

Adsorption and Photo-Degradation of Organophosphates on Sulfate-Terminated Anatase TiO2 Nanoparticles



Abstract
1. Introduction
2. Results
2.1. Materials Characterization
2.1.1. Powder X-ray Diffraction and Elemental Analysis
2.1.2. Electron Microscopy
2.1.3. Bandgap Determination
2.2. Adsorption of TMP and TEP
2.2.1. Adsorption of Trimethyl Phosphate (TMP)
2.2.2. Adsorption of Triethyl Phosphate
2.3. Photodegradation of TMP and TEP
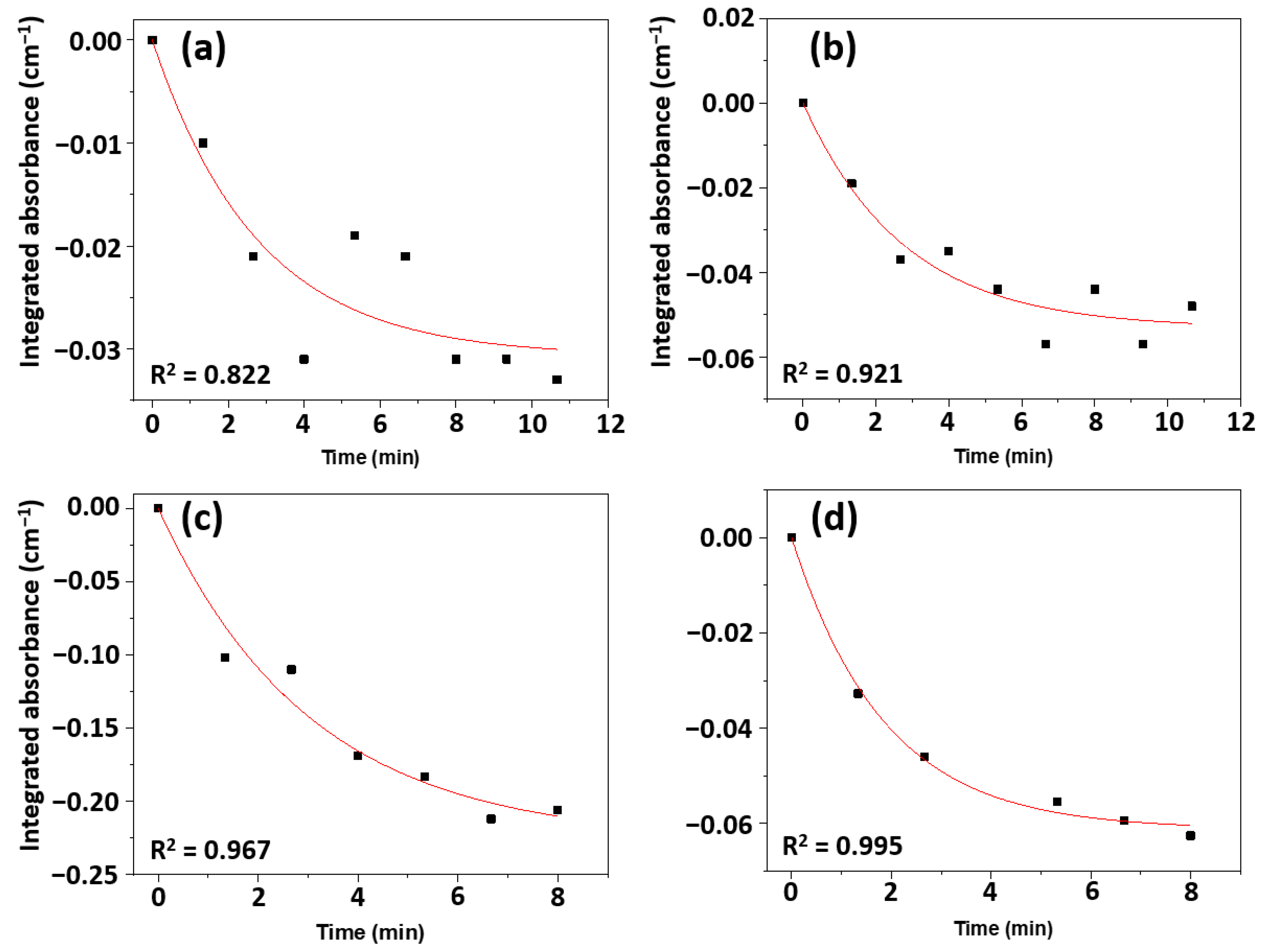
2.3.1. Photodegradation of TMP
2.3.2. Photodegradation of TEP
2.4. Reaction Kinetics
3. Materials and Methods
3.1. Photocatalysts Preparation
3.2. Materials Characterization
3.3. DRIFTS
3.4. 2D Correlation Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sidhu, G.K.; Singh, S.; Kumar, V.; Dhanjal, D.S.; Datta, S.; Singh, J. Toxicity, monitoring and biodegradation of organophosphate pesticides: A review. Crit. Rev. Environ. Sci. Technol. 2019, 49, 1135–1187. [Google Scholar] [CrossRef]
- Greaves, A.K.; Letcher, R.J. A Review of Organophosphate esters in the environment from biological effects to distribution and fate. Bull. Environ. Contam. Toxcol. 2017, 98, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, H.; Nilsson, U.; Becker, G.; Östman, C. Organophosphate ester flame retardants and plasticizers in the indoor environment: Analytical methodology and occurrence. Environ. Sci. Technol. 1997, 31, 2931–2936. [Google Scholar] [CrossRef]
- Mali, H.; Shah, C.D.; Raghunandan, B.H.; Prajapati, A.S.; Patel, D.H.; Trivedi, U.; Subramanian, R.B. Organophosphate pesticides an emerging environmental contaminant: Pollution, toxicity, bioremediation progress, and remaining challenges. J. Environ. Sci. 2023, 127, 234–250. [Google Scholar] [CrossRef]
- Salamova, A.; Ma, Y.N.; Venier, M.; Hites, R.A. High levels of organophosphate flame retardants in the Great Lakes atmosphere. Environ. Sci. Technol. Lett. 2014, 1, 8–14. [Google Scholar] [CrossRef]
- Gusain, R.; Gupta, K.; Joshi, P.; Khatri, O.P. Adsorptive removal and photocatalytic degradation of organic pollutants using metal oxides and their composites: A comprehensive review. Adv. Colloid Interface Sci. 2019, 272, 102009. [Google Scholar] [CrossRef] [PubMed]
- Sud, D.; Kaur, P. Heterogeneous Photocatalytic degradation of selected organophosphate pesticides: A review. Crit. Rev. Environ. Sci. Technol. 2012, 42, 2365–2407. [Google Scholar] [CrossRef]
- Paz, Y. Application of TiO2 photocatalysis for air treatment: Patents’ overview. Appl. Catal. B 2010, 99, 448–460. [Google Scholar] [CrossRef]
- Mäkie, P.; Persson, P.; Österlund, L. Solar light degradation of trimethyl phosphate and triethyl phosphate on dry and water-precovered hematite and goethite nanoparticles. J. Phys. Chem. C 2012, 116, 14917–14929. [Google Scholar] [CrossRef]
- Henych, J.; Janos, P.; Kormunda, M.; Tolasz, J.; Stengl, V. Reactive adsorption of toxic organophosphates parathion methyl and DMMP on nanostructured Ti/Ce oxides and their composites. Arab. J. Chem. 2019, 12, 4258–4269. [Google Scholar] [CrossRef]
- Chen, D.A.; Ratliff, J.S.; Hu, X.F.; Gordon, W.O.; Senanayake, S.D.; Mullins, D.R. Dimethyl methylphosphonate decomposition on fully oxidized and partially reduced ceria thin films. Surf. Sci. 2010, 604, 574–587. [Google Scholar] [CrossRef]
- Holdren, S.; Tsyshevsky, R.; Fears, K.; Owrutsky, J.; Wu, T.; Wang, X.Z.; Eichhorn, B.W.; Kuklja, M.M.; Zachariah, M.R. Adsorption and destruction of the g-series nerve agent simulant dimethyl methylphosphonate on zinc oxide. ACS Catal. 2019, 9, 902–911. [Google Scholar] [CrossRef]
- Korake, P.V.; Sridharkrishna, R.; Hankare, P.P.; Garadkar, K.M. Photocatalytic degradation of phosphamidon using Ag-doped ZnO nanorods. Toxicol. Environ. Chem. 2012, 94, 1075–1085. [Google Scholar] [CrossRef]
- Moss, J.A.; Szczepankiewicz, S.H.; Park, E.; Hoffmann, M.R. Adsorption and photodegradation of dimethyl methylphosphonate vapor at TiO2 surfaces. J. Phys. Chem. B 2005, 109, 19779–19785. [Google Scholar] [CrossRef] [PubMed]
- Rusu, C.N.; Yates, J.T. Photooxidation of dimethyl methylphosphonate on TiO2 powder. J. Phys. Chem. B 2000, 104, 12299–12305. [Google Scholar] [CrossRef]
- Bachinger, A.; Kickelbick, G. Photocatalytic stability of organic phosphonates and phosphates on TiO2 nanoparticles. Appl. Catal. A 2011, 409, 122–132. [Google Scholar] [CrossRef]
- Serpone, N.; Emeline, A.V. Semiconductor photocatalysis—Past, present, and future outlook. J. Phys. Chem. Lett. 2012, 3, 673–677. [Google Scholar] [CrossRef]
- Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.L.; Horiuchi, Y.; Anpo, M.; Bahnemann, D.W. Understanding TiO2 photocatalysis: Mechanisms and materials. Chem. Rev. 2014, 114, 9919–9986. [Google Scholar] [CrossRef]
- Low, J.X.; Yu, J.G.; Jaroniec, M.; Wageh, S.; Al-Ghamdi, A.A. Heterojunction photocatalysts. Adv. Mater. 2017, 29, 1601694. [Google Scholar] [CrossRef]
- Huang, Q.L.; Zhang, Q.T.; Yuan, S.S.; Zhang, Y.C.; Zhang, M. One-pot facile synthesis of branched Ag-ZnO heterojunction nanostructure as highly efficient photocatalytic catalyst. Appl. Surf. Sci. 2015, 353, 949–957. [Google Scholar] [CrossRef]
- Im, J.K.; Sohn, E.J.; Kim, S.; Jang, M.; Son, A.; Zoh, K.D.; Yoon, Y. Review of MXene-based nanocomposites for photocatalysis. Chemosphere 2021, 270, 129478. [Google Scholar] [CrossRef]
- Li, J.L.; Zhang, Q.; Zou, Y.Z.; Cao, Y.H.; Cui, W.; Dong, F.; Zhou, Y. Ti3C2 MXene modified g-C3N4 with enhanced visible-light photocatalytic performance for NO purification. J. Colloid. Interf. Sci. 2020, 575, 443–451. [Google Scholar] [CrossRef]
- Morris, M.R.; Pendlebury, S.R.; Hong, J.; Dunn, S.; Durrant, J.R. Effect of internal electric fields on charge carrier dynamics in a ferroelectric material for solar energy conversion. Adv. Mater. 2016, 28, 7123–7128. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Jeon, W.; Choi, W. Photocatalytic air purification mimicking the self-cleaning process of the atmosphere. Nat. Commun. 2021, 12, 2528. [Google Scholar] [CrossRef]
- Mamaghani, A.H.; Haghighat, F.; Lee, C.S. Photocatalytic oxidation technology for indoor environment air purification: The state-of-the-art. Appl. Catal. B 2017, 203, 247–269. [Google Scholar] [CrossRef]
- Topalian, Z.; Stefanov, B.I.; Granqvist, C.G.; Österlund, L. Adsorption and photo-oxidation of acetaldehyde on TiO2 and sulfate-modified TiO2: Studies by in situ FTIR spectroscopy and micro-kinetic modeling. J. Catal. 2013, 307, 265–274. [Google Scholar] [CrossRef]
- Svensson, F.G.; Österlund, L. One-step synthesis of sulfate-modified titania nanoparticles with surface acidic and sustained photocatalytic properties via solid-state thermolysis of titanyl sulfate. Chemcatchem 2022, 14, e202200682. [Google Scholar] [CrossRef]
- Barraud, E.; Bosc, F.; Edwards, D.; Keller, N.; Keller, V. Gas phase photocatalytic removal of toluene effluents on sulfated titania. J. Catal. 2005, 235, 318–326. [Google Scholar] [CrossRef]
- Li, M.; Xing, Z.P.; Jiang, J.J.; Li, Z.Z.; Kuang, J.Y.; Yin, J.W.; Wan, N.; Zhu, Q.; Zhou, W. In-situ Ti3+/S doped high thermostable anatase TiO2 nanorods as efficient visible-light-driven photocatalysts. Mater. Chem. Phys. 2018, 219, 303–310. [Google Scholar] [CrossRef]
- Mattsson, A.; Lejon, C.; Stengl, V.; Bakardjieva, S.; Oplustil, F.; Andersson, P.O.; Österlund, L. Photodegradation of DMMP and CEES on zirconium doped titania nanoparticles. Appl. Catal. B 2009, 92, 401–410. [Google Scholar] [CrossRef]
- Kim, C.S.; Lad, R.J.; Tripp, C.P. Interaction of organophosphorous compounds with TiO2 and WO3 surfaces probed by vibrational spectroscopy. Sens. Actuator B 2001, 76, 442–448. [Google Scholar] [CrossRef]
- Stengl, V.; Henych, J.; Grygar, T.; Perez, R. Chemical degradation of trimethyl phosphate as surrogate for organo-phosporus pesticides on nanostructured metal oxides. Mater. Res. Bull. 2015, 61, 259–269. [Google Scholar] [CrossRef]
- Oh, Y.C.; Bao, Y.; Jenks, W.S. Isotope studies of photocatalysis TiO2-mediated degradation of dimethyl phenylphosphonate. J. Photochem. Photobiol. A 2003, 161, 69–77. [Google Scholar] [CrossRef]
- Mutin, P.H.; Guerrero, G.; Vioux, A. Hybrid materials from organophosphorous coupling molecules. J. Mater. Chem. 2005, 15, 3761–3768. [Google Scholar] [CrossRef]
- Krosley, K.W.; Collard, D.M.; Adamson, J.; Fox, M.A. Degradation of organophosphonic acids catalyzed by irradiated titanium-dioxide. J. Photochem. Photobiol. A 1993, 69, 357–360. [Google Scholar] [CrossRef]
- Noda, I. Determination of two-dimensional correlation spectra using the Hilbert transform. Appl. Spectrosc. 2000, 54, 994–999. [Google Scholar] [CrossRef]
- Noda, I. Graphical representation of two-dimensional correlation in vector space. Vib. Spectrosc. 2004, 36, 261–266. [Google Scholar] [CrossRef]
- Lebedev, V.A.; Kozlov, D.A.; Kolesnik, I.V.; Poluboyarinov, A.S.; Becerikli, A.E.; Grunert, W.; Garshev, A.V. The amorphous phase in titania and its influence on photocatalytic properties. Appl. Catal. B 2016, 195, 39–47. [Google Scholar] [CrossRef]
- Alonso-Tellez, A.; Masson, R.; Robert, D.; Keller, N.; Keller, V. Comparison of Hombikat UV100 and P25 TiO2 performance in gas-phase photocatalytic oxidation reactions. J. Photochem. Photobiol. A 2012, 250, 58–65. [Google Scholar] [CrossRef]
- Topalian, Z.; Niklasson, G.A.; Granqvist, C.G.; Österlund, L. Photo-fixation of SO2 in nanocrystalline TiO2 films prepared by reactive DC magnetron sputtering. Thin Solid Films 2009, 518, 1341–1344. [Google Scholar] [CrossRef]
- Österlund, L.; Stengl, V.; Mattsson, A.; Bakardjieva, S.; Andersson, P.O.; Oplustil, F. Effect of sample preparation and humidity on the photodegradation rate of CEES on pure and Zn doped anatase TiO2 nanoparticles prepared by homogeneous hydrolysis. Appl. Catal. B 2009, 88, 194–203. [Google Scholar] [CrossRef]
- Mäkie, P.; Persson, P.; Österlund, L. Adsorption of trimethyl phosphate and triethyl phosphoate on dry and water pre-covered hematite, maghemite, and goethite nanoparticles. J. Colloid Interface Sci. 2013, 392, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Groff, R.P.; Manogue, W.H. An Infrared Study of Formate Formation and Reactivity on TiO2 Surfaces. J. Catal. 1983, 79, 462–465. [Google Scholar] [CrossRef]
- Kiselev, A.; Mattson, A.; Andersson, M.; Palmqvist, A.E.C.; Österlund, L. Adsorption and photocatalytic degradation of diisopropyl fluorophosphate and dimethyl methylphosphonate over dry and wet rutile TiO2. J. Photochem. Photobiol. A 2006, 184, 125–134. [Google Scholar] [CrossRef]
- Stefanov, B.I.; Topalian, Z.; Granqvist, C.G.; Österlund, L. Acetaldehyde adsorption and condensation on anatase TiO2: Influence of acetaldehyde dimerization. J. Mol. Catal. A 2014, 381, 77–88. [Google Scholar] [CrossRef]
- Busca, G.; Lamotte, J.; Lavalley, J.C.; Lorenzelli, V. Ft-Ir study of the adsorption and transformation of formaldehyde on oxide surfaces. J. Am. Chem. Soc. 1987, 109, 5197–5202. [Google Scholar] [CrossRef]
- Seki, T.; Onaka, M. Sulfated mesoporous alumina: A highly effective solid strong base catalyst for the Tishchenko reaction in supercritical carbon dioxide. J. Phys. Chem. B 2006, 110, 1240–1248. [Google Scholar] [CrossRef]
- Topalian, Z.; Niklasson, G.A.; Granqvist, C.G.; Österlund, L. Spectroscopic study of the photofixation of so2 on anatase TiO2 thin films and their oleophobic properties. ACS Appl. Mater. Interfaces 2012, 4, 672–679. [Google Scholar] [CrossRef]
- Lin, X.H.; Yin, X.J.; Liu, J.Y.; Li, S.F.Y. Elucidation of structures of surface sulfate species on sulfated titania and mechanism of improved activity. Appl. Catal. B 2017, 203, 731–739. [Google Scholar] [CrossRef]
- Langhammer, D.; Kullgren, J.; Österlund, L. Photoinduced adsorption and oxidation of SO2 on anatase TiO2(101). J. Am. Chem. Soc. 2020, 142, 21767–21774. [Google Scholar] [CrossRef]
- Daash, L.W.; Smith, D.C. Infrared spectra of phosphorous compounds. Anal. Chem. 1951, 23, 853–868. [Google Scholar] [CrossRef]
- Li, Y.X.; Schlup, J.R.; Klabunde, K.J. Fourier transform infrared photoacoustic spectroscopy study of the adsorption of organophosphorus compounds on heat-treated magnesium oxide. Langmuir 1991, 7, 1394–1399. [Google Scholar] [CrossRef]
- Zehr, R.T.; Henderson, M.A. Acetaldehyde photochemistry on TiO2(110). Surf. Sci. 2008, 602, 2238–2249. [Google Scholar] [CrossRef]
- Harrison, G.; Katsiev, K.; Alsalik, Y.; Thornton, G.; Idriss, H. Switch in photocatalytic reaction selectivity: The effect of oxygen partial pressure on carbon-carbon bond dissociation over hydroxylated TiO2(110) surfaces. J. Catal. 2018, 363, 117–127. [Google Scholar] [CrossRef]
- Wijaya, K.; Putri, A.R.; Sudiono, S.; Mulijani, S.; Patah, A.; Wibowo, A.C.; Saputri, W.D. Effectively synthesizing SO4/TiO2 catalyst and its performance for converting ethanol into diethyl ether (DEE). Catalysts 2021, 11, 1492. [Google Scholar] [CrossRef]
- Yu, Z.Q.; Chuang, S.S.C. In situ IR study of adsorbed species and photogenerated electrons during photocatalytic oxidation of ethanol on TiO2. J. Catal. 2007, 246, 118–126. [Google Scholar] [CrossRef]
- Murcia, J.J.; Hidalgo, M.C.; Navio, J.A.; Arana, J.; Dona-Rodriguez, J.M. In situ FT-IR study of the adsorption and photocatalytic oxidation of ethanol over sulfated and metallized TiO2. Appl. Catal. B 2013, 142, 205–213. [Google Scholar] [CrossRef]
- Idriss, H.; Barteau, M.A. Selectivity and mechanism shifts in the reactions of acetaldehyde on oxidized and reduced TiO2(001) surfaces. Catal. Lett. 1996, 40, 147–153. [Google Scholar] [CrossRef]
- Singh, M.; Zhou, N.; Paul, D.K.; Klabunde, K.J. IR spectral evidence of aldol condensation: Acetaldehyde adsorption over TiO2 surface. J. Catal. 2008, 260, 371–379. [Google Scholar] [CrossRef]
- Niu, Y.X.; Xing, M.Y.; Tian, B.Z.; Zhang, J.L. Improving the visible light photocatalytic activity of nano-sized titanium dioxide via the synergistic effects between sulfur doping and sulfation. Appl. Catal. B 2012, 115, 253–260. [Google Scholar] [CrossRef]
- van der Meulen, T.; Mattson, A.; Österlund, L. A comparative study of the photocatalytic oxidation of propane on anatase, rutile, and mixed-phase anatase-rutile TiO2 nanoparticles: Role of surface intermediates. J. Catal. 2007, 251, 131–144. [Google Scholar] [CrossRef]
- Mattsson, A.; Leideborg, M.; Larsson, K.; Westin, G.; Österlund, L. Adsorption and solar light decomposition of acetone on anatase TiO2 and niobium doped TiO2 thin films. J. Phys. Chem. B 2006, 110, 1210–1220. [Google Scholar] [CrossRef] [PubMed]
- Morita, S. 2Dshige; Kwansei-Gakuin University: Nishinomiya, Japan, 2004–2005. [Google Scholar]
- Yu, Y.; Ozaki, Y.; Noda, I.; Jung, Y. Molecular and Laser Spectroscopy: Advances and Applications; Gupta, V.G., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 10. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Rate Constant (×10−3) [min−1 cm−2 g] | R2 |
|---|---|---|
| UV100_TMP | 0.377 | 0.953 |
| UV100_TEP | 0.608 | 0.926 |
| Ti-600_TMP | 5.26 | 0.964 |
| Ti-600_TEP | 5.05 | 0.997 |
| Rate Constant (×10−3) [min−1 cm−2 g] | Aldehyde Build-Up | OP Decomposition | Carboxylate/(bi)Carbonate Build-Up | Carboxylate Decomposition |
|---|---|---|---|---|
| UV100_TMP | 1.21 | 1.44 | 0.18 | 1.10 |
| UV100_TEP | 0.44 | 0.57 | 1.10 | 1.10 |
| Ti-600_TMP | 12.9 | 4.10 | 15.9 | 3.42 |
| Ti-600_TEP | 13.3 | 2.86 | n/a * | 5.61 |
| TMP | TEP | |
|---|---|---|
| Ti-600 | 37.0% | 33.0% |
| UV100 | 32.3% | 23.6% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Svensson, F.G.; Österlund, L. Adsorption and Photo-Degradation of Organophosphates on Sulfate-Terminated Anatase TiO2 Nanoparticles. Catalysts 2023, 13, 526. https://doi.org/10.3390/catal13030526
Svensson FG, Österlund L. Adsorption and Photo-Degradation of Organophosphates on Sulfate-Terminated Anatase TiO2 Nanoparticles. Catalysts. 2023; 13(3):526. https://doi.org/10.3390/catal13030526
Chicago/Turabian StyleSvensson, Fredric G., and Lars Österlund. 2023. "Adsorption and Photo-Degradation of Organophosphates on Sulfate-Terminated Anatase TiO2 Nanoparticles" Catalysts 13, no. 3: 526. https://doi.org/10.3390/catal13030526
APA StyleSvensson, F. G., & Österlund, L. (2023). Adsorption and Photo-Degradation of Organophosphates on Sulfate-Terminated Anatase TiO2 Nanoparticles. Catalysts, 13(3), 526. https://doi.org/10.3390/catal13030526