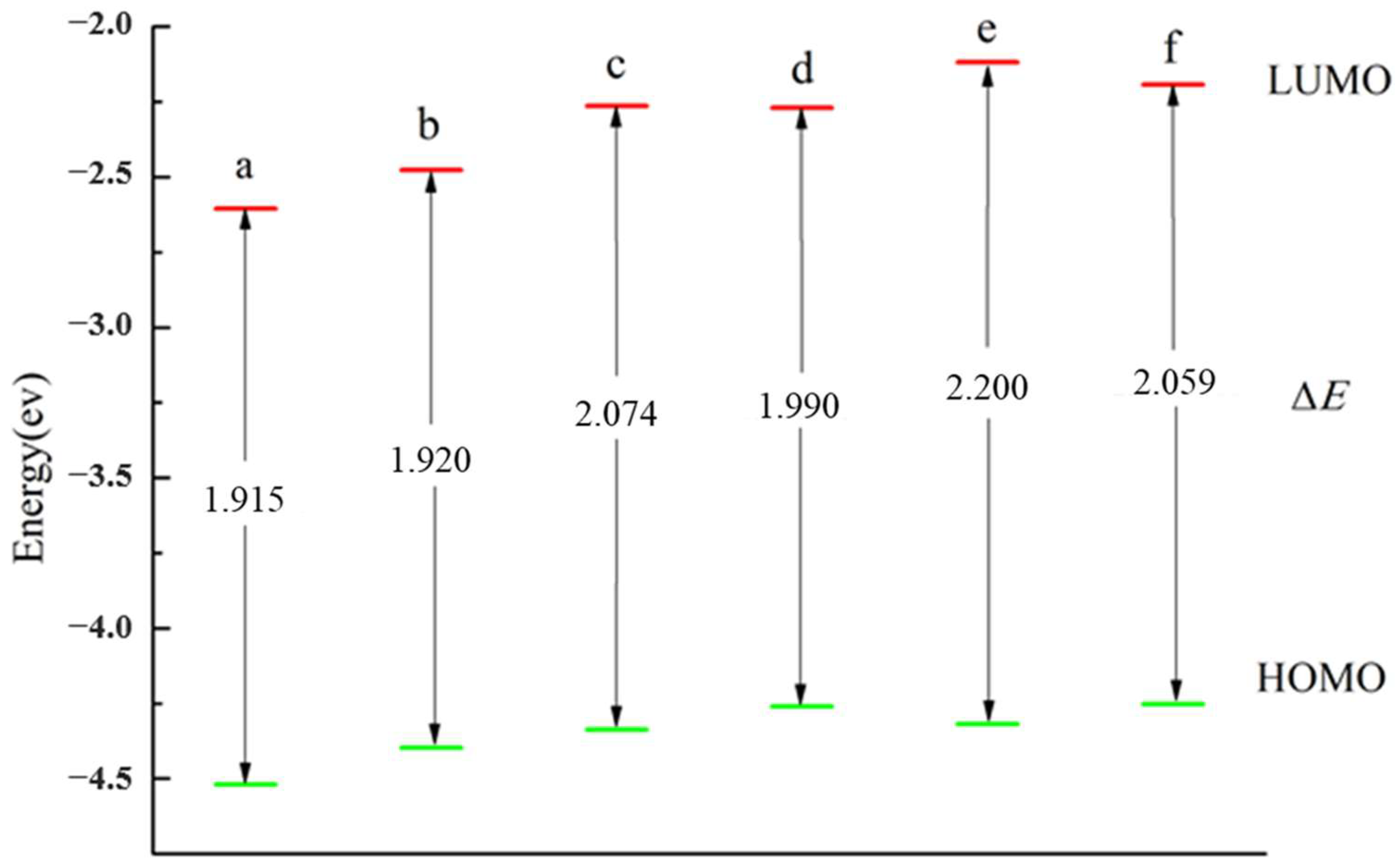
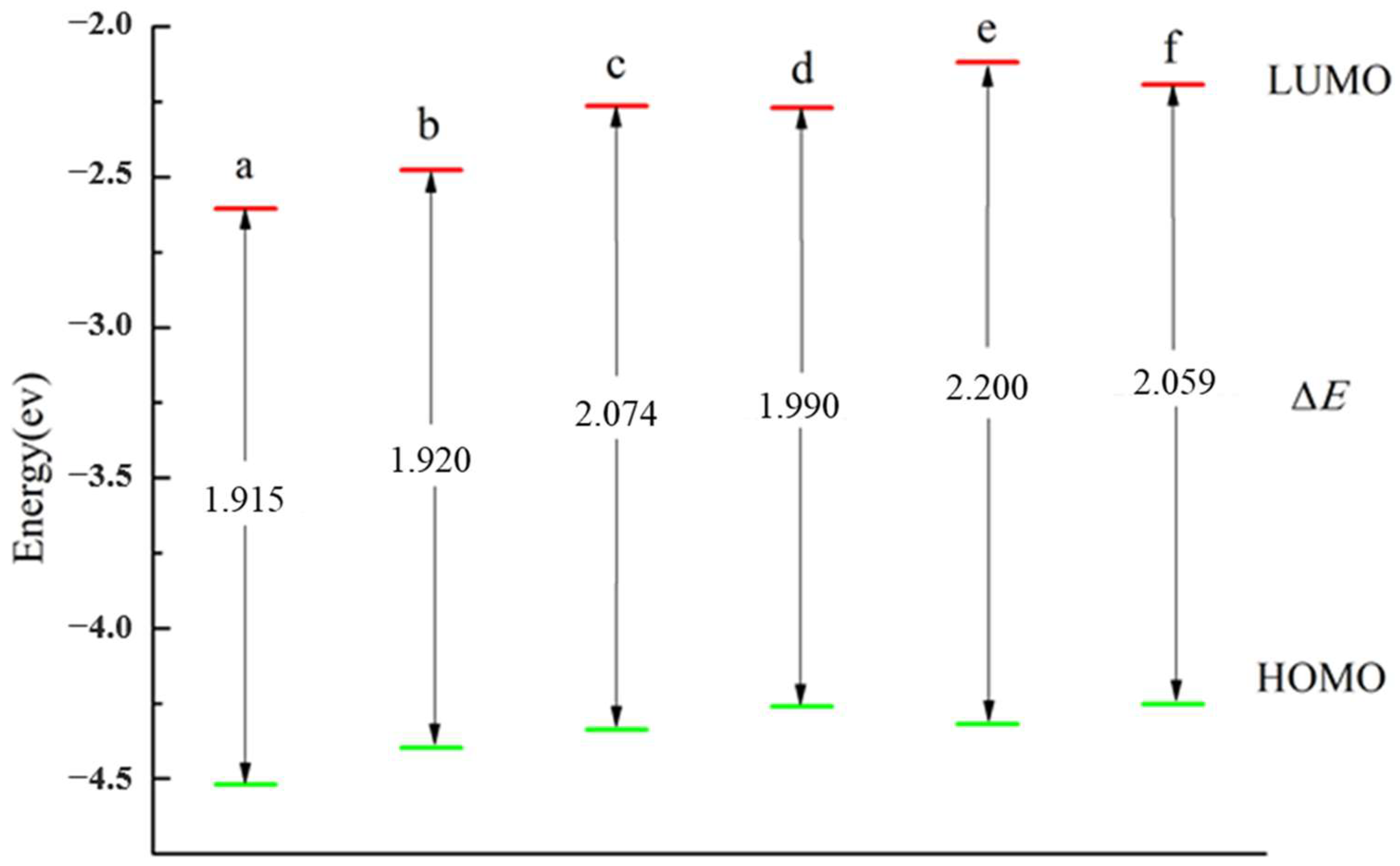
3.3. The HOMO, LUMO and ΔE of the Cluster
3.4. Calculation of the Fukui Index of Atoms in the Cluster
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ooi, R.E.; Foo, D.C.; Ng, D.K.; Tan, R.R. Planning of carbon capture and storage with pinch analysis techniques. Chem. Eng. Res. Des. 2013, 91, 2721–2731. [Google Scholar] [CrossRef]
- Hospital-Benito, D.; Lemus, J.; Moya, C.; Santiago, R.; Palomar, J. Process analysis overview of ionic liquids on CO2 chemical capture. Chem. Eng. J. 2020, 390, 124509. [Google Scholar] [CrossRef]
- Shi, Y.; Hou, S.; Qiu, X.; Zhao, B. MOFs-Based Catalysts Supported Chemical Conversion of CO2. Top. Curr. Chem. 2020, 378, 11. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Qu, Y.; Zhang, X.; Ma, H.; Xu, P.; Sun, J. A novel water-stable MOF Zn(Py)(Atz) as heterogeneous catalyst for chemical conversion of CO2 with various epoxides under mild conditions. J. CO2 Util. 2020, 35, 216–224. [Google Scholar] [CrossRef]
- Singh, A.K.; Singh, S.; Kumar, A. Hydrogen energy future with formic acid: A renewable chemical hydrogen storage system. Catal. Sci. Technol. 2016, 6, 12–40. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A.; Angelini, A. Catalysis for the Valorization of Exhaust Carbon: From CO2 to Chemicals, Materials, and Fuels. Technological Use of CO2. Chem. Rev. 2014, 114, 1709–1742. [Google Scholar] [CrossRef] [PubMed]
- Leitner, W. Carbon-Dioxide as a Raw-Material—The Synthesis of Formic-Acid and Its Derivatives from CO2. Angew. Chem. -Int. Ed. 1995, 34, 2207–2221. [Google Scholar] [CrossRef]
- Kortlever, R.; Shen, J.; Schouten, K.J.P.; Calle-Vallejo, F.; Koper, M.T.M. Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide. J. Phys. Chem. Lett. 2015, 6, 4073–4082. [Google Scholar] [CrossRef]
- Hori, Y.; Wakebe, H.; Tsukamoto, T.; Koga, O. Electrocatalytic Process of Co Selectivity in Electrochemical Reduction of CO2 at Metal-Electrodes in Aqueous-Media. Electrochim. Acta 1994, 39, 1833–1839. [Google Scholar] [CrossRef]
- Whipple, D.T.; Kenis, P.J.A. Prospects of CO2 Utilization via Direct Heterogeneous Electrochemical Reduction. J. Phys. Chem. Lett. 2010, 1, 3451–3458. [Google Scholar] [CrossRef]
- Gao, D.; Zhou, H.; Cai, F.; Wang, J.-G.; Wang, G.; Bao, X. Pd-Containing Nanostructures for Electrochemical CO2 Reduction Reaction. Acs Catal. 2018, 8, 1510–1519. [Google Scholar] [CrossRef]
- Yoo, J.S.; Christensen, R.; Vegge, T.; Nørskov, J.K.; Studt, F. Theoretical Insight into the Trends that Guide the Electrochemical Reduction of Carbon Dioxide to Formic Acid. Chemsuschem 2016, 9, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Back, S.; Kim, H.; Jung, Y. Selective Heterogeneous CO2 Electroreduction to Methanol. Acs Catal. 2015, 5, 965–971. [Google Scholar] [CrossRef]
- Peterson, A.A.; Abild-Pedersen, F.; Studt, F.; Rossmeisl, J.; Nørskov, J.K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 2010, 3, 1311–1315. [Google Scholar] [CrossRef]
- Dinh, C.-T.; Burdyny, T.; Kibria, M.G.; Seifitokaldani, A.; Gabardo, C.M.; de Arquer, F.P.G.; Kiani, A.; Edwards, J.P.; De Luna, P.; Bushuyev, O.S.; et al. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 2018, 360, 783–787. [Google Scholar] [CrossRef]
- Gattrell, M.; Gupta, N.; Co, A. A review of the aqueous electrochemical reduction of CO2 to hydrocarbons at copper. J. Electroanal. Chem. 2006, 594, 1–19. [Google Scholar] [CrossRef]
- Nie, X.; Luo, W.; Janik, M.J.; Asthagiri, A. Reaction mechanisms of CO2 electrochemical reduction on Cu(111) determined with density functional theory. J. Catal. 2014, 312, 108–122. [Google Scholar] [CrossRef]
- Ren, D.; Fong, J.; Yeo, B.S. The effects of currents and potentials on the selectivities of copper toward carbon dioxide electroreduction. Nat. Commun. 2018, 9, 925. [Google Scholar] [CrossRef]
- Reske, R.; Mistry, H.; Behafarid, F.; Cuenya, B.R.; Strasser, P. Particle Size Effects in the Catalytic Electroreduction of CO2 on Cu Nanoparticles. J. Am. Chem. Soc. 2014, 136, 6978–6986. [Google Scholar] [CrossRef]
- Choi, C.; Cheng, T.; Espinosa, M.F.; Fei, H.; Duan, X.; Goddard, W.A.; Huang, Y. A Highly Active Star Decahedron Cu Nanocatalyst for Hydrocarbon Production at Low Overpotentials. Adv. Mater. 2019, 31, e1805405. [Google Scholar] [CrossRef]
- Ma, M.; Djanashvili, K.; Smith, W.A. Controllable Hydrocarbon Formation from the Electrochemical Reduction of CO2 over Cu Nanowire Arrays. Angew. Chem. Int. Ed. 2016, 55, 6680–6684. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cui, F.; Ross, M.B.; Kim, D.; Sun, Y.; Yang, P. Structure-Sensitive CO2 Electroreduction to Hydrocarbons on Ultrathin 5-fold Twinned Copper Nanowires. Nano Lett. 2017, 17, 1312–1317. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Jiang, K.; Fan, S.; Kanan, M.W. A Direct Grain-Boundary-Activity Correlation for CO Electroreduction on Cu Nanoparticles. ACS Cent. Sci. 2016, 2, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Hirunsit, P.; Soodsawang, W.; Limtrakul, J. CO2 Electrochemical Reduction to Methane and Methanol on Copper-Based Alloys: Theoretical Insight. J. Phys. Chem. C 2015, 119, 8238–8249. [Google Scholar] [CrossRef]
- Zheng, X.; Ji, Y.; Tang, J.; Wang, J.; Liu, B.; Steinrück, H.-G.; Lim, K.; Li, Y.; Toney, M.F.; Chan, K.; et al. Theory-guided Sn/Cu alloying for efficient CO2 electroreduction at low overpotentials. Nat. Catal. 2019, 2, 55–61. [Google Scholar] [CrossRef]
- Kim, D.; Resasco, J.; Yu, Y.; Asiri, A.M.; Yang, P. Synergistic geometric and electronic effects for electrochemical reduction of carbon dioxide using gold-copper bimetallic nanoparticles. Nat. Commun. 2014, 5, 4948. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Mensi, M.; Oveisi, E.; Mantella, V.; Buonsanti, R. Structural Sensitivities in Bimetallic Catalysts for Electrochemical CO2 Reduction Revealed by Ag-Cu Nanodimers. J. Am. Chem. Soc. 2019, 141, 2490–2499. [Google Scholar] [CrossRef]
- Zhu, W.; Tackett, B.M.; Chen, J.G.; Jiao, F. Bimetallic Electrocatalysts for CO2 Reduction. Top. Curr. Chem. 2018, 376, 41. [Google Scholar] [CrossRef]
- Kim, D.; Xie, C.; Becknell, N.; Yu, Y.; Karamad, M.; Chan, K.; Crumlin, E.J.; Nørskov, J.K.; Yang, P. Electrochemical Activation of CO2 through Atomic Ordering Transformations of AuCu Nanoparticles. J. Am. Chem. Soc. 2017, 139, 8329–8336. [Google Scholar] [CrossRef]
- Zhang, S.; Kang, P.; Bakir, M.; Lapides, A.M.; Dares, C.J.; Meyer, T.J. Polymer-supported CuPd nanoalloy as a synergistic catalyst for electrocatalytic reduction of carbon dioxide to methane. Proc. Natl. Acad. Sci. USA 2015, 112, 15809–15814. [Google Scholar] [CrossRef]
- Sarfraz, S.; Garcia-Esparza, A.T.; Jedidi, A.; Cavallo, L.; Takanabe, K. Cu-Sn Bimetallic Catalyst for Selective Aqueous Electroreduction of CO2 to CO. ACS Catal. 2016, 6, 2842–2851. [Google Scholar] [CrossRef]
- Choi, C.; Cai, J.; Lee, C.; Lee, H.M.; Xu, M.; Huang, Y. Intimate atomic Cu-Ag interfaces for high CO2RR selectivity towards CH4 at low over potential. Nano Res. 2021, 14, 3497–3501. [Google Scholar] [CrossRef]
- Guan, W.; Zhu, H.; Zhang, Y.; Ren, X.; Ma, T.; Liu, A. Molecular structure design and interface behavior of ionic liquids on metal surfaces: A theoretical study. Surf. Interfaces 2022, 34, 102314. [Google Scholar] [CrossRef]
- Liu, A.; Yang, Y.; Kong, D.; Ren, X.; Gao, M.; Liang, X.; Yang, Q.; Zhang, J.; Gao, L.; Ma, T. DFT study of the defective carbon materials with vacancy and heteroatom as catalyst for NRR. Appl. Surf. Sci. 2021, 536, 147851. [Google Scholar] [CrossRef]
- Liu, A.; Guan, W.; Zhao, X.; Ren, X.; Liang, X.; Gao, L.; Li, Y.; Ma, T. Investigation on the interfacial behavior of polyorganic inhibitors on a metal surface by DFT study and MD simulation. Appl. Surf. Sci. 2021, 541, 148570. [Google Scholar] [CrossRef]
- Liu, A.; Guan, W.; Wu, K.; Ren, X.; Gao, L.; Ma, T. Density functional theory study of nitrogen-doped graphene as a high-performance electrocatalyst for CO2RR. Appl. Surf. Sci. 2021, 540, 148319. [Google Scholar] [CrossRef]
- Liang, X.; Ren, X.; Guo, M.; Li, Y.; Xiong, W.; Guan, W.; Gao, L.; Liu, A. CO2 electroreduction by AuCu bimetallic clusters: A first principles study. Int. J. Energy Res. 2021, 45, 18684–18694. [Google Scholar] [CrossRef]
- Zhu, H.; Liang, Z.; Xue, S.; Ren, X.; Liang, X.; Xiong, W.; Gao, L.; Liu, A. DFT practice in MXene-based materials for electrocatalysis and energy storage: From basics to applications. Ceram. Int. 2022, 48, 27217–27239. [Google Scholar] [CrossRef]
- Huang, X.; Tang, J.; Luo, B.; Knibbe, R.; Lin, T.; Hu, H.; Wang, L. Sandwich-Like Ultrathin TiS2 Nanosheets Confined within N, S Codoped Porous Carbon as an Effective Polysulfide Promoter in Lithium-Sulfur Batteries. Adv. Energy Mater. 2019, 9, 1901872. [Google Scholar] [CrossRef]
- Tang, Q.; Lee, Y.; Li, D.-Y.; Choi, W.; Liu, C.W.; Lee, D.; Jiang, D.-E. Lattice-Hydride Mechanism in Electrocatalytic CO2 Reduction by Structurally Precise Copper-Hydride Nanoclusters. J. Am. Chem. Soc. 2017, 139, 9728–9736. [Google Scholar] [CrossRef]
- Nie, X.; Jiang, X.; Wang, H.; Luo, W.; Janik, M.J.; Chen, Y.; Guo, X.; Song, C. Mechanistic Understanding of Alloy Effect and Water Promotion for Pd-Cu Bimetallic Catalysts in CO2 Hydrogenation to Methanol. ACS Catal. 2018, 8, 4873–4892. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Configuration | Site | Value | Site | Value | Site | Value | Site | Value |
|---|---|---|---|---|---|---|---|---|
| a | Cu (4/6) | +0.237 | Ag (1) | −0.228 | Cu (4/6) | −0.204 | Ag (1) | +0.175 |
| b | Cu (6) | +0.276 | Ag (1/4) | −0.229 | Cu (6) | −0.185 | Ag (1/4) | +0.183 |
| c | Ag (1) | +0.233 | Ag (4/6) | +0.221 | Ag (1) | −0.219 | Ag (4/6) | −0.217 |
| d | Ag (1) | +0.238 | Ag (4/6) | −0.252 | Ag (4/6) | +0.211 | Ag (1) | −0.182 |
| e | Ag (1/4) | −0.252 | Ag (6) | +0.241 | Ag (1/4) | +0.210 | Ag (6) | −0.206 |
| f | Ag (1/4/6) | −0.234 | Ag (1/4/6) | −0.231 | Ag (2/3/5) | +0.103 | Ag (2/3/5) | −0.102 |
| Isomers | ∠O1CO2/° | dC−O1/Å | dC−O2/Å | ECuAg−CO2/Ha | Eobs/eV |
|---|---|---|---|---|---|
| a | 175.605 | 1.264 | 1.449 | −13,589.920 | −0.555 |
| b | 171.015 | 1.330 | 1.332 | −13,589.911 | −0.299 |
| c | 177.676 | 1.270 | 1.469 | −13,589.905 | −0.1439 |
| d | 170.699 | 1.322 | 1.332 | −13,589.911 | −0.301 |
| e | 173.680 | 1.335 | 1.346 | −13,589.906 | −0.163 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, S.; Liang, X.; Zhang, Q.; Ren, X.; Gao, L.; Ma, T.; Liu, A. Density Functional Theory Study of CuAg Bimetal Electrocatalyst for CO2RR to Produce CH3OH. Catalysts 2024, 14, 7. https://doi.org/10.3390/catal14010007
Xue S, Liang X, Zhang Q, Ren X, Gao L, Ma T, Liu A. Density Functional Theory Study of CuAg Bimetal Electrocatalyst for CO2RR to Produce CH3OH. Catalysts. 2024; 14(1):7. https://doi.org/10.3390/catal14010007
Chicago/Turabian StyleXue, Sensen, Xingyou Liang, Qing Zhang, Xuefeng Ren, Liguo Gao, Tingli Ma, and Anmin Liu. 2024. "Density Functional Theory Study of CuAg Bimetal Electrocatalyst for CO2RR to Produce CH3OH" Catalysts 14, no. 1: 7. https://doi.org/10.3390/catal14010007
APA StyleXue, S., Liang, X., Zhang, Q., Ren, X., Gao, L., Ma, T., & Liu, A. (2024). Density Functional Theory Study of CuAg Bimetal Electrocatalyst for CO2RR to Produce CH3OH. Catalysts, 14(1), 7. https://doi.org/10.3390/catal14010007