
Bimetallic (or Multimetallic) Synthesis of N-Heterocycles



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
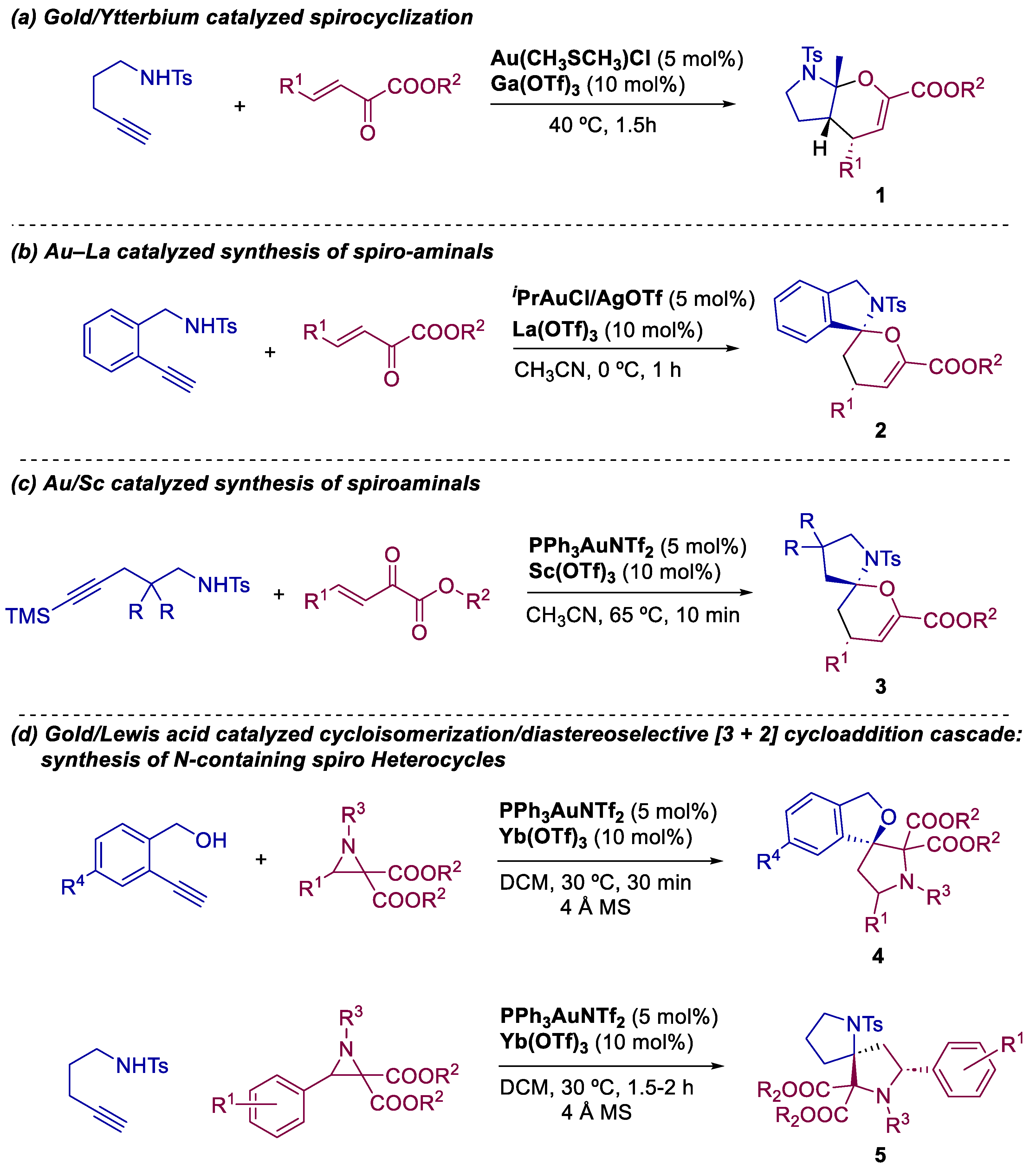
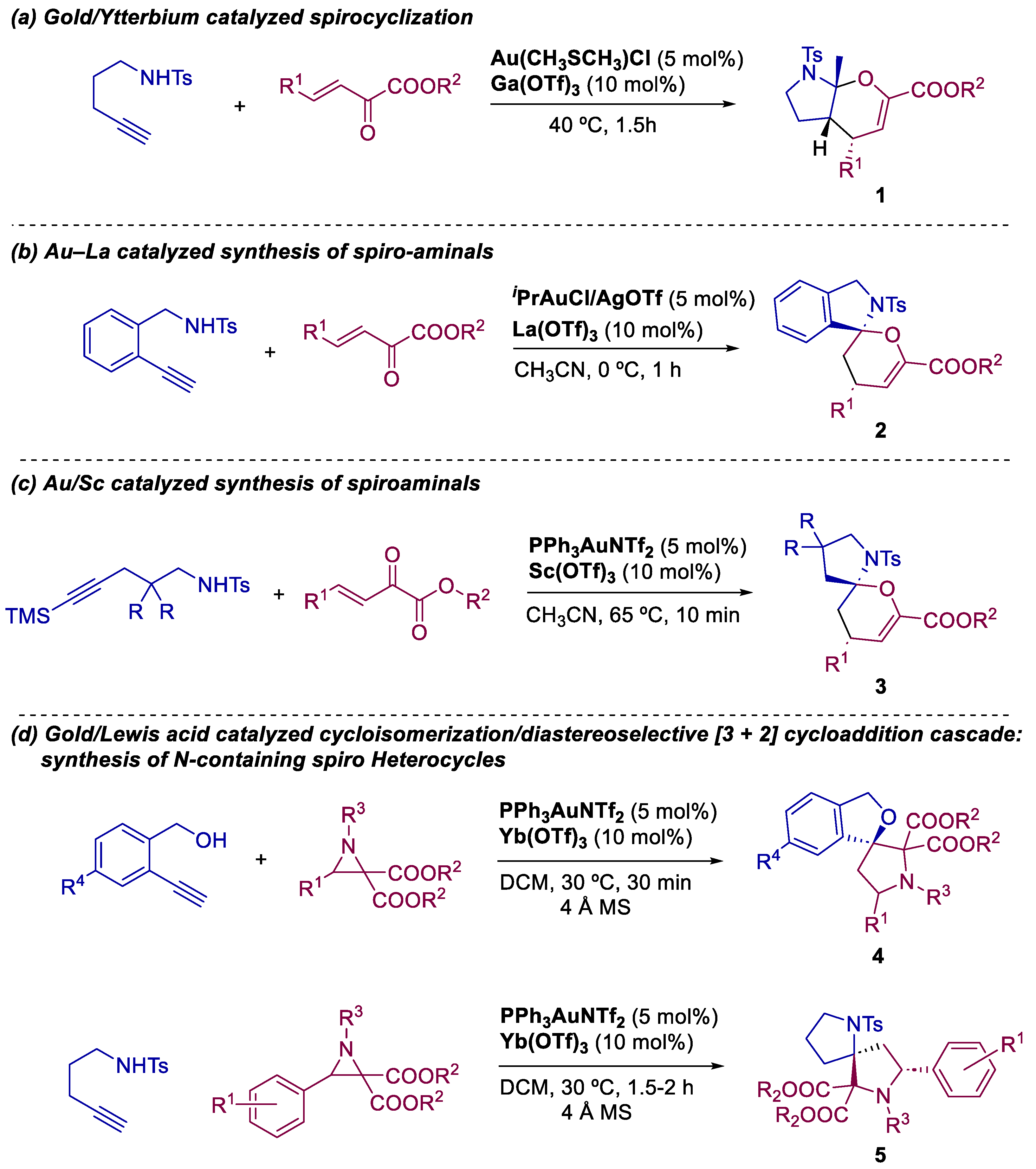
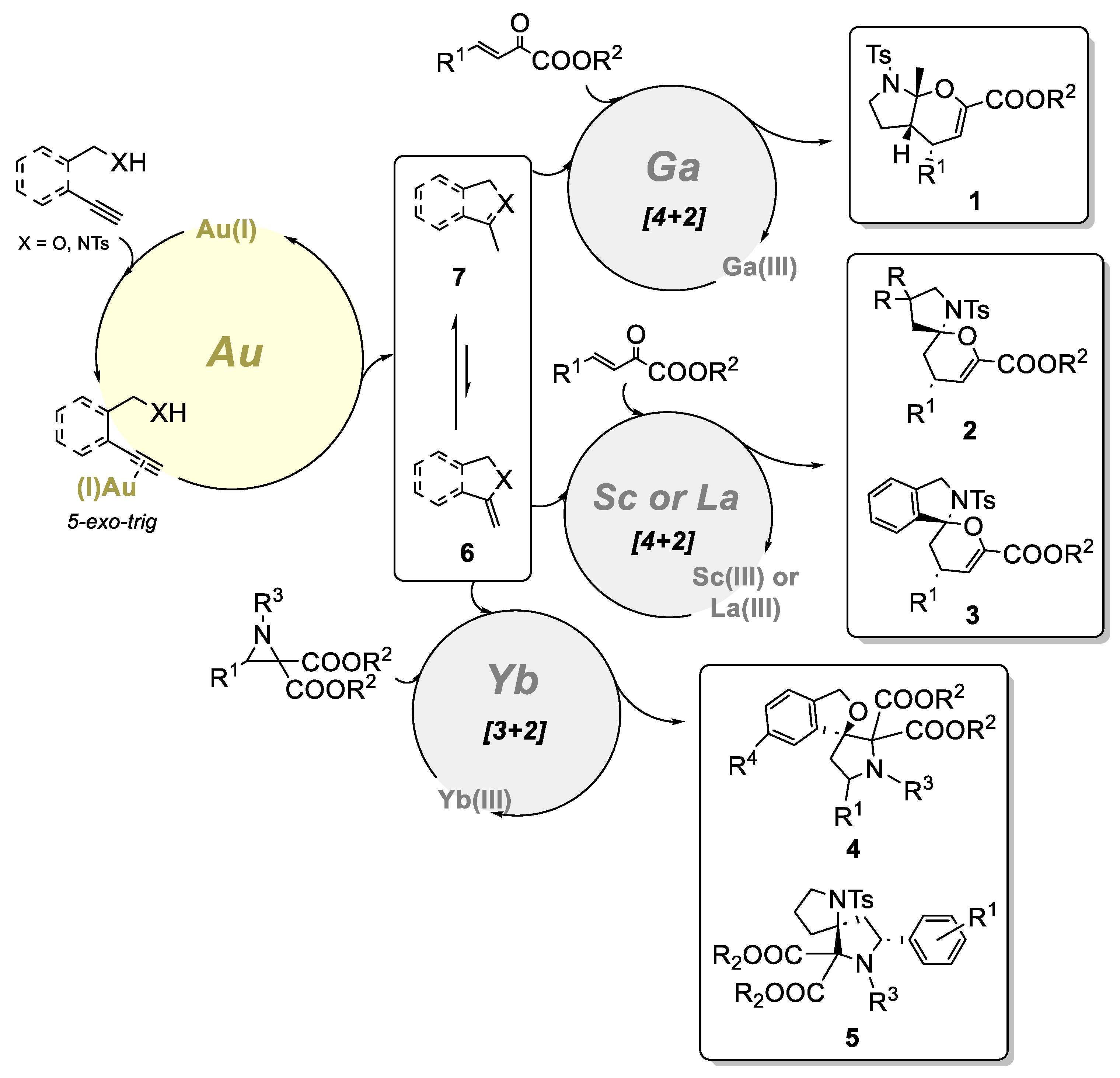
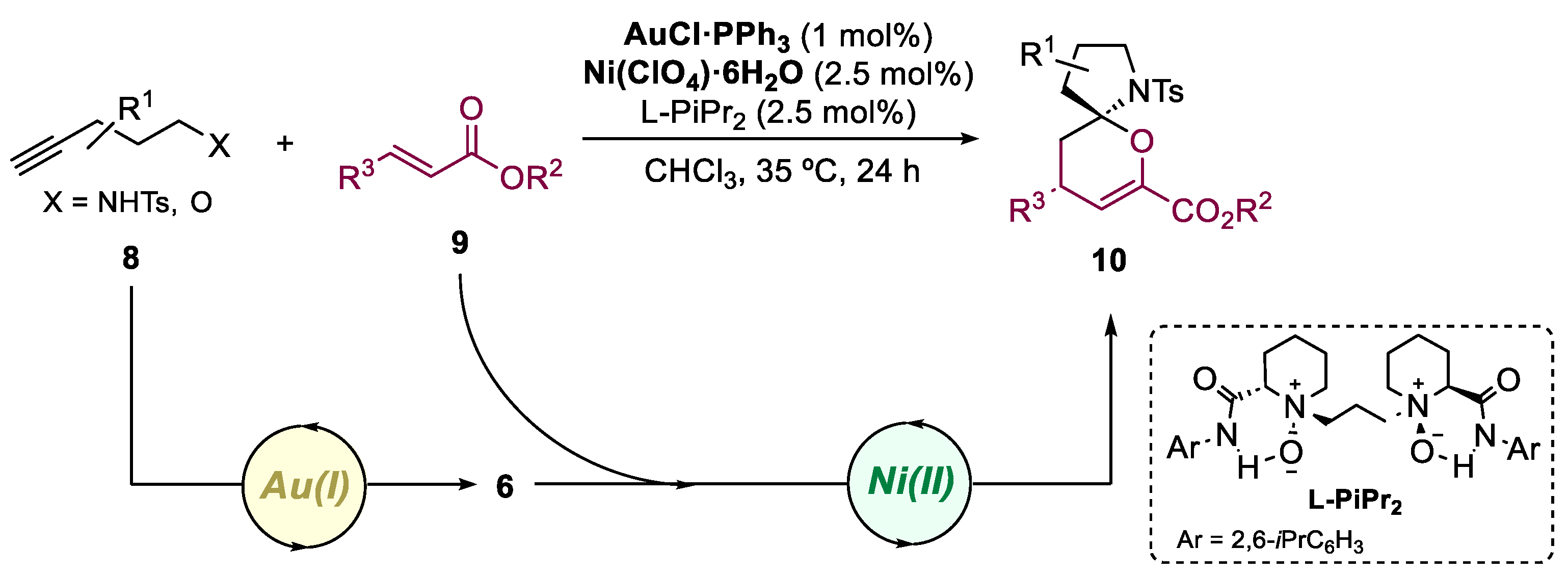
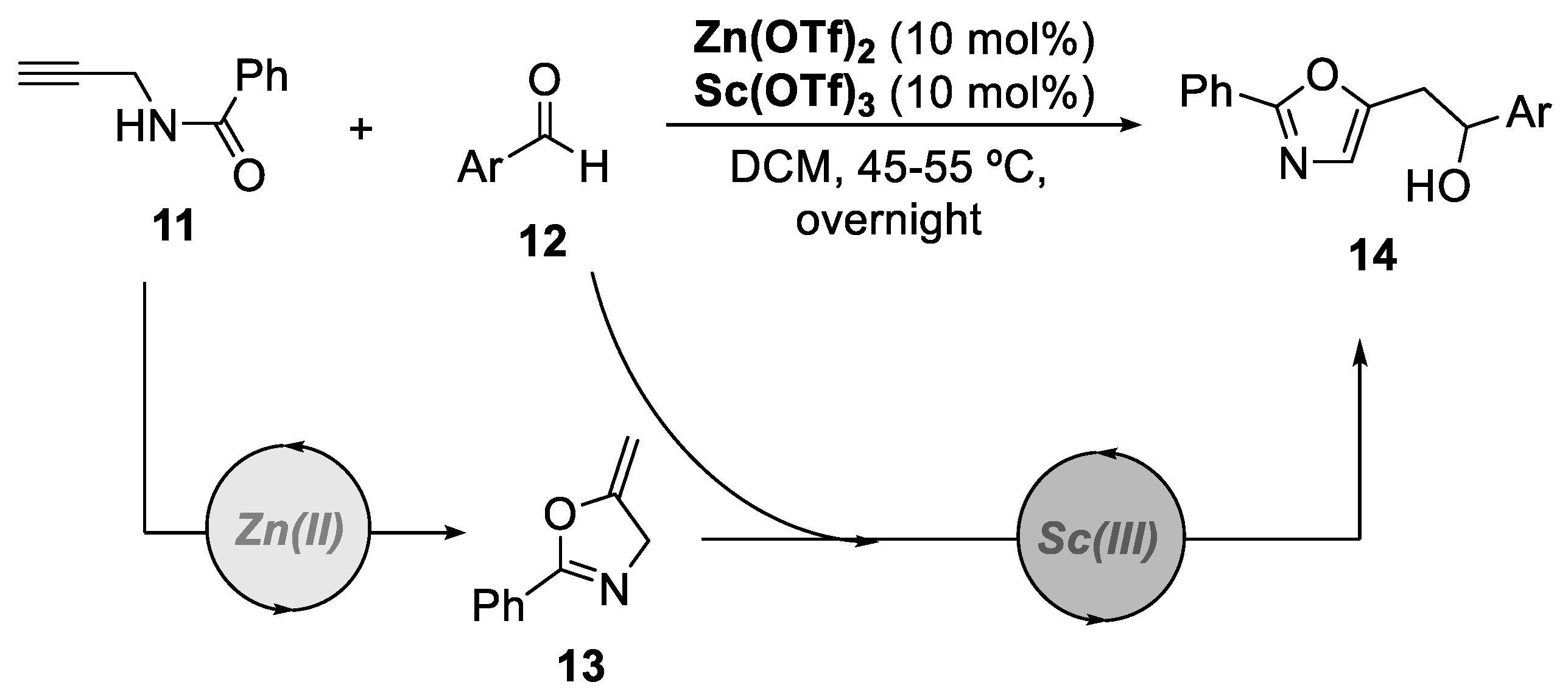
2. Bimetallic Approaches to N,O-Aminals and Related Spiro-N,O-Heterocycles
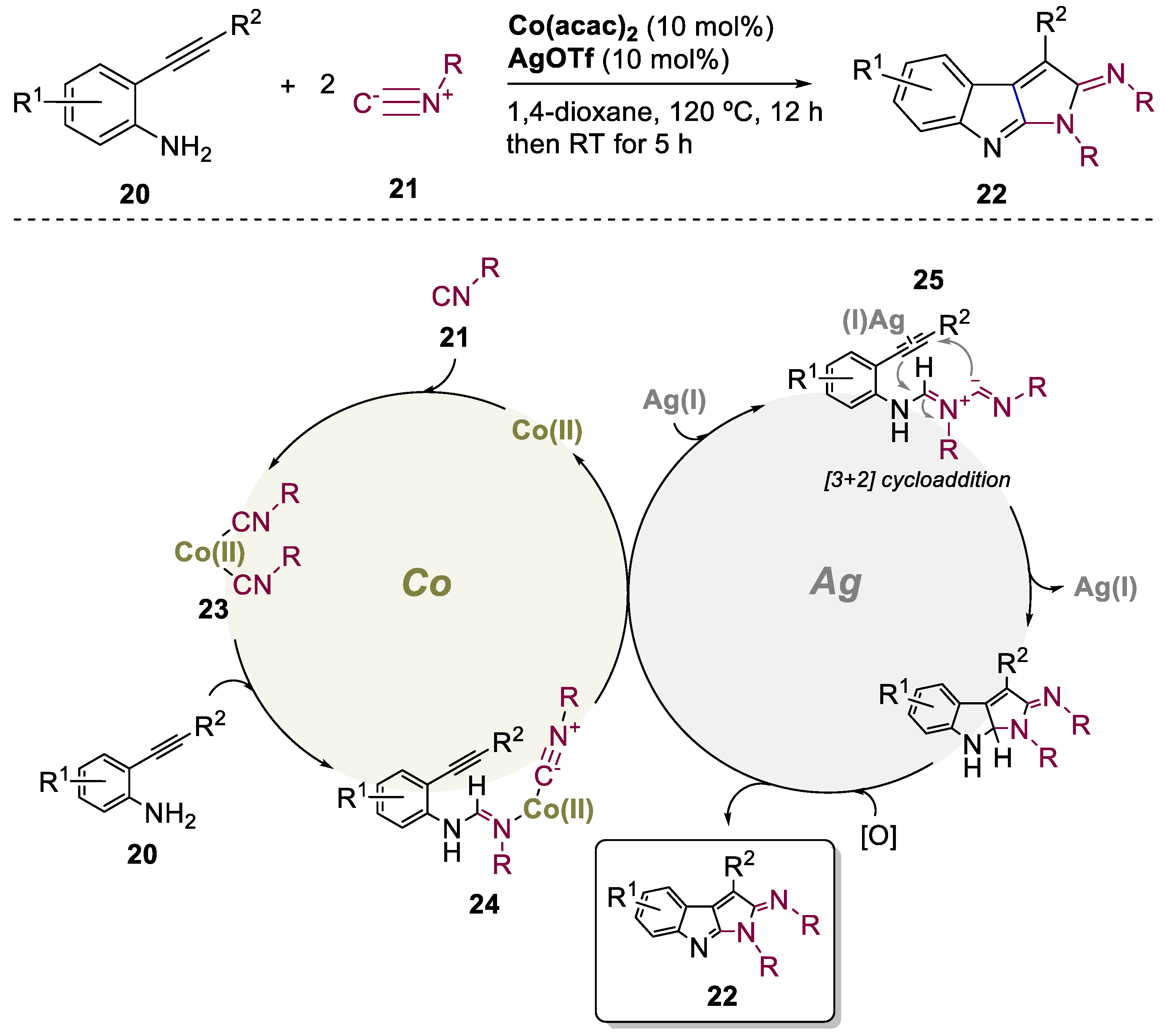
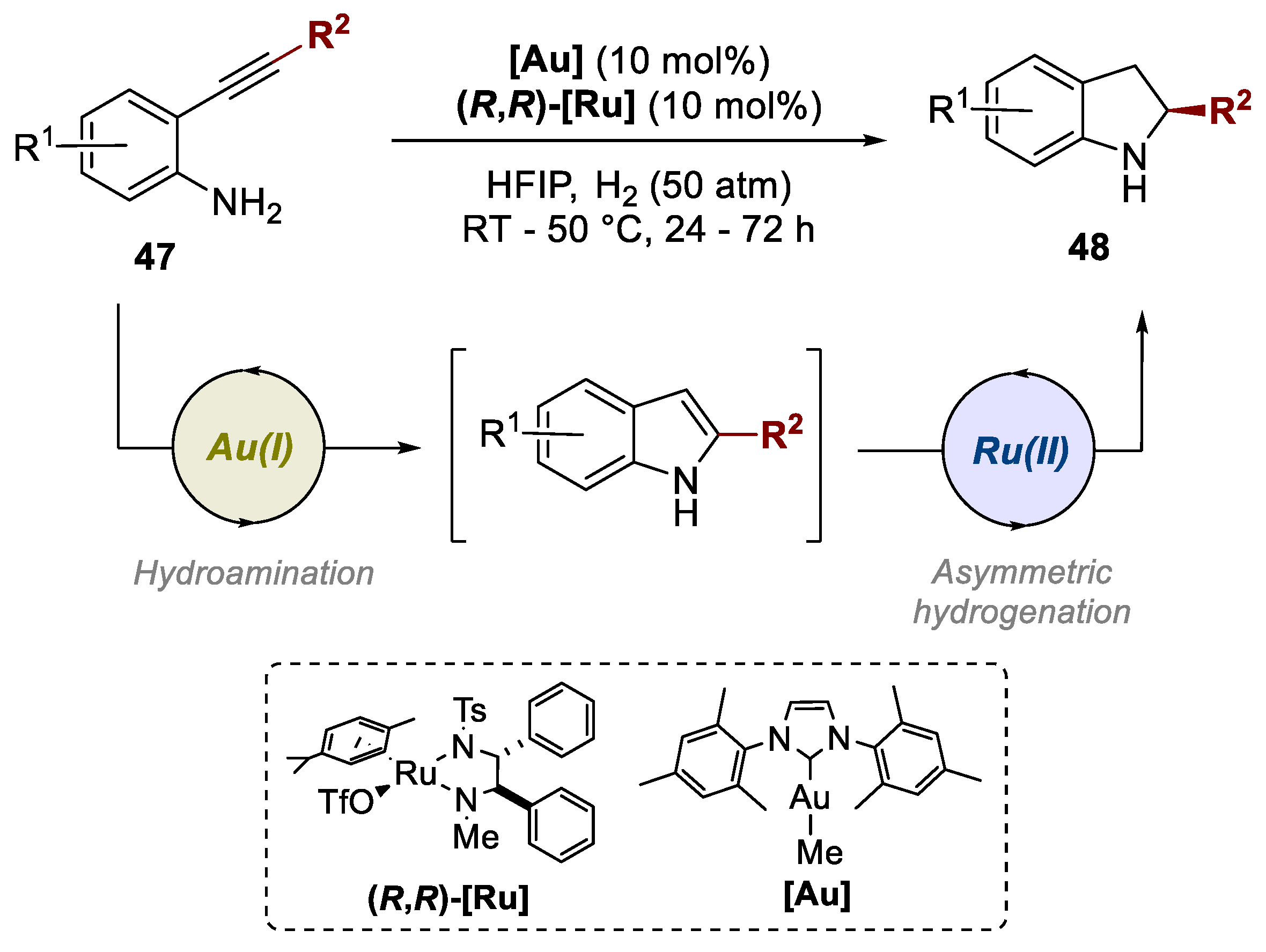
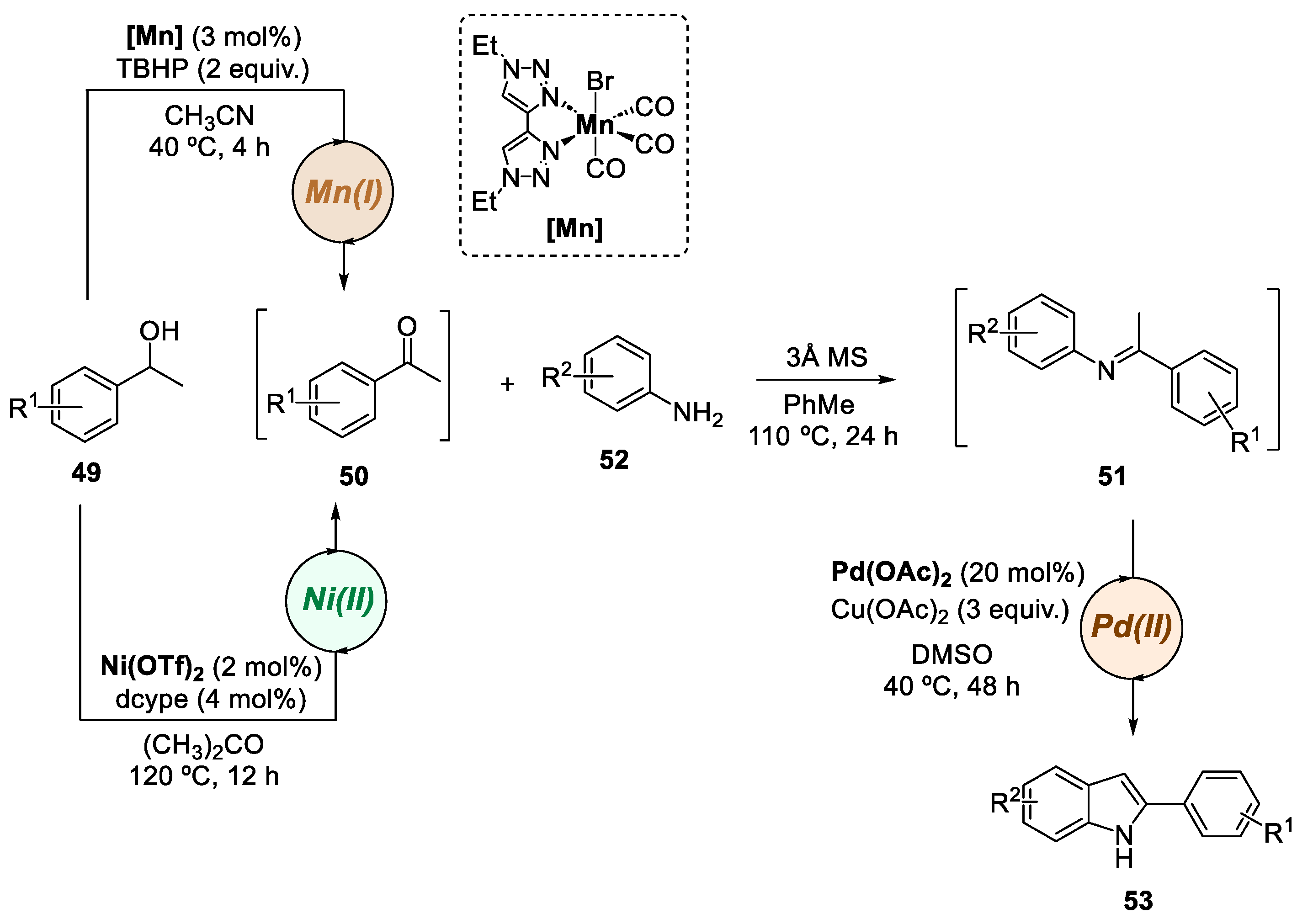
3. Bimetallic Approaches Involving Indole Scaffolds
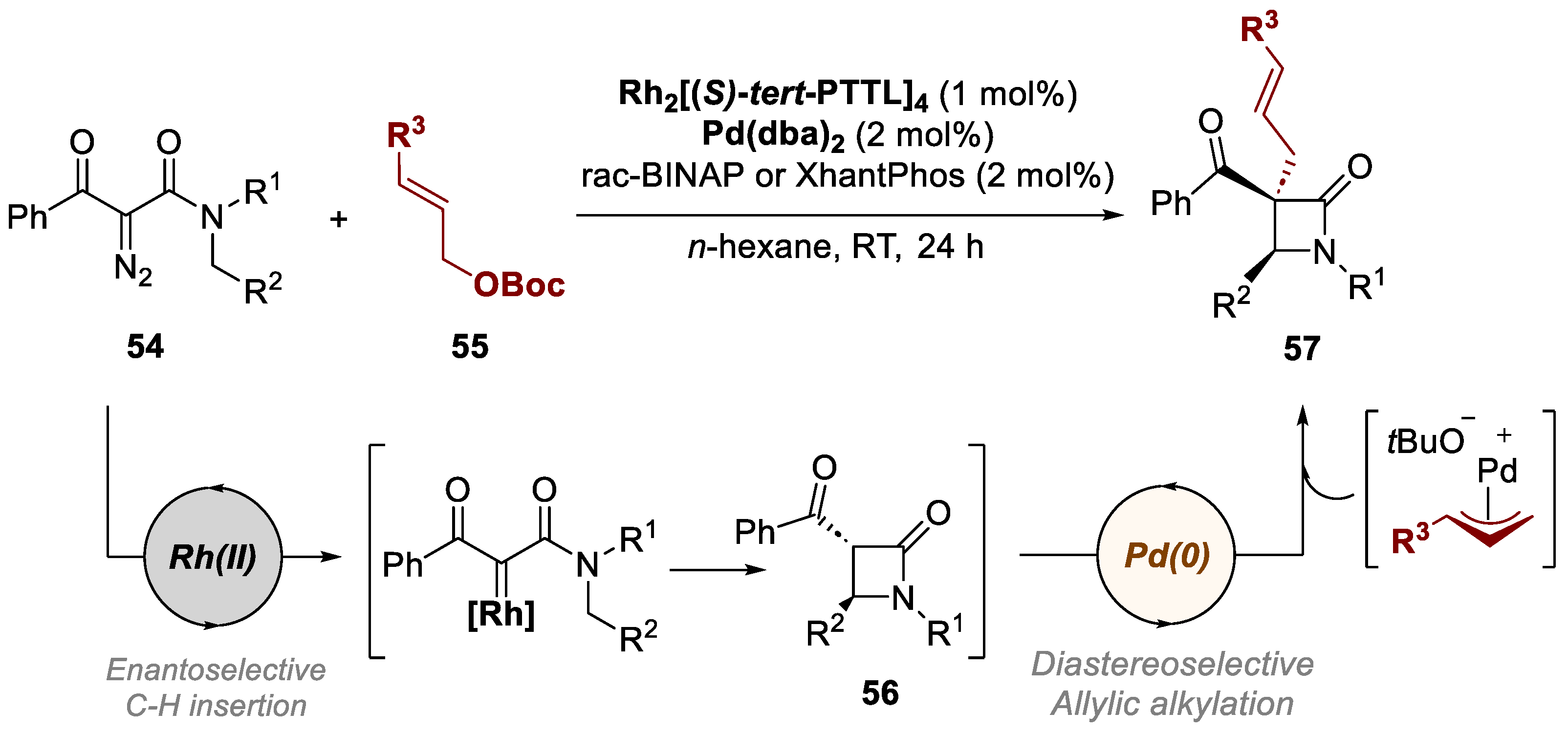
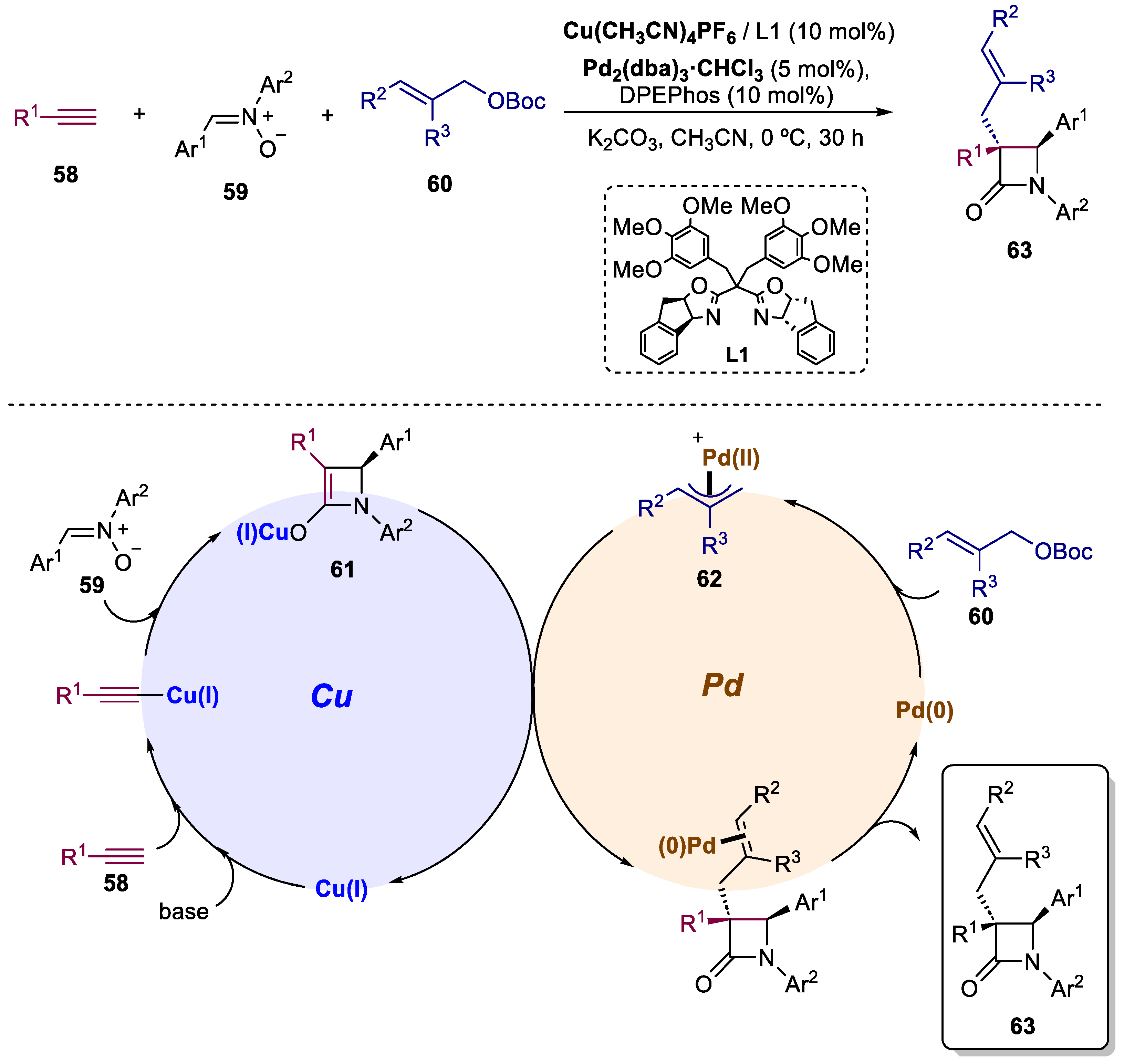
4. Bimetallic Approaches Involving Lactam Scaffolds
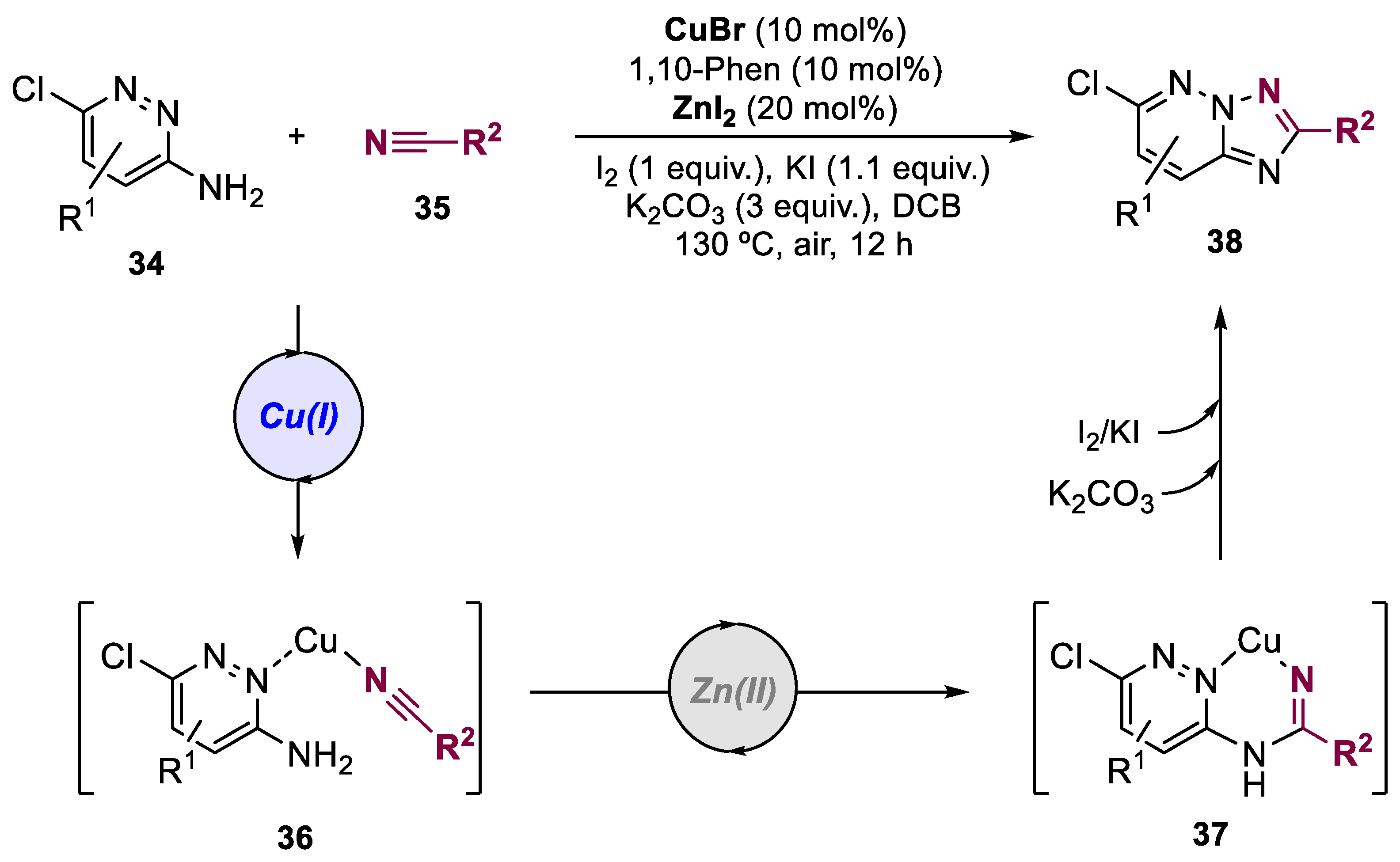
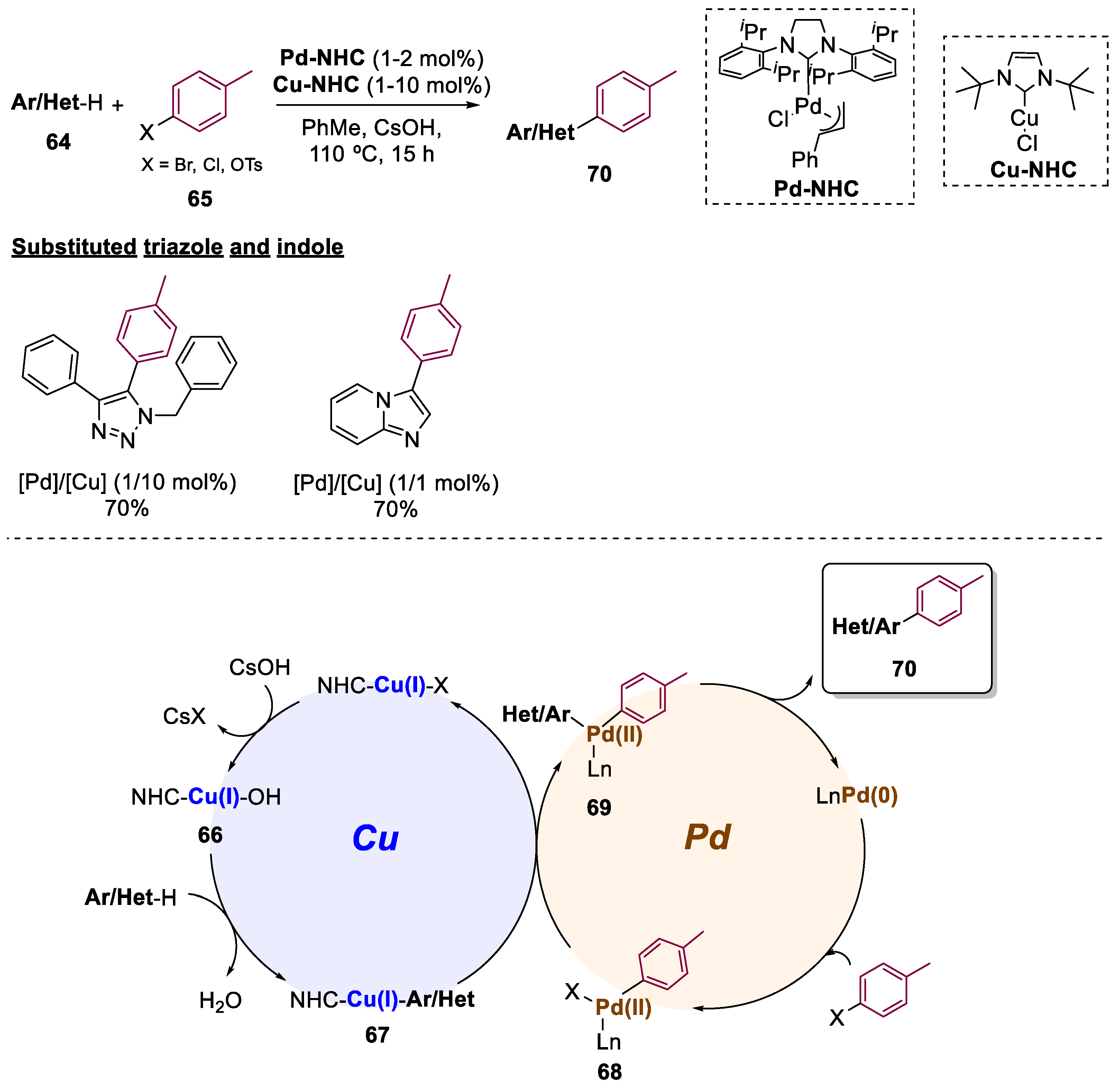
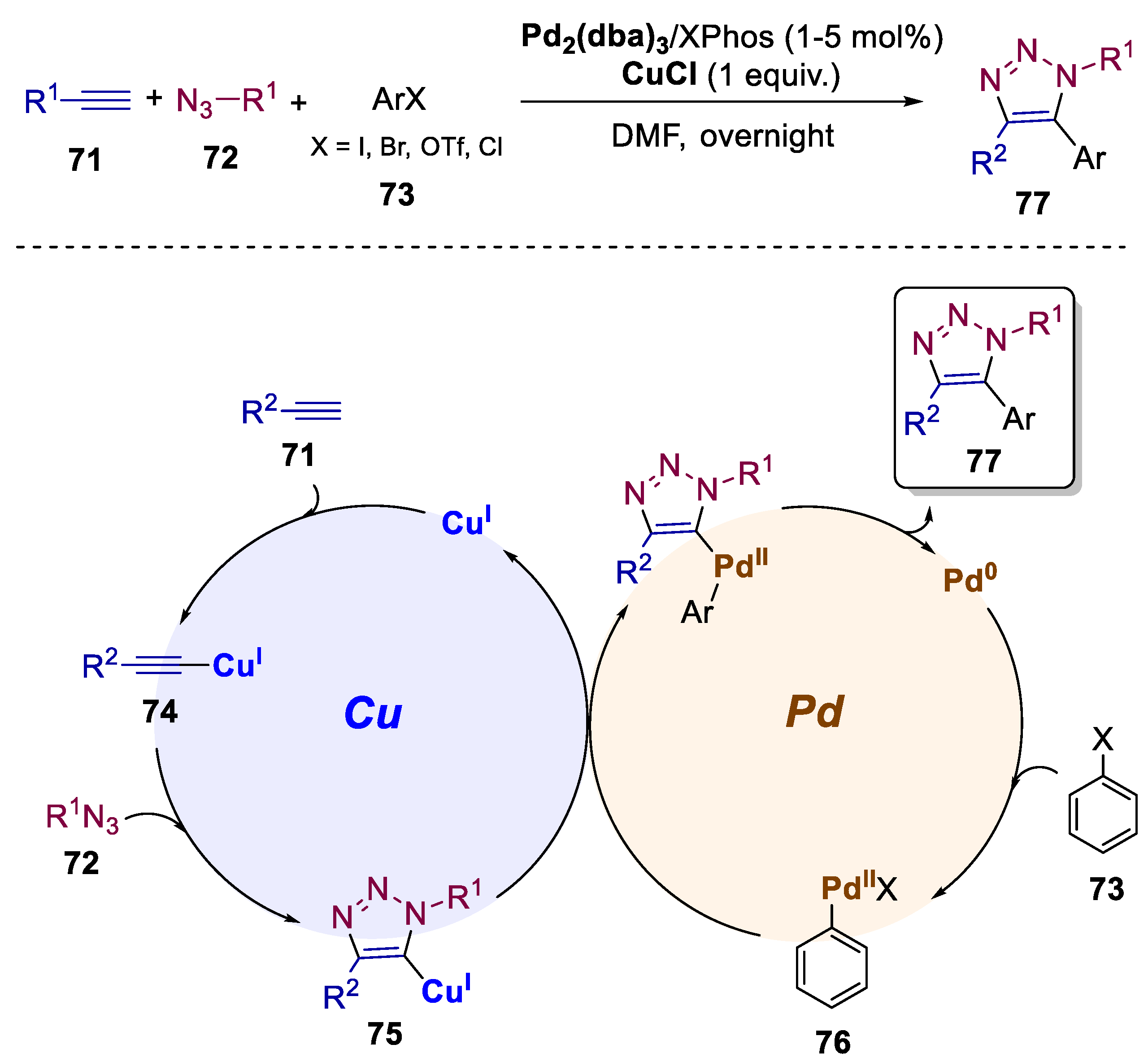
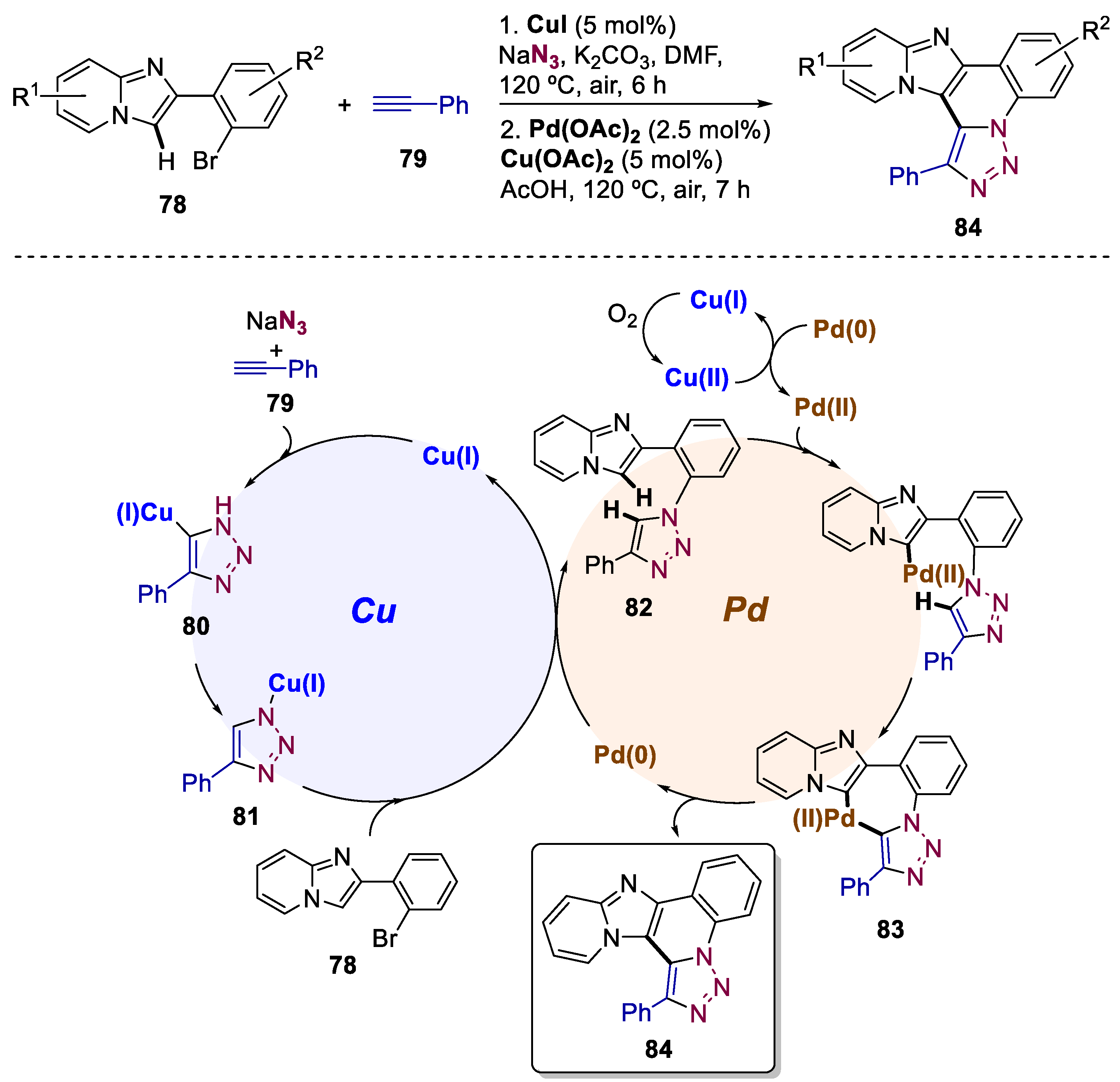
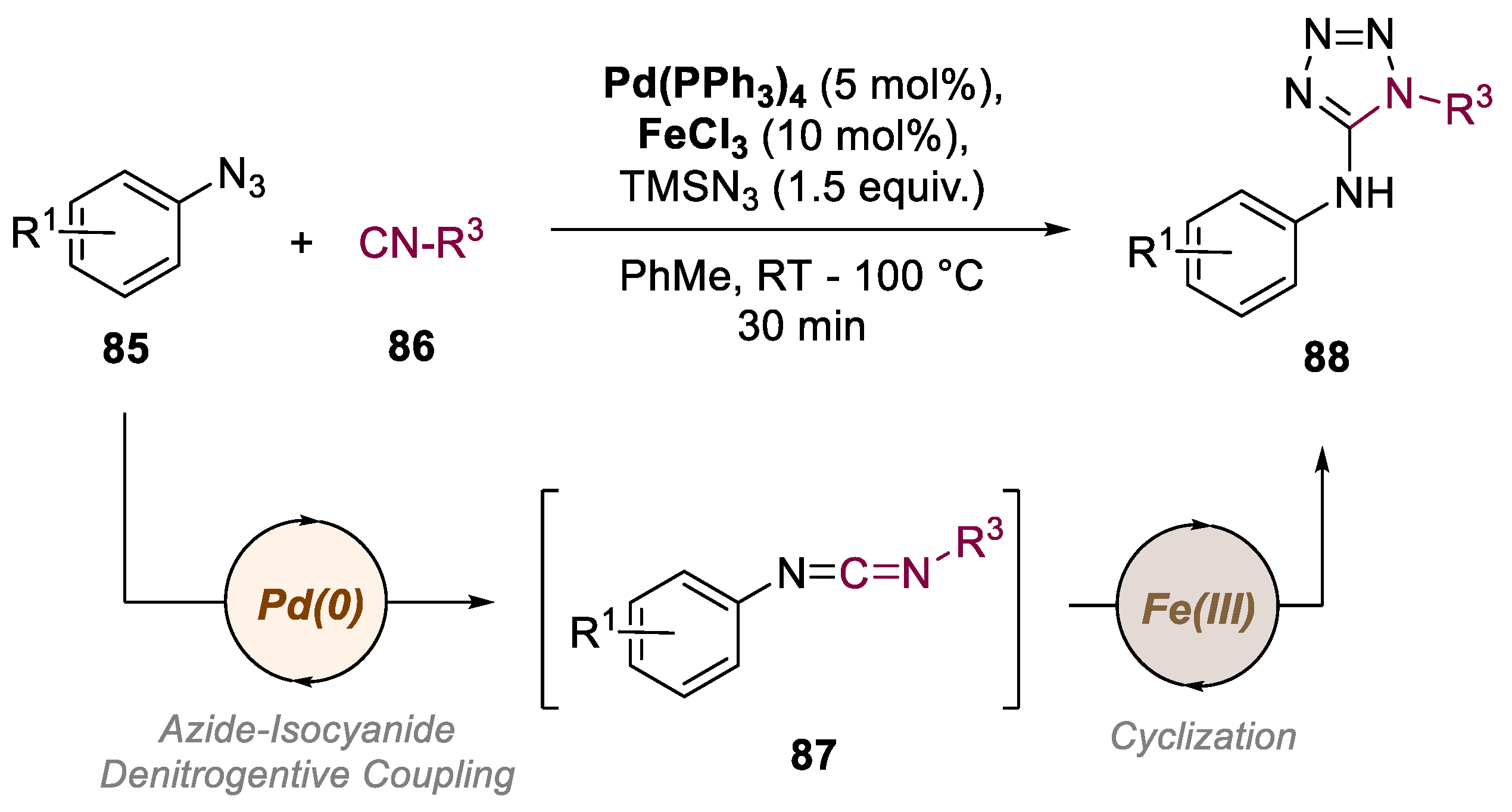
5. Bimetallic Approaches Involving Triazole and Tetrazole Scaffolds
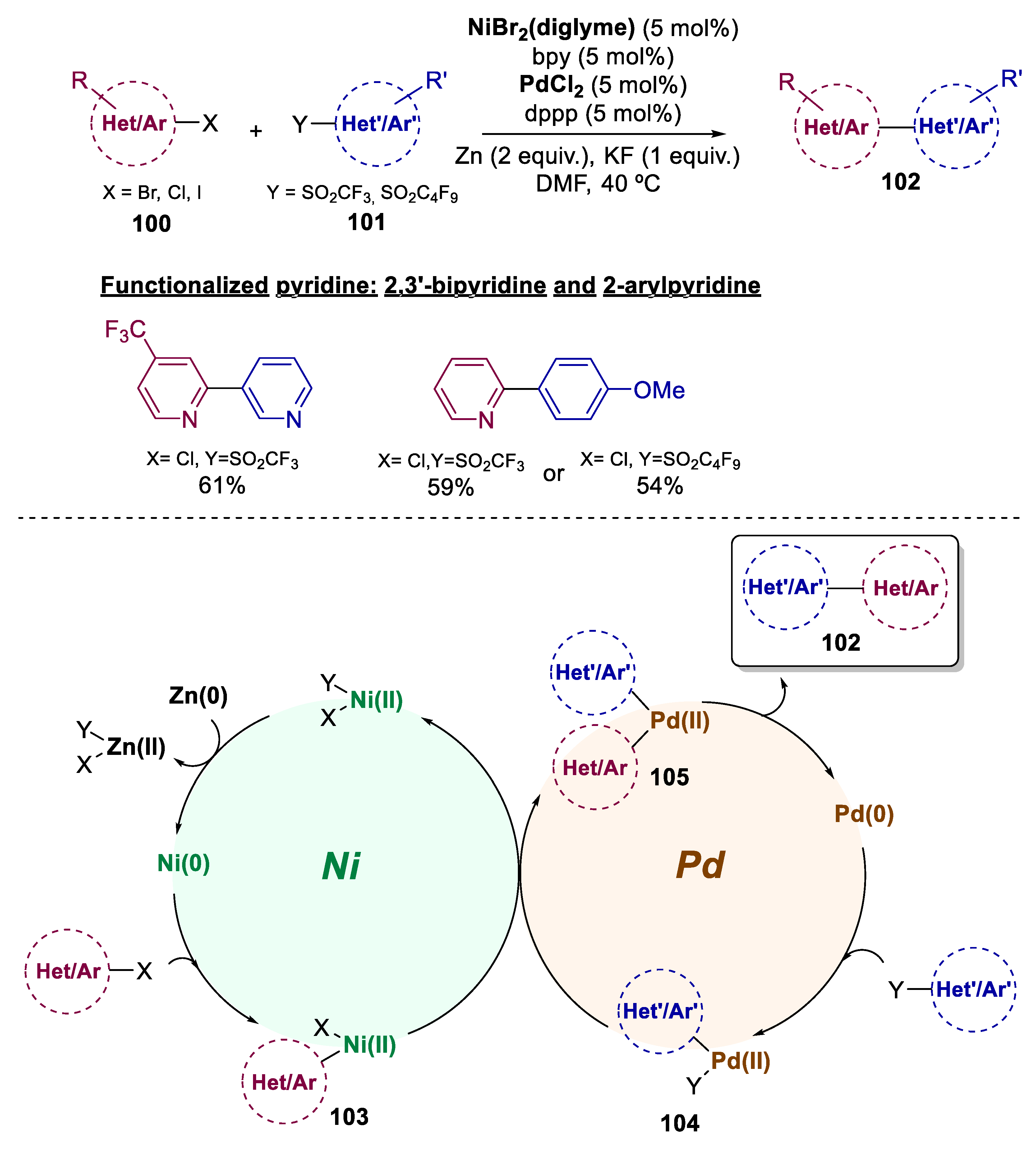
6. Bimetallic Approaches Involving Pyridine, Pyrimidine, and Related Scaffolds
7. Bimetallic Approaches Involving Other N-Heterocyclic Scaffolds
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huo, X.; Li, G.; Wang, X.; Zhang, W. Bimetallic Catalysis in Stereodivergent Synthesis. Angew. Chemie-Int. Ed. 2022, 61, e202210086. [Google Scholar] [CrossRef] [PubMed]
- Mankad, N.P. Selectivity Effects in Bimetallic Catalysis. Chem.-A Eur. J. 2016, 22, 5822–5829. [Google Scholar] [CrossRef] [PubMed]
- Pye, D.R.; Mankad, N.P. Bimetallic Catalysis for C–C and C–X Coupling Reactions. Chem. Sci. 2017, 8, 1705–1718. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Hong, S. Cooperative Bimetallic Catalysis in Asymmetric Transformations. Chem. Soc. Rev. 2012, 41, 6931–6943. [Google Scholar] [CrossRef]
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef]
- Reetz, M.T.; Breinbauer, R.; Wanninger, K. Suzuki and Heck Reactions Catalyzed by Preformed Palladium Clusters and Palladium/Nickel Bimetallic Clusters. Tetrahedron Lett. 1996, 37, 4499–4502. [Google Scholar] [CrossRef]
- Gupta, G.R.; Shah, J.; Vadagaonkar, K.S.; Lavekar, A.G.; Kapdi, A.R. Hetero-Bimetallic Cooperative Catalysis for the Synthesis of Heteroarenes. Org. Biomol. Chem. 2019, 17, 7596–7631. [Google Scholar] [CrossRef]
- Ferro, R.; Viduedo, N.; Santos, A.S.; Silva, A.M.S.; Royo, B.; Marques, M.M.B. Bimetallic Catalyzed Synthesis of 2-Arylindoles. Synthesis 2023, 25, A–G. [Google Scholar] [CrossRef]
- Malakar, C.C.; Dell’Amico, L.; Zhang, W. Dual Catalysis in Organic Synthesis: Current Challenges and New Trends. Eur. J. Org. Chem. 2023, 26, e202201114. [Google Scholar] [CrossRef]
- Huang, Y.Y.; Cai, C.; Yang, X.; Lv, Z.C.; Schneider, U. Catalytic Asymmetric Reactions with N,O-Aminals. ACS Catal. 2016, 6, 5747–5763. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Pernak, J.; Fan, W.Q.; Saczewski, F. N-(1-Benzotriazol-1-Ylalkyl)Amides, Versatile α-Amidoalkylation Reagents. 1. α-Amidoalkylation of CH Acids. J. Org. Chem. 1991, 56, 4439–4443. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Fan, W.Q.; Black, M.; Pernak, J. N-[1-(Benzotriazol-1-Yl)Alkyl]Amides, Versatile Amidoalkylation Reagents. 5. A General and Convenient Route to N-(α-Alkoxyalkyl)Amides. J. Org. Chem. 1992, 57, 547–549. [Google Scholar] [CrossRef]
- Gizecki, P.; Ait Youcef, R.; Poulard, C.; Dhal, R.; Dujardin, G. Diastereoselective Preparation of Novel Tetrahydrooxazinones via Heterocycloaddition of N-Boc, O-Me-Acetals. Tetrahedron Lett. 2004, 45, 9589–9592. [Google Scholar] [CrossRef]
- Harding, K.E.; Todd Coleman, M.; Liu, L.T. Stereoselective Reactions of N-Acyl-N,O-Acetals with 2-(Trimethysilyloxy)Furan. Tetrahedron Lett. 1991, 32, 3795–3798. [Google Scholar] [CrossRef]
- Yin, B.; Zhang, Y.; Xu, L.W. Recent Applications of α-Amido Sulfones as in Situ Equivalents of Activated Imines for Asymmetric Catalytic Nucleophilic Addition Reactions. Synthesis 2010, 2010, 3583–3595. [Google Scholar] [CrossRef]
- Li, H.; Belyk, K.M.; Yin, J.; Chen, Q.; Hyde, A.; Ji, Y.; Oliver, S.; Tudge, M.T.; Campeau, L.C.; Campos, K.R. Enantioselective Synthesis of Hemiaminals via Pd-Catalyzed C-N Coupling with Chiral Bisphosphine Mono-Oxides. J. Am. Chem. Soc. 2015, 137, 13728–13731. [Google Scholar] [CrossRef]
- Zhang, W.Z.; Chu, J.C.K.; Oberg, K.M.; Rovis, T. Enantioselective Rhodium-Catalyzed Isomerization of 4-Iminocrotonates: Asymmetric Synthesis of a Unique Chiral Synthon. J. Am. Chem. Soc. 2015, 137, 553–555. [Google Scholar] [CrossRef]
- Wang, T.; Yu, Z.; Hoon, D.L.; Phee, C.Y.; Lan, Y.; Lu, Y. Regiodivergent Enantioselective γ-Additions of Oxazolones to 2,3-Butadienoates Catalyzed by Phosphines: Synthesis of α,α-Disubstituted α-Amino Acids and N,O-Acetal Derivatives. J. Am. Chem. Soc. 2016, 138, 265–271. [Google Scholar] [CrossRef]
- Kim, H.; Rhee, Y.H. Stereodefined N,O-Acetals: Pd-Catalyzed Synthesis from Homopropargylic Amines and Utility in the Flexible Synthesis of 2,6-Substituted Piperidines. J. Am. Chem. Soc. 2012, 134, 4011–4014. [Google Scholar] [CrossRef]
- Rueping, M.; Antonchick, A.P.; Sugiono, E.; Grenader, K. Asymmetric Brønsted Acid Catalysis: Catalytic Enantioselective Synthesis of Highly Biologically Active Dihydroquinazolinones. Angew. Chemie-Int. Ed. 2009, 48, 908–910. [Google Scholar] [CrossRef]
- Vellalath, S.; Čorić, I.; List, B. N-Phosphinyl Phosphoramide-A Chiral Brønsted Acid Motif for the Direct Asymmetric N,O-Acetalization of Aldehydes. Angew. Chemie-Int. Ed. 2010, 49, 9749–9752. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Fronczek, F.R.; Antilla, J.C. Catalytic Asymmetric Addition of Alcohols to Imines: Enantioselective Preparation of Chiral N,O-Aminals. J. Am. Chem. Soc. 2008, 130, 12216–12217. [Google Scholar] [CrossRef]
- Li, T.Z.; Wang, X.B.; Sha, F.; Wu, X.Y. Catalytic Enantioselective Addition of Alcohols to Isatin-Derived N-Boc Ketimines. Tetrahedron 2013, 69, 7314–7319. [Google Scholar] [CrossRef]
- Hiramatsu, K.; Honjo, T.; Rauniyar, V.; Toste, F.D. Enantioselective Synthesis of Fluoro-Dihydroquinazolones and -Benzooxazinones by Fluorination-Initiated Asymmetric Cyclization Reactions. ACS Catal. 2016, 6, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Dong, S.; Yao, Z.; Feng, L.; Daka, P.; Wang, H.; Xu, Z. Synthesis of Spiroaminals and Spiroketals with Bimetallic Relay Catalysis. Org. Lett. 2014, 16, 22–25. [Google Scholar] [CrossRef]
- Wang, X.; Yao, Z.; Dong, S.; Wei, F.; Wang, H.; Xu, Z. Synthesis of Fused Bicyclic Aminals through Sequential Gold/Lewis Acid Catalysis. Org. Lett. 2013, 15, 2234–2237. [Google Scholar] [CrossRef]
- Zhang, S.; Xu, Z.; Jia, J.; Tung, C.H.; Xu, Z. Synthesis of Spiroaminals by Bimetallic Au/Sc Relay Catalysis: TMS as a Traceless Controlling Group. Chem. Commun. 2014, 50, 12084–12087. [Google Scholar] [CrossRef]
- Wang, B.; Liang, M.; Tang, J.; Deng, Y.; Zhao, J.; Sun, H.; Tung, C.H.; Jia, J.; Xu, Z. Gold/Lewis Acid Catalyzed Cycloisomerization/Diastereoselective [3+2] Cycloaddition Cascade: Synthesis of Diverse Nitrogen-Containing Spiro Heterocycles. Org. Lett. 2016, 18, 4614–4617. [Google Scholar] [CrossRef]
- Li, J.; Lin, L.; Hu, B.; Lian, X.; Wang, G.; Liu, X.; Feng, X. Bimetallic Gold(I)/Chiral N,N′-Dioxide Nickel(II) Asymmetric Relay Catalysis: Chemo- and Enantioselective Synthesis of Spiroketals and Spiroaminals. Angew. Chemie-Int. Ed. 2016, 55, 6075–6078. [Google Scholar] [CrossRef]
- Wang, B.; Chen, Y.; Zhou, L.; Wang, J.; Xu, Z. Zn/Sc Bimetallic Relay Catalysis: One Pot Cycloisomerization/Carbonyl-Ene Reaction toward Oxazole Derivatives. Org. Biomol. Chem. 2016, 14, 826–829. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Li, J.; Cao, W.; Lin, Q.; Yang, J.; Lin, L.; Liu, X.; Feng, X. Asymmetric Synthesis of Fused Bicyclic N,O- and O,O-Acetals via Cascade Reaction by Gold(I)/N,N′-Dioxide-Nickel(II) Bimetallic Relay Catalysis. Adv. Synth. Catal. 2018, 360, 2831–2835. [Google Scholar] [CrossRef]
- Taber, D.F.; Tirunahari, P.K. Indole Synthesis: A Review and Proposed Classification. Tetrahedron 2011, 67, 7195–7210. [Google Scholar] [CrossRef]
- Tobisu, M.; Ito, S.; Kitajima, A.; Chatani, N. GaCI3- and TiCl4-Catalyzed Insertion of Isocyanides into a C-S Bond of Dithioacetals. Org. Lett. 2008, 10, 5223–5225. [Google Scholar] [CrossRef]
- Kong, W.; Wang, Q.; Zhu, J. Synthesis of Diversely Functionalized Oxindoles Enabled by Migratory Insertion of Isocyanide to a Transient σ-Alkylpalladium(II) Complex. Angew. Chemie-Int. Ed. 2016, 55, 9714–9718. [Google Scholar] [CrossRef]
- Vlaar, T.; Cioc, R.C.; Mampuys, P.; Maes, B.U.W.; Orru, R.V.A.; Ruijter, E. Sustainable Synthesis of Diverse Privileged Heterocycles by Palladium-Catalyzed Aerobic Oxidative Isocyanide Insertion. Angew. Chemie-Int. Ed. 2012, 51, 13058–13061. [Google Scholar] [CrossRef]
- Liu, B.; Yin, M.; Gao, H.; Wu, W.; Jiang, H. Synthesis of 2-Aminobenzoxazoles and 3-Aminobenzoxazines via Palladium-Catalyzed Aerobic Oxidation of o -Aminophenols with Isocyanides. J. Org. Chem. 2013, 78, 3009–3020. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Zhou, P.; Liu, F.; Hao, W.J.; Yao, C.; Jiang, B.; Tu, S.J. Cobalt(II)/Silver Relay Catalytic Isocyanide Insertion/Cycloaddition Cascades: A New Access to Pyrrolo[2,3-b]Indoles. Chem. Commun. 2015, 51, 9519–9522. [Google Scholar] [CrossRef] [PubMed]
- Manisha; Dhiman, S.; Mathew, J.; Ramasastry, S.S.V. One-Pot Relay Catalysis: Divergent Synthesis of Furo[3,4-: B] Indoles and Cyclopenta [b] Indoles from 3-(2-Aminophenyl)-1,4-Enynols. Org. Biomol. Chem. 2016, 14, 5563–5568. [Google Scholar] [CrossRef]
- Dhiman, S.; Mishra, U.K.; Ramasastry, S.S.V. One-Pot Trimetallic Relay Catalysis: A Unified Approach for the Synthesis of β-Carbolines and Other [ c ]-Fused Pyridines. Angew. Chemie 2016, 128, 7868–7872. [Google Scholar] [CrossRef]
- Mu, Q.C.; Lv, J.Y.; Chen, M.Y.; Bai, X.F.; Chen, J.; Xia, C.G.; Xu, L.W. Bimetallic Copper and Zinc-Catalyzed Oxidative Cycloaddition of 3-Aminopyridazines and Nitriles: A Direct Synthesis of 1,2,4-Triazolo[1,5-: B] Pyridazines via C–N and N–N Bond-Forming Process. RSC Adv. 2017, 7, 37208–37213. [Google Scholar] [CrossRef]
- Ji, W.W.; Lin, E.; Li, Q.; Wang, H. Heteroannulation Enabled by a Bimetallic Rh(III)/Ag(I) Relay Catalysis: Application in the Total Synthesis of Aristolactam BII. Chem. Commun. 2017, 53, 5665–5668. [Google Scholar] [CrossRef]
- Zhang, D.; Lin, L.; Yang, J.; Liu, X.; Feng, X. Asymmetric Synthesis of Tetrahydroindolizines by Bimetallic Relay Catalyzed Cycloaddition of Pyridinium Ylides. Angew. Chemie-Int. Ed. 2018, 57, 12323–12327. [Google Scholar] [CrossRef]
- Xu, C.; Feng, Y.; Li, F.; Han, J.; He, Y.M.; Fan, Q.H. A Synthetic Route to Chiral Benzo-Fused N-Heterocycles via Sequential Intramolecular Hydroamination and Asymmetric Hydrogenation of Anilino-Alkynes. Organometallics 2019, 38, 3979–3990. [Google Scholar] [CrossRef]
- Von Nussbaum, F.; Brands, M.; Hinzen, B.; Weigand, S.; Häbich, D. Antibacterial Natural Products in Medicinal Chemistry-Exodus or Revival? Angew. Chemie-Int. Ed. 2006, 45, 5072–5129. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.; Floyd, D.M.; Georgopapadakou, N.H.; Koster, W.H.; Liu, W.C.; Parker, W.L.; Principe, P.A.; Rathnum, M.L.; Slusarchyk, W.A.; Trejo, W.H.; et al. Monocyclic β-lactam antibiotics produced by bacteria. Nature 1971, 291, 489–491. [Google Scholar] [CrossRef]
- Imada, A.; Kitano, K.; Kintaka, K.; Muroi, M.; Asai, M. Sulfazecin and Isosulfazecin, Novel β-Lactam Antibiotics of Bacterial Origin. Nature 1981, 289, 590–591. [Google Scholar] [CrossRef]
- Galletti, P.; Giacomini, D. Monocyclic β-Lactams: New Structures for New Biological Activities. Curr. Med. Chem. 2011, 18, 4265–4283. [Google Scholar] [CrossRef] [PubMed]
- Pitts, C.R.; Lectka, T. Chemical Synthesis of β-Lactams: Asymmetric Catalysis and Other Recent Advances. Chem. Rev. 2014, 114, 7930–7953. [Google Scholar] [CrossRef] [PubMed]
- France, S.; Weatherwax, A.; Taggi, A.E.; Lectka, T. Advances in the Catalytic, Asymmetric Synthesis of β-Lactams. Acc. Chem. Res. 2004, 37, 592–600. [Google Scholar] [CrossRef]
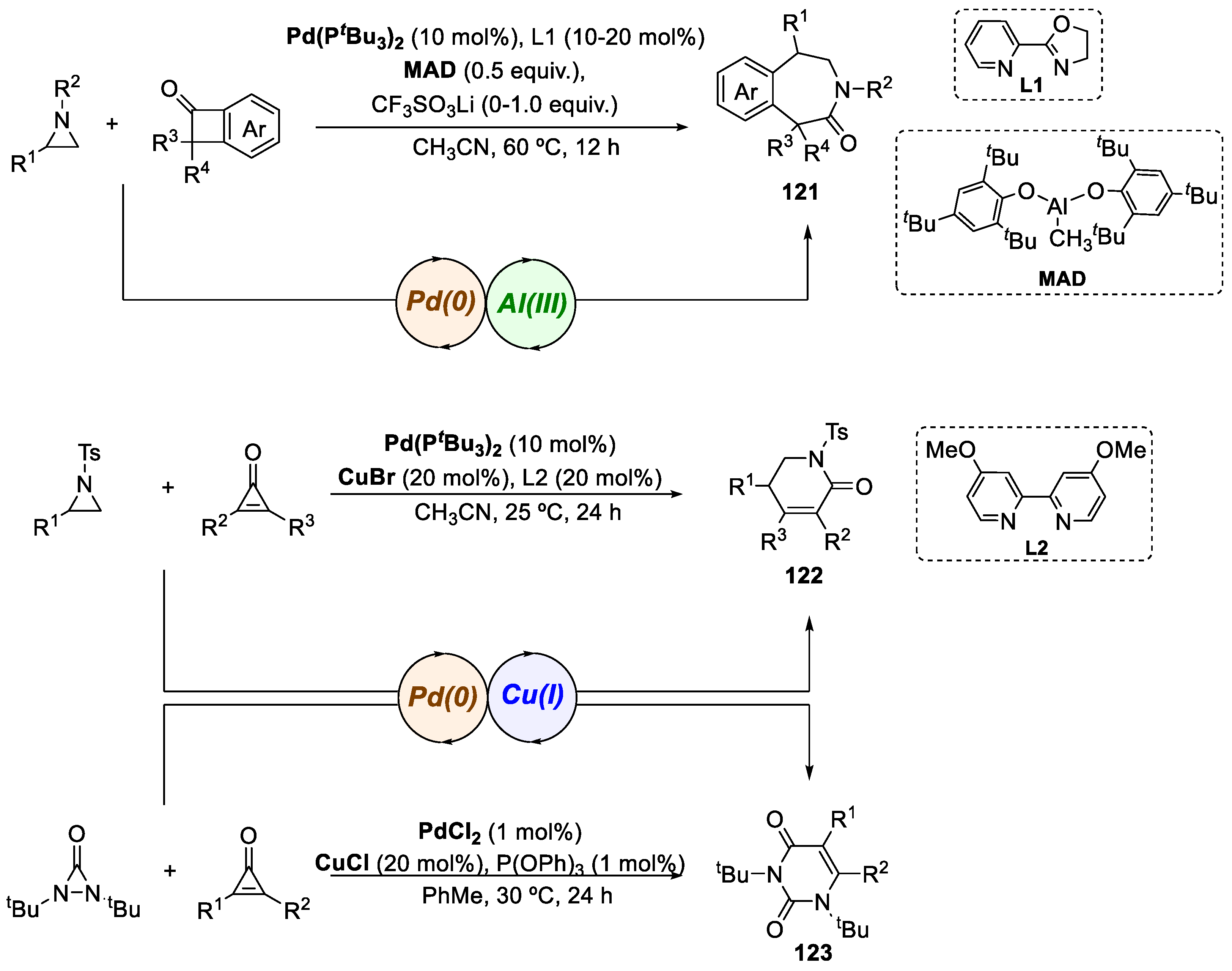
- Huang, L.Z.; Xuan, Z.; Jeon, H.J.; Du, Z.T.; Kim, J.H.; Lee, S.G. Asymmetric Rh(II)/Pd(0) Relay Catalysis: Synthesis of α-Quaternary Chiral β-Lactams through Enantioselective C−H Insertion/Diastereoselective Allylation of Diazoamides. ACS Catal. 2018, 8, 7340–7345. [Google Scholar] [CrossRef]
- Qi, J.; Wei, F.; Tung, C.H.; Xu, Z. Modular Synthesis of α-Quaternary Chiral β-Lactams by a Synergistic Copper/Palladium-Catalyzed Multicomponent Reaction. Angew. Chemie-Int. Ed. 2021, 60, 13814–13818. [Google Scholar] [CrossRef]
- Qi, J.; Song, T.; Yang, Z.; Sun, S.; Tung, C.H.; Xu, Z. Simultaneous Dual Cu/Ir Catalysis: Stereodivergent Synthesis of Chiral β-Lactams with Adjacent Tertiary/Quaternary/Tertiary Stereocenters. ACS Catal. 2023, 13, 2555–2564. [Google Scholar] [CrossRef]
- Matin, M.M.; Matin, P.; Rahman, M.R.; Ben Hadda, T.; Almalki, F.A.; Mahmud, S.; Ghoneim, M.M.; Alruwaily, M.; Alshehri, S. Triazoles and Their Derivatives: Chemistry, Synthesis, and Therapeutic Applications. Front. Mol. Biosci. 2022, 9, 864286. [Google Scholar] [CrossRef]
- Dalton, T.; Faber, T.; Glorius, F. C−H Activation: Toward Sustainability and Applications. ACS Cent. Sci. 2021, 7, 245–261. [Google Scholar] [CrossRef]
- Lesieur, M.; Lazreg, F.; Cazin, C.S.J. A Cooperative Pd-Cu System for Direct C−H Bond Arylation. Chem. Commun. 2014, 50, 8927–8929. [Google Scholar] [CrossRef]
- Spiteri, C.; Moses, J.E. Copper-Catalyzed Azide-Alkyne Cycloaddition: Regioselective Synthesis of 1,4,5-Trisubstituted 1,2,3-Triazoles. Angew. Chemie-Int. Ed. 2010, 49, 31–33. [Google Scholar] [CrossRef]
- Wei, F.; Li, H.; Song, C.; Ma, Y.; Zhou, L.; Tung, C.H.; Xu, Z. Cu/Pd-Catalyzed, Three-Component Click Reaction of Azide, Alkyne, and Aryl Halide: One-Pot Strategy toward Trisubstituted Triazoles. Org. Lett. 2015, 17, 2860–2863. [Google Scholar] [CrossRef]
- Verma, Y.K.; Reddy, B.S.; Pawar, M.S.; Bhunia, D.; Sampath Kumar, H.M. Design, Synthesis, and Immunological Evaluation of Benzyloxyalkyl-Substituted 1,2,3-Triazolyl α-GalCer Analogues. ACS Med. Chem. Lett. 2016, 7, 172–176. [Google Scholar] [CrossRef]
- Meldal, M.; Tomøe, C.W. Cu-Catalyzed Azide-Alkyne Cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef]
- Xiao, X.; Xie, Y.; Bai, S.; Deng, Y.; Jiang, H.; Zeng, W. Transition-Metal-Free Tandem Chlorocyclization of Amines with Carboxylic Acids: Access to Chloroimidazo[1,2-α]Pyridines. Org. Lett. 2015, 17, 3998–4001. [Google Scholar] [CrossRef]
- Monir, K.; Bagdi, A.K.; Ghosh, M.; Hajra, A. Regioselective Oxidative Trifluoromethylation of Imidazoheterocycles via C(Sp2)-H Bond Functionalization. J. Org. Chem. 2015, 80, 1332–1337. [Google Scholar] [CrossRef]
- Cai, Q.; Yan, J.; Ding, K. A CuAAC/Ullmann C–C Coupling Tandem Reaction: Copper-Catalyzed Reactions of Organic Azides with N-(2-Iodoaryl)Propiolamides or 2-Iodo-N-(Prop-2-Ynyl)Benzenamines. Org. Lett. 2012, 14, 3332–3335. [Google Scholar] [CrossRef] [PubMed]
- Pericherla, K.; Jha, A.; Khungar, B.; Kumar, A. Copper-Catalyzed Tandem Azide-Alkyne Cycloaddition, Ullmann Type C–N Coupling, and Intramolecular Direct Arylation. Org. Lett. 2013, 15, 4304–4307. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, B.; Zhang, X.; Fan, X. One-Pot Cascade Reactions Leading to Pyrido[2′,1′:2,3]Imidazo[4,5-c][1,2,3]Triazolo[1,5-a]Quinolines under Bimetallic Relay Catalysis with Air as the Oxidant. J. Org. Chem. 2016, 81, 6357–6363. [Google Scholar] [CrossRef] [PubMed]
- Pathare, R.S.; Ansari, A.J.; Verma, S.; Maurya, A.; Maurya, A.K.; Agnihotri, V.K.; Sharon, A.; Pardasani, R.T.; Sawant, D.M. Sequential Pd(0)/Fe(III) Catalyzed Azide-Isocyanide Coupling/Cyclization Reaction: One-Pot Synthesis of Aminotetrazoles. J. Org. Chem. 2018, 83, 9530–9537. [Google Scholar] [CrossRef]
- Wang, H.; Grohmann, C.; Nimphius, C.; Glorius, F. Mild Rh(III)-Catalyzed C−H Activation and Annulation with Alkyne MIDA Boronates: Short, Efficient Synthesis of Heterocyclic Boronic Acid Derivatives. J. Am. Chem. Soc. 2012, 134, 19592–19595. [Google Scholar] [CrossRef] [PubMed]
- Guimond, N.; Gorelsky, S.I.; Fagnou, K. Rhodium(III)-Catalyzed Heterocycle Synthesis Using an Internal Oxidant: Improved Reactivity and Mechanistic Studies. J. Am. Chem. Soc. 2011, 133, 6449–6457. [Google Scholar] [CrossRef]
- Hyster, T.K.; Rovis, T. An Improved Catalyst Architecture for Rhodium(Iii) Catalyzed C−H Activation and Its Application to Pyridone Synthesis. Chem. Sci. 2011, 2, 1606–1610. [Google Scholar] [CrossRef]
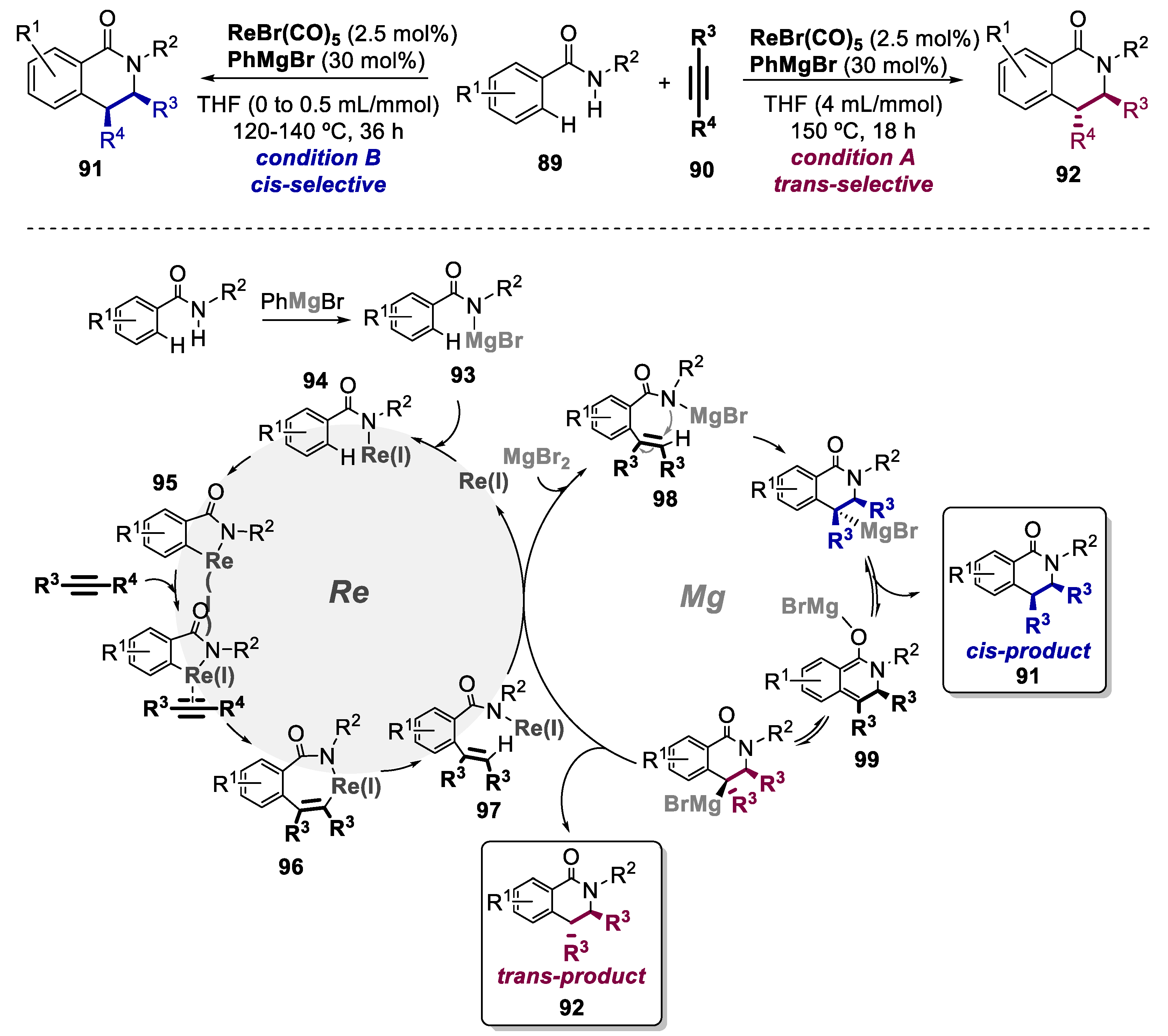
- Tang, Q.; Xia, D.; Jin, X.; Zhang, Q.; Sun, X.Q.; Wang, C. Re/Mg Bimetallic Tandem Catalysis for [4+2] Annulation of Benzamides and Alkynes via C−H/N−H Functionalization. J. Am. Chem. Soc. 2013, 135, 4628–4631. [Google Scholar] [CrossRef]
- Kang, K.; Huang, L.; Weix, D.J. Sulfonate Versus Sulfonate: Nickel and Palladium Multimetallic Cross-Electrophile Coupling of Aryl Triflates with Aryl Tosylates. J. Am. Chem. Soc. 2020, 142, 10634–10640. [Google Scholar] [CrossRef]
- Ackerman, L.K.G.; Lovell, M.M.; Weix, D.J. Multimetallic Catalysed Cross-Coupling of Aryl Bromides with Aryl Triflates. Nature 2015, 524, 454–457. [Google Scholar] [CrossRef]
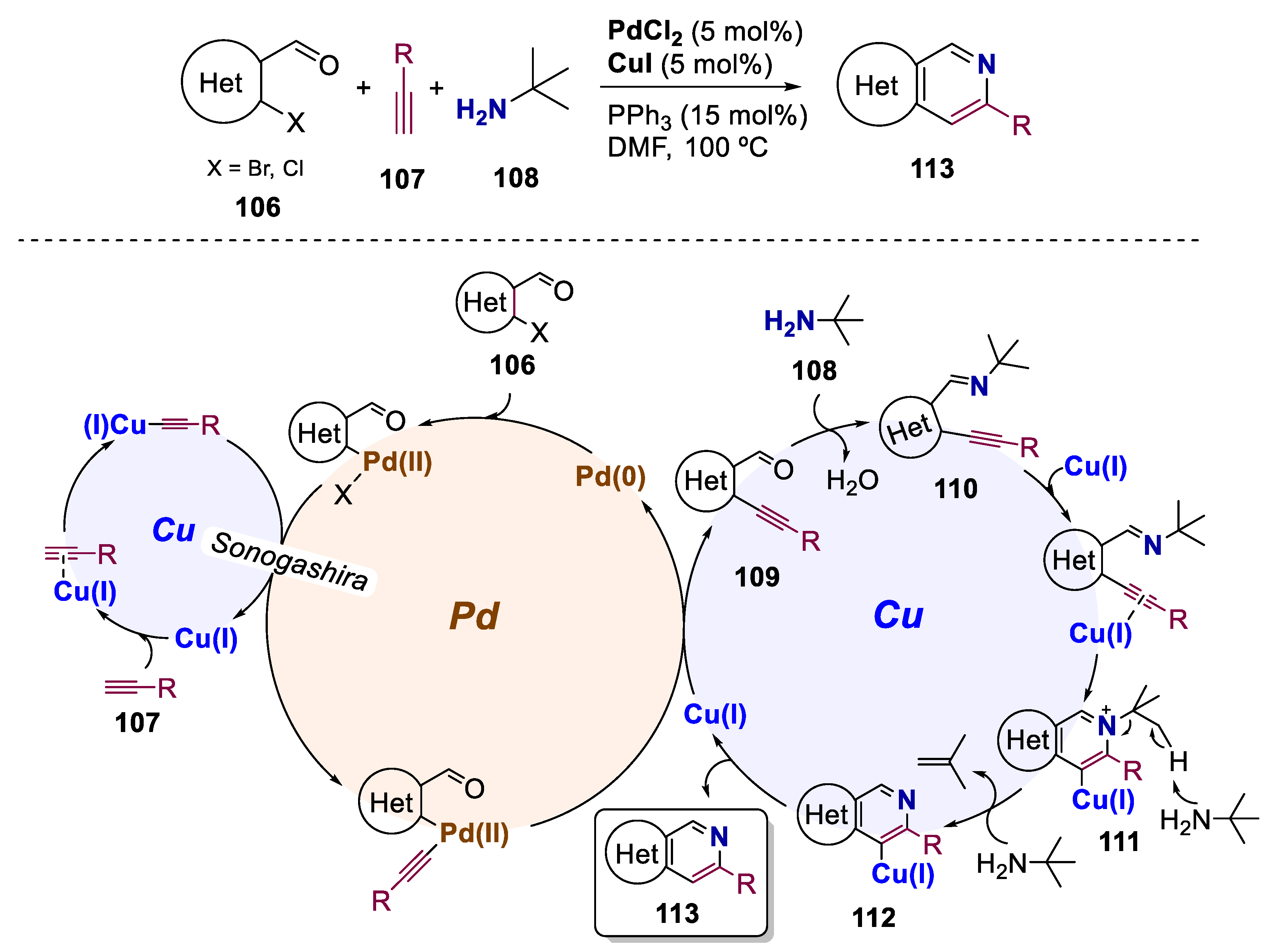
- Kumar, S.; Saunthwal, R.K.; Aggarwal, T.; Kotla, S.K.R.; Verma, A.K. Palladium Meets Copper: One-Pot Tandem Synthesis of Pyrido Fused Heterocycles: Via Sonogashira Conjoined Electrophilic Cyclization. Org. Biomol. Chem. 2016, 14, 9063–9071. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Koester, D.C.; Boultadakis-Arapinis, M.; Glorius, F. Rh(III)-Catalyzed Synthesis of Multisubstituted Isoquinoline and Pyridine N-Oxides from Oximes and Diazo Compounds. J. Am. Chem. Soc. 2013, 135, 12204–12207. [Google Scholar] [CrossRef] [PubMed]
- Phatake, R.S.; Patel, P.; Ramana, C.V. Ir(III)-Catalyzed Synthesis of Isoquinoline N-Oxides from Aryloxime and α-Diazocarbonyl Compounds. Org. Lett. 2016, 18, 292–295. [Google Scholar] [CrossRef] [PubMed]
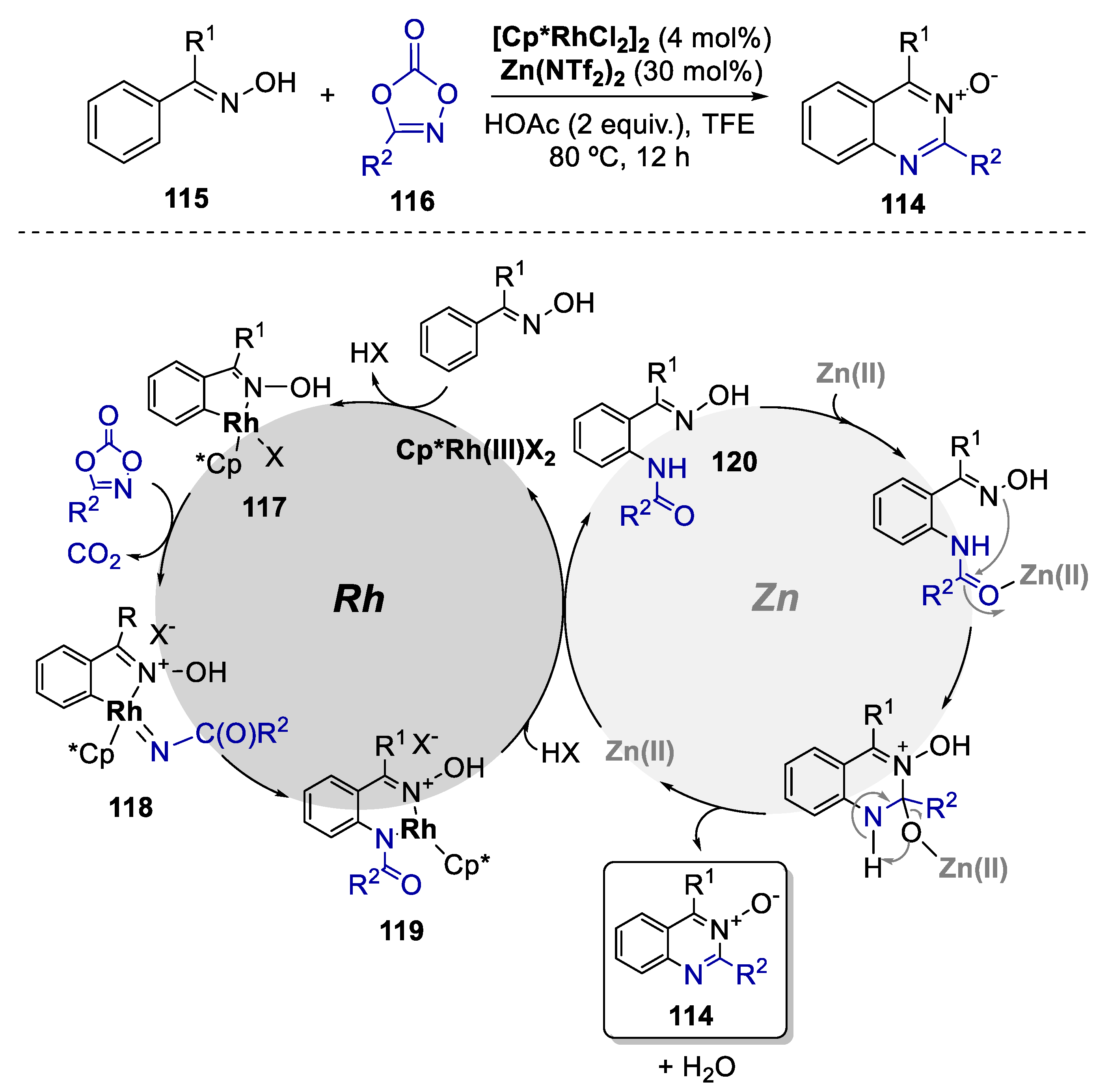
- Wang, Q.; Wang, F.; Yang, X.; Zhou, X.; Li, X. Rh(III)- and Zn(II)-Catalyzed Synthesis of Quinazoline N-Oxides via C–H Amidation-Cyclization of Oximes. Org. Lett. 2016, 18, 6144–6147. [Google Scholar] [CrossRef]
- Li, R.; Li, B.; Zhang, H.; Ju, C.W.; Qin, Y.; Xue, X.S.; Zhao, D. A Ring Expansion Strategy towards Diverse Azaheterocycles. Nat. Chem. 2021, 13, 1006–1016. [Google Scholar] [CrossRef]
- Gao, K.; Yoshikai, N. Low-Valent Cobalt Catalysis: New Opportunities for C–H Functionalization. Acc. Chem. Res. 2014, 47, 1208–1219. [Google Scholar] [CrossRef]
- Xie, P.; Xie, Y.; Qian, B.; Zhou, H.; Xia, C.; Huang, H. Palladium-Catalyzed Oxidative Carbonylation of Benzylic C–H Bonds via Nondirected C(Sp3)–H Activation. J. Am. Chem. Soc. 2012, 134, 9902–9905. [Google Scholar] [CrossRef]
- Hasegawa, N.; Shibata, K.; Charra, V.; Inoue, S.; Fukumoto, Y.; Chatani, N. Ruthenium-Catalyzed Cyclocarbonylation of Aliphatic Amides through the Regioselective Activation of Unactivated C(Sp3)-H Bonds. Tetrahedron 2013, 69, 4466–4472. [Google Scholar] [CrossRef]
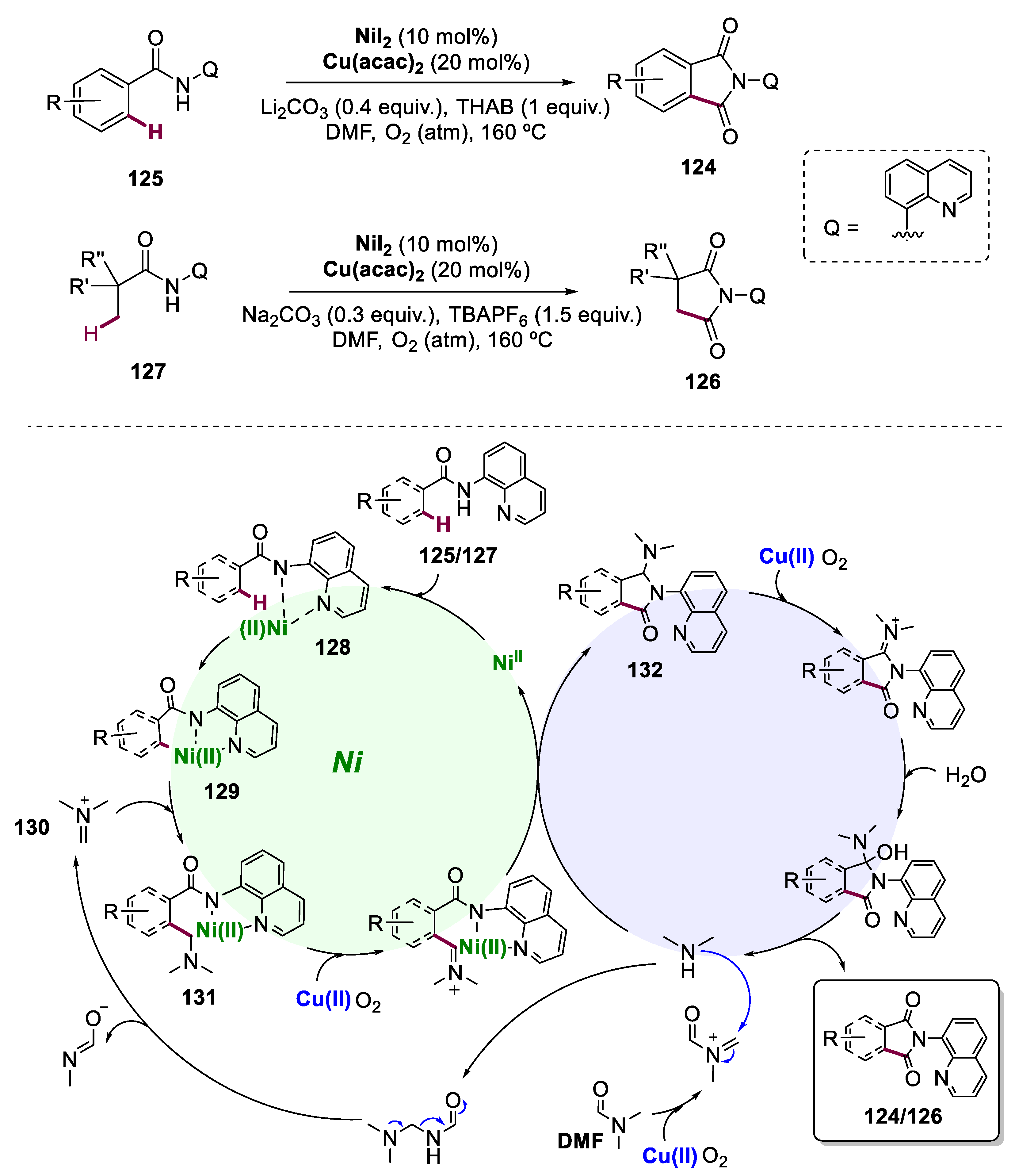
- Wu, X.; Zhao, Y.; Ge, H. Direct Aerobic Carbonylation of C(Sp2)–H and C(Sp3)–H Bonds through Ni/Cu Synergistic Catalysis with DMF as the Carbonyl Source. J. Am. Chem. Soc. 2015, 137, 4924–4927. [Google Scholar] [CrossRef]
- Vrancken, E.; Alexakis, A.; Mangeney, P. Organolithium/Chiral Lewis Base/BF3: A Versatile Combination for the Enantioselective Desymmetrization of Meso-Epoxides. Eur. J. Org. Chem. 2005, 2005, 1354–1366. [Google Scholar] [CrossRef]
- Oguni, N.; Miyagi, Y.; Itoh, K. Highly Enantioselective Arylation of Symmetrical Epoxides with Phenyllithium Promoted by Chiral Schiff Bases and Salens. Tetrahedron Lett. 1998, 39, 9023–9026. [Google Scholar] [CrossRef]
- Zhao, Y.; Weix, D.J. Enantioselective Cross-Coupling of Meso-Epoxides with Aryl Halides. J. Am. Chem. Soc. 2015, 137, 3327–3340. [Google Scholar] [CrossRef] [PubMed]
- Cesarotti, E.; Kagan, H.B.; Goddard, R.; Krüger, C. Synthesis of New Ligands for Transition Metal Complexes: Menthyl- and Neomenthyl-Cyclopentadienes. J. Organomet. Chem. 1978, 162, 297–309. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reis, A.R.; Viduedo, N.; Raydan, D.; Marques, M.M.B. Bimetallic (or Multimetallic) Synthesis of N-Heterocycles. Catalysts 2023, 13, 1268. https://doi.org/10.3390/catal13091268
Reis AR, Viduedo N, Raydan D, Marques MMB. Bimetallic (or Multimetallic) Synthesis of N-Heterocycles. Catalysts. 2023; 13(9):1268. https://doi.org/10.3390/catal13091268
Chicago/Turabian StyleReis, Ana Rita, Nuno Viduedo, Daniel Raydan, and Maria Manuel B. Marques. 2023. "Bimetallic (or Multimetallic) Synthesis of N-Heterocycles" Catalysts 13, no. 9: 1268. https://doi.org/10.3390/catal13091268
APA StyleReis, A. R., Viduedo, N., Raydan, D., & Marques, M. M. B. (2023). Bimetallic (or Multimetallic) Synthesis of N-Heterocycles. Catalysts, 13(9), 1268. https://doi.org/10.3390/catal13091268