
Feedback Inhibition of DszC, a Crucial Enzyme for Crude Oil Biodessulfurization



Abstract
1. Introduction
1.1. Engineering the 4S Pathway
1.2. The Feedback Inhibition Mechanism of the 4S Pathway
2. Results and Discussion
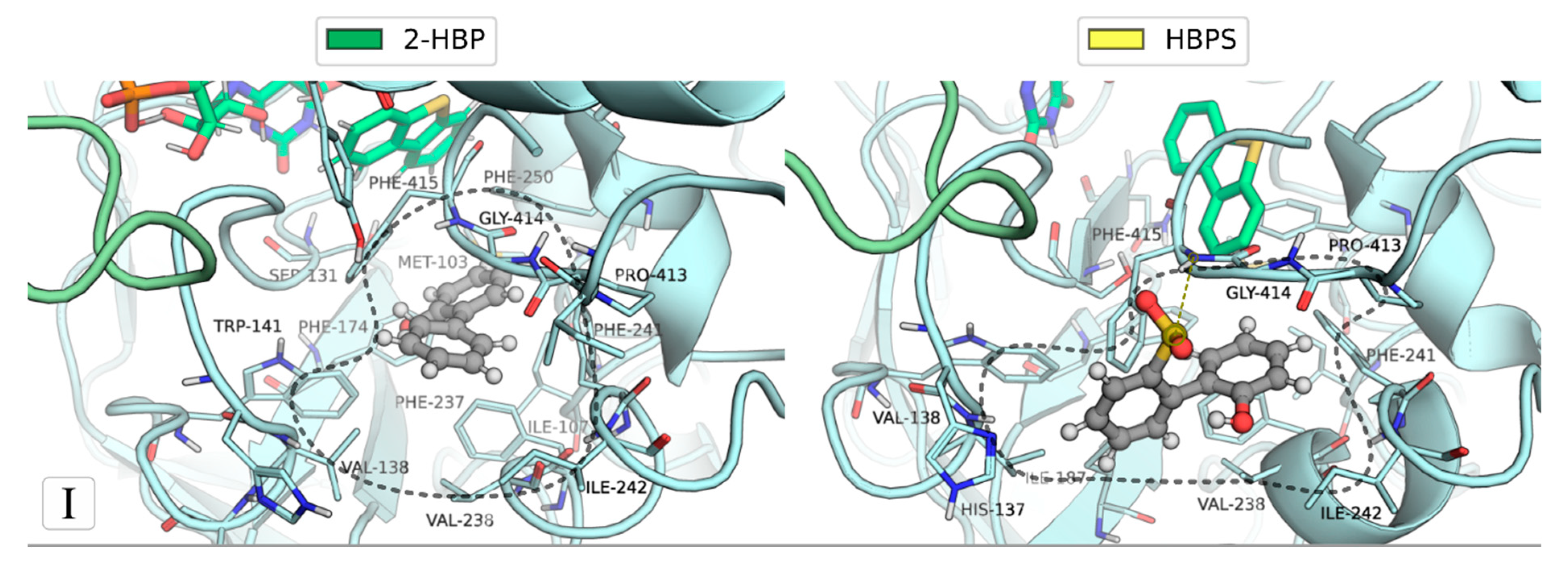
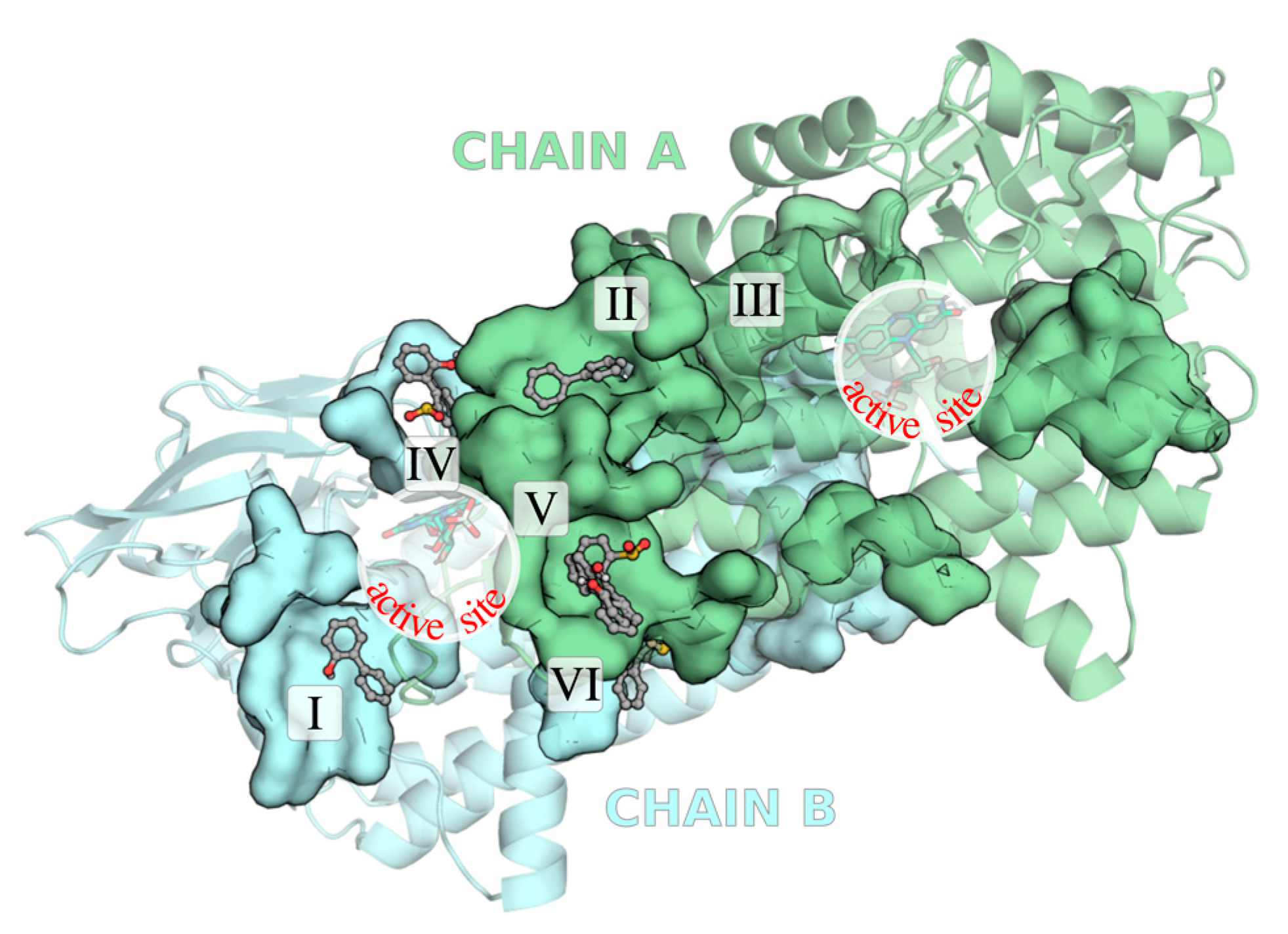
2.1. Identification of the Binding Sites of 2-HBP and HBPS
2.2. Ranking of the Binding Sites
2.3. Mechanism of Feedback Inhibition by 2-HBT and HBPS
3. Materials and Methods
3.1. Structure Selection and Preparation
3.2. Docking of 2-HBP and HBPS
3.3. System Parameterization and Simulation Setup
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- US Energy Information Administration. International Energy Outlook 2019 with Projections to 2050; U.S. Department of Energy: Washington, DC, USA, 2019. [Google Scholar]
- IRENA. Renewable Energy Statistics 2019; The International Renewable Energy Agency: Abu Dhabi, United Arab Emirates, 2019. [Google Scholar]
- Javadli, R.; de Klerk, A. Desulfurization of heavy oil. Appl. Petrochem. Res. 2012, 1, 3–19. [Google Scholar] [CrossRef]
- Boniek, D.; Figueiredo, D.; dos Santos, A.F.B.; Stoianoff, M.A.D. Biodesulfurization: A mini review about the immediate search for the future technology. Clean Technol. Environ. Policy 2015, 17, 29–37. [Google Scholar] [CrossRef]
- Ali, S.H.; Hamad, D.M.; Albusairi, B.H.; Fahim, M.A. Removal of Dibenzothiophenes from Fuels by Oxy-desulfurization. Energy Fuels 2009, 23, 5986–5994. [Google Scholar] [CrossRef]
- Hernandez-Maldonado, A.J.; Yang, R.T. Desulfurization of transportation fuels by adsorption. Catal. Rev. 2004, 46, 111–150. [Google Scholar] [CrossRef]
- Shafi, R.; Hutchings, G.J. Hydrodesulfurization of hindered dibenzothiophenes: An overview. Catal. Today 2000, 59, 423–442. [Google Scholar] [CrossRef]
- Environmental Protection Agency. Technical Amendments to the Highway and Nonroad Diesel Regulations. Fed. Regist. 2006, 71, 25706–25726. [Google Scholar]
- Environmental Protection Agency. Diesel Fuel Standards and Rulemakings. Available online: https://www.epa.gov/diesel-fuel-standards/diesel-fuel-standards-and-rulemakings (accessed on 9 April 2023).
- Agency, E.E. Directive 2003/17/EC of the European Parliament and of the Council of 3 March 2003. Off. J. Eur. Union 2003, L76, 10–19. [Google Scholar]
- Agency, E.E. Directive 2009/30/EC of the European Parliament and of the Council of 23 April 2009. Off. J. Eur. Union 2009, L140, 88–113. [Google Scholar]
- Kilbane, J.J.; Le Borgne, S. Petroleum biorefining: The selective removal of sulfur, nitrogen, and metals. In Studies in Surface Science and Catalysis; Vazquez-Duhalt, R., Quintero-Ramirez, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2004; Volume 151, pp. 29–65. [Google Scholar]
- Mohebali, G.; Ball, A.S. Biodesulfurization of diesel fuels—Past, present and future perspectives. Int. Biodeterior. Biodegrad. 2016, 110, 163–180. [Google Scholar] [CrossRef]
- Ismagilov, Z.; Yashnik, S.; Kerzhentsev, M.; Parmon, V.; Bourane, A.; Al-Shahrani, F.M.; Hajji, A.A.; Koseoglu, O.R. Oxidative Desulfurization of Hydrocarbon Fuels. Catal. Rev. 2011, 53, 199–255. [Google Scholar] [CrossRef]
- Hossain, M.N.; Park, H.C.; Choi, H.S. A Comprehensive Review on Catalytic Oxidative Desulfurization of Liquid Fuel Oil. Catalysts 2019, 9, 229. [Google Scholar] [CrossRef]
- Le Borgne, S.; Quintero, R. Biotechnological processes for the refining of petroleum. Fuel Process. Technol. 2003, 81, 155–169. [Google Scholar] [CrossRef]
- Montiel, C.; Rodolfo, Q.; Aburto, J. Petroleum biotechnology: Technology trends for the future. Afr. J. Biotechnol. 2009, 8, 2653–2666. [Google Scholar]
- Gallagher, J.R.; Olson, E.S.; Stanley, D.C. Microbial desulfurization of dibenzothiophene: A sulfur-specific pathway. FEMS Microbiol. Lett. 1993, 107, 31–35. [Google Scholar] [CrossRef]
- Gray, K.A.; Mrachko, G.T.; Squires, C.H. Biodesulfurization of fossil fuels. Curr. Opin. Microbiol. 2003, 6, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Roychoudhury, P.K.; Deb, J.K. Biotechnology of desulfurization of diesel: Prospects and challenges. Appl. Microbiol. Biotechnol. 2005, 66, 356–366. [Google Scholar] [CrossRef]
- Kilbane, J.J. Biodesulfurization: How to Make it Work? Arab. J. Sci. Eng. 2017, 42, 1–9. [Google Scholar] [CrossRef]
- Kamali, N.; Tavallaie, M.; Bambai, B.; Karkhane, A.A.; Miri, M. Site-directed mutagenesis enhances the activity of NADH-FMN oxidoreductase (DszD) activity of Rhodococcus erythropolis. Biotechnol. Lett. 2010, 32, 921–927. [Google Scholar] [CrossRef]
- Abin-Fuentes, A.; Mohamed, M.E.; Wang, D.I.C.; Prathera, K.L.J. Exploring the Mechanism of Biocatalyst Inhibition in Microbial Desulfurization. Appl. Environ. Microbiol. 2013, 79, 7807–7817. [Google Scholar] [CrossRef]
- Pan, J.; Wu, F.; Wang, J.; Yu, L.Q.; Khayyat, N.H.; Stark, B.C.; Kilbane, J.J. Enhancement of desulfurization activity by enzymes of the Rhodococcus dsz operon through coexpression of a high sulfur peptide and directed evolution. Fuel 2013, 112, 385–390, Corrigendum in Fuel 2013, 113, 766. [Google Scholar] [CrossRef]
- Li, L.; Liao, Y.; Luo, Y.; Zhang, G.; Liao, X.; Zhang, W.; Zheng, S.; Han, S.; Lin, Y.; Liang, S. Improved Efficiency of the Desulfurization of Oil Sulfur Compounds in Escherichia coli Using a Combination of Desensitization Engineering and DszC Overexpression. ACS Synth. Biol. 2019, 8, 1441–1451. [Google Scholar] [CrossRef]
- Ohshiro, T.; Ohkita, R.; Takikawa, T.; Manabe, M.; Lee, W.C.; Tanokura, M.; Izumi, Y. Improvement of 2′-Hydroxybiphenyl-2-sulfinate Desulfinase, an Enzyme Involved in the Dibenzothiophene Desulfurization Pathway, from Rhodococcus erythropolis KA2-5-1 by Site-Directed Mutagenesis. Biosci. Biotechnol. Biochem. 2007, 71, 2815–2821. [Google Scholar] [CrossRef]
- Kilbane, J.J. Microbial biocatalyst developments to upgrade fossil fuels. Curr. Opin. Biotechnol. 2006, 17, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Tokuriki, N.; Stricher, F.; Serrano, L.; Tawfik, D.S. How Protein Stability and New Functions Trade Off. PLoS Comput. Biol. 2008, 4, e1000002. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, A.C.C.; Neves, R.P.P.; Sousa, S.F.; Ramos, M.J.; Fernandes, P.A. Mechanistic Studies of a Flavin Monooxygenase: Sulfur Oxidation of Dibenzothiophenes by DszC. ACS Catal. 2018, 8, 9298–9311. [Google Scholar] [CrossRef]
- Sousa, J.P.M.; Neves, R.P.P.; Sousa, S.F.; Ramos, M.J.; Fernandes, P.A. Reaction Mechanism and Determinants for Efficient Catalysis by DszB, a Key Enzyme for Crude Oil Bio-desulfurization. ACS Catal. 2020, 10, 9545–9554. [Google Scholar] [CrossRef]
- Neves, R.P.P.; Ramos, M.J.; Fernandes, P.A. Engineering DszC Mutants from Transition State Macrodipole Considerations and Evolutionary Sequence Analysis. J. Chem. Inf. Model. 2022, 63, 20. [Google Scholar] [CrossRef]
- Chen, M.M.Y.; Snow, C.D.; Vizcarra, C.L.; Mayo, S.L.; Arnold, F.H. Comparison of random mutagenesis and semi-rational designed libraries for improved cytochrome P450 BM3-catalyzed hydroxylation of small alkanes. Protein Eng. Des. Sel. 2012, 25, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Welborn, V.V.; Head-Gordon, T. Computational Design of Synthetic Enzymes. Chem. Rev. 2019, 119, 6613–6630. [Google Scholar] [CrossRef]
- Gray, K.A.; Pogrebinsky, O.S.; Mrachko, G.T.; Xi, L.; Monticello, D.J.; Squires, C.H. Molecular mechanisms of biocatalytic desulfurization of fossil fuels. Nat. Biotechnol. 1996, 14, 1705–1709. [Google Scholar] [CrossRef]
- Watkins, L.M.; Rodriguez, R.; Schneider, D.; Broderick, R.; Cruz, M.; Chambers, R.; Ruckman, E.; Cody, M.; Mrachko, G.T. Purification and characterization of the aromatic desulfinase, 2-(2′-hydroxyphenyl)benzenesulfinate desulfinase. Arch. Biochem. Biophys. 2003, 415, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Ohshiro, T.; Nishina, Y.; Izumi, Y. Purification, Characterization, and Overexpression of Flavin Reductase Involved in Dibenzothiophene Desulfurization by<em>Rhodococcus erythropolis</em> D-1. Appl. Environ. Microbiol. 2001, 67, 1179. [Google Scholar] [CrossRef] [PubMed]
- Lei, B.F.; Tu, S.C. Gene overexpression, purification, and identification of a desulfurization enzyme from Rhodococcus sp strain IGTS8 as a sulfide/sulfoxide monooxygenase. J. Bacteriol. 1996, 178, 5699–5705. [Google Scholar] [CrossRef]
- Liu, S.H.; Zhang, C.G.; Su, T.T.; Wei, T.D.; Zhu, D.Y.; Wang, K.; Huang, Y.; Dong, Y.H.; Yin, K.; Xu, S.J.; et al. Crystal structure of DszC from Rhodococcus sp. XP at 1.79 angstrom. Proteins 2014, 82, 1708–1720. [Google Scholar] [CrossRef]
- Zhang, L.; Duan, X.L.; Zhou, D.M.; Dong, Z.; Ji, K.H.; Meng, W.Y.; Li, G.Q.; Li, X.; Yang, H.T.; Ma, T.; et al. Structural insights into the stabilization of active, tetrameric DszC by its C-terminus. Proteins 2014, 82, 2733–2743. [Google Scholar] [CrossRef]
- Guan, L.J.; Lee, W.C.; Wang, S.; Ohshiro, T.; Izumi, Y.; Ohtsuka, J.; Tanokura, M. Crystal structures of apo-DszC and FMN-bound DszC from Rhodococcus erythropolis D-1. FEBS J. 2015, 282, 3126–3135. [Google Scholar] [CrossRef] [PubMed]
- Doerr, S.; Harvey, M.J.; Noe, F.; De Fabritiis, G. HTMD: High-Throughput Molecular Dynamics for Molecular Discovery. J. Chem. Theory Comput. 2016, 12, 1845–1852. [Google Scholar] [CrossRef]
- Pan, A.C.; Jacobson, D.; Yatsenko, K.; Sritharan, D.; Weinreich, T.M.; Shaw, D.E. Atomic-level characterization of protein-protein association. Proc. Natl. Acad. Sci. USA 2019, 116, 4244–4249. [Google Scholar] [CrossRef]
- Ahmad, K.; Rizzi, A.; Capelli, R.; Mandelli, D.; Lyu, W.P.; Carloni, P. Enhanced-Sampling Simulations for the Estimation of Ligand Binding Kinetics: Current Status and Perspective. Front. Mol. Biosci. 2022, 9, 899805. [Google Scholar] [CrossRef]
- Stevens, J.A.; Grunewald, F.; van Tilburg, P.A.M.; Konig, M.; Gilbert, B.R.; Brier, T.A.; Thornburg, Z.R.; Luthey-Schulten, Z.; Marrink, S.J. Molecular dynamics simulation of an entire cell. Front. Chem. 2023, 11, 1106495. [Google Scholar] [CrossRef]
- Hino, T.; Hamamoto, H.; Suzuki, H.; Yagi, H.; Ohshiro, T.; Nagano, S. Crystal structures of TdsC, a dibenzothiophene monooxygenase from the thermophile Paenibacillus sp A11-2, reveal potential for expanding its substrate selectivity. J. Biol. Chem. 2017, 292, 15804–15813. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.M.; Sondergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. Software News and Update AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding Site | Ligand (no. poses) | Residues |
|---|---|---|
| I | 2-HBP (6) | Asn134; Ser136; Val138; Trp141; Ile187; Phe237; Val238; Phe241; Ile242; Pro413; Gly414; Phe415; Thr416 |
| II * | 2-HBP (6) | Arg32; Val36; Ala272; Glu275; Tyr276; Thr279; Gln280; Tyr376; Phe378; Arg380; Phe381 |
| III * | 2-HBP (3)/HBPS (1) | Arg40 AB; Ala206 A; Ala207 A; Ile208 A; Arg211 AB; Pro374 B; Asp379 B |
| IV | HBPS (5) | Arg370 A; His373 A; Arg375 A; His160 B; Phe161 B; Asp204 B; Trp205 B; Ser217 B |
| V * | 2-HBP (2)/HBPS (10) | Arg274; Thr277; Arg278; Thr293; Thr298; Ile299; Tyr302; Thr306 |
| VI | 2-HBP (1)/HBPS (2) | Ser301 A; Glu304 A; Glu365 A; Thr353 B; Asn354 B; Val394 B; Ser395 B; Ile398 B |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neves, R.P.P.; Araújo, B.; Ramos, M.J.; Fernandes, P.A. Feedback Inhibition of DszC, a Crucial Enzyme for Crude Oil Biodessulfurization. Catalysts 2023, 13, 736. https://doi.org/10.3390/catal13040736
Neves RPP, Araújo B, Ramos MJ, Fernandes PA. Feedback Inhibition of DszC, a Crucial Enzyme for Crude Oil Biodessulfurization. Catalysts. 2023; 13(4):736. https://doi.org/10.3390/catal13040736
Chicago/Turabian StyleNeves, Rui P. P., Bruno Araújo, Maria J. Ramos, and Pedro A. Fernandes. 2023. "Feedback Inhibition of DszC, a Crucial Enzyme for Crude Oil Biodessulfurization" Catalysts 13, no. 4: 736. https://doi.org/10.3390/catal13040736
APA StyleNeves, R. P. P., Araújo, B., Ramos, M. J., & Fernandes, P. A. (2023). Feedback Inhibition of DszC, a Crucial Enzyme for Crude Oil Biodessulfurization. Catalysts, 13(4), 736. https://doi.org/10.3390/catal13040736