

Nitrogen Reduction Reaction Catalyzed by Diatomic Metals Supported by N-Doped Graphite


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Calculation Detail
3. Discussion and Results
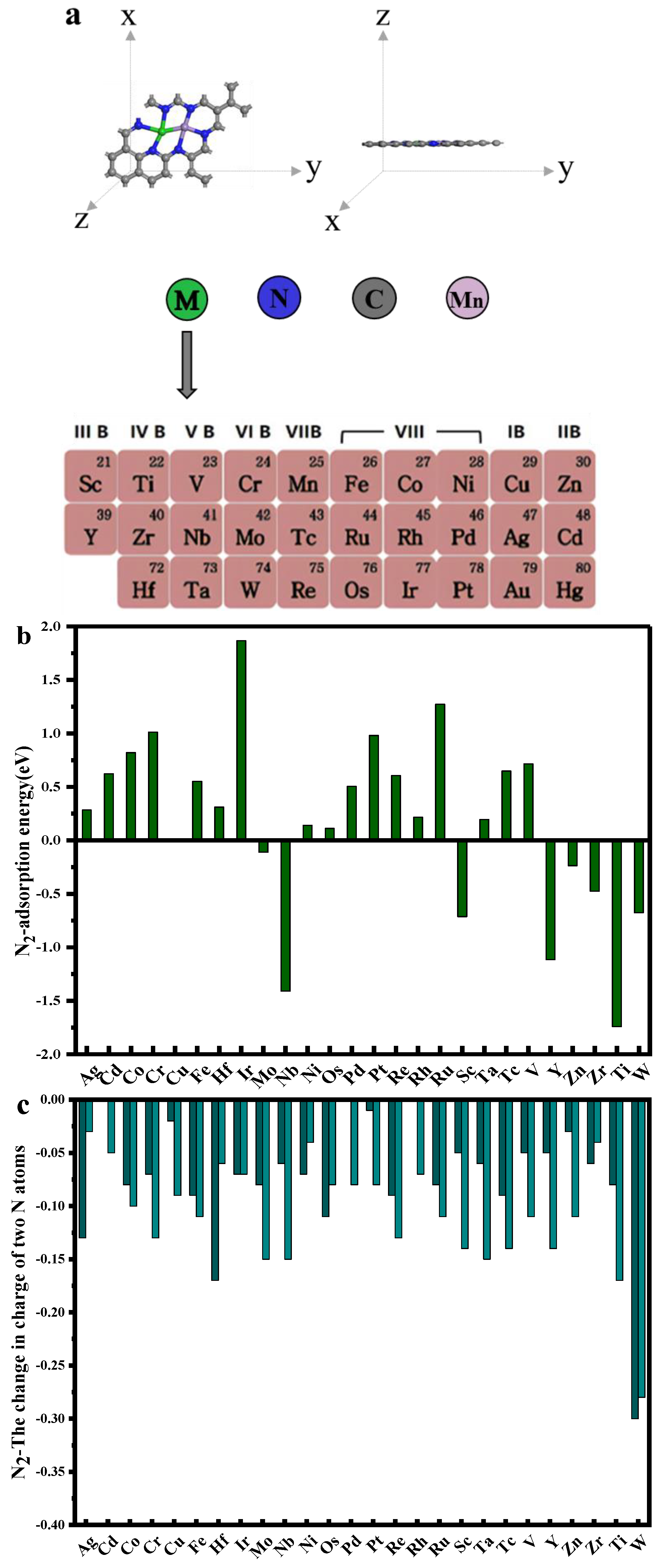
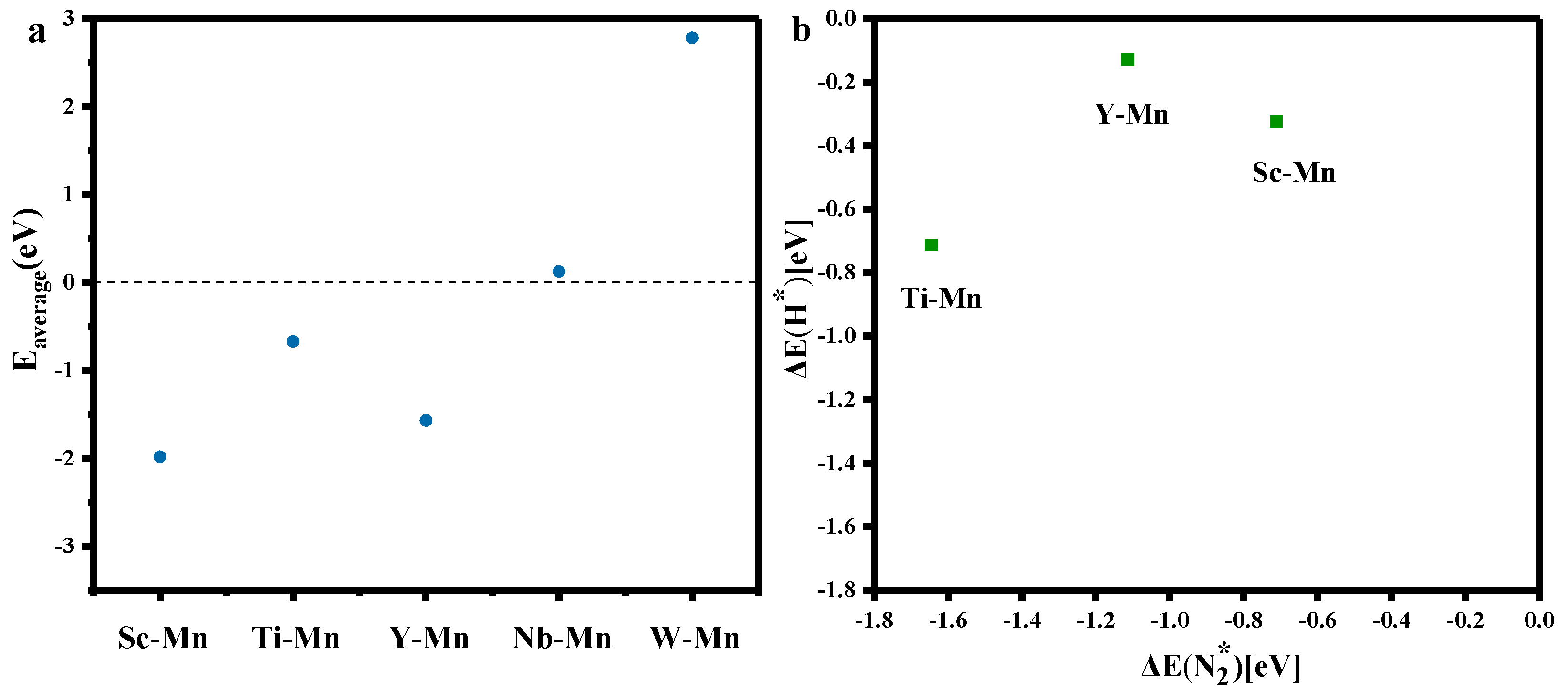
3.1. The Structures of Mn-M@N6-C (M = Sc, Ti, Y, Nb)
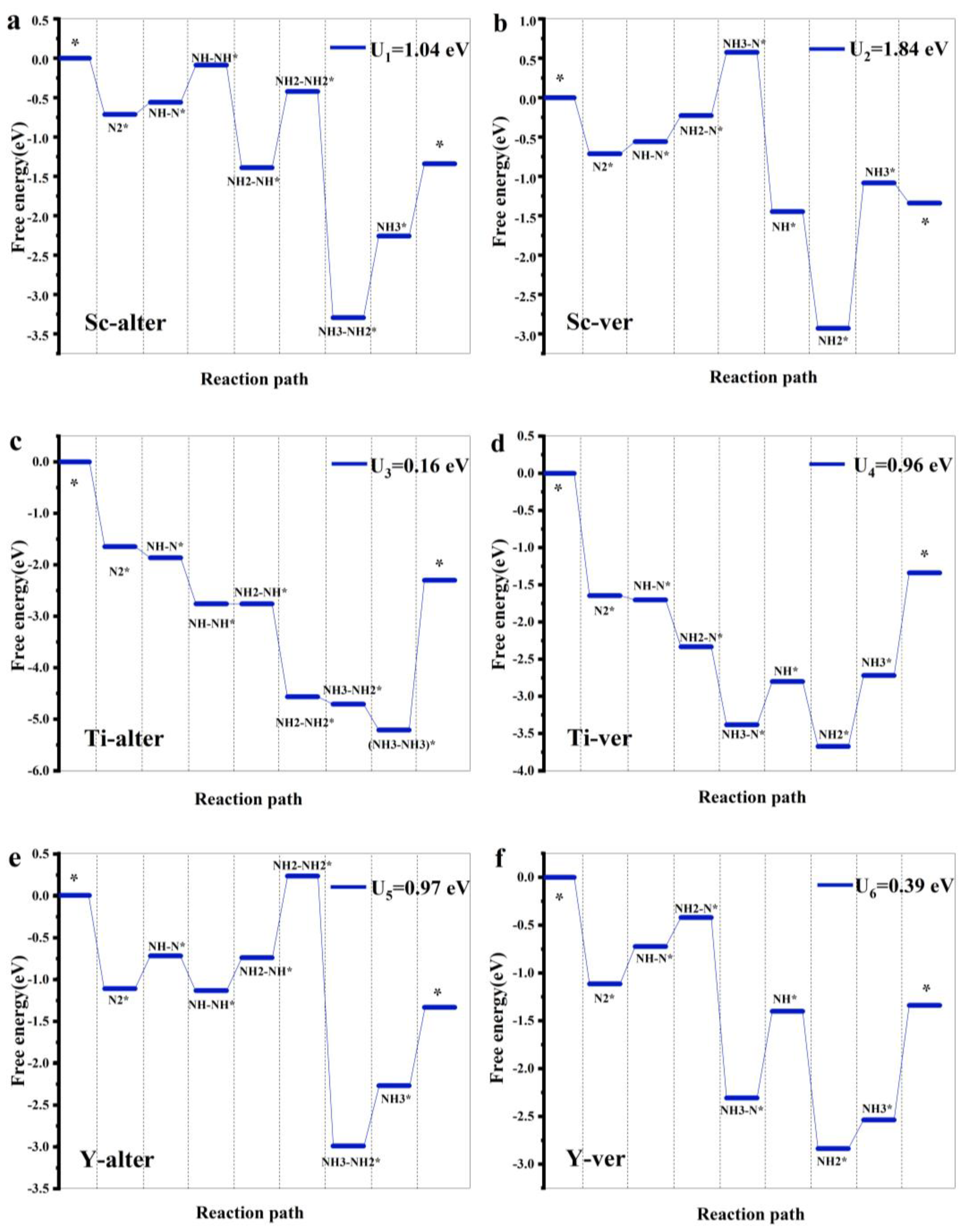
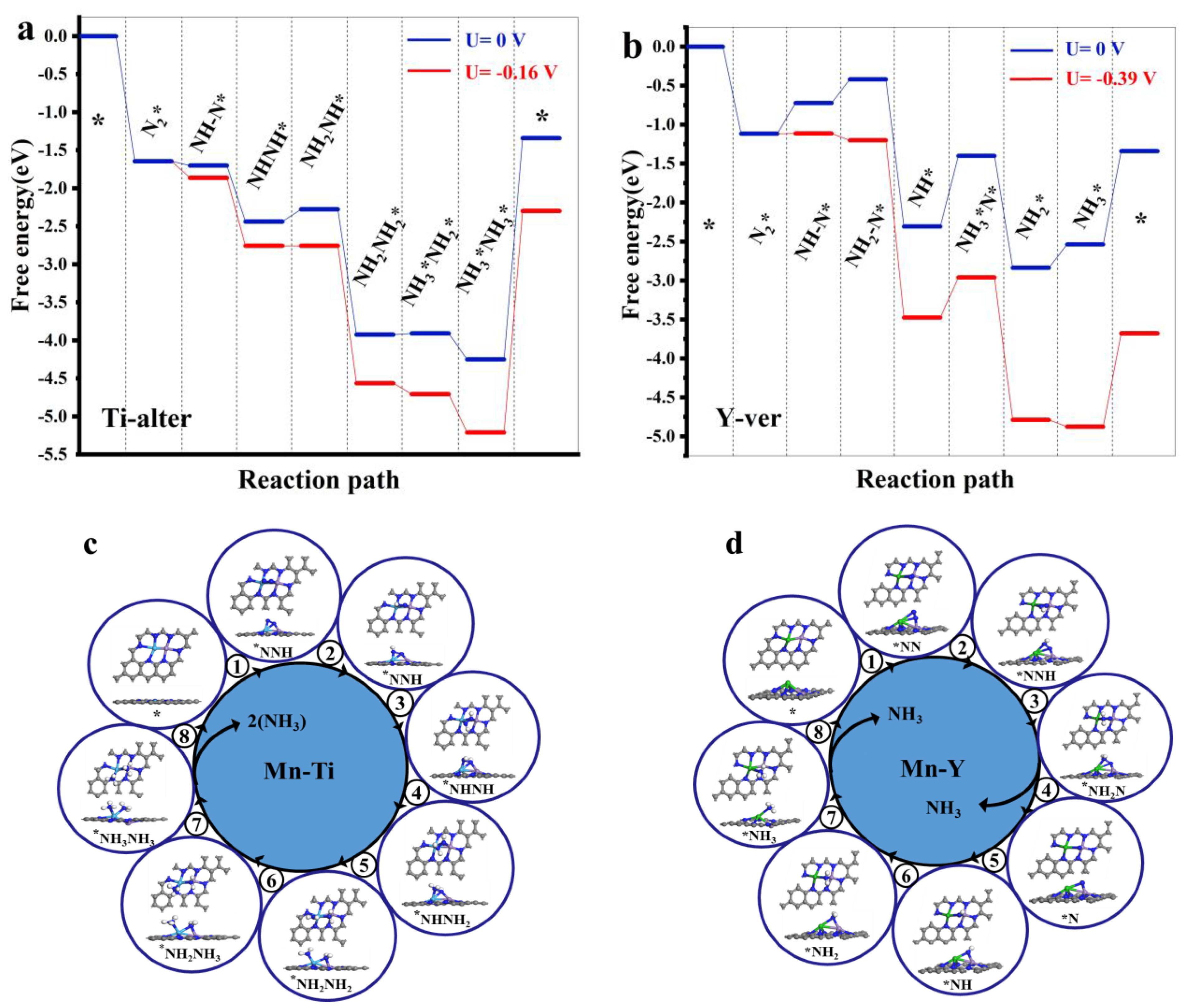
3.2. HER and NRR Path
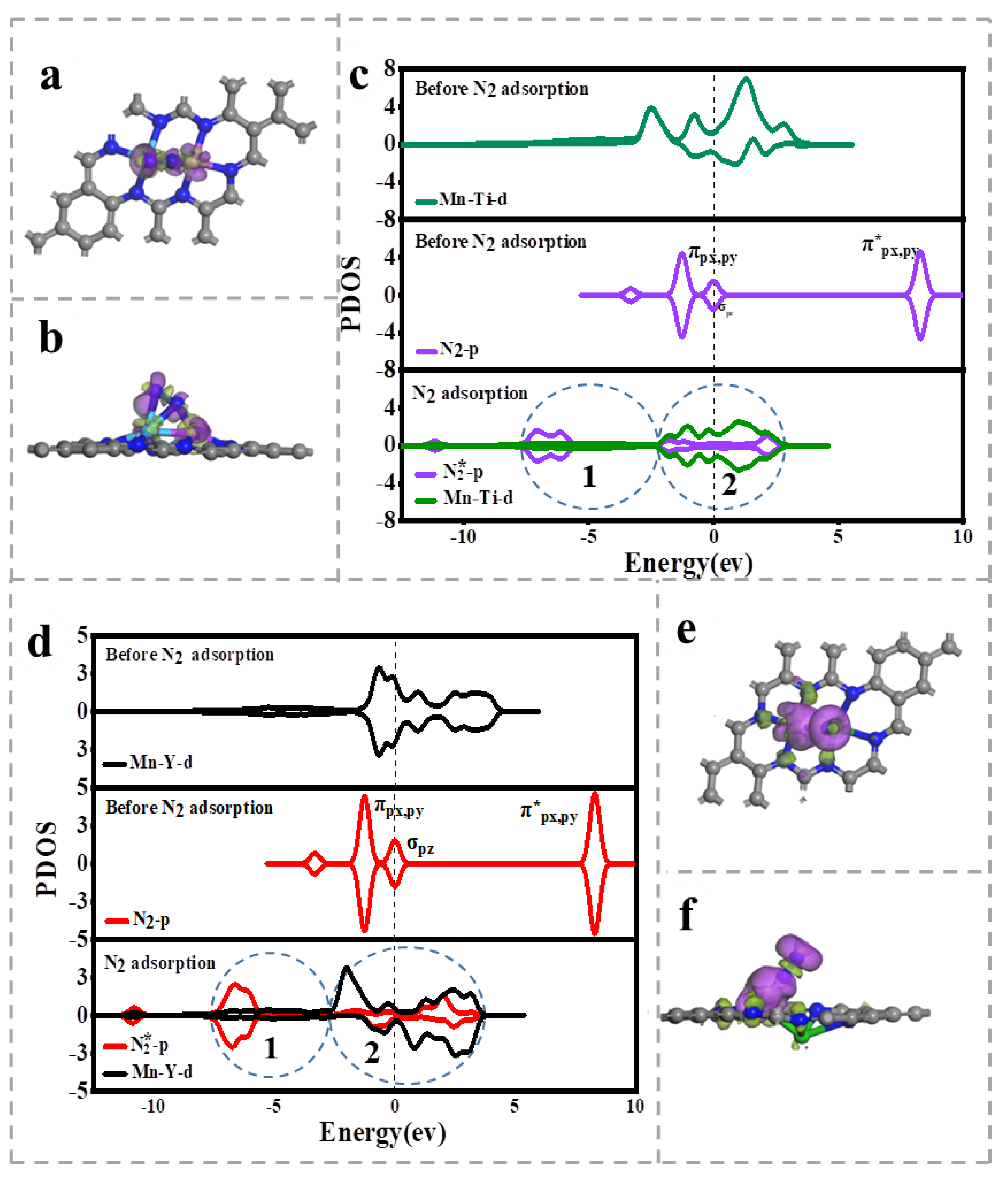
3.3. Charge Density Difference (CDD) and Projected Electron State Density (PDOS)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhang, Y.L.; Liu, H.; Li, J.J.; Deng, Y.L.; Miao, X.P.; Xu, D.P.; Liu, S.Q.; Xie, K.C.; Tian, Y.J. Life cycle assessment of ammonia synthesis in China. Int. J. Life Cycle Assess. 2022, 27, 50–61. [Google Scholar] [CrossRef]
- Cai, X.X.; Luo, G.Y.; Li, Z.Q.; He, Y. Optical conductivity of twisted bilayer graphene under heterostrain. Acta Phys. Sin. 2021, 70, 187301. [Google Scholar] [CrossRef]
- Tiwari, S.K.; Sahoo, S.; Wang, N.; Huczko, A. Graphene research and their outputs: Status and prospect. J. Sci. Adv. Mater. Devices 2020, 5, 10–29. [Google Scholar] [CrossRef]
- Hu, X.; Xiong, L.; Fang, W.-H.; Su, N.Q. Computational Insight into Metallated Graphynes as Single Atom Electrocatalysts for Nitrogen Fixation. ACS Appl. Mater. Interfaces 2022, 14, 27861–27872. [Google Scholar] [CrossRef]
- Yang, W.; Xu, S.; Ma, K.; Wu, C.; Gates, I.D.; Ding, X.; Meng, W.; Gao, Z. Geometric structures, electronic characteristics, stabilities, catalytic activities, and descriptors of graphene-based single-atom catalysts. Nano Mater. Sci. 2020, 2, 120–131. [Google Scholar] [CrossRef]
- Tang, Y.; Chen, W.; Zhou, J.; Chai, H.; Li, Y.; Cui, Y.; Feng, Z.; Dai, X. Mechanistic insight into the selective catalytic oxidation for NO and CO on co-doping graphene sheet: A theoretical study. Fuel 2019, 253, 1531–1544. [Google Scholar] [CrossRef]
- Tang, Y.; Chen, W.; Shen, Z.; Chang, S.; Zhao, M.; Dai, X. Nitrogen coordinated silicon-doped graphene as a potential alternative metal-free catalyst for CO oxidation. Carbon 2017, 111, 448–458. [Google Scholar] [CrossRef]
- Zhou, Y.; Yu, X.; Sun, F.; Zhang, J. MoP supported on reduced graphene oxide for high performance electrochemical nitrogen reduction. Dalton Trans. 2020, 49, 988–992. [Google Scholar] [CrossRef]
- Wang, W.; Wang, X.; Sun, Y.; Tian, Y.; Liu, X.; Chu, K.; Li, J. Ultrasmall iridium nanoparticles on graphene for efficient nitrogen reduction reaction. New J. Chem. 2022, 46, 5464–5469. [Google Scholar] [CrossRef]
- Wang, F.; Wei, X.; Mao, J. Graphene with SiC3 ligand for N2 to NH3 reduction with an ultralow overpotential of 0.03 V. J. Electroanal. Chem. 2022, 904, 115953. [Google Scholar] [CrossRef]
- Wang, F.; Mao, L.; Xie, H.; Mao, J. Graphene Derivatives and Graphene Composite Electrocatalysts for N-2 Reduction Reaction. Small Struct. 2021, 2, 2000075. [Google Scholar] [CrossRef]
- Huang, H.; Gong, F.; Wang, Y.; Wang, H.; Wu, X.; Lu, W.; Zhao, R.; Chen, H.; Shi, X.; Asiri, A.M.; et al. Mn3O4 nanoparticles@reduced graphene oxide composite: An efficient electrocatalyst for artificial N-2 fixation to NH3 at ambient conditions. Nano Res. 2019, 12, 1093–1098. [Google Scholar] [CrossRef]
- Chen, Z.W.; Yan, J.M.; Jiang, Q. Single or Double: Which Is the Altar of Atomic Catalysts for Nitrogen Reduction Reaction? Small Methods 2018, 3, 1800291. [Google Scholar] [CrossRef]
- Wang, J.; He, C.; Huo, J.; Fu, L.; Zhao, C. A Theoretical Evaluation of Possible N2 Reduction Mechanism on Mo2B2. Adv. Theory Simul. 2021, 4, 2100003. [Google Scholar] [CrossRef]
- Yu, J.; He, C.; Huo, J.; Zhao, C.; Yu, L. Electric field controlled CO2 capture and activation on BC6N monolayers: A first-principles study. Surf. Interfaces 2022, 30, 101885. [Google Scholar] [CrossRef]
- Zhang, Q.; He, C.; Huo, J. Mechanism of complete dehydrogenation of ammonia borane in electrochemical alkaline environment. Comput. Mater. Sci. 2022, 207, 111306. [Google Scholar] [CrossRef]
- He, C.; Wang, J.; Fu, L.; Zhao, C.; Huo, J. Associative vs. dissociative mechanism: Electrocatalysis of nitric oxide to ammonia. Chin. Chem. Lett. 2022, 33, 1051–1057. [Google Scholar] [CrossRef]
- Wang, R.; He, C.; Chen, W.; Fu, L.; Zhao, C.; Huo, J.; Sun, C. Design strategies of two-dimensional metal-organic frameworks toward efficient electrocatalysts for N2 reduction: Cooperativity of transition metals and organic linkers. Nanoscale 2021, 13, 19247–19254. [Google Scholar] [CrossRef]
- He, C.; Wang, H.; Fu, L.; Huo, J.; Zheng, Z.; Zhao, C.; An, M. Principles for designing CO2 adsorption catalyst: Serving thermal conductivity as the determinant for reactivity. Chin. Chem. Lett. 2022, 33, 990–994. [Google Scholar] [CrossRef]
- Zheng, G.; Li, L.; Tian, Z.; Zhang, X.; Chen, L. Heterogeneous single-cluster catalysts (Mn3, Fe3, Co3, and Mo3) supported on nitrogen-doped graphene for robust electrochemical nitrogen reduction. J. Energy Chem. 2021, 54, 612–619. [Google Scholar] [CrossRef]
- Song, W.; Xie, K.; Wang, J.; Guo, Y.; He, C.; Fu, L. Density functional theory study of transition metal single-atoms anchored on graphyne as efficient electrocatalysts for the nitrogen reduction reaction. Phys. Chem. Chem. Phys. 2021, 23, 10418–10428. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Sun, R.; Fu, L.; Huo, J.; Zhao, C.; Li, X.; Song, Y.; Wang, S. Defect engineering for high-selection-performance of NO reduction to NH3 over CeO2 (111) surface: A DFT study. Chin. Chem. Lett. 2022, 33, 527–532. [Google Scholar] [CrossRef]
- Zhao, C.; Xi, M.; Huo, J.; He, C. B-Doped 2D-InSe as a bifunctional catalyst for CO2/CH4 separation under the regulation of an external electric field. Phys. Chem. Chem. Phys. 2021, 23, 23219–23224. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Xi, M.; Huo, J.; He, C.; Fu, L. Electro-reduction of N2 on nanostructured materials and the design strategies of advanced catalysts based on descriptors. Mater. Today Phys. 2022, 22, 100609. [Google Scholar] [CrossRef]
- Qin, Y.; Zhang, S.; Gao, G.; Ding, S.; Su, Y. A theoretical study on molybdenum and sulfur co-doped graphene for electrocatalytic nitrogen reduction. Mol. Catal. 2022, 517, 112048. [Google Scholar] [CrossRef]
- Liu, G.; Niu, L.; Ma, Z.; An, L.; Qu, D.; Wang, D.; Wang, X.; Sun, Z. Fe2Mo3O8/XC-72 electrocatalyst for enhanced electrocatalytic nitrogen reduction reaction under ambient conditions. Nano Res. 2022, 15, 5940–5945. [Google Scholar] [CrossRef]
- Liao, Y.; Ye, W.; Zhu, Y.; Wang, L. Ultrathin MoS2 Nanosheets for Electrocatalytic N-2-To-NH3 Fixation Under Ambient Conditions. Int. J. Electrochem. Sci. 2020, 15, 11555–11566. [Google Scholar] [CrossRef]
- Ball, B.; Das, P.; Sarkar, P. Molybdenum Atom-Mediated Salphen-Based Covalent Organic Framework as a Promising Electrocatalyst for the Nitrogen Reduction Reaction: A First-Principles Study. J. Phys. Chem. C 2021, 125, 26061–26072. [Google Scholar] [CrossRef]
- Huo, J.; Wei, H.; Fu, L.; Zhao, C.; He, C. Highly active Fe36Co44 bimetallic nanoclusters catalysts for hydrolysis of ammonia borane: The first-principles study. Chin. Chem. Lett. 2022. [Google Scholar] [CrossRef]
- Song, W.; Fu, Z.; Liu, X.; Guo, Y.; He, C.; Fu, L. Density functional theory study of a two-atom active site transition-metal/iridium electrocatalyst for ammonia synthesis. J. Mater. Chem. A 2022, 10, 13946–13957. [Google Scholar] [CrossRef]
- Guo, X.; Gu, J.; Lin, S.; Zhang, S.; Chen, Z.; Huang, S. Tackling the Activity and Selectivity Challenges of Electrocatalysts toward the Nitrogen Reduction Reaction via Atomically Dispersed Biatom Catalysts. J. Am. Chem. Soc. 2020, 142, 5709–5721. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.; Zhang, X.; Qin, J.; Liu, R. Anchoring Mo on C9N4 monolayers as an efficient single atom catalyst for nitrogen fixation. J. Energy Chem. 2021, 57, 443–450. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huo, J.; Wei, H.; Zhang, K.; Zhao, C.; He, C. Nitrogen Reduction Reaction Catalyzed by Diatomic Metals Supported by N-Doped Graphite. Catalysts 2023, 13, 49. https://doi.org/10.3390/catal13010049
Huo J, Wei H, Zhang K, Zhao C, He C. Nitrogen Reduction Reaction Catalyzed by Diatomic Metals Supported by N-Doped Graphite. Catalysts. 2023; 13(1):49. https://doi.org/10.3390/catal13010049
Chicago/Turabian StyleHuo, Jinrong, Haocong Wei, Kai Zhang, Chenxu Zhao, and Chaozheng He. 2023. "Nitrogen Reduction Reaction Catalyzed by Diatomic Metals Supported by N-Doped Graphite" Catalysts 13, no. 1: 49. https://doi.org/10.3390/catal13010049
APA StyleHuo, J., Wei, H., Zhang, K., Zhao, C., & He, C. (2023). Nitrogen Reduction Reaction Catalyzed by Diatomic Metals Supported by N-Doped Graphite. Catalysts, 13(1), 49. https://doi.org/10.3390/catal13010049