

Tailoring of Three-Atom Metal Cluster Catalysts for Ammonia Synthesis

Abstract
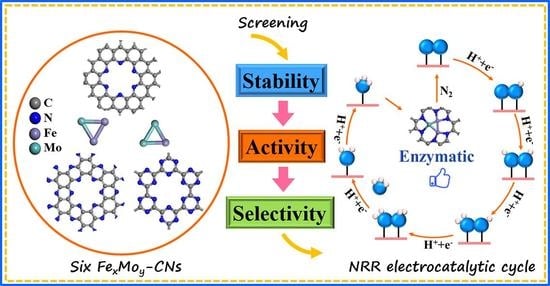
1. Introduction
2. Results and Discussion
2.1. Structure and Stability
2.2. N2 Adsorption
2.3. NRR Mechanism
2.4. Origin of NRR Activity on Fe2Mo-CNs
2.5. NRR Selectivity of the FexMoy–CNs
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, X.; Wu, T.; Wang, H.; Zhao, R.; Chen, H.; Wang, T.; Wei, P.; Luo, Y.; Zhang, Y.; Sun, X. Boron nanosheet: An elemental two-dimensional (2D) material for ambient electrocatalytic N2-to-NH3 fixation in neutral media. ACS Catal. 2019, 9, 4609–4615. [Google Scholar] [CrossRef]
- Ren, Y.; Yu, C.; Tan, X.; Wei, Q.; Wang, Z.; Ni, L.; Wang, L.; Qiu, J. Strategies to activate inert nitrogen molecules for efficient ammonia electrosynthesis: Current status, challenges, and perspectives. Energy Environ. Sci. 2022, 15, 2776–2805. [Google Scholar] [CrossRef]
- Lv, C.; Qian, Y.; Yan, C.; Ding, Y.; Liu, Y.; Chen, G.; Yu, G. Defect engineering metal-free polymeric carbon nitride electrocatalyst for effective nitrogen fixation under ambient conditions. Angew. Chem. Int. Ed. 2018, 57, 10246–10250. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Su, C.; Jia, G.; Xu, L.; Shao, Z. Emerging two-dimensional nanomaterials for electrochemical nitrogen reduction. Chem. Soc. Rev. 2021, 50, 12744–12787. [Google Scholar] [CrossRef]
- Hao, Y.C.; Guo, Y.; Chen, L.W.; Shu, M.; Wang, X.Y.; Bu, T.A.; Gao, W.Y.; Zhang, N.; Su, X.; Feng, X. Promoting nitrogen electroreduction to ammonia with bismuth nanocrystals and potassium cations in water. Nat. Catal. 2019, 2, 448–456. [Google Scholar] [CrossRef]
- Zhang, S.; Han, M.; Shi, T.; Zhang, H.; Lin, Y.; Zheng, X.; Zheng, L.R.; Zhou, H.; Chen, C.; Zhang, Y. Atomically dispersed bimetallic Fe–Co electrocatalysts for green production of ammonia. Nat. Sustain. 2023, 6, 169–179. [Google Scholar] [CrossRef]
- Ahmed, M.I.; Arachchige, L.J.; Su, Z.; Hibbert, D.B.; Sun, C.; Zhao, C. Nitrogenase-inspired atomically dispersed Fe–S–C linkages for improved electrochemical reduction of dinitrogen to ammonia. ACS Catal. 2022, 12, 1443–1451. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Q.; Liu, D.X.; Wen, Z.; Yao, J.X.; Shi, M.M.; Zhu, Y.F.; Yan, J.M.; Jiang, Q. High spin polarization ultrafine Rh nanoparticles on CNT for efficient electrochemical N2 fixation to ammonia. Appl. Catal. B 2021, 298, 120592. [Google Scholar] [CrossRef]
- Chu, K.; Li, X.; Tian, Y.; Li, Q.; Guo, Y. Boron nitride quantum dots/Ti3C2Tx-MXene heterostructure for efficient electrocatalytic nitrogen fixation. Energy Environ. Mater. 2022, 5, 1303–1309. [Google Scholar] [CrossRef]
- Choi, C.; Back, S.; Kim, N.Y.; Lim, J.; Kim, Y.H.; Jung, Y. Suppression of hydrogen evolution reaction in electrochemical N2 reduction using single-atom catalysts: A computational guideline. ACS Catal. 2018, 8, 7517–7525. [Google Scholar] [CrossRef]
- Li, L.S.; Martirez, J.M.P.; Carter, E.A. Prediction of Highly Selective Electrocatalytic Nitrogen Reduction at Low Overpotential on a Mo-Doped g-GaN Monolayer. ACS Catal. 2020, 10, 12841–12857. [Google Scholar] [CrossRef]
- Huang, C.X.; Li, G.L.; Yang, L.M.; Ganz, E. Ammonia Synthesis Using Single-Atom Catalysts Based on Two-Dimensional Organometallic Metal Phthalocyanine Monolayers under Ambient Conditions. ACS Appl. Mater. Interfaces 2021, 13, 608–621. [Google Scholar] [CrossRef]
- Sahoo, S.K.; Heske, J.L.; Antonietti, M.; Qin, Q.; Oschatz, M.; Kühne, T.D. Electrochemical N2 Reduction to Ammonia Using Single Au/Fe Atoms Supported on Nitrogen-Doped Porous Carbon. ACS Appl. Energy Mater. 2020, 3, 10061–10069. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Jiang, Q.; Lin, W.; Zhang, Y.; Huang, S.; Ding, K. Density functional theory study of single-atom V, Nb, and Ta catalysts on graphene and carbon nitride for selective nitrogen reduction. ACS Appl. Nano Mater. 2020, 3, 5149–5159. [Google Scholar] [CrossRef]
- Chen, Z.W.; Yan, J.M.; Jiang, Q. Single or Double: Which is the altar of atomic catalysts for nitrogen reduction reaction? Small Methods 2019, 3, 1800291. [Google Scholar] [CrossRef]
- Wang, S.; Wei, W.; Lv, X.; Huang, B.; Dai, Y. W supported on g-CN manifests high activity and selectivity for N2 electroreduction to NH3. J. Mater. Chem. A 2020, 8, 1378–1385. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, Z. Single Mo atom supported on defective boron nitride monolayer as an efficient electrocatalyst for nitrogen fixation: A computational study. J. Am. Chem. Soc. 2017, 139, 12480–12487. [Google Scholar] [CrossRef]
- Yan, H.; Cheng, H.; Yi, H.; Lin, Y.; Yao, T.; Wang, C.; Li, J.; Wei, S.; Lu, J. Single-atom Pd1/graphene catalyst achieved by atomic layer deposition: Remarkable performance in selective hydrogenation of 1,3-Butadiene. J. Am. Chem. Soc. 2015, 137, 10484–10487. [Google Scholar] [CrossRef]
- Qiu, H.J.; Ito, Y.; Cong, W.; Tan, Y.; Liu, P.; Hirata, A.; Fujita, T.; Tang, Z.; Chen, M. Nanoporous graphene with single-atom nickel dopants: An efficient and stable catalyst for electrochemical hydrogen production. Angew. Chem. Int. Ed. 2015, 54, 14031–14035. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, G.; Gauquelin, N.; Chen, N.; Zhou, J.; Yang, S.; Chen, W.; Meng, X.; Geng, D.; Banis, M.N. Single-atom catalysis using Pt/graphene achieved through atomic layer deposition. Sci. Rep. 2013, 3, 1775. [Google Scholar] [CrossRef]
- Kong, Y.; Wu, L.; Yang, X.; Li, Y.; Zheng, S.; Yang, B.; Li, Z.; Zhang, Q.; Zhou, S.; Lei, L. Accelerating protonation kinetics for ammonia electrosynthesis on single Iron sites embedded in carbon with intrinsic defects. Adv. Funct. Mater. 2022, 32, 2205409. [Google Scholar] [CrossRef]
- Wu, Y.; He, C.; Zhang, W. Novel design strategy of high activity electrocatalysts toward nitrogen reduction reaction via boron-transition-metal hybrid double-atom catalysts. ACS Appl. Mater. Interfaces 2021, 13, 47520–47529. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Wu, Y.; Chen, B.; Qian, Y.; Zhou, N.; Li, N. Transition-metal-atom-pairs deposited on g-CN monolayer for nitrogen reduction reaction: Density functional theory calculations. Chin. J. Catal. 2021, 42, 1160–1167. [Google Scholar] [CrossRef]
- Zheng, G.; Li, L.; Tian, Z.; Zhang, X.; Chen, L. Heterogeneous single-cluster catalysts (Mn3, Fe3, Co3, and Mo3) supported on nitrogen-doped graphene for robust electrochemical nitrogen reduction. J. Energy Chem. 2021, 54, 612–619. [Google Scholar] [CrossRef]
- Cui, X.; An, W.; Liu, X.; Wang, H.; Men, Y.; Wang, J. C2N-graphene supported single-atom catalysts for CO2 electrochemical reduction reaction: Mechanistic insight and catalyst screening. Nanoscale 2018, 10, 15262–15272. [Google Scholar] [CrossRef]
- He, M.; An, W.; Wang, Y.; Men, Y.; Liu, S. Hybrid Metal–Boron Diatomic Site Embedded in C2N Monolayer Promotes C–C Coupling in CO2 Electroreduction. Small 2021, 17, 2104445. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.; Dar, M.A. Understanding the activity of single atom catalysts for CO2 reduction to C2 products: A high throughput computational screening. New J. Chem. 2023, 47, 7225–7231. [Google Scholar] [CrossRef]
- Hassan, A.; Anis, I.; Shafi, S.; Assad, A.; Rasool, A.; Khanam, R.; Bhat, G.A.; Krishnamurty, S.; Dar, M.A. First-Principles Investigation of the Electrocatalytic Reduction of CO2 on Zirconium-Based Single-, Double-, and Triple-Atom Catalysts Anchored on a Graphitic Carbon Nitride Monolayer. ACS Appl. Nano Mater. 2022, 5, 15409–15417. [Google Scholar] [CrossRef]
- Wu, C.; Yang, W.; Wang, J.J.; Li, H.; Gates, I.D. Methane activation on dual-atom catalysts supported on graphene. Chem. Commun. 2021, 57, 12127–12130. [Google Scholar] [CrossRef]
- Pei, W.; Zhou, S.; Zhao, J.; Xu, X.; Du, Y.; Dou, S.X. Immobilized trimeric metal clusters: A family of the smallest catalysts for selective CO2 reduction toward multi-carbon products. Nano Energy 2020, 76, 105049. [Google Scholar] [CrossRef]
- Han, B.; Meng, H.; Li, F. Supported bimetallic trimers Fe2M@NG: The triple-atom catalysts for CO2 electro-reduction. ACS Omega 2022, 7, 16080–16086. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Wang, Q.; Yang, J.; Ye, K.; Li, X.; Zhong, W.; Shen, H.; Sharman, E.; Luo, Y.; Jiang, J. Toward rational design of dual-metal-site catalysts: Catalytic descriptor exploration. ACS Catal. 2022, 12, 3420–3429. [Google Scholar] [CrossRef]
- Yang, H.; Wang, L.; Xu, S.; Hui, X.; Cao, Y.; He, P.; Li, Y.; Li, H. Highly dispersed Ru anchored on nanosheet N-doped carbon for efficient and chemoselective hydrogenation of nitroaromatics to aromatic amines under mild conditions. Chem. Eng. J. 2022, 431, 133863. [Google Scholar] [CrossRef]
- Zhao, J.; Han, M.; Wang, Z.; Ling, L.; Zhang, R.; Wang, B. The regulating effect of doping Cu on the catalytic performance of CO oxidative coupling to DMO on PdxCuy/GDY: A DFT study. Green Energy Environ. 2022, 7, 742–754. [Google Scholar] [CrossRef]
- Wang, X.; Qiu, S.; Feng, J.; Tong, Y.; Zhou, F.; Li, Q.; Song, L.; Chen, S.; Wu, K.H.; Su, P. Confined Fe-Cu clusters as sub-nanometer reactors for efficiently regulating the electrochemical nitrogen reduction reaction. Adv. Mater. 2020, 32, e2004382. [Google Scholar] [CrossRef]
- Ma, D.W.; Zeng, Z.P.; Liu, L.L.; Huang, X.W.; Jia, Y. Computational evaluation of electrocatalytic nitrogen reduction on TM Single-, Double-, and Triple-Atom Catalysts (TM = Mn, Fe, Co, Ni) based on graphdiyne monolayers. J. Phys. Chem. C 2019, 123, 19066–19076. [Google Scholar] [CrossRef]
- Chen, Z.W.; Chen, L.X.; Jiang, M.; Chen, D.; Wang, Z.L.; Yao, X.; Singh, C.V.; Jiang, Q. A triple atom catalyst with ultrahigh loading potential for nitrogen electrochemical reduction. J. Mater. Chem. A 2020, 8, 15086–15093. [Google Scholar] [CrossRef]
- Rasool, A.; Anis, I.; Dixit, M.; Maibam, A.; Hassan, A.; Krishnamurty, S.; Dar, M.A. Tantalum based single, double, and triple atom catalysts supported on g-C2N monolayer for effective nitrogen reduction reaction: A comparative DFT investigation. Catal. Sci. Technol. 2022, 12, 310–319. [Google Scholar] [CrossRef]
- Li, W.J.; Lou, Z.X.; Yuan, H.Y.; Yang, H.G.; Wang, H.F. Oriented design of triple atom catalysts for electrocatalytic nitrogen reduction with the genetic-algorithm-based global optimization method driven by first principles calculations. J. Mater. Chem. A 2022, 10, 16106–16114. [Google Scholar] [CrossRef]
- Zhang, J.; An, W. Single-, double-, and triple-atom catalysts on graphene-like C2N enable electrocatalytic nitrogen reduction: Insight from first principles. Catal. Sci. Technol. 2022, 12, 2604–2617. [Google Scholar] [CrossRef]
- Han, X.Q.; Lang, Z.L.; Zhang, F.Y.; Xu, H.L.; Su, Z.M. Computational evaluation of FeMo heteroatom coeffect induced high electroreduction activity of N2-to-NH3. Appl. Surf. Sci. 2022, 579, 152214. [Google Scholar] [CrossRef]
- Gao, S.; Liu, X.; Wang, Z.; Lu, Y.; Sa, R.; Li, Q.; Sun, C.; Chen, X.; Ma, Z. Spin regulation for efficient electrocatalytic N2 reduction over diatomic Fe-Mo catalyst. J. Colloid. Interface Sci. 2022, 630, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Q.; Li, C.; Fan, H.N.; Luo, W.B.; Liu, H.K.; Dou, S.X. Atomically dispersed metal dimer species with selective catalytic activity for nitrogen electrochemical reduction. J. Mater. Chem. A 2019, 7, 22242–22247. [Google Scholar] [CrossRef]
- Liu, W.; Han, L.; Wang, H.T.; Zhao, X.; Boscoboinik, J.A.; Liu, X.; Pao, C.W.; Sun, J.; Zhuo, L.; Luo, J. FeMo sub-nanoclusters/single atoms for neutral ammonia electrosynthesis. Nano Energy 2020, 77, 105078. [Google Scholar] [CrossRef]
- Van der Ham, C.J.M.; Koper, M.T.M.; Hetterscheid, D.G.H. Challenges in reduction of dinitrogen by proton and electron transfer. Chem. Soc. Rev. 2014, 43, 5183–5191. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Z.; Li, Y.; Qu, Y.; Li, Y.; Li, W.; Zhao, M. Screening of transition-metal single-atom catalysts anchored on covalent-organic frameworks for efficient nitrogen fixation. ACS Appl. Mater. Interfaces 2022, 14, 1024–1033. [Google Scholar] [CrossRef]
- Yuan, S.; Meng, G.; Liu, D.; Zhao, W.; Zhu, H.; Chi, Y.; Ren, H.; Guo, W. Synergy of substrate chemical environments and single-atom catalysts promotes catalytic performance: Nitrogen reduction on chiral and defected carbon nanotubes. ACS Appl. Mater. Interfaces 2022, 14, 52544–52552. [Google Scholar] [CrossRef]
- Gao, S.; Ma, Z.; Xiao, C.; Cui, Z.; Du, W.; Sun, X.; Li, Q.; Sa, R.; Sun, C. TM3 (TM = V, Fe, Mo, W) single-cluster catalyst confined on porous BN for electrocatalytic nitrogen reduction. J. Mater. Sci. Technol. 2022, 108, 46–53. [Google Scholar] [CrossRef]
- Ma, Z.; Cui, Z.; Xiao, C.; Dai, W.; Lv, Y.; Li, Q.; Sa, R. Theoretical screening of efficient single-atom catalysts for nitrogen fixation based on a defective BN monolayer. Nanoscale 2020, 12, 1541–1550. [Google Scholar] [CrossRef]
- Zheng, X.; Liu, Y.; Yao, Y. Trimetallic single-cluster catalysts for electrochemical nitrogen reduction reaction: Activity prediction, mechanism, and electronic descriptor. Chem. Eng. J. 2021, 426, 130745. [Google Scholar] [CrossRef]
- Zhai, X.; Li, L.; Liu, X.; Li, Y.; Yang, J.; Yang, D.; Zhang, J.; Yan, H.; Ge, G. A DFT screening of single transition atoms supported on MoS2 as highly efficient electrocatalysts for the nitrogen reduction reaction. Nanoscale 2020, 12, 10035–10043. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jiao, Y.; Zheng, Y.; Qiao, S.Z. Isolated boron sites for electroreduction of dinitrogen to ammonia. ACS Catal. 2020, 10, 1847–1854. [Google Scholar] [CrossRef]
- Ma, D.; Wang, Y.; Liu, L.; Jia, Y. Electrocatalytic nitrogen reduction on the transition-metal dimer anchored N-doped graphene: Performance prediction and synergetic effect. Phys. Chem. Chem. Phys. 2021, 23, 4018–4029. [Google Scholar] [CrossRef]
- Liu, Y.; Song, B.; Huang, C.X.; Yang, L.M. Dual transition metal atoms embedded in N-doped graphene for electrochemical nitrogen fixation under ambient conditions. J. Mater. Chem. A 2022, 10, 13527–13543. [Google Scholar] [CrossRef]
- Li, H.; Zhao, Z.; Cai, Q.; Yin, L.; Zhao, J. Nitrogen electroreduction performance of transition metal dimers embedded into N-doped graphene: A theoretical prediction. J. Mater. Chem. A 2020, 8, 4533–4543. [Google Scholar] [CrossRef]
- Maibam, A.; Krishnamurty, S.; Ahmad Dar, M. Electrocatalytic nitrogen reduction directed through the p-band center of boron on BSAC@Mo2C. Mater. Adv. 2022, 3, 592–598. [Google Scholar] [CrossRef]
- Cui, C.; Zhang, H.; Luo, Z. Nitrogen reduction reaction on small iron clusters supported by N-doped graphene: A theoretical study of the atomically precise active-site mechanism. Nano Res. 2020, 13, 2280–2288. [Google Scholar] [CrossRef]
- Yang, X.; Shang, C.; Zhou, S.; Zhao, J. MBenes: Emerging 2D materials as efficient electrocatalysts for the nitrogen reduction reaction. Nanoscale Horiz. 2020, 5, 1106–1115. [Google Scholar] [CrossRef]
- Luo, Y.; Li, M.; Dai, Y.; Zhang, X.; Zhao, R.; Jiang, F.; Ling, C.; Huang, Y. Screening of effective NRR electrocatalysts among the Si-based MSi2N4 (M = Ti, Zr, Hf, V, Nb, Ta, Cr, Mo, and W) monolayers. J. Mater. Chem. A 2021, 9, 15217–15225. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B Condens Matter 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B Condens. Matter 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Peterson, A.A.; Abild-Pedersen, F.; Studt, F.; Rossmeisl, J.; Nørskov, J.K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 2010, 3, 1311–1315. [Google Scholar] [CrossRef]
- Mathew, K.; Sundararaman, R.; Letchworth-Weaver, K.; Arias, T.A.; Hennig, R.G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 2014, 140, 084106. [Google Scholar] [CrossRef]
- Mathew, K.; Kolluru, V.C.; Mula, S.; Steinmann, S.N.; Hennig, R.G. Implicit self-consistent electrolyte model in plane-wave density-functional theory. J. Chem. Phys. 2019, 151, 234101. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Bader Charge (e−) | Bader Charge (e−) * N2 | |||||
|---|---|---|---|---|---|---|---|
| M1 | M2 | M3 | M1 | M2 | M3 | N2 | |
| Fe2Mo–C6N6 | −0.35 | −0.50 | −0.88 | −0.52 | −0.68 | −1.23 | +0.75 |
| FeMo2–C6N6 | −0.47 | −0.80 | −0.65 | −0.62 | −1.03 | −1.15 | +0.85 |
| Fe2Mo–C2N | −0.45 | −0.50 | −0.78 | −0.64 | −0.52 | −1.18 | +0.70 |
| FeMo2–C2N | −0.61 | −0.70 | −0.74 | −0.52 | −1.12 | −1.04 | +0.83 |
| Fe2Mo–NG | −0.80 | −0.50 | −0.51 | −1.24 | −0.54 | −0.62 | +0.73 |
| FeMo2–NG | −0.64 | −0.78 | −0.61 | −1.08 | −1.10 | −0.53 | +0.86 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Zhao, T.; Yan, L. Tailoring of Three-Atom Metal Cluster Catalysts for Ammonia Synthesis. Catalysts 2023, 13, 869. https://doi.org/10.3390/catal13050869
Wang S, Zhao T, Yan L. Tailoring of Three-Atom Metal Cluster Catalysts for Ammonia Synthesis. Catalysts. 2023; 13(5):869. https://doi.org/10.3390/catal13050869
Chicago/Turabian StyleWang, Shuo, Tingting Zhao, and Likai Yan. 2023. "Tailoring of Three-Atom Metal Cluster Catalysts for Ammonia Synthesis" Catalysts 13, no. 5: 869. https://doi.org/10.3390/catal13050869
APA StyleWang, S., Zhao, T., & Yan, L. (2023). Tailoring of Three-Atom Metal Cluster Catalysts for Ammonia Synthesis. Catalysts, 13(5), 869. https://doi.org/10.3390/catal13050869