
Density Functional Theory Study of Mechanism of Reduction of N2O by CO over Fe-ZSM-5 Zeolites

Abstract

1. Introduction
2. Results and Discussion
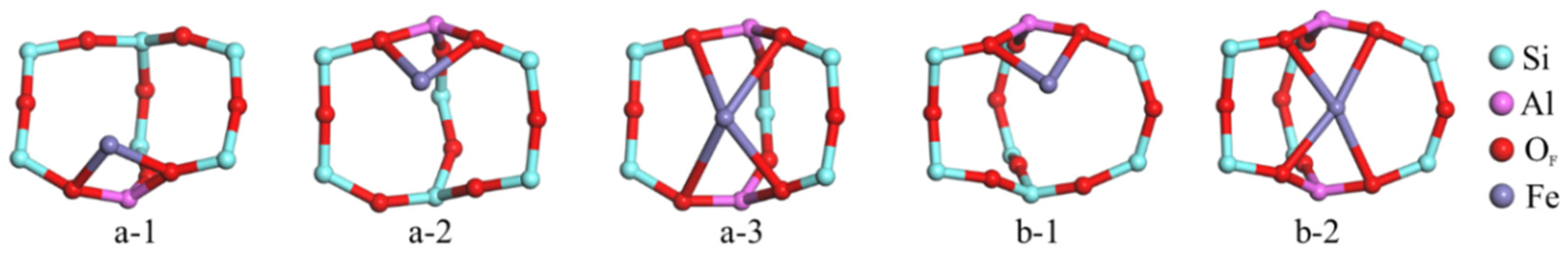
2.1. Location and Bonding Energy of Mononuclear Fe Species in Fe-ZSM-5
2.1.1. Optimization Configuration of Fe-ZSM-5
2.1.2. Prediction of Reactive Active Sites on Fe-ZSM-5
2.2. Proposed Catalytic Paths over Fe-ZSM-5
| Part 1: | ||
| Step 1 | Z-Fe + N2O→ Z-Fe-N2O | |
| Step 2 | Z-Fe-N2O → Z-Fe-N2O-TS | |
| Step 3 | Z-Fe-N2O-TS → Z-Fe-O-N2 | |
| Step 4 | Z-Fe-O-N2 → Z-Fe-O + N2 | |
| Part 2-1: | ||
| Step 5 | Z-Fe-O + N2O → Z-Fe-O-N2O | |
| Step 6 | Z-Fe-O-N2O → Z-Fe-O-N2O-TS | |
| Step 7 | Z-Fe-O-N2O-TS → Z-Fe-O-O-N2 | |
| Step 8 | Z-Fe-O-O-N2 → Z-Fe-O-O + N2 | |
| Step 9 | Z-Fe-O-O → Z-Fe-O2 | |
| Step 10 | Z-Fe-O2 → Z-Fe + O2 | |
| Part 2-2: | ||
| Step 11 | Z-Fe-O + CO → Z-Fe-O-CO | |
| Step 12 | Z-Fe-O-CO → Z-Fe-O-CO-TS | |
| Step 13 | Z-Fe-O-CO-TS → Z-Fe-CO2 | |
| Step 14 | Z-Fe-CO2 → Z-Fe + CO2 |
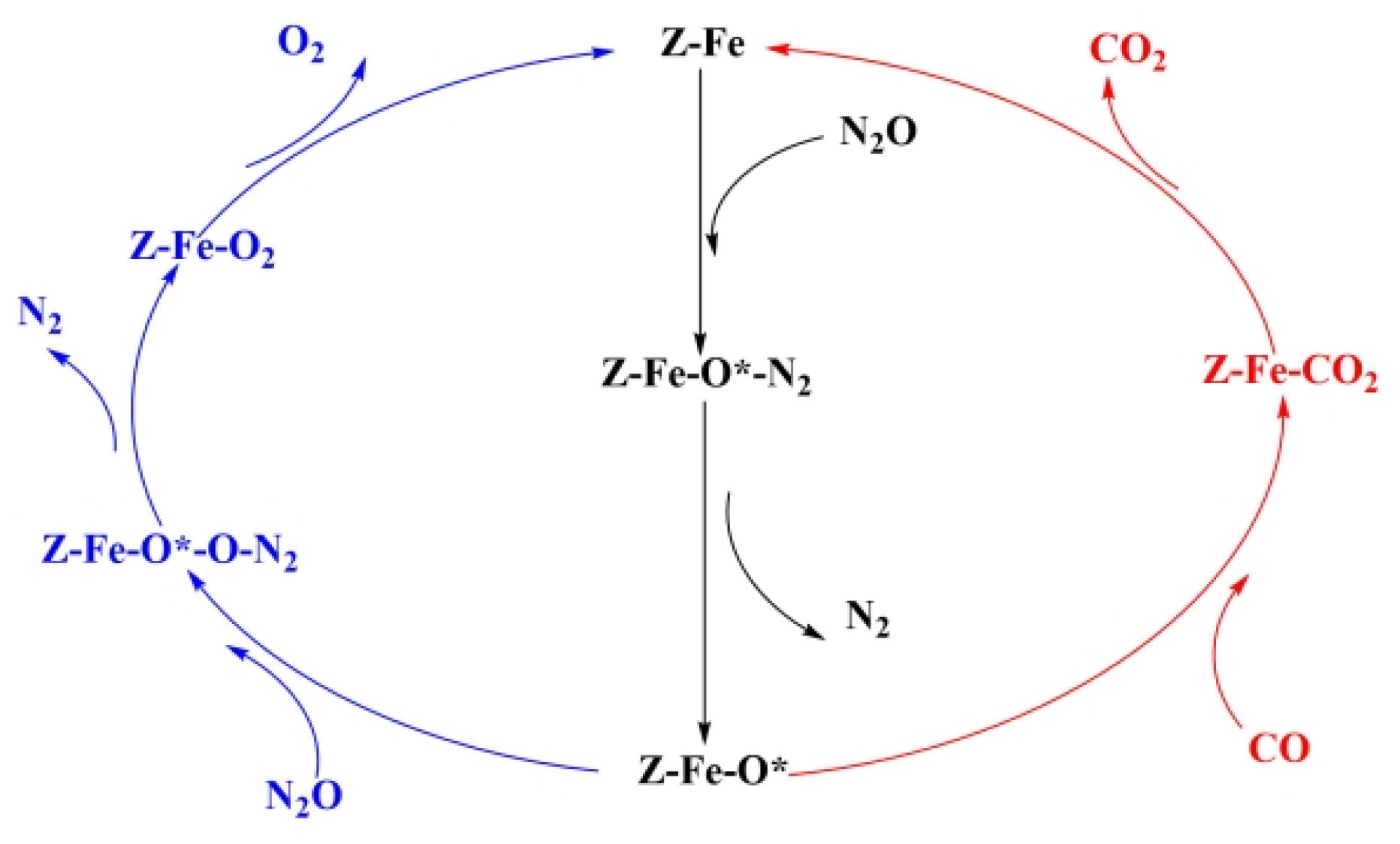
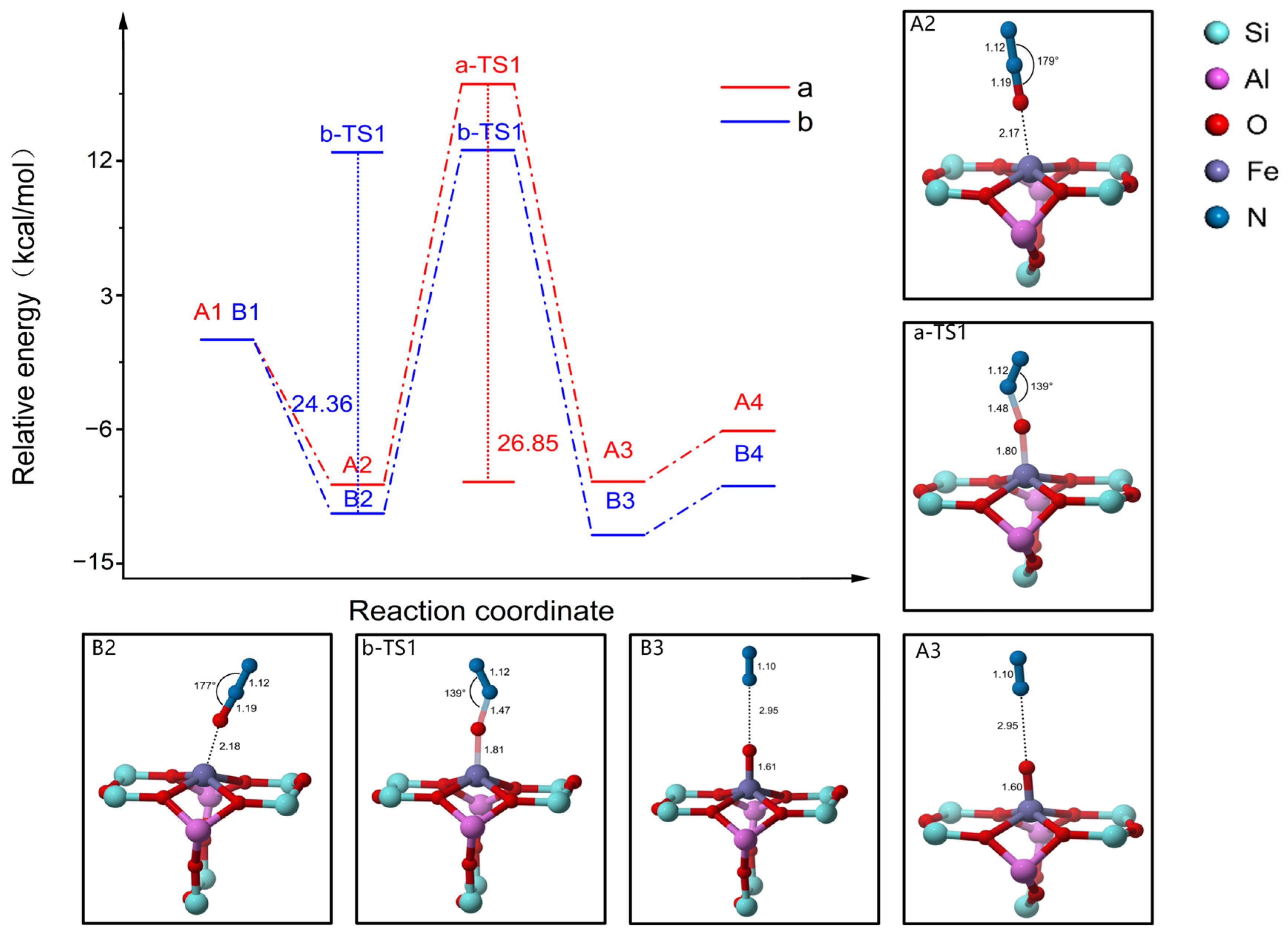
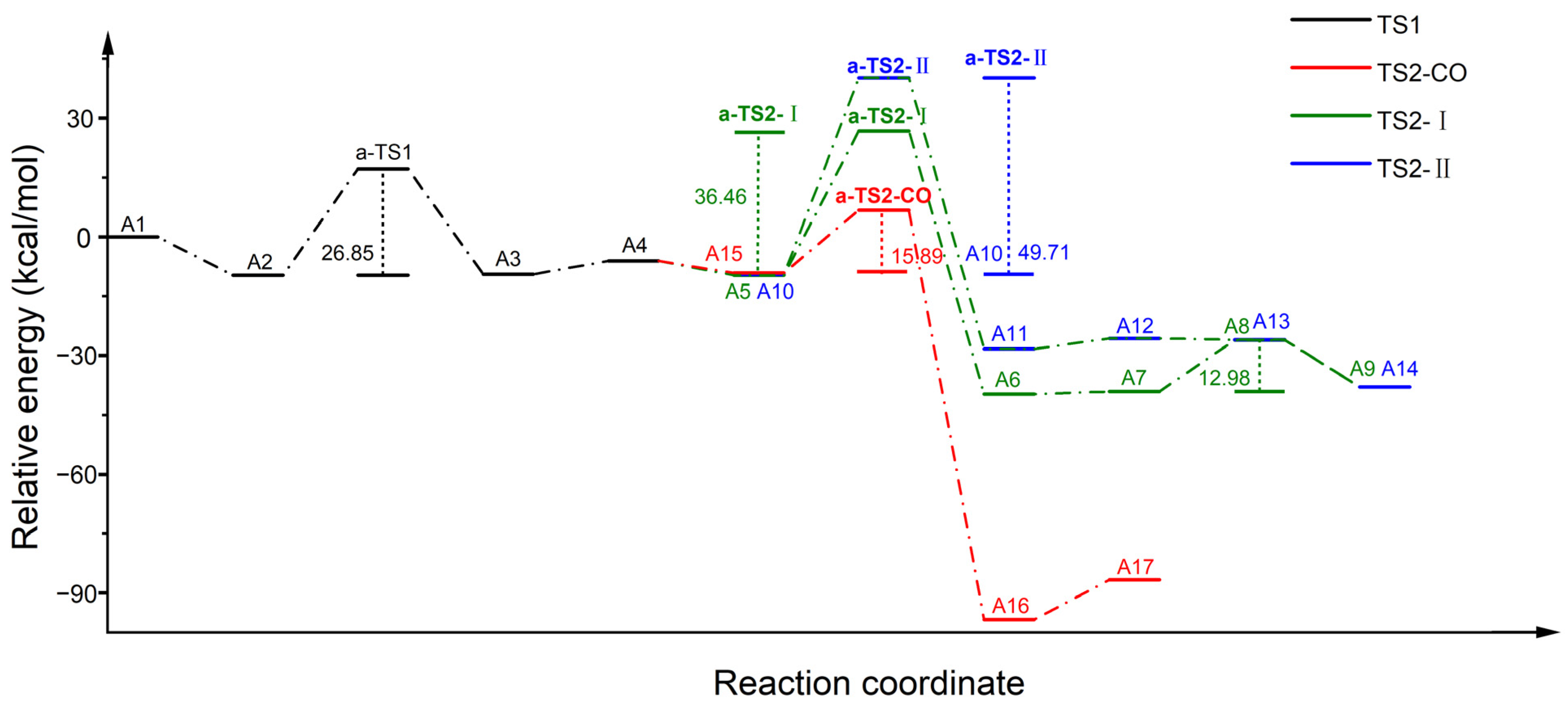
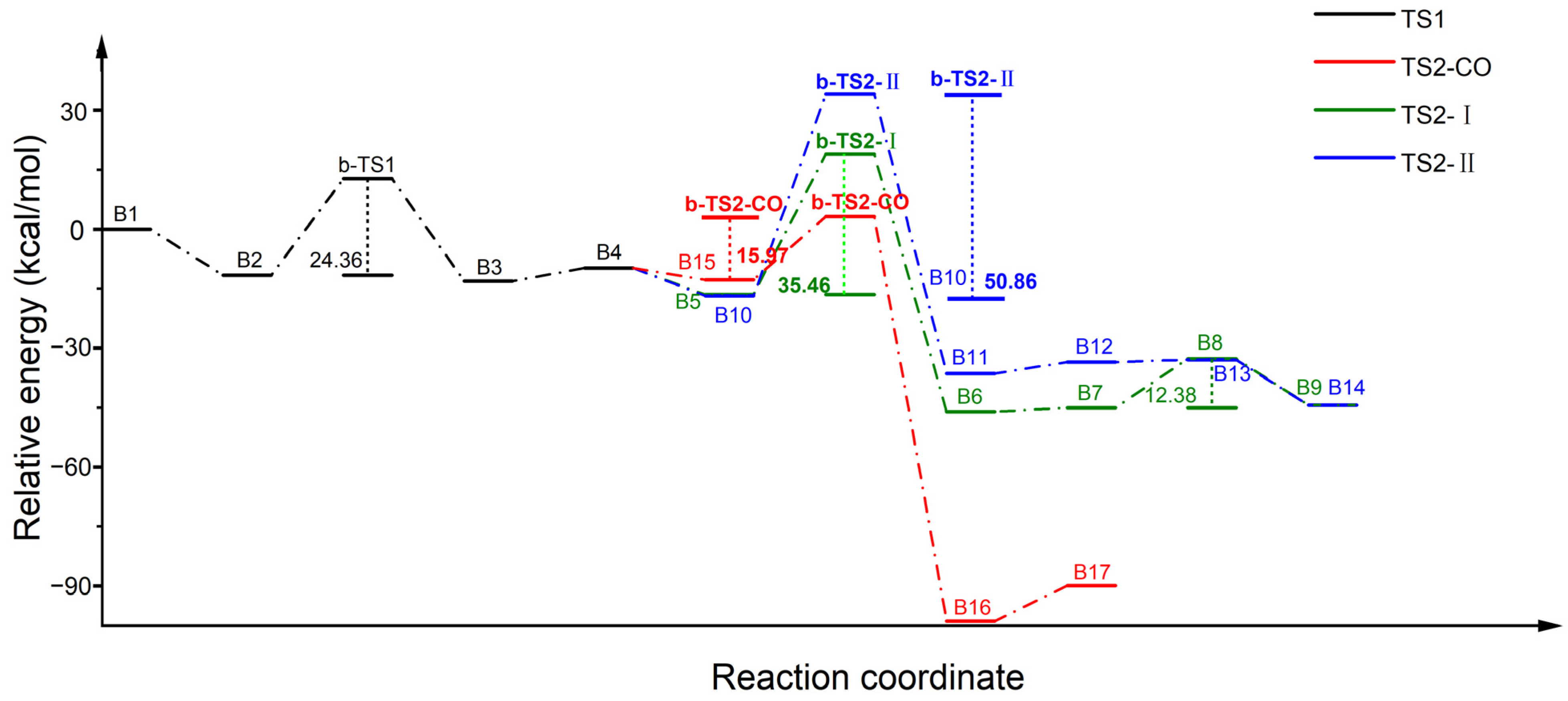
2.3. Reaction Mechanism of N2O Decomposition over Fe-ZSM-5
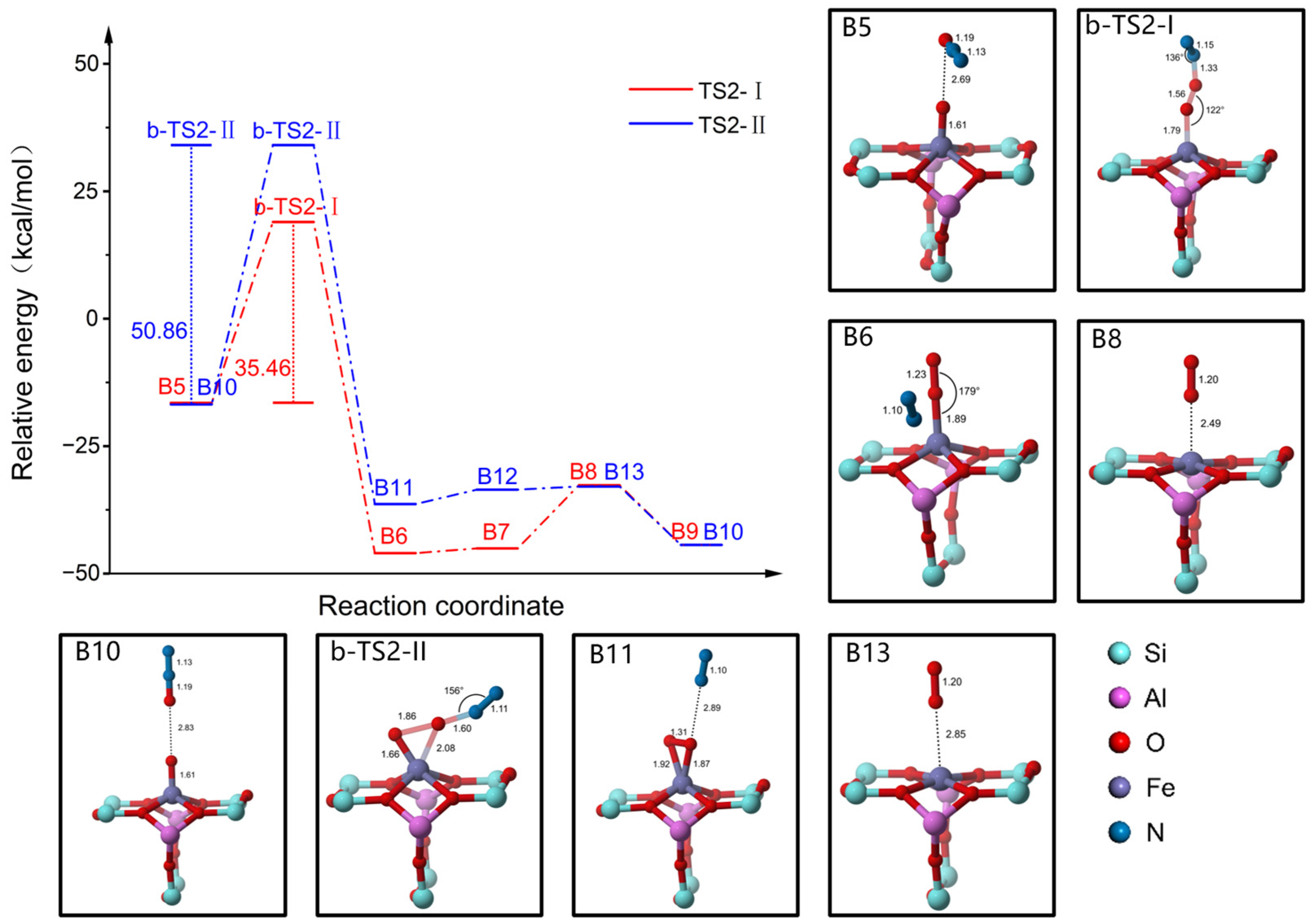
2.4. Reaction Mechanism of N2O Decomposition over Z(Fe-O*)
2.5. Reaction Mechanism of CO Participation over Z(Fe-O*)
2.6. Discussion
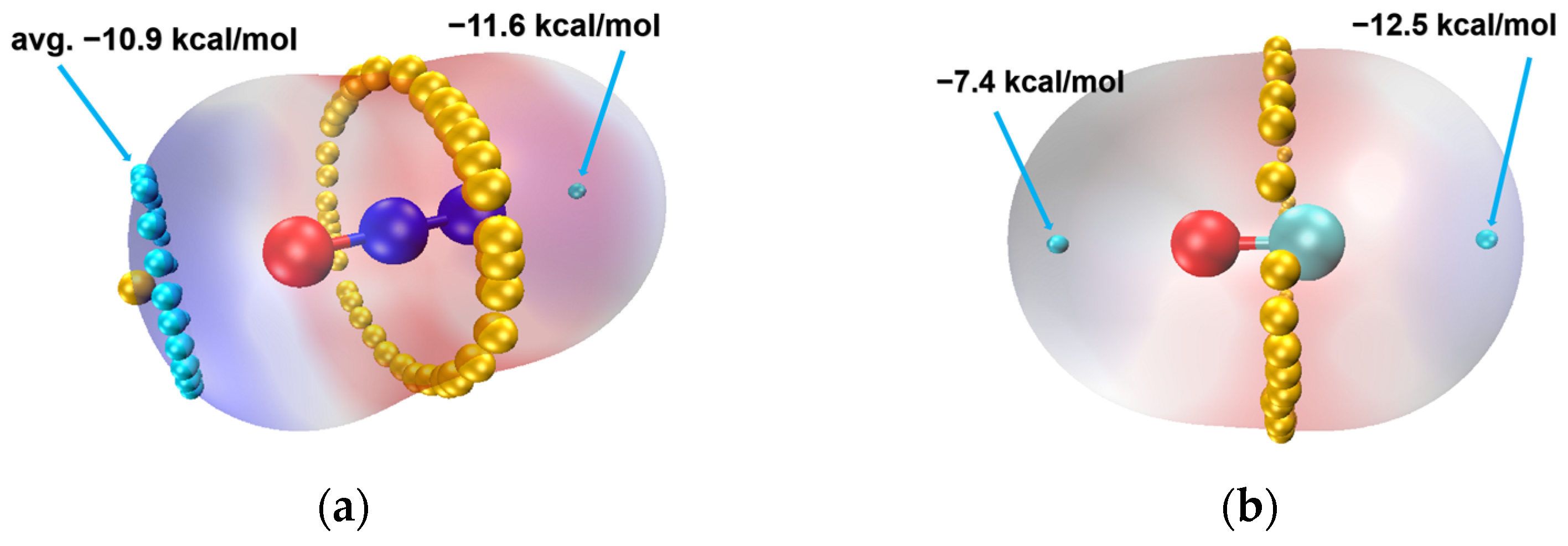
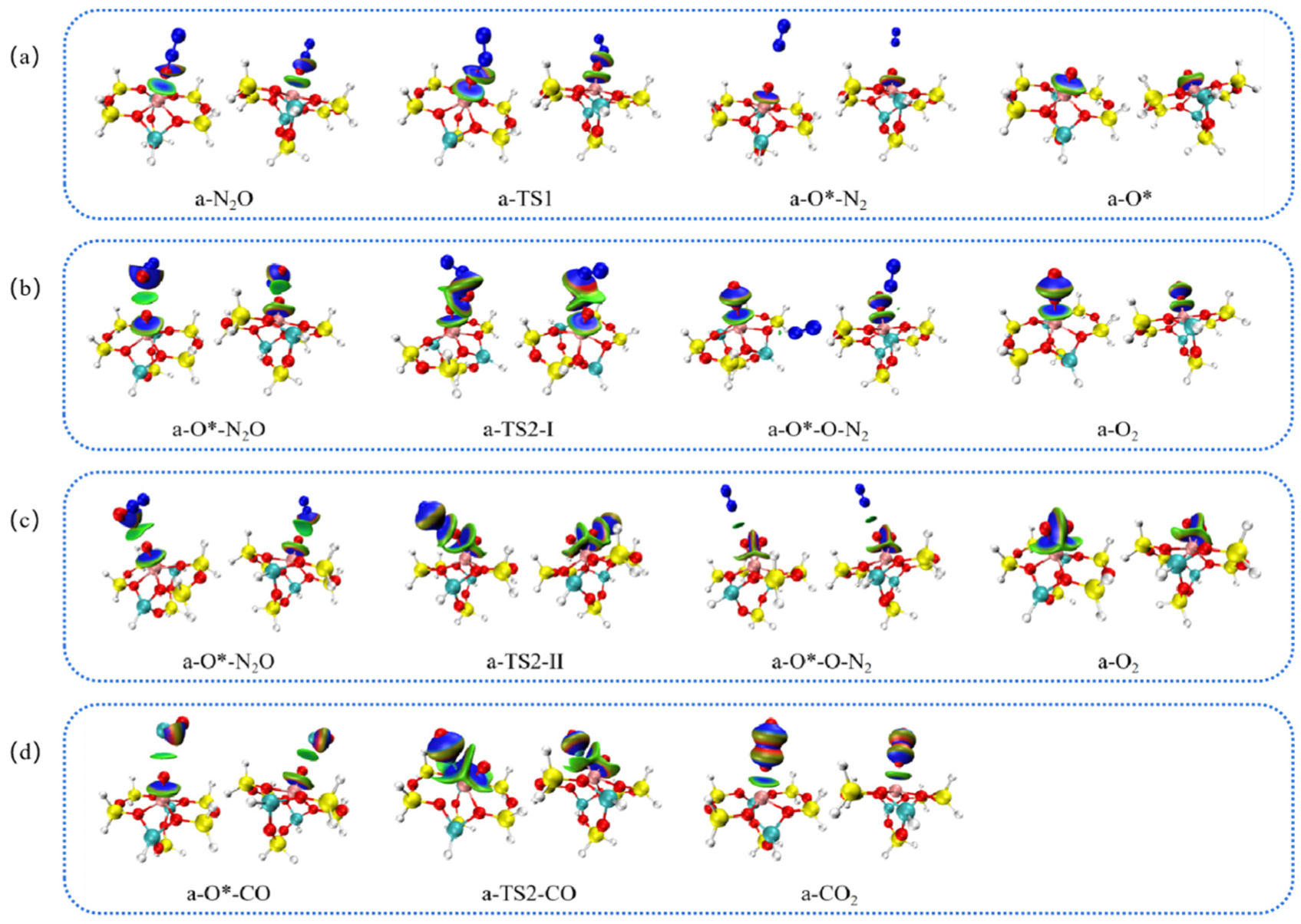
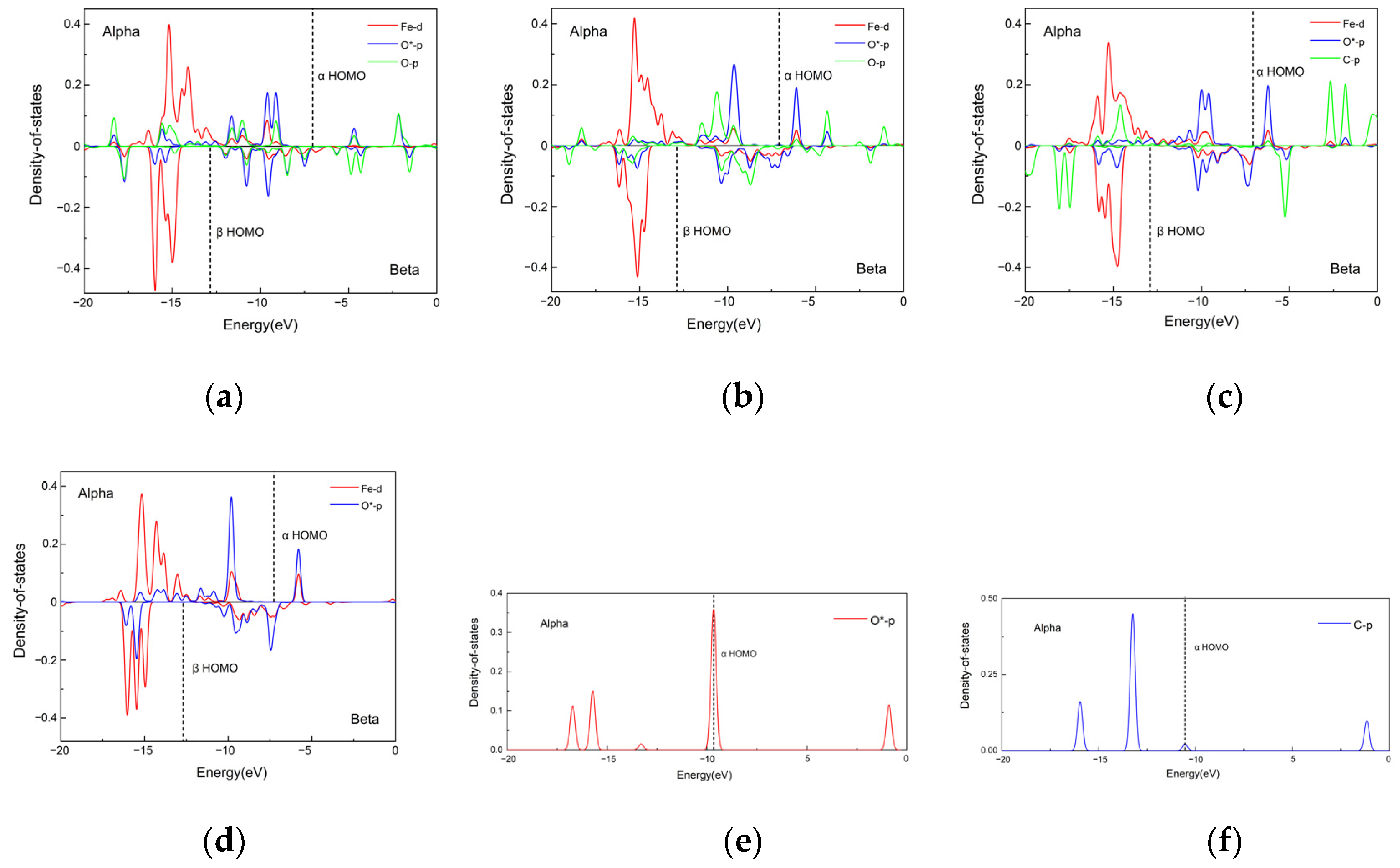
2.7. Electronic Analysis
2.7.1. Analysis of Interatomic Interactions
2.7.2. Analysis of Hirshfeld Charge
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Roy, S.; Hegde, M.S.; Madras, G. Catalysis for NOx abatement. Appl. Energy 2009, 86, 2283–2297. [Google Scholar] [CrossRef]
- Yan, W.; Zhang, G.; Wang, J.; Liu, M.; Sun, Y.; Zhou, Z.; Zhang, W.; Zhang, S.; Xu, X.; Shen, J.; et al. Recent Progress in Adipic Acid Synthesis Over Heterogeneous Catalysts. Front. Chem. 2020, 8, 185. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, F.; Wunderlin, P.; Udert, K.M.; Wells, G.F. Nitric oxide and nitrous oxide turnover in natural and engineered microbial communities: Biological pathways, chemical reactions, and novel technologies. Front. Microbiol. 2012, 3, 372. [Google Scholar] [CrossRef] [PubMed]
- Ritz, B.; Hoffmann, B.; Peters, A. The Effects of Fine Dust, Ozone, and Nitrogen Dioxide on Health. Dtsch. Arztebl. Int. 2019, 116, 881. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xu, J.; Hu, J.; Han, J. Reducing Nitrous Oxide Emissions to Mitigate Climate Change and Protect the Ozone Layer. Environ. Sci. Technol. 2014, 48, 5290–5297. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhu, J.; Zhang, Y.; Chen, X. An Optimal Pollution Control Model for Environmental Protection Cooperation between Developing and Developed Countries. Int. J. Environ. Res. Public Health 2020, 17, 3868. [Google Scholar] [CrossRef] [PubMed]
- Biturini, N.F.; Santos, A.P.N.M.; Batista, M.S. Influence of co-fed gases (O2, CO2, CH4, and H2O) on the N2O decomposition over (Co, Fe)-ZSM-5 and (Co, Fe)-BETA catalysts. React. Kinet. Mech. Catal. 2019, 126, 341–352. [Google Scholar] [CrossRef]
- Erisman, J.W.; Galloway, J.; Seitzinger, S.; Bleeker, A.; Butterbach-Bahl, K. Reactive nitrogen in the environment and its effect on climate change. Curr. Opin. Environ. Sustain. 2011, 3, 281–290. [Google Scholar] [CrossRef]
- Galle, M.; Agar, D.W.; Watzenberger, O. Thermal N2O decomposition in regenerative heat exchanger reactors. Chem. Eng. Sci. 2001, 56, 1587–1595. [Google Scholar] [CrossRef]
- Pérez-Ramírez, J.; Kapteijn, F.; Schöffel, K.; Moulijn, J.A. Formation and control of N2O in nitric acid production: Where do we stand today? Appl. Catal. B Environ. 2003, 44, 117–151. [Google Scholar] [CrossRef]
- Konsolakis, M. Recent Advances on Nitrous Oxide (N2O) Decomposition over Non-Noble-Metal Oxide Catalysts: Catalytic Performance, Mechanistic Considerations, and Surface Chemistry Aspects. ACS Catal. 2015, 5, 6397–6421. [Google Scholar] [CrossRef]
- Pietrogiacomi, D.; Campa, M.C.; Carbone, L.R.; Occhiuzzi, M. N2O decomposition and reduction on Co-MOR, Fe-MOR and Ni-MOR catalysts: In situ UV–vis DRS and operando FTIR investigation. An insight on the reaction pathways. Appl. Catal. B Environ. 2019, 240, 19–29. [Google Scholar] [CrossRef]
- Wang, R.; Zou, C.; Zhang, Y. A DFT Study of N2O Homogeneous and Heterogeneous Reduction Reaction by the Carbon Monoxide. Combust. Sci. Technol. 2022, 194, 963–976. [Google Scholar] [CrossRef]
- Boissel, V.; Tahir, S.; Koh, C.A. Catalytic decomposition of N2O over monolithic supported noble metal-transition metal oxides. Appl. Catal. B-Environ. 2006, 64, 234–242. [Google Scholar] [CrossRef]
- Grünert, W.; Kydala Ganesha, P.; Ellmers, I.; Pérez Vélez, R.; Huang, H.; Bentrup, U.; Schünemann, V.; Brückner, A. Active Sites of the Selective Catalytic Reduction of NO by NH3 over Fe-ZSM-5: Combining Reaction Kinetics with Postcatalytic Mössbauer Spectroscopy at Cryogenic Temperatures. ACS Catal. 2020, 10, 3119–3130. [Google Scholar] [CrossRef]
- Liu, H.; You, C.; Wang, H. Experimental and Density Functional Theory Studies on the Zeolite-Based Fe–Ni–W Trimetallic Catalyst for High-Temperature NOx Selective Catalytic Reduction: Identification of Active Sites Suppressing Ammonia Over-oxidation. ACS Catal. 2021, 11, 1189–1201. [Google Scholar] [CrossRef]
- Melian-Cabrera, I.; van Eck, E.R.H.; Espinosa, S.; Siles-Quesada, S.; Falco, L.; Kentgens, A.P.M.; Kapteijn, F.; Moulijn, J.A. Tail gas catalyzed N2O decomposition over Fe-beta zeolite. On the promoting role of framework connected AlO6 sites in the vicinity of Fe by controlled dealumination during exchange. Appl. Catal. B-Environ. 2017, 203, 218–226. [Google Scholar] [CrossRef]
- Senamart, N.; Buttha, S.; Pantupho, W.; Koleva, I.Z.; Loiha, S.; Aleksandrov, H.A.; Wittayakun, J.; Vayssilov, G.N. Characterization and temperature evolution of iron-containing species in HZSM-5 zeolite prepared from different iron sources. J. Porous Mater. 2019, 26, 1227–1240. [Google Scholar] [CrossRef]
- Zhang, T.; Qin, X.; Peng, Y.; Wang, C.; Chang, H.; Chen, J.; Li, J. Effect of Fe precursors on the catalytic activity of Fe/SAPO-34 catalysts for N2O decomposition. Catal. Commun. 2019, 128, 105706. [Google Scholar] [CrossRef]
- Jiao, A.; Zhang, H.; Liu, J.; Jiang, X. Quantum chemical and kinetics calculations for the NO reduction with char(N): Influence of the carbon monoxide. Combust. Flame 2018, 196, 377–385. [Google Scholar] [CrossRef]
- Jin, X.; Huai, L.-y.; Wen, H.; Yi, W.-c.; Liu, J.-y. Reduction of NO with CO on the Co3O4(110)-B and CoO(110) Surfaces: A First-Principles Study. J. Phys. Chem. C 2019, 123, 1770–1778. [Google Scholar] [CrossRef]
- Konsolakis, M.; Drosou, C.; Yentekakis, I.V. Support mediated promotional effects of rare earth oxides (CeO2 and La2O3) on N2O decomposition and N2O reduction by CO or C3H6 over Pt/Al2O3 structured catalysts. Appl. Catal. B-Environ. 2012, 123, 405–413. [Google Scholar] [CrossRef]
- Bols, M.L.; Snyder, B.E.R.; Rhoda, H.M.; Cnudde, P.; Fayad, G.; Schoonheydt, R.A.; Van Speybroeck, V.; Solomon, E.I.; Sels, B.F. Coordination and activation of nitrous oxide by iron zeolites. Nat. Catal. 2021, 4, 332. [Google Scholar] [CrossRef]
- Wannakao, S.; Nongnual, T.; Khongpracha, P.; Maihom, T.; Limtrakul, J. Reaction Mechanisms for CO Catalytic Oxidation by N2O on Fe-Embedded Graphene. J. Phys. Chem. C 2012, 116, 16992–16998. [Google Scholar] [CrossRef]
- You, Y.; Chen, S.; Li, J.; Zeng, J.; Chang, H.; Ma, L.; Li, J. Low-temperature selective catalytic reduction of N2O by CO over Fe-ZSM-5 catalysts in the presence of O2. J. Hazard. Mater. 2020, 383, 121117. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Lei, Z.; Wang, Y.; Zhang, R.; Chen, B. Reduction of N2O by CO over Fe- and Cu-BEA zeolites: An experimental and computational study of the mechanism. Microporous Mesoporous Mater. 2013, 167, 254–266. [Google Scholar] [CrossRef]
- Zhou, H.; Li, Y.; Li, N.; Qiu, R.; Meng, S.; Cen, K. Experimental study of the NO and N2O emissions during devolatilization and char combustion of a single biomass particle in O2/N2 and O2/H2O under low temperature condition. Fuel 2017, 206, 162–170. [Google Scholar] [CrossRef]
- Li, F.F.; Shi, C.M.; Wang, X.F.; Cui, G.L.; Wang, D.C.; Chen, L. The important role of oxygen defect for NO gas-sensing behavior of α-Fe2O3 surface: Predicted by density functional theory. Comput. Mater. Sci. 2018, 146, 1–8. [Google Scholar] [CrossRef]
- Smeets, P.J.; Sels, B.F.; van Teeffelen, R.M.; Leeman, H.; Hensen, E.J.M.; Schoonheydt, R.A. The catalytic performance of Cu-containing zeolites in N2O decomposition and the influence of O2, NO and H2O on recombination of oxygen. J. Catal. 2008, 256, 183–191. [Google Scholar] [CrossRef]
- Li, G.; Pidko, E.A.; van Santen, R.A.; Feng, Z.; Li, C.; Hensen, E.J.M. Stability and reactivity of active sites for direct benzene oxidation to phenol in Fe/ZSM-5: A comprehensive periodic DFT study. J. Catal. 2011, 284, 194–206. [Google Scholar] [CrossRef]
- Cheng, J.; Zheng, D.H.; Dai, C.N.; Xu, R.N.A.; Liu, N.; Yu, G.Q.; Wang, N.; Chen, B.A.H. Constructing active copper species in Cu-zeolites for coal-gas-SCR and elucidating the synergistic catalytic function of CuO and Cu2+ ion species. Environ. Sci. Nano 2022, 9, 2372–2387. [Google Scholar] [CrossRef]
- Cheng, J.; Zheng, D.; Yu, G.; Xu, R.; Dai, C.; Liu, N.; Wang, N.; Chen, B. N2O Catalytic Decomposition and NH3-SCR Coupling Reactions over Fe-SSZ-13 Catalyst: Mechanisms and Interactions Unraveling via Experiments and DFT Calculations. ACS Catal. 2023, 13, 934–947. [Google Scholar] [CrossRef]
- Benco, L.; Bucko, T.; Grybos, R.; Hafner, J. Free energy calculation of the reaction path of the N2O decomposition over Fe(II)-ferrierite. In Studies in Surface Science and Catalysis; Gédéon, A., Massiani, P., Babonneau, F., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; Volume 174, pp. 689–694. [Google Scholar]
- Gao, C.R.; Li, J.W.; Zhang, J.; Sun, X.L. DFT Study on the Combined Catalytic Removal of N2O, NO, and NO2 over Binuclear Cu-ZSM-5. Catalysts 2022, 12, 438. [Google Scholar] [CrossRef]
- Ryder, J.A.; Chakraborty, A.K.; Bell, A.T. Density Functional Theory Study of Nitrous Oxide Decomposition over Fe- and Co-ZSM-5. J. Phys. Chem. B 2002, 106, 7059–7064. [Google Scholar] [CrossRef]
- Heyden, A. Theoretical Investigation of the Nitrous Oxide Decomposition over Iron Zeolite Catalysts. Doctoral Dissertation, Technische Universität Hamburg, Hamburg, Germany, 2005. [Google Scholar]
- Li, G.N.; Pidko, E.A.; Filot, I.A.W.; van Santen, R.A.; Li, C.; Hensen, E.J.M. Catalytic properties of extraframework iron-containing species in ZSM-5 for N2O decomposition. J. Catal. 2013, 308, 386–397. [Google Scholar] [CrossRef]
- Sklenak, S.; Andrikopoulos, P.C.; Boekfa, B.; Jansang, B.; Nováková, J.; Benco, L.; Bucko, T.; Hafner, J.; Dědeček, J.; Sobalík, Z. N2O decomposition over Fe-zeolites: Structure of the active sites and the origin of the distinct reactivity of Fe-ferrierite, Fe-ZSM-5, and Fe-beta. A combined periodic DFT and multispectral study. J. Catal. 2010, 272, 262–274. [Google Scholar] [CrossRef]
- Kapteijn, F.; Rodriguez-Mirasol, J.; Moulijn, J.A. Heterogeneous catalytic decomposition of nitrous oxide. Appl. Catal. B Environ. 1996, 9, 25–64. [Google Scholar] [CrossRef]
- Gao, C.R.; Li, J.W.; Zhang, J.; Sun, X.L. DFT Study on Mechanisms of the N2O Direct Catalytic Decomposition over Cu-ZSM-5: The Detailed Investigation on NO Formation Mechanism. Catalysts 2020, 10, 646. [Google Scholar] [CrossRef]
- Chen, B.; Liu, N.; Liu, X.; Zhang, R.; Li, Y.; Li, Y.; Sun, X. Study on the direct decomposition of nitrous oxide over Fe-beta zeolites: From experiment to theory. Catal. Today 2011, 175, 245–255. [Google Scholar] [CrossRef]
- Peng, Y.; Si, W.; Li, X.; Luo, J.; Li, J.; Crittenden, J.; Hao, J. Comparison of MoO3 and WO3 on arsenic poisoning V2O5/TiO2 catalyst: DRIFTS and DFT study. Appl. Catal. B Environ. 2016, 181, 692–698. [Google Scholar] [CrossRef]
- Li, G.N.; Pidko, E.A.; van Santen, R.A.; Li, C.; Hensen, E.J.M. Stability of Extraframework Iron-Containing Complexes in ZSM-5 Zeolite. J. Phys. Chem. C 2013, 117, 413–426. [Google Scholar] [CrossRef]
- Kachurovskaya, N.A.; Zhidomirov, G.M.; Hensen, E.J.M.; van Santen, R.A. Cluster model DFT study of the intermediates of benzene to phenol oxidation by N2O on FeZSM-5 zeolites. Catal. Lett. 2003, 86, 25–31. [Google Scholar] [CrossRef]
- Fellah, M.F. CO and NO Adsorptions on Different Iron Sites of Fe-ZSM-5 Clusters: A Density Functional Theory Study. J. Phys. Chem. C 2011, 115, 1940–1951. [Google Scholar] [CrossRef]
- Sazama, P.; Wichterlová, B.; Tábor, E.; Šťastný, P.; Sathu, N.K.; Sobalík, Z.; Dědeček, J.; Sklenák, Š.; Klein, P.; Vondrová, A. Tailoring of the structure of Fe-cationic species in Fe-ZSM-5 by distribution of Al atoms in the framework for N2O decomposition and NH3-SCR-NOx. J. Catal. 2014, 312, 123–138. [Google Scholar] [CrossRef]
- van Koningsveld, H.; Jansen, J.C. Single crystal structure analysis of zeolite H-ZSM-5 loaded with naphthalene. Microporous Mater. 1996, 6, 159–167. [Google Scholar] [CrossRef]
- Chao, K.-J.; Lin, J.-C.; Wang, Y.; Lee, G.H. Single crystal structure refinement of TPA ZSM-5 zeolite. Zeolites 1986, 6, 35–38. [Google Scholar] [CrossRef]
- Lee, S.K.; Stebbins, J.F. Al-O-Al and Si-O-Si sites in framework aluminosilicate glasses with Si/Al = 1: Quantification of framework disorder. J. Non-Cryst. Solids 2000, 270, 260–264. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2009.
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A Gen. Phys. 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Mota, A.J.; de Cienfuegos, L.Á.; Robles, R.; Perea-Buceta, J.E.; Colacio, E.; Timón, V.; Hernández-Laguna, A. DFT approach to reaction mechanisms through molecular complexes. The case of an organo-catalysed nucleosidation reaction. J. Mol. Struct. Theochem 2010, 944, 43–52. [Google Scholar] [CrossRef]
- Hehre, W.J. Ab initio molecular orbital theory. Acc. Chem. Res. 1976, 9, 399–406. [Google Scholar] [CrossRef]
- Leininger, T.; Nicklass, A.; Stoll, H.; Dolg, M.; Schwerdtfeger, P. The accuracy of the pseudopotential approximation. II. A comparison of various core sizes for indium pseudopotentials in calculations for spectroscopic constants of InH, InF, and InCl. J. Chem. Phys. 1996, 105, 1052–1059. [Google Scholar] [CrossRef]
- García-Melchor, M.; Gorelsky, S.I.; Woo, T.K. Mechanistic Analysis of Iridium(III) Catalyzed Direct C-H Arylations: A DFT Study. Chem. A Eur. J. 2011, 17, 13847–13853. [Google Scholar] [CrossRef] [PubMed]
- Fukui, K. The path of chemical reactions-the IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Cao, F.; Su, S.; Xiang, J.; Sun, L.; Hu, S.; Zhao, Q.; Wang, P.; Lei, S. Density functional study of adsorption properties of NO and NH3 over CuO/γ-Al2O3 catalyst. Appl. Surf. Sci. 2012, 261, 659–664. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Independent gradient model based on Hirshfeld partition: A new method for visual study of interactions in chemical systems. J. Comput. Chem. 2022, 43, 539–555. [Google Scholar] [CrossRef]
- Legault, C.Y. CYLview, 1.0b: Université de Sherbrooke. 2009. Available online: http://www.cylview.org (accessed on 12 December 2023).
- Mendelsohn, L.D. ChemDraw 8 Ultra, Windows and Macintosh Versions. J. Chem. Inf. Comput. Sci. 2004, 44, 2225–2226. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Configuration | Fe-O Bond Length | Fe-O (avg.) | Binding Energy of Doped Fe (kcal/mol) | ||||
|---|---|---|---|---|---|---|---|
| Structure | T-Site of Al/Si | ||||||
| a-Site | |||||||
| a-1 | T1 | 1.957 | 1.972 | 1.965 | −142.72 | ||
| a-2 | T7 | 1.974 | 1.960 | 1.967 | −151.07 | ||
| a-3 | T1, T7 | 1.979 | 1.966 | 1.986 | 2.003 | 1.984 | −271.15 |
| b-Site | |||||||
| b-1 | T11 | 2.049 | 2.005 | 2.027 | −173.01 | ||
| b-2 | T11, T11 | 1.936 | 1.925 | 1.981 | 1.966 | 1.952 | −272.26 |
| Configuration | SM | Relative Stability (kcal/mol) | |
|---|---|---|---|
| Structure | T-Site of Al/Si | ||
| a-Site | |||
| a-3 | T1, T7 | 1 | 40 |
| 3 | 15 | ||
| 5 | 0 | ||
| 7 | 111 | ||
| b-Site | |||
| b-2 | T11, T11 | 1 | 79 |
| 3 | 31 | ||
| 5 | 0 | ||
| 7 | 122 | ||
| Number | Process | State | Atom | ||||
|---|---|---|---|---|---|---|---|
| Fe | O* | O | N | N | |||
| 5 | a-O* | 0.4985 | –0.1733 | - | - | - | |
| 6 | TS2-I | a-O*-N2O | 0.5016 | –0.1842 | –0.1349 | 0.2349 | –0.0819 |
| 7 | a-O*-N2O-TS | 0.5163 | –0.2105 | 0.0046 | 0.1182 | –0.0161 | |
| 8 | a-O*-O-N2 | 0.4587 | 0.0120 | 0.0168 | 0.0133 | 0.0354 | |
| 9 | a-O*-O | 0.4602 | 0.0143 | 0.0201 | - | - | |
| 10 | a-O2 | 0.2676 | 0.0742 | 0.0491 | - | - | |
| Number | Process | State | Atom | ||||
|---|---|---|---|---|---|---|---|
| Fe | O* | O | N | N | |||
| 5 | a-O* | 0.4985 | –0.1733 | - | - | - | |
| 11 | TS2-II | a-O*-N2O | 0.5372 | –0.1653 | –0.1475 | 0.2291 | –0.0874 |
| 12 | a-O*-N2O-TS | 0.4882 | –0.1423 | –0.1225 | 0.1747 | 0.0670 | |
| 13 | a-O*-O-N2 | 0.4848 | –0.0396 | –0.0314 | 0.0064 | 0.0032 | |
| 14 | a-O*-O | 0.4848 | –0.0403 | –0.0420 | - | - | |
| 15 | a-O2 | 0.2676 | 0.0491 | 0.0742 | - | - | |
| Number | Process | State | Atom | |||
|---|---|---|---|---|---|---|
| Fe | O* | C | O | |||
| 5 | a-O* | 0.4985 | –0.1733 | - | - | |
| 16 | TS2-CO | a-O*-CO | 0.5033 | –0.1788 | 0.0936 | –0.0839 |
| 17 | a-O*-CO-TS | 0.4826 | –0.1821 | 0.2270 | –0.0266 | |
| 18 | a-CO2 | 0.3790 | –0.0731 | 0.3869 | –0.1100 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, N.; Gao, C.; Sun, X.; Li, J. Density Functional Theory Study of Mechanism of Reduction of N2O by CO over Fe-ZSM-5 Zeolites. Catalysts 2024, 14, 49. https://doi.org/10.3390/catal14010049
Yuan N, Gao C, Sun X, Li J. Density Functional Theory Study of Mechanism of Reduction of N2O by CO over Fe-ZSM-5 Zeolites. Catalysts. 2024; 14(1):49. https://doi.org/10.3390/catal14010049
Chicago/Turabian StyleYuan, Ning, Congru Gao, Xiuliang Sun, and Jianwei Li. 2024. "Density Functional Theory Study of Mechanism of Reduction of N2O by CO over Fe-ZSM-5 Zeolites" Catalysts 14, no. 1: 49. https://doi.org/10.3390/catal14010049
APA StyleYuan, N., Gao, C., Sun, X., & Li, J. (2024). Density Functional Theory Study of Mechanism of Reduction of N2O by CO over Fe-ZSM-5 Zeolites. Catalysts, 14(1), 49. https://doi.org/10.3390/catal14010049