Abstract
Manganese has attracted significant recent attention due to its abundance, low toxicity, and versatility in catalysis. In the present study, a series of manganese (III) complexes supported by salan ligands have been synthesized and characterized, and their activity as catalysts in the hydrosilylation of carbonyl compounds was examined. While manganese (III) chloride complexes exhibited minimal catalytic efficacy without activation of silver perchlorate, manganese (III) azide complexes showed good activity in the hydrosilylation of carbonyl compounds. Under optimized reaction conditions, several types of aldehydes and ketones could be reduced with good yields and tolerance to a variety of functional groups. The possible mechanisms of silane activation and hydrosilylation were discussed in light of relevant experimental observations.
1. Introduction
There has been an emerging shift towards first-row transition metals in catalysis [1,2,3,4]. Conventionally, the precious metal-based catalysts, represented by palladium, have been a dominating force, particularly in the pharmaceutical industry. However, the toxicity and the increasing scarcity of these elements have raised health and sustainability concerns and stimulated the search for alternative catalytic systems. Consequently, first-row metals such as Fe [5,6,7,8], Cu [9,10], and Zn [11,12,13,14] have attracted growing interest in various catalytic reactions due to their abundance and low toxicity. The distinct reactivity of the first-row metals may enable new transformations inaccessible with the second- and third-row metals. As one of the most abundant transition metals, Mn has seen a recent resurgence of research beyond traditional oxidation/oxygenation catalysis [15,16]. A wide variety of chemical transformations have been achieved with manganese catalysis, such as hydrogenation of alkenes [17], CO2, esters, ketones, nitriles [18,19,20,21,22,23,24], hydroboration of carbonyls, nitriles carboxylic acids and CO2 [25,26,27,28,29,30], electrochemical hydrogen production [31], dehydrogenative olefination of alkyl-substituted heteroarenes and sulfones with alcohols [32,33,34,35], alkylation of nitriles, esters, and amides [36,37], dehydrogenation of alcohols [38,39,40] and its application in the synthesis of pyrroles and imides from amines and diols [41,42], and dehydrogenative cross-coupling of hydrosilanes and alcohols [43,44,45,46].
Among these developments, manganese-catalyzed hydrosilylation occupies a special place, thanks to the early work by the Cutler group, showing that simple manganese carbonyl complexes were effective catalysts for the hydrosilylation of various unsaturated substrates [47,48,49,50,51,52]. Hydrosilylation of C=O-containing compounds represents a mild and versatile approach for the reduction of these compounds and various metal and nonmetal-catalyzed reactions have been reported using commercial hydrosilane reagents [53,54,55,56]. Not surprisingly, recent research in manganese also provides a wide range of catalysts for the hydrosilylation of aldehydes, ketones, esters, amides, and carboxylic acids [57,58,59,60,61,62,63,64,65,66,67,68,69,70]. Efficient hydrosilylation of C=C double bonds by manganese catalysts has also been reported [71,72]. A few representative examples of manganese (pre)catalysts for hydrosilylation are shown in Scheme 1. Although a direct comparison of the catalytic activity of these catalysts across different groups is difficult due to the varied reaction conditions and the choice of substrates, a brief summary of typical carbonyl substrates is compiled in Table S1.
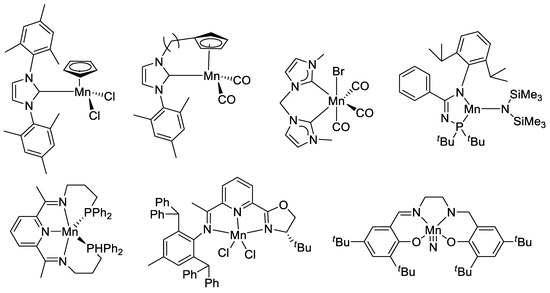
Scheme 1.
Selected examples of Mn (pre)catalysts for hydrosilylation.
Metals supported by salen ligands have been widely reported as catalysts in different transformations. Our previous work has shown a manganese salen complex is highly efficient in catalyzing the hydrosilylation and hydroboration of carbonyl groups with high yields and broad tolerance of functional groups [28,68]. Mechanistic investigations suggest that the availability of two cis-coordination sites at the Mn center is important for the catalytic activity. Although such a cis-coordination mode has been observed with salen complexes [73], it is conceivably difficult to achieve without some transformation on the salen ligand, since such ligands typically need four planar coordination sites on the metal center. Thus our attention has turned to the reduced salen, or salan ligands (where H2salan = N,N′-dimethyl-N,N′-bis(o-hydroxybenzyl)-1,2-diaminoethane), which are derived from the hydrogenation of the imine bonds in salen. The replacement of the C=N double bonds by the C-N single bonds provides a more flexible coordination environment around the metal center, and this may allow ready access to the cis-coordination mode and facilitate the catalytic reaction. It has been illustrated that salan ligands are a good platform that could support various transition metals and provide catalytic reactivity as well as selectivity [74,75,76]. Herein we report our studies of manganese salan complexes for catalytic hydrosilylation of carbonyl compounds.
2. Results and Discussion
2.1. Synthesis and Characterization of (Salan)Mn Complexes
The salan ligands were obtained by following the literature procedures [77]. Briefly, the reaction of salicyaldehyde derivatives with diamine afforded the parent H2salen ligand, which was first reduced with excess NaBH4 and subsequently methylated by reductive amination with NaBH4/CH2O. The metallation was carried out with MnCl2 or Mn(OAc)2/LiCl to first obtain the chloride compounds. The substitution with NaN3 in the presence of AgClO4 afforded the azido complex (Scheme 2) [78]. The presence of the azido ligand in (salan-tBu2)Mn(N3) (5b) was confirmed by the peak at 2050 cm−1 in the FT-IR spectrum (Figure 1). Similarly, the parent (salan)Mn(N3) (5a) featured an azide-stretching peak at 2037 cm−1. For comparison, the starting NaN3 shows an IR peak at 2104 cm−1. The known salen analogue, (salen-tBu2)Mn(N3) (6) [79], was also obtained in a similar procedure, and the IR peak for the azido ligand as a thin film is observed at 2063 cm−1. The vibrational mode of a series of (salen)Mn(N3) derivatives appeared in the range of 2046–2048 cm−1 in a solution phase determination, typical for the apical azido group in five-coordinate square pyramidal Mn(III) complexes. Efforts were made to prepare the nitrido manganese salan complexes since this would offer a direct comparison with the salen analogue [68]. However, experimental attempts, either by photolysis [80,81,82,83,84] of the azido precursor (salan-tBu2)Mn(N3) or by bleach oxidation of (salan-tBu2)MnCl in the presence of NH4OH [85], were not successful. Thus, we focused on the (salan)Mn(N3) complexes which showed reasonable activity in the hydrosilylation reaction.
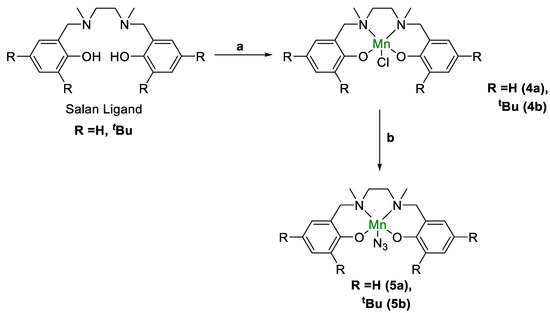
Scheme 2.
Synthesis of salan Mn complexes. (a) MnCl2 or Mn(OAc)2/LiCl; (b) AgClO4/NaN3.
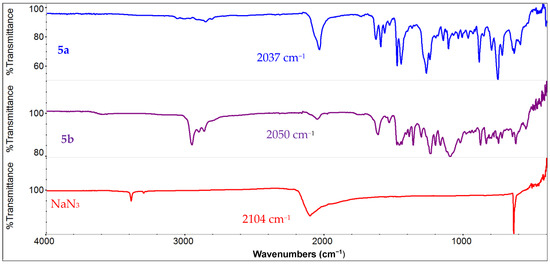
Figure 1.
Comparison of FT-IR spectra of (salan)Mn(N3) (5a) (top), (salan-tBu2)Mn(N3) (5b) (middle), and NaN3 (bottom).
2.2. Catalytic Hydrosilylation
Having a series of manganese (III) complexes in hand, we first examined the efficacy of complex 5b in the hydrosilylation under a variety of reaction conditions, with benzaldehyde PhCHO as a model substrate and phenylsilane PhSiH3 as the hydrogen source, and the results are compiled in Table 1. Screening different solvents in the reaction illustrated that benzene was an optimal solvent in this system, while the hydrosilylation in other common solvents such as CD3CN and CDCl3 was slower or with low conversions. The reaction with benzene as a solvent at elevated temperature exhibited a high conversion of >99 % in less than 1 h with 0.5 mol % loading of the catalyst (entry 1, Table 1). Reaction in CD3CN under identical conditions resulted in a 94 % conversion within 6.5 h (entry 2). Unlike C6D6 and CD3CN, CDCl3 as a solvent showed negligible conversion after a long reaction time (entry 4), though increasing the catalyst loading to 1 mol % at a longer reaction time did lead to some conversion of PhCHO (entry 5). Reaction in toluene-d8 showed comparable reactivity, as in benzene, reaching a >99% conversion within 40 min (entry 3). Other reaction parameters were also explored. The reaction with C6D6 could be carried out at a lower temperature (80 °C or at room temperature), and longer reaction times were needed for reasonable conversion (entries 6 & 7). For example, the reaction could still reach a 90% conversion after a few days at 80 °C. However, when the reaction was run under air, only minimal conversion was obtained within 5 h (entry 8), which was less active than the (salen)MnN system in the presence of air [68]. So the rest of the reactions were carried out under N2. Among other common hydrosilanes, a tertiary hydrosilane, triethoxysilane, was also active for the hydrosilylation in CD3CN, and a 54% conversion of PhCHO was observed after 8 h of reaction (entry 9). At this point, the triethoxysilane was completely consumed, though the side reaction product was not identified. When the loading of the triethoxysilane was increased to two equivalents of PhCHO, the complete conversion of PhCHO could be achieved within 8 h (entry 10). Under the same conditions, a reaction with a ketone, acetophenone, was slower but reached a 99% conversion within 25 h (entry 11). Secondary silanes, such as Ph2SiH2, showed lower activity than PhSiH3 and tertiary silanes such as Et3SiH showed no activity.

Table 1.
Hydrosilylation of PhCHO under different conditions a.
After exploration of the reaction conditions, we examined the hydrosilyation of PhCHO with different salan Mn(III) complexes (entries 12–15, Table 1). The parent salen azido complex 5a was capable of catalyzing the reaction, though with somewhat lower activity (entry 14). On the other hand, the chloride complex 4b on its own showed little activity in the hydrosilylation of PhCHO (entry 12). However, it could be activated by treatment with an equimolar amount of AgClO4. The hydrosilylation of PhCHO took place after an induction period and up to 90% conversion was achieved after 48 h (entry 13). Figure 2 depicts the conversion-time profiles of these catalytic systems and it is notable that the (salan-tBu2)Mn(N3) complex (5b) was the most active among these complexes. For further comparison, the salen analogue (salen-tBu2)Mn(N3) (6) was also tested under the same conditions, though it displayed lower activity than 5a and 5b (entry 15).
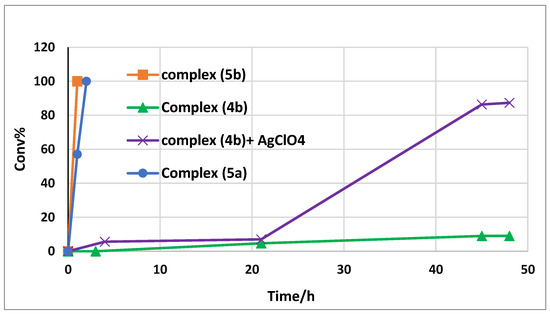
Figure 2.
The conversion-time profile with different catalysts (0.5 mol%) in the hydrosilylation of PhCHO.
With the reaction condition established above, we next examined the scope of carbonyl substrates with 5b. Hydrosilylations of a variety of aldehydes and ketones occurred at 120 °C with low loadings of the catalyst 5b (0.5 mol %) in benzene-d6 (as it is cheaper than toluene-d8) in the presence of PhSiH3 (Table 2). The purified corresponding alcohols were obtained after an acidic workup of the reaction mixtures. High conversions and good isolated yields were observed in most cases. The reaction generally proceeded faster with aldehydes when compared to ketones, as typically observed in the hydrosilylation of carbonyl compounds [53,54,55,56]. In an intermolecular competition reaction between PhCHO and PhCOMe with PhSiH3 (1:1:1 molar ratio), PhCHO conversion reached 89% within 3 h while only <5% PhCOMe reacted. Common functional groups, including halides, nitro, and methoxy, were tolerated under the reaction conditions. The benzaldehyde derivatives bearing the electron-withdrawing groups Br and NO2 tended to react more rapidly than those with electron-donating OMe (entries 2–3 vs. 4). The progress of the reaction of 4-methoxyacetophenone (entry 7, Table 2) monitored by 1H NMR spectroscopy was shown in Figure S5. In addition, the reaction of 4-nitroacetophenone was peculiar under this condition in that no further conversion was observed at 76% after 72 h (entry 6, Table 2). The reaction of cinnamaldehyde led to the reduction of the carbonyl group without any reduction of the C=C double bond, suggesting that the reaction was selective toward carbonyl groups over alkenes (entry 9). The nearly exclusive 1,2-hydrosilylation of the α,β-unsaturated carbonyl substrates was different from that of the (salen)MnN-catalyzed reaction, in which the 1,4-addition product seemed to dominate [68]. Furthermore, aliphatic carbonyl substrates also underwent hydrosilylation reactions under the same conditions (entries 10–11), and the conversions were generally high (>95%).
Table 2.
Hydrosilylation of Aldehydes and Ketones Catalyzed by (salan)MnN3 a.
In a reusability test of catalyst 5b, we carried out the hydrosilylation of PhCHO with PhSiH3 under standard conditions. After the complete consumption of PhCHO, a second batch of PhCHO and PhSiH3 (1:1 molar ratio) was added and the complete reaction of PhCHO was observed within 2 h. A third and a fourth batch were added in the subsequent runs and gave comparable results. These observations indicated that the catalyst could be reused without much loss of reactivity. At a practical level, a gram-scale reaction with PhCHO was carried out with 0.5 mol% of 5b under solvent-free conditions, and >97% conversion of PhCHO was observed in 4 h. After hydrolysis and purification, a benzyl alcohol product was obtained in good yield.
2.3. Mechanistic Consideration
In our previous study with a (salen)MnVN catalyst, [68] it was observed that the reduction of MnVN by hydrosilanes, as indicated by the color change and the NMR observations, was the first step of catalysis. Though the exact nature of the reduced Mn species was not confirmed, it was thought to be a Mn(II) or Mn(III) species that could interact with and activate hydrosilanes for the subsequent reactions. There was also evidence that the salen ligand might have undergone some transformation. In the current study, we employed Mn(III)-salan species as the (pre)catalyst so the reduction of Mn might not be necessary, though the reduction to a Mn(II) species as the active catalyst was possible under the current conditions. It has been shown that several manganese(III) catalysts could be efficient catalysts in hydrosilylation reactions [61,62,63,64,65,66,67,68,69,70]. However, (salan)MnCl 4b showed minimum catalytic activity in the hydrosilylation of PhCHO, indicating that Mn(III) alone was not a determining factor. Activation of 4b by AgClO4 suggested that it was critical to open up the coordination site around the metal center for catalysis. The comparatively higher activity of 5b (salan)Mn(N3) vs. 4b (salan)MnCl could be attributed to the weaker azido-metal interaction than M-Cl due to the larger size of N3−. In an analogous case, (salan)Cr(N3) was active in the ring-opening reaction of epoxides, while (salan)CrCl was considered a precatalyst that required further activation [79]. By the same consideration, the reduction of MnVN was supposedly required to open up the coordination site in the (salen)MnN catalysis. It should also be pointed out that the metal center in (salen)MnN and (salan)MnCl presumably adopts a square pyramidal geometry with an open site trans to the nitrido or chloride ligands. This indicated that a second open site, preferably cis to the first one, around the metal center, might be needed for hydrosilylation, i.e. to accommodate the coordination of both hydrosilane (or a hydride) and the carbonyl substrate. The observation that (salan-tBu2)Mn(N3) is more active than (salen-tBu2)Mn(N3) in catalysis (Table 1, entry 1 vs. 13) lends support to this notion since the salan ligands are more flexible to accommodate two cis open sites. As to the further activation of the hydrosilanes, a radical mechanism seems unlikely, since no ring-opening product was observed with cyclopropyl phenyl ketone [86,87]. The reaction progress of the cyclopropyl phenyl ketone monitored by the NMR spectroscopy was shown in Figure 3, and the straight conversion of the ketone to silyl ethers could be noted with the cyclopropyl ring intact during the process. The electronic effect of 4-substituted benzaldehydes indicated that the electron-withdrawing groups tended to give faster reactions than the electron-donating groups, which was comparable to the (salen)MnN-catalyzed hydrosilylation reactions [68], suggesting a similar activation mechanism. Taken together, we speculated a reaction pathway involving the formation of a hydrosilane-Mn adduct that generated an electrophilic silicon center, the coordination of a carbonyl substrate cis to the silane, followed by a subsequent nucleophilic attack of oxygen on the silicon for a silyl ether bond, though further details may vary.
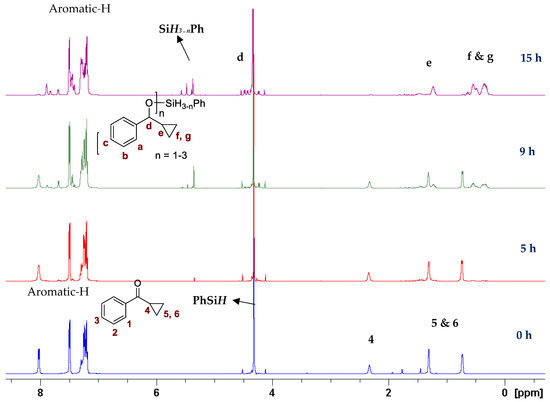
Figure 3.
Hydrosilylation of cyclopropyl phenyl ketone monitored by the NMR spectroscopy.
3. Materials and Methods
All the substrates and reagents were obtained commercially, and the liquid substrates were degasified and dried over activated molecular sieves (4 Å) prior to the reaction. The 1H and 13C NMR spectra were recorded on a Bruker AVANCE 500 (Billerica, MA, USA) or AVANCE NEO 400 (Billerica, MA, USA) NMR spectrometer and referenced to the residue peaks in CDCl3 (7.26), CD3CN (1.94), or C6D6 (7.16).
General procedures for hydrosilylation. To a J Young NMR tube, the catalyst (0.5 mol %), substrate (one equivalent), C6D6 (0.35 mL), and PhSiH3 (one equivalent) were added in order under nitrogen in a glovebox. The sealed tube was heated in an oil heating bath preset at 120 °C and the reaction was monitored by the NMR spectroscopy. When the reaction was completed, the reaction mixture was transferred to a vial with diethyl ether (2 mL). The mixture was then hydrolyzed using HCl (2 mL, 1 M) and extracted with diethyl ether. The organic phase was combined and dried over anhydrous Na2SO4. After the removal of the solvent, the alcohol product was isolated by column chromatography using silica with hexane-EtOAc as an eluent. The final products were confirmed by 1H NMR and comparison with the literature’s data.
4. Conclusions
A series of well-defined manganese salan complexes have been synthesized and shown to exhibit catalytic activity in the hydrosilylation of various aldehydes and ketones. Decent-to-high yields of the corresponding alcohols were obtained after an acidic workup, and a variety of functional groups in the carbonyl compounds, such as the methoxy, halides, and nitro groups, were tolerated. Future efforts will focus on the mechanistic elucidation of the silane activation and on additional ligand systems that may improve the catalytic activity of manganese compounds.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal13040665/s1, Detailed experimental procedural, and characterization data for catalysts and the hydrosilylation products; Figures S1–S5: 1H NMR spectra of catalytic hydrosilylation reactions of selected substrates with PhSiH3. Table S1: Comparison of catalytic activity of selective Mn complexes [76,77,88,89,90,91,92].
Author Contributions
Conceptualization, S.V. and G.D.; investigation, N.A. and S.V.; writing—original draft preparation, N.A.; writing—review and editing, G.D.; supervision, G.D.; funding acquisition, G.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was based upon work supported by the National Science Foundation under Grant No. NSF EPSCoR Award IIA-1355466. We thank the NSF MRI award # 2117699 for the acquisition of the NMR instrument. N.A. thanks SACM for a scholarship.
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.
Acknowledgments
This manuscript is based on the M.S. thesis work of N.A. at the University of North Dakota (https://commons.und.edu/theses/2444/, accessed on 27 February 2023).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Obligacion, J.V.; Chirik, P.J. Earth-abundant transition metal catalysts for alkene hydrosilylation and hydroboration. Nat. Rev. Chem. 2018, 2, 15–34. [Google Scholar] [CrossRef] [PubMed]
- Filonenko, G.A.; van Putten, R.; Hensen, E.J.M.; Pidko, E.A. Catalytic (de)hydrogenation promoted by non-precious metals–Co, Fe and Mn: Recent advances in an emerging field. Chem. Soc. Rev. 2018, 47, 1459–1483. [Google Scholar] [CrossRef] [PubMed]
- Bullock, R.M. Abundant metals give precious hydrogenation performance. Science 2013, 342, 1054–1055. [Google Scholar] [CrossRef] [PubMed]
- Zell, T.; Langer, R. From Ruthenium to Iron and Manganese—A Mechanistic View on Challenges and Design Principles of Base-Metal Hydrogenation Catalysts. ChemCatChem 2018, 10, 1930–1940. [Google Scholar] [CrossRef]
- Rana, S.; Biswas, J.P.; Paul, S.; Paik, A.; Maiti, D. Organic synthesis with the most abundant transition metal–iron: From rust to multitasking catalysts. Chem. Soc. Rev. 2021, 50, 243–472. [Google Scholar] [CrossRef]
- Guðmundsson, A.; Bäckvall, J.-E. On the use of iron in organic chemistry. Molecules 2020, 25, 1349. [Google Scholar] [CrossRef]
- Batista, V.F.; Pinto, D.C.G.A.; Silva, A.M.S. Iron: A worthy contender in metal carbene chemistry. ACS Catal. 2020, 10, 10096–10116. [Google Scholar] [CrossRef]
- Fürstner, A. Iron Catalysis in Organic Synthesis: A Critical Assessment of What It Takes To Make This Base Metal a Multitasking Champion. ACS Cent. Sci. 2016, 2, 778–789. [Google Scholar] [CrossRef]
- Cheng, L.-J.; Mankad, N.P. C–C and C–X coupling reactions of unactivated alkyl electrophiles using copper catalysis. Chem. Soc. Rev. 2020, 49, 8036–8064. [Google Scholar] [CrossRef]
- Yang, Y.; Gao, W.; Wang, Y.; Wang, X.; Cao, F.; Shi, T.; Wang, Z. Recent Advances in Copper Promoted Inert C(sp3)–H Functionalization. ACS Catal. 2021, 11, 967–984. [Google Scholar] [CrossRef]
- Colonna, P.; Bezzenine, S.; Gil, R.; Hannedouche, J. Alkene Hydroamination via Earth-Abundant Transition Metal (Iron, Cobalt, Copper and Zinc) Catalysis: A Mechanistic Overview. Adv. Synth. Catal. 2020, 362, 1550–1563. [Google Scholar] [CrossRef]
- Enthaler, S. Rise of the zinc age in homogeneous catalysis? ACS Catal. 2013, 3, 150–158. [Google Scholar] [CrossRef]
- Szewczyk, M.; Bezłada, A.; Mlynarski, J. Zinc-Catalyzed Enantioselective Hydrosilylation of Ketones and Imines under Solvent-Free Conditions. ChemCatChem 2016, 8, 3575–3579. [Google Scholar] [CrossRef]
- Lortie, J.L.; Dudding, T.; Gabidullin, B.M.; Nikonov, G.I. Zinc-catalyzed hydrosilylation and hydroboration of N-heterocycles. ACS Catal. 2017, 7, 8454–8459. [Google Scholar] [CrossRef]
- Das, K.; Waiba, S.; Jana, A.; Maji, B. Manganese-catalyzed hydrogenation, dehydrogenation, and hydroelementation reactions. Chem. Soc. Rev. 2022, 51, 4386–4464. [Google Scholar] [CrossRef]
- Mukherjee, A.; Milstein, D. Homogeneous catalysis by cobalt and manganese pincer complexes. ACS Catal. 2018, 8, 11435–11469. [Google Scholar] [CrossRef]
- Weber, S.; Stöger, B.; Veiros, L.F.; Kirchner, K. Rethinking basic concepts—Hydrogenation of alkenes catalyzed by bench-stable alkyl Mn(I) complexes. ACS Catal. 2019, 9, 9715–9720. [Google Scholar] [CrossRef]
- Elangovan, S.; Topf, C.; Fischer, S.; Jiao, H.; Spannenberg, A.; Baumann, W.; Ludwig, R.; Junge, K.; Beller, M. Selective catalytic hydrogenations of nitriles, ketones, and aldehydes by well-defined manganese pincer complexes. J. Am. Chem. Soc. 2016, 138, 8809–8814. [Google Scholar] [CrossRef]
- Wu, Y.; Song, X.; Zhang, J.; Xu, S.; Gao, L.; Zhang, J.; Xiao, G. Mn-based MOFs as efficient catalysts for catalytic conversion of carbon dioxide into cyclic carbonates and DFT studies. Chem. Eng. Sci. 2019, 201, 288–297. [Google Scholar] [CrossRef]
- Rawat, K.S.; Garg, P.; Bhauriyal, P.; Pathak, B. Metal-ligand bifunctional based Mn-catalysts for CO2 hydrogenation reaction. Mol. Catal. 2019, 468, 109–116. [Google Scholar] [CrossRef]
- Zhang, L.; Tang, Y.; Han, Z.; Ding, K. Lutidine-based chiral pincer manganese catalysts for enantioselective hydrogenation of ketones. Angew. Chem. Int. Ed. 2019, 58, 4973–4977. [Google Scholar] [CrossRef] [PubMed]
- Schneekönig, J.; Junge, K.; Beller, M. Manganese catalyzed asymmetric transfer hydrogenation of ketones using chiral oxamide ligands. Synlett 2019, 30, 503–507. [Google Scholar] [CrossRef]
- Qian, F.; Chen, X.; Yang, X. DFT and AIMD prediction of a SNS manganese pincer complex for hydrogenation of acetophenone. Chem. Phys. Lett. 2019, 714, 37–44. [Google Scholar] [CrossRef]
- Van Putten, R.; Uslamin, E.A.; Garbe, M.; Liu, C.; Gonzalez-de-Castro, A.; Lutz, M.; Junge, K.; Hensen, E.J.M.; Beller, M.; Lefort, L.; et al. Non-pincer-type manganese complexes as efficient catalysts for the hydrogenation of esters. Angew. Chem. Int. Ed. 2017, 56, 7531–7534. [Google Scholar] [CrossRef] [PubMed]
- Barman, M.K.; Das, K.; Maji, B. Selective hydroboration of carboxylic acids with a homogeneous manganese catalyst. J. Org. Chem. 2019, 84, 1570–1579. [Google Scholar] [CrossRef]
- Erken, C.; Kaithal, A.; Sen, S.; Weyhermüller, T.; Hölscher, M.; Werlé, C.; Leitner, W. Manganese-catalyzed hydroboration of carbon dioxide and other challenging carbonyl groups. Nat. Commun. 2018, 9, 4521. [Google Scholar] [CrossRef]
- Vasilenko, V.; Blasius, C.K.; Gade, L.H. One-pot sequential kinetic profiling of a highly reactive manganese catalyst for ketone hydroboration: Leveraging σ-bond metathesis via alkoxide exchange steps. J. Am. Chem. Soc. 2018, 140, 9244–9254. [Google Scholar] [CrossRef]
- Vijjamarri, S.; O’Denius, T.M.; Yao, B.; Kubatova, A.; Du, G. Highly Selective Hydroboration of Carbonyls by a Manganese Catalyst: Insight into the Reaction Mechanism. Organometallics 2020, 39, 3375–3383. [Google Scholar] [CrossRef]
- Kostera, S.; Peruzzini, M.; Kirchner, K.; Gonsalvi, L. Mild and Selective Carbon Dioxide Hydroboration to Methoxyboranes Catalyzed by Mn(I) PNP Pincer Complexes. ChemCatChem 2020, 12, 4625–4631. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Kim, J.-H.; Kim, S.; Oh, C.; Flores, M.; Groy, T.L.; Baik, M.-H.; Trovitch, R.J. Scope and mechanism of nitrile dihydroboration mediated by a β-diketiminate manganese hydride catalyst. Chem. Commun. 2020, 56, 3959–3962. [Google Scholar] [CrossRef]
- Mukhopadhyay, T.K.; MacLean, N.L.; Flores, M.; Groy, T.L.; Trovitch, R.J. Isolation of Mn (I) compounds featuring a reduced bis(imino)pyridine chelate and their relevance to electrocatalytic hydrogen production. Inorg. Chem. 2018, 57, 6065–6075. [Google Scholar] [CrossRef] [PubMed]
- Bruneau-Voisine, A.; Pallova, L.; Bastin, S.; Césarb, V.; Sortais, J.-B. Manganese catalyzed α-methylation of ketones with methanol as a C1 source. Chem. Commun. 2019, 55, 314–317. [Google Scholar] [CrossRef] [PubMed]
- Barman, M.K.; Waiba, S.; Maji, B. Manganese-Catalyzed Direct Olefination of Methyl-Substituted Heteroarenes with Primary Alcohols. Angew. Chem. Int. Ed. 2018, 57, 9126–9130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Irrgang, T.; Dietel, T.; Kallmeier, F.; Kempe, R. Manganese-Catalyzed Dehydrogenative Alkylation or α-Olefination of Alkyl-Substituted N-Heteroarenes with Alcohols. Angew. Chem. Int. Ed. 2018, 57, 9131–9135. [Google Scholar] [CrossRef]
- Waiba, S.; Barman, M.K.; Maji, B. Manganese-catalyzed acceptorless dehydrogenative coupling of alcohols with sulfones: A tool to access highly substituted vinyl sulfones. J. Org. Chem. 2019, 84, 973–982. [Google Scholar] [CrossRef]
- Jana, A.; Reddy, C.B.; Maji, B. Manganese catalyzed α-alkylation of nitriles with primary alcohols. ACS Catal. 2018, 8, 9226–9231. [Google Scholar] [CrossRef]
- Jang, Y.K.; Krückel, T.; Rueping, M.; El-Sepelgy, O. Sustainable alkylation of unactivated esters and amides with alcohols enabled by manganese catalysis. Org. Lett. 2018, 20, 7779–7783. [Google Scholar] [CrossRef]
- Samuelsen, S.V.; Santilli, C.; Ahlquist, M.S.G.; Madsen, R. Development and mechanistic investigation of the manganese(III) salen-catalyzed dehydrogenation of alcohols. Chem. Sci. 2019, 10, 1150–1157. [Google Scholar] [CrossRef]
- Mukherjee, A.; Nerush, A.; Leitus, G.; Shimon, L.J.W.; Ben-David, Y.; Jalapa, N.A.E.; Milstein, D. Manganese-Catalyzed Environmentally Benign Dehydrogenative Coupling of Alcohols and Amines to Form Aldimines and H2: A Catalytic and Mechanistic Study. J. Am. Chem. Soc. 2016, 138, 4298–4301. [Google Scholar] [CrossRef]
- Das, U.K.; Ben-David, Y.; Leitus, G.; Diskin-Posner, Y.; Milstein, D. Dehydrogenative cross-coupling of primary alcohols to form cross-esters catalyzed by a manganese pincer complex. ACS Catal. 2019, 9, 479–484. [Google Scholar] [CrossRef]
- Borghs, J.C.; Lebedev, Y.; Rueping, M.; El-Sepelgy, O. Sustainable manganese-catalyzed solvent-free synthesis of pyrroles from 1, 4-diols and primary amines. Org. Lett. 2019, 21, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Jalapa, N.A.; Kumar, A.; Leitus, G.; Diskin-Posner, Y.; Milstein, D. Synthesis of cyclic imides by acceptorless dehydrogenative coupling of diols and amines catalyzed by a manganese pincer complex. J. Am. Chem. Soc. 2017, 139, 11722–11725. [Google Scholar] [CrossRef] [PubMed]
- Vijjamarri, S.; Chidara, V.K.; Rousova, J.; Du, G. Dehydrogenative coupling of alcohols and carboxylic acids with hydrosilanes catalyzed by a salen–Mn (V) complex. Catal. Sci. Technol. 2016, 6, 3886–3892. [Google Scholar] [CrossRef]
- Vijjamarri, S.; Chidara, V.K.; Du, G. Versatile Manganese Catalysis for the Synthesis of Poly(silylether)s from Diols and Dicarbonyls with Hydrosilanes. ACS Omega 2017, 2, 582–591. [Google Scholar] [CrossRef]
- Vijjamarri, S.; Streed, S.; Serum, E.M.; Sibi, M.P.; Du, G. Polymers from bioderived resources: Synthesis of poly (silylether) s from furan derivatives catalyzed by a salen–Mn (V) complex. ACS Sustain. Chem. Eng. 2018, 6, 2491–2497. [Google Scholar] [CrossRef]
- Vijjamarri, S.; Hull, M.; Kolodka, E.; Du, G. Renewable Isohexide-Based, Hydrolytically Degradable Poly (silyl ether) s with High Thermal Stability. ChemSusChem 2018, 11, 2881–2888. [Google Scholar] [CrossRef]
- Gregg, B.T.; Hanna, P.K.; Crawford, E.J.; Cutler, A.R. Hydrosilation of manganese acyls (CO)5MnCOR (R= CH3, Ph). J. Am. Chem. Soc. 1991, 113, 384–385. [Google Scholar] [CrossRef]
- Hanna, P.K.; Gregg, B.T.; Cutler, A.R. Manganese carbonyl compounds as hydrosilation catalysts for organoiron acyl complexes. Organometallics 1991, 10, 31–33. [Google Scholar] [CrossRef]
- Mao, Z.; Gregg, B.T.; Cutler, A.R. Catalytic hydrosilylation of organic esters using manganese carbonyl acetyl complexes. J. Am. Chem. Soc. 1995, 117, 10139–10140. [Google Scholar] [CrossRef]
- Gregg, B.T.; Cutler, A.R. Hydrosilation of the Manganese Acetyl (CO)5MnC(O)CH3 with Monohydrosilanes. J. Am. Chem. Soc. 1996, 118, 10069–10084. [Google Scholar] [CrossRef]
- Cavanaugh, M.D.; Gregg, B.T.; Cutler, A.R. Manganese Carbonyl Complexes as Catalysts for the Hydrosilation of Ketones: Comparison with RhCl(PPh3)3. Organometallics 1996, 15, 2764–2769. [Google Scholar] [CrossRef]
- Mao, Z.; Gregg, B.T.; Cutler, A.R. Manganese- and Rhodium-Catalyzed Phenylsilane Hydrosilation−Deoxygenation of Iron Acyl Complexes Cp(L)(CO)FeC(O)R (L = CO, PPh3, P(OMe)3, P(OPh)3; R = CH3, Ph, CHMe2, CMe3). Organometallics 1998, 17, 1993–2002. [Google Scholar] [CrossRef]
- Fang, F.; Zhang, J. Notable Catalytic Activity of Transition Metal Thiolate Complexes against Hydrosilylation and Hydroboration of Carbon-Heteroatom Bonds. Chem. Asian J. 2023, 18, e202201181. [Google Scholar] [CrossRef]
- Sousa, S.C.A.; Cabritaa, I.; Fernandes, A.C. High-valent oxo-molybdenum and oxo-rhenium complexes as efficient catalysts for X–H (X= Si, B, P and H) bond activation and for organic reductions. Chem. Soc. Rev. 2012, 41, 5641–5653. [Google Scholar] [CrossRef] [PubMed]
- Du, G.; Abu-Omar, M.M. Oxo and imido complexes of rhenium and molybdenum in catalytic reductions. Cur. Org. Chem. 2008, 12, 1185–1198. [Google Scholar] [CrossRef]
- Truong, T.V.; Kastl, E.A.; Du, G. Cationic nitridoruthenium(VI) catalyzed hydrosilylation of ketones and aldehydes. Tetrahedron Lett. 2011, 52, 1670–1672. [Google Scholar] [CrossRef]
- Yang, X.; Wang, C. Manganese-catalyzed hydrosilylation reactions. Chem. Asian J. 2018, 13, 2307–2315. [Google Scholar] [CrossRef]
- Trovitch, R.J. The emergence of manganese-based carbonyl hydrosilylation catalysts. Acc. Chem. Res. 2017, 50, 2842–2852. [Google Scholar] [CrossRef]
- Valyaev, D.A.; Lavigne, G.; Lugan, N. Manganese organometallic compounds in homogeneous catalysis: Past, present, and prospects. Coord. Chem. Rev. 2016, 308, 191–235. [Google Scholar] [CrossRef]
- Trovitch, R.J. Comparing well-defined manganese, iron, cobalt, and nickel ketone hydrosilylation catalysts. Synlett 2014, 25, 1638–1642. [Google Scholar] [CrossRef]
- Pinto, M.; Friães, S.; Franco, F.; Lloret-Fillol, J.; Royo, B. Manganese N-Heterocyclic Carbene Complexes for Catalytic Reduction of Ketones with Silanes. ChemCatChem 2018, 10, 2734–2740. [Google Scholar] [CrossRef]
- Kelly, C.M.; McDonald, R.; Sydora, O.L.; Stradiotto, M.; Turculet, L. A manganese pre-catalyst: Mild reduction of amides, ketones, aldehydes, and esters. Angew. Chem. Int. Ed. 2017, 56, 15901–15904. [Google Scholar] [CrossRef]
- Ma, X.; Zuo, Z.; Liu, G.; Huang, Z. Manganese-catalyzed asymmetric hydrosilylation of aryl ketones. ACS Omega 2017, 2, 4688–4692. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, T.K.; Ghosh, C.; Flores, M.; Groy, T.L.; Trovitch, R.J. Hydrosilylation of aldehydes and formates using a dimeric manganese precatalyst. Organometallics 2017, 36, 3477–3483. [Google Scholar] [CrossRef]
- Zheng, J.; Elangovan, S.; Valyaev, D.A.; Brousses, R.; Cesar, V.; Sortais, J.-B.; Darcel, C.; Lugan, N.; Lavigne, G. Hydrosilylation of Aldehydes and Ketones Catalyzed by Half-Sandwich Manganese(I) N-Heterocyclic Carbene Complexes. Adv. Synth. Catal. 2014, 356, 1093–1097. [Google Scholar] [CrossRef]
- Mukhopadhyay, T.K.; Flores, M.; Groy, T.L.; Trovitch, R.J. A highly active manganese precatalyst for the hydrosilylation of ketones and esters. J. Am. Chem. Soc. 2014, 136, 882–885. [Google Scholar] [CrossRef]
- Mukhopadhyay, T.K.; Rock, C.L.; Hong, M.; Ashley, D.C.; Groy, T.L.; Baik, M.-H.; Trovitch, R.J. Mechanistic investigation of bis(imino)pyridine manganese catalyzed carbonyl and carboxylate hydrosilylation. J. Am. Chem. Soc. 2017, 139, 4901–4915. [Google Scholar] [CrossRef]
- Chidara, V.K.; Du, G. An efficient catalyst based on manganese salen for hydrosilylation of carbonyl compounds. Organometallics 2013, 32, 5034–5037. [Google Scholar] [CrossRef]
- Zheng, J.; Chevance, S.; Darcel, C.; Sortais, J.-B. Selective reduction of carboxylic acids to aldehydes through manganese catalysed hydrosilylation. Chem. Commun. 2013, 49, 10010–10012. [Google Scholar] [CrossRef]
- Valyaev, D.A.; Wei, D.; Elangovan, S.; Cavailles, M.; Dorcet, V.; Sortais, J.-B.; Darcel, C.; Lugan, N. Half-Sandwich Manganese Complexes Bearing Cp Tethered N-Heterocyclic Carbene Ligands: Synthesis and Mechanistic Insights into the Catalytic Ketone Hydrosilylation. Organometallics 2016, 35, 4090–4098. [Google Scholar] [CrossRef]
- Carney, J.; Dillon, B.; Campbell, L.; Thomas, S.P. Manganese-Catalyzed Hydrofunctionalization of Alkenes. Angew. Chem. Int. Ed. 2018, 57, 10620–10624. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, T.K.; Flores, M.; Groy, T.L.; Trovitch, R.J. A β-diketiminate manganese catalyst for alkene hydrosilylation: Substrate scope, silicone preparation, and mechanistic insight. Chem. Sci. 2018, 9, 7673–7680. [Google Scholar] [CrossRef]
- Xu, Z.-J.; Fang, R.; Zhao, C.; Huang, J.-S.; Li, G.-Y.; Zhu, N.; Che, C.-M. cis-β-Bis(carbonyl) Ruthenium−Salen Complexes: X-ray Crystal Structures and Remarkable Catalytic Properties toward Asymmetric Intramolecular Alkene Cyclopropanation. J. Am. Chem. Soc. 2009, 131, 4405–4417. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, M.; Leo, V.; Tedesco, C.; Mazzeo, M.; Lamberti, M. Salen, salan and salalen iron(iii) complexes as catalysts for CO2/epoxide reactions and ROP of cyclic esters. Dalton Trans. 2018, 47, 13229–13238. [Google Scholar] [CrossRef] [PubMed]
- Dixit, A.; Kumar, P.; Yadav, G.D.; Singh, S. Asymmetric Henry reaction catalyzed by chiral Cu(II) salalen and salan complexes derived from (S)-proline. Inorg. Chim. Acta 2018, 479, 240–246. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Ulusoy, M.; Karroonnirum, O.; Poland, R.R.; Reibenspies, J.H.; Cetinkaya, B. Highly selective and reactive (salan)CrCl catalyst for the copolymerization and block copolymerization of epoxides with carbon dioxide. Macromolecules 2009, 42, 6992–6998. [Google Scholar] [CrossRef]
- Balsells, J.; Carroll, J.P.; Walsh, P.J. Achiral Tetrahydrosalen Ligands for the Synthesis of C2-Symmetric Titanium Complexes: A Structure and Diastereoselectivity Study. Inorg. Chem. 2001, 40, 5568–5574. [Google Scholar] [CrossRef]
- Leighton, J.L.; Jacobsen, E.N. Efficient Synthesis of (R)-4-((Trimethylsilyl)oxy)-2-cyclopentenone by Enantioselective Catalytic Epoxide Ring Opening. J. Org. Chem. 1996, 61, 389–390. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Frantz, E.B. X-Ray crystal structures of five-coordinate (salen)MnN3 derivatives and their binding abilities towards epoxides: Chemistry relevant to the epoxide–CO2 copolymerization process. Dalton Trans. 2008, 37, 5031–5036. [Google Scholar] [CrossRef]
- Barluzzi, L.; Scopelliti, R.; Mazzanti, M.M. Photochemical synthesis of a stable terminal uranium(VI) nitride. J. Am. Chem. Soc. 2020, 142, 19047–19051. [Google Scholar] [CrossRef]
- Scheibel, M.G.; Askevold, B.; Heinemann, F.W.; Reijerse, E.J.; de Bruin, B.; Schneider, S. Closed-shell and open-shell square-planar iridium nitrido complexes. Nat. Chem. 2012, 4, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Zolnhofer, E.M.; Kaess, M.; Khusniyarov, M.M.; Heinemann, F.W.; Maron, L.; van Gastel, M.; Bill, E.; Meyer, K. An intermediate cobalt(IV)nitrido complex and its N-migratory insertion product. J. Am. Chem. Soc. 2014, 136, 15072–15078. [Google Scholar] [CrossRef] [PubMed]
- Camp, C.; Grant, L.N.; Bergman, R.G.; Arnold, J. Photo-activation of d0 niobium imido azides: En route to nitrido complexes. Chem. Commun. 2016, 52, 5538–5541. [Google Scholar] [CrossRef] [PubMed]
- Scepaniak, J.J.; Fulton, M.D.; Bontchev, R.P.; Duesler, E.N.; Kirk, M.L.; Smith, J.M. Structural and Spectroscopic Characterization of an Electrophilic Iron Nitrido Complex. J. Am. Chem. Soc. 2008, 130, 10515–10516. [Google Scholar] [CrossRef]
- Bois, J.D.; Hong, J.; Carreira, E.M.; Day, M.W. Nitrogen transfer from a nitridomanganese(V) complex: Amination of silyl enol ethers. J. Am. Chem. Soc. 1996, 118, 915–916. [Google Scholar] [CrossRef]
- Yang, J.; Tilley, T.D. Efficient Hydrosilylation of Carbonyl Compounds with the Simple Amide Catalyst [Fe{N(SiMe3)2}2]. Angew. Chem. Int. Ed. 2010, 49, 10186–10188. [Google Scholar] [CrossRef]
- Yang, D.; Tanner, D.D. Mechanism of the reduction of ketones by trialkylsilane. Hydride transfer, SET-hydrogen atom abstraction, or free radical addition. J. Org. Chem. 1986, 51, 2267–2270. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Mackiewicz, R.M.; Rodgers, J.L.; Fang, C.C.; Billodeaux, D.R.; Reibenspies, J.H. Cyclohexene oxide/CO2 copolymerization catalyzed by chromium(III)salen complexes and N-methylimidazole: Effects of varying salen ligand substituents and relative cocatalyst loading. Inorg. Chem. 2004, 43, 6024–6034. [Google Scholar] [CrossRef]
- Jakhar, V.K.; Barman, M.K.; Nembenna, S. Aluminum monohydride catalyzed selective hydroboration of carbonyl compounds. Org. Lett. 2016, 18, 4710–4713. [Google Scholar] [CrossRef]
- Zeng, H.; Wu, J.; Li, S.; Hui, C.; Ta, A.; Cheng, S.Y.; Zheng, S.; Zhang, G. Copper(II)-Catalyzed Selective Hydroboration of Ketones and Aldehydes. Org. Lett. 2019, 21, 401–406. [Google Scholar] [CrossRef]
- Qi, X.; Zheng, T.; Zhou, J.; Dong, Y.; Zuo, X.; Li, X.; Sun, H.; Fuhr, O.; Fenske, D. Synthesis and Catalytic Activity of Iron Hydride Ligated with Bidentate N-Heterocyclic Silylenes for Hydroboration of Carbonyl Compounds. Organometallics 2019, 38, 268–277. [Google Scholar] [CrossRef]
- Clarke, Z.E.; Maragh, P.T.; Dasgupta, T.P.; Gusev, D.G.; Lough, A.J.; Abdur-Rashid, K. A family of active iridium catalysts for transfer hydrogenation of ketones. Organometallics 2006, 25, 4113–4117. [Google Scholar] [CrossRef]




























Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).