Abstract
Asymmetric Michael additions of carbonyl compounds to N-substituted maleimides are among the most convenient reactions to prepare optically pure succinimide building blocks. Although a few β-amino acids were found to be highly efficient organocatalysts in the addition of α-branched aldehydes, the effect of their structure on the results of these reactions has not yet been investigated. In the present study, we disclose several unexpected and interesting structural effects of aliphatic and cycloaliphatic β-amino acids obtained in the enantioselective conjugate addition of isobutyraldehyde to N-benzylmaleimide. The dependence of the sense of the enantioselectivity on the bulkiness of the substituent on the β-carbon atom, the beneficial spatial arrangements of the functional groups in cis isomers with cyclohexane scaffold and the inversion of the enantioselectivity depending on the absence of a base additive observed with some trans isomers are unprecedented findings. The minor influence of the nitrogen substituent of the maleimide ring on both the reaction rate and the enantioselectivity was also evidenced using alicyclic β-amino acid prepared from an easily available terpene derivative.
1. Introduction
Asymmetric catalytic reactions are among convenient methods for preparing optically pure organic intermediates needed in the pharmaceutical and fine chemical industries [1,2,3,4]. Accordingly, the development of chiral catalysts is a main research area of the most recent half-century, leading to various favourable solutions applicable in numerous chemical transformations designed to create chiral centres in organic molecules [5,6,7,8,9,10]. At present, in addition to the widely applied enantioselective metal complexes, optically pure organic molecules are frequently employed as stereoselective catalysts [11,12,13,14,15]. These organocatalysts are competitive alternatives of the metal complexes, though their application is often associated with increased catalyst amounts and costs, difficulties in recovery and prolonged reaction times. Accordingly, searching for simple, highly active, stereoselective and less expensive chiral organocatalysts is still in the focus of the investigations.
Asymmetric organocatalytic conjugate additions are among the synthetically most useful reactions to create molecular complexity along with the generation of novel chiral centres in organic molecules [16,17,18]. The application of N-substituted maleimides as Michael acceptors affords chiral succinimides, which are valuable pharmaceutical intermediates [19,20,21,22,23,24]. Hence, several organocatalysts have been developed to promote the enantioselective addition of aldehydes, ketones, 1,3-dicarbonyl compounds and other nucleophiles to maleimides [25]. Among the most efficient catalysts, cinchona alkaloids [26,27,28], pyrrolidine derivatives [29,30], a variety of C2-symmetric 1,2-diamine derivatives [31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46], oligopeptides [47,48] and primary amino acids have been utilised, the latter as salts formed in situ or deposited on solid supports [49,50,51,52,53,54,55,56]. Primary amino acid derivatives afforded excellent enantioselectivities in the addition of α,α-disubstituted aldehydes to various N-substituted maleimides. The most efficient amino acids used as chiral sources in these asymmetric Michael additions are O-tert-butyl-l-threonine (O-tBu-l-Thr) and l-isoleucine (l-Ile) described by Nugent and co-workers [49,52], S-β-phenylalanine (β-Phe) and aspartic acid α-tert-butyl ester (H-Asp-OtBu) as well as several natural α-amino acids reported by Kokotos [50,51]. Substituted phenylglycines in combination with cinchona alkaloid derivatives were found to be efficient in the addition of aldehydes and ketones to maleimides by Zhao and co-workers [54]. Moreover, natural α-amino acids, such as l-phenylalanine (l-Phe), were intercalated into layered double hydroxides or adsorbed on inorganic oxides, leading to heterogeneous, recyclable chiral hybrid materials [55,56]. The structure of selected amino acids applied as catalysts is presented in Figure 1.
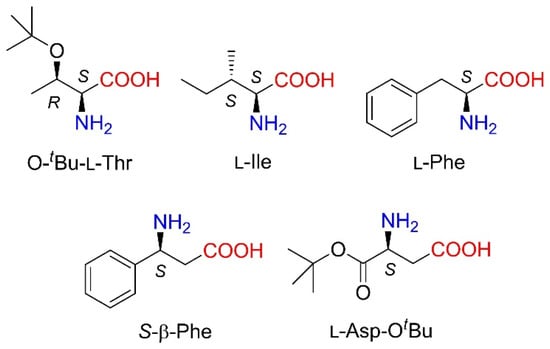
Figure 1.
Amino acid organocatalysts found to be highly efficient in Michael additions of carbonyl compounds to N-substituted maleimides.
The above reports indicate that, in addition to α-amino acids, β-amino acids may also afford excellent results in the asymmetric addition of aldehydes to maleimides. The application of β-amino acids as chiral organocatalysts in other stereoselective transformations has rarely been reported [57,58,59]. Because of the easy access to natural α-amino acids, the effect of their structure could be studied in detail. However, the influence of the substitution pattern of β-amino acids on their performance in asymmetric organocatalytic reactions is yet poorly explored. Accordingly, our aim in the present study is to investigate the effect of the structure of these compounds on the results obtained in the asymmetric organocatalytic Michael addition of an aldehyde to N-substituted maleimides.
2. Results
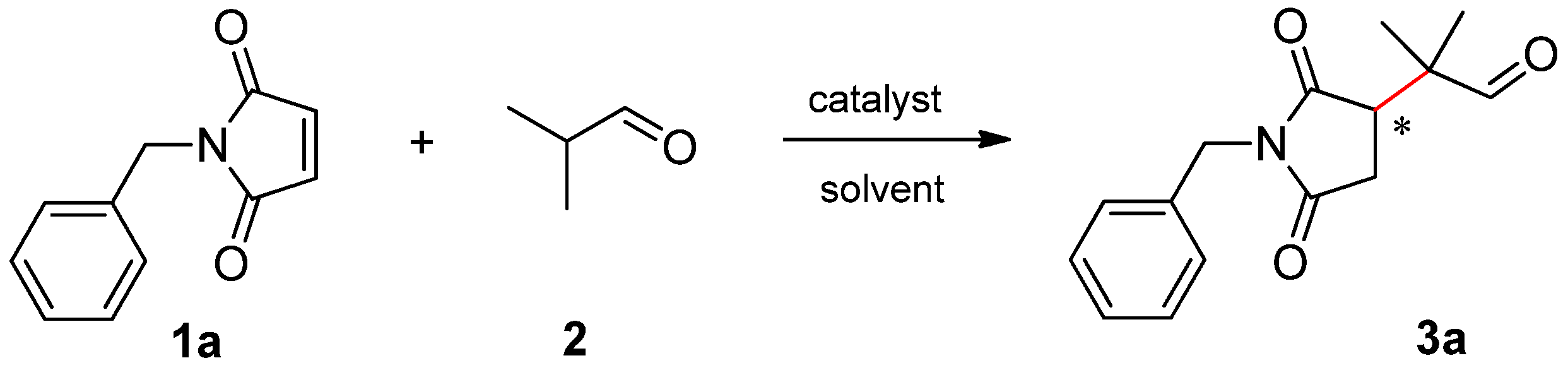
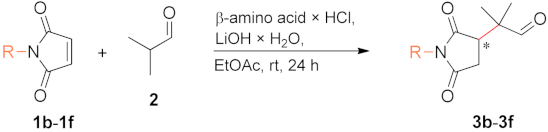
To evaluate the effect of the structure of β-amino acids, we selected the addition of isobutyraldehyde (2) to N-benzylmaleimide (1a) as a test reaction (see Scheme 1).
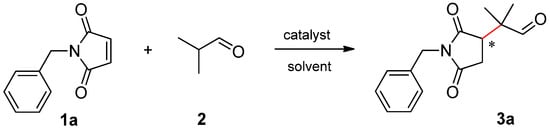
Scheme 1.
Michael addition of isobutyraldehyde (2) to N-benzylmaleimide (1a).
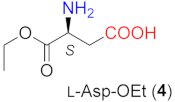
A short screening of the influence of the reaction conditions was carried out using the easily available l-Asp-OEt (4) as the catalyst, derived from natural l-Asp. The results of these experiments are presented in Table 1. The examination of the effect of the basic additive in the ethyl acetate (EtOAc) solvent showed that the bases used in previous studies (Cs2CO3 and KOH [50,52]) afforded lower conversions and/or enantiomeric excesses (ee) compared to LiOH, which offered full conversion and the highest ee (95%, entry 4). l-Asp, the parent natural amino acid, was inefficient in catalysing the reaction (in 6 h at 50 °C: conversion <1%) either using 1.5 or 3 eq of LiOH × H2O (not included in Table 1). High conversion and ee values were obtained in other aprotic solvents as well (entries 5–8). However, EtOAc, a recommended organic solvent in the pharmaceutical industry [60], was found to be the most appropriate under these reaction conditions. Protic solvents provided decreased enantioselectivities (entries 9, 10). Accordingly, further reactions were carried out in EtOAc. Decreasing the amount of both the amino acid and the base led to a decrease in the conversion, without altering enantioselectivity (entries 11, 12). A shorter reaction time (6 h) did not afford a complete transformation of 1a, provided the temperature was increased to 50 °C. This, however, resulted in a drop of the ee to 92% (entry 14). Increasing the amount of the solvent decreased the conversion without affecting the ee (entries 15 and 16 compared to entries 4 and 11, respectively). Furthermore, lowering the EtOAc amount slightly increased the conversion in parallel with a negligible decrease in the ee (94%, entry 17).

Table 1.
Michael addition of 2 to 1a catalysed by l-Asp-OEt (4): effect of reaction conditions a.
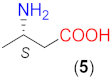
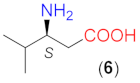
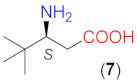
On the basis of the above results, we carried out reactions with three commercially available optically pure aliphatic β-amino acids using ethyl acetate (EtOAc) solvent and LiOH × H2O as base additive both at room temperature (rt, 24 °C) and 50 °C. Selected results are summarised in Table 2. Without the addition of LiOH × H2O, all these amino acids provided low conversions. However, the bulkiness of the substituent on the β-carbon had a marked effect on both the conversion and ee, i.e., the conversions afforded by these catalysts decreased in the order 5 > 6 > 7, whereas ee values increased in the same sequence (entries 4, 7, 11). Accordingly, the bulkiness of this substituent, in addition to hindering the reaction, also had a beneficial effect on the stereocontrol of the addition, affording 91% ee with catalyst 7 without the addition of a base additive (entry 11).
Table 2.
Asymmetric Michael addition of 2 to 1a catalysed by β-amino acids 4–7 a.
The structure of the amino acids had a similar effect on the conversion when they were transformed in situ to their lithium salts by the addition of 1.5 equivalent (eq, compared to the amount of the β-amino acid) LiOH × H2O. Under these conditions, the reactions were faster. As a result, close to complete transformations could be reached with 6 and 7, having iPr and tBu groups in the β position. Note, however, that these catalysts needed longer reactions (entries 9, 13) and an increased amount of catalyst 7 (entry 13). The presence of the base increased the enantioselectivities as well, thus high ee values (94–95%) were obtained with 6 or 7. High conversions were reached in less time by increasing the reaction temperature to 50 °C without a significant decrease in the ee values (entries 10, 14). It is interesting that the configuration of the excess enantiomer afforded by 7 (i.e., S) was opposite to that obtained using 4 or 6. Compound 5 also provided an excess of the S enantiomer; however, the chiral centre in this compound had the opposite stereo-arrangement compared to the other amino acids. Thus, the bulkiness of the substituent of the β-carbon atom influenced both the magnitude and the sense of stereodifferentiation.
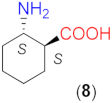
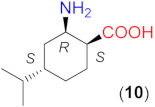
The above-detected effects of the β-amino acid structure motivated us to examine compounds with more rigid backbone, such as cycloaliphatic derivatives. The results obtained using β-amino acids having cyclohexane structural motifs in the molecule are presented in Table 3. Initially, we tested the commercially available optically pure (1S,2S)-2-aminocyclohexane-1-carboxylic acid (8). Our results show a peculiar behaviour (entries 1–5). Although in the absence of LiOH × H2O, 8 afforded the same conversion as 5, possibly due to the lack of branching next to the C2 atom, the ee value was higher and of the opposite sense compared to that provided by 5 (entry 1). The sense of the enantioselectivity can be considered inverted as compared to 6 as well, as the opposite arrangement of the NH2 group in 6 and 8 should provide opposite enantiomers in excess. Still, both compounds provided the R product as the major enantiomer. Both the observed value and the sense of the enantioselectivity may be either due to the rigidity of the ring or to the additional C1 (α-C) chiral centre of the molecule compared to the aliphatic α-unsubstituted 5 or 6. The moderate conversion reached with 8 in the absence of the base could be increased by decreasing the amount of solvent (0.5 cm3) or increasing the temperature (50 °C) and extending the reactions to 24 h (entries 2, 3). Meanwhile, the ee values remained close to the one determined previously (R enantiomer in excess).
Table 3.
Asymmetric Michael addition of 2 to 1a catalysed by β-amino acids 8–15 having a cyclohexane backbone a.
By the addition of LiOH × H2O, in addition to a substantial increase in the rate shown by the close to full conversions reached at both temperatures, ee values increased. Furthermore, the opposite product enantiomers (S) were formed in excess compared to reactions without the additive (entries 4, 5). Previously, in Michael additions of 2 to maleimides, a solvent-dependent switch of the enantioselectivity direction was reported with certain cyclohexane-1,2-diamine derivatives [41,61]. A turn in the sense of enantioselection was also observed when l-Asp-OtBu was replaced with S-β-Phe, which differs in the substituent on the β-carbon (COOtBu vs. phenyl, see Figure 1) [50]. However, to the best of our knowledge, the change in the excess enantiomer configuration by the addition of a base has not yet been reported in reactions of aldehydes and maleimides. We note that, in the presence of LiOH × H2O, both catalysts, i.e., 5 and 8, provided the same S enantiomer in excess (ee 81–82%). Thus, the unusual enantiodiscrimination occurs with 8 without being transformed into its Li salt.
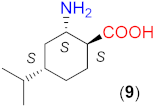
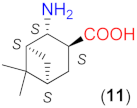
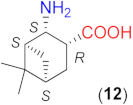
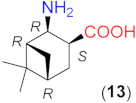
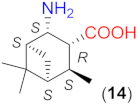
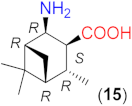
Due to the above observations, we set out to study the effect of the structure of other β-amino acids with cyclohexane backbone. Although a relatively good ee value was obtained with the use of the Li salt of 8 (82%), it did not reach those afforded by the more β-hindered compounds 6 and 7. Thus, we focused on 2-aminocyclohexane-1-carboxylic acids bearing additional substituents on the ring to increase the enantioselectivity. The preparation of substituted cycloaliphatic β-amino acid series has been reported using monoterpenes as chiral, easily available natural starting materials [62,63,64,65]. Thus, stereoisomers of 2-amino-4(S)-isopropylcyclohexane-1-carboxylic acid (9, 10) were obtained from S-perillaldehyde [65], whereas bicyclic stereoisomers could be prepared from apopinenes (11–13) [63,64] and cis-δ-pinenes (14, 15) [62]. These compounds were stored and used as hydrochloride salts. Consequently, larger amounts of the base were needed for their in situ transformation to lithium carboxylates. Based on the results obtained in reactions catalysed by 8, the solvent amount was decreased to 0.5 cm3. Selected results achieved with these β-amino acids are also presented in Table 3.
The compound having the COOH and the NH2 groups in trans arrangement (9) afforded low enantioselectivity, which increased in the presence of the base to moderate values (entries 6–8). Although the configurations of the C1 and C2 chiral centres in 8 and 9 were the same, the inversion observed with 8 was not detected when 9 was applied. Thus, without the addition of LiOH × H2O, the S enantiomer formed in excess. The stereoisomer with the cis arrangement of the functional groups (10) afforded a high conversion and ee when LiOH × H2O was added to the reaction slurry (entries 10, 11). The R product enantiomer was formed in excess with this compound. Accordingly, the configuration of the C2 atom determined the sense of the enantioselectivity, whereas that of the C1 did not influence the ee direction. Since 8 and 9 provided significantly lower ee values than 10, the good performance obtained with the latter compound may be attributed to the close spatial arrangement of the cis functional groups.
To further test the effect of the substitution pattern of the cycloaliphatic β-amino acids, next we used bicyclic derivatives 11–15 (see Table 3). Compound 11 having trans orientation of the NH2 and COOH groups showed a similar inversion of the sense of the enantioselectivity in the absence of LiOH × H2O, as observed with 8 (entries 12–16).
Comparing the performances reached with the three compounds having S-C1 and S-C2 configurations (8, 9 and 11), one can see that the enantioselectivity direction in the absence of base varies. Thus, under these conditions, the effect of the configurations of these centres may be overwritten by other geometrical characteristics of the molecule. The in situ formation of the lithium salt of 11 by the addition of LiOH × H2O resulted in a product mixture in which the S adduct was in slight excess. Accordingly, using the lithium salts, the configuration of the centre bearing the amino group (C2) determines the sense of enantioselectivity, as mentioned before. Compounds 12 and 13 with the functional groups in cis arrangements offered low conversion and ee values in the absence of base (entries 17, 20) and both values increased significantly by the addition of LiOH × H2O (entries 18, 19 and 21, 22). These two enantiomeric amino acids provided the opposite Michael adduct enantiomers in excess, in accordance with the configuration of the C2 chiral centre. The other β-amino acid enantiomeric pair (14 and 15) having both cis NH2 and COOH groups also allowed the formation of the opposite product enantiomers in excess, affording high conversion and ee values, when they were transformed to lithium salts. Interestingly, these derivatives, although bearing an additional methyl group near the carboxylate moiety, provided better conversions in one-day reactions than 13, which has a similar structure, but lacks the methyl group bonded to C6.
Finally, we examined the effect of the nitrogen substituent of maleimide on the results obtained with β-amino acids 15 and 14, which provided high conversion and ee values at rt in 24 h in the addition of 2 to 1a (Table 3, entries 24, 26). The results found with other maleimide derivatives (1b–1f) are presented in Table 4. A decrease in the size of the substituent, i.e., a change to methyl group (1b), afforded enantioselectivities similar to those obtained with the N-benzyl derivative 1a, however with slightly decreased conversions (entries 1, 2). An increased conversion was obtained with the α-branched isopropyl derivative (1c). Moreover, in reactions of N-phenyl or its ring-substituted derivatives (1d–1f), complete transformations and similar ee values were attained, even when the CF3 substituent was in the ortho position. This functional group may have a hindering effect, as observed previously with l-Phe catalyst adsorbed on an inorganic oxide surface [56].
Table 4.
Asymmetric Michael addition of 2 to N-substituted maleimides catalysed by β-amino acids 15 a or 14 b.
3. Discussion
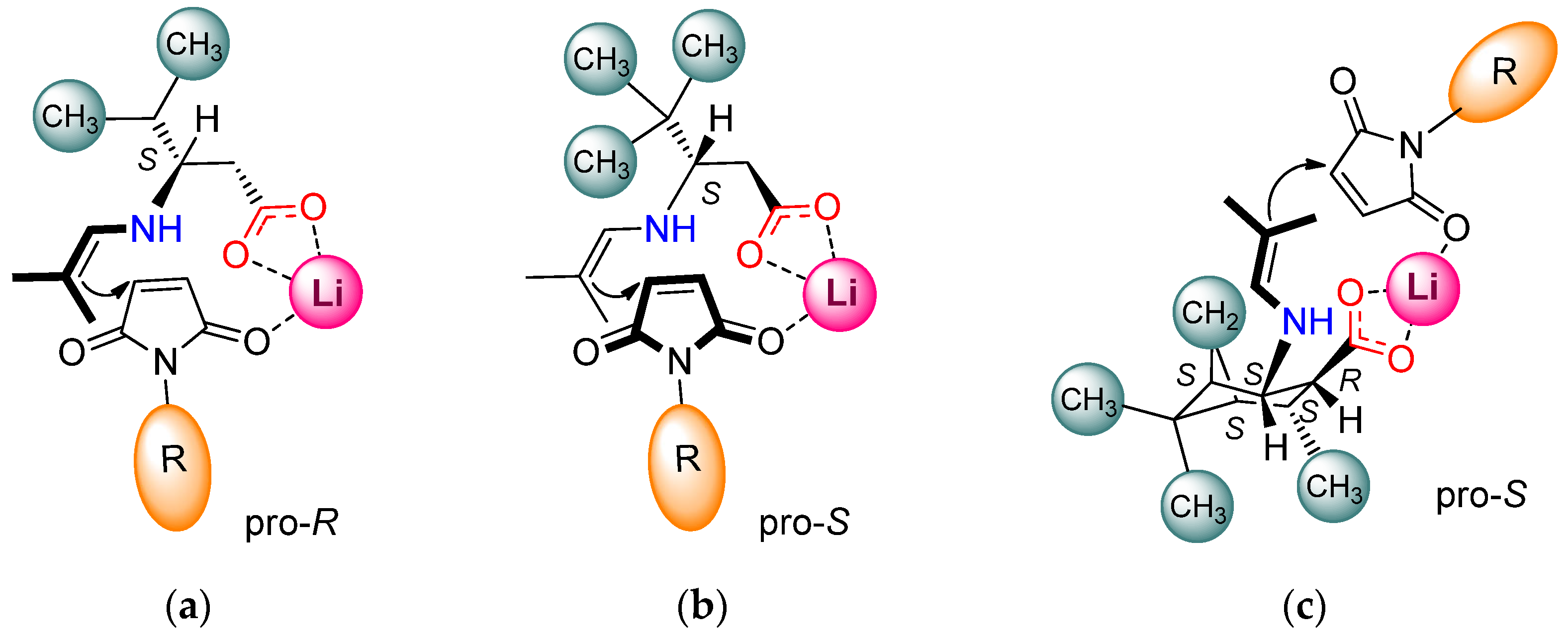
In the present study, exploring the effect of the structure of chiral β-amino acids applied as catalysts in the asymmetric addition of isobutyraldehyde to N-benzylmaleimide, several remarkable results were observed. For interpreting these observations, we recall that the conjugate additions of aldehydes and ketones to maleimides catalysed by primary amine–hydrogen-bond donor groups containing bifunctional organocatalysts occur through the formation of an enamine intermediate [25,31,32,34,39,40,41,42,43,45]. The maleimide is activated by hydrogen bonding, whereas the stereochemical outcome of the reaction is determined by the preferential direction of the approach of the enamine found in the proximity of the chiral centre to the activated electrophile. In reactions catalysed by salts of primary amino acids, the maleimide interacts via electrostatic forces or by hydrogen bonding with other additives (sulfamides, thioureas, surface Brønsted or Lewis acidic sites) [49,50,54,55,56]. During catalysis by β-amino acid salts, similar transition states are likely to operate, as envisaged by Kokotos [50]. The opposite sense of enantioselectivities obtained using 6 compared to 7 may be due to the significant steric hindrance of the tert-butyl group, which may have a strong shielding effect. The bulkiness of this group may change the orientation of the carboxylate anion compared to the smaller substituents (iPr), as illustrated in Figure 2a,b, leading to a lower reaction rate and inversion of the enantioselectivity.
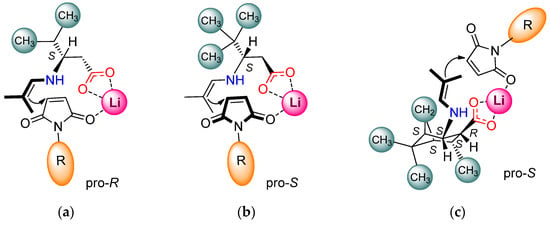
Figure 2.
Proposed transition states formed in the asymmetric addition of 2 to N-substituted maleimides catalysed by β-amino acids in the presence of LiOH × H2O: (a) 6; (b) 7; (c) 14.
β-Amino acids with cyclohexane framework with the COOH and NH2 groups in the 1,2-positions following their in situ transformation to lithium salts showed that the configuration of the excess enantiomer is determined by the chirality of C2 (i.e., β-carbon), both when the functional groups are in trans or cis arrangement. However, the ee values were low to moderate with the trans derivatives (up to 82% with 8), with a strongly detrimental effect of additional substituents on the ring (compare 8, 9, 11). In contrast, the cis stereoisomers (10, 12, 13, 14, 15) provided high ee values (88–95%). Moreover, a comparison of the performances of the identically substituted stereoisomers (9 vs. 10; 11 vs. 12 or 13) showed that the cis isomers also afford better reaction rates.
A plausible reason of this behaviour is the closer spatial arrangement of the cis functional groups compared to that of the trans, leading to a more directed and efficient interaction of the enamine and the activated electrophile. Additional substituents on the cyclohexane scaffold had a limited influence on the stereoselectivity, having a more significant effect on the rate, probably affecting the formation of the enamine intermediates. Thus, the bicyclic structure of 13 or 12, presumably due to branching on C3 (next to the amine group), allowed slower reactions compared to the 4-iPr-substituted 10, similar to the effect of γ-branching in 6. However, the trans-methyl group substitution on C6 (next to the carboxylate group, 14 and 15) increased the rate compared to 12 or 13, attributable to the beneficial steric constraints of the opposite side of the molecule compared to the position of the functional groups (Figure 2c), thus contributing to the favourable positioning of the activated electrophile. The nitrogen substituent of maleimide also had a small steric effect on the rate of the additions, as indicated by comparing the reactions of maleimides substituted with Me, Bn, iPr, 2-CF3-Ph, 4-F-Ph and Ph groups, which provided increasing conversions in this order.
Finally, an interesting inversion of the sense of enantioselectivity was noted with two trans-cyclohexane-2-amino-1-carboxylic acids in the absence of the base additive. This change in the configuration of the excess enantiomer may be caused by the stronger attachment of the electrophile to the catalysts. This, in combination with the less efficient arrangement of the functional groups (trans), leads to the preferential attack at the opposite enantioface of the electrophile compared to that interacting with the lithium cation. It must be noted that this effect is strongly affected by the substitution pattern of the cyclohexane ring, as the unsubstituted 8 and the heavily substituted 11 offered inversions, in contrast to the C4-substituted 9 derivative.
4. Materials and Methods
The optically pure β-amino acids were either commercial products used as received (4–8) or prepared in previous studies (9–15) and stored and used as hydrochlorides [60,61,62,63]. Isobutyraldehyde (2) and N-substituted maleimides 1a, 1b and 1d were purchased from commercial sources (Sigma-Aldrich, St. Louis, MO, USA). Maleimides 1c, 1e and 1f were prepared during our previous study [56]. Solvents of analytical grade and high-purity additives were commercial products and used as received (Merck KGaA, Darmstadt, Germany).
Products formed in catalytic reactions were analysed by gas chromatography using Agilent Techn. 6890 N GC-5973 MSD (GC-MSD, Agilent Co., Santa Clara, CA, USA) equipped with a 30 m long DB-1MS UI (J&W, Agilent) capillary column for mass spectrometric identification of the compounds and Agilent 7890A GC-FID (GC-FID, Agilent Co., Santa Clara, CA, USA) equipped with chiral capillary columns (Cyclosil-B, 30 m × 0.25 mm, J&W, Agilent or Hydrodex g-TBDAc, 25 m × 0.25 mm, Macherey-Nagel) for quantitative analysis. The adducts were purified by flash chromatography using silica gel 60, 40–63 μm. 1H- and 13C-NMR spectra were recorded on a Bruker DRX-500 spectrometer (Bruker Corp., Millerica, MA, USA) at 500 (1H) and 125 (13C) MHz in CDCl3 solvent.
Asymmetric Michael additions were carried out in 4 cm3 closed glass vials using magnetic agitation (800 rpm). In a typical run, 0.03 mmol β-amino acid was suspended by stirring in the solvent (EtOAc) followed by the addition of LiOH × H2O. After 10 min stirring, 0.3 mmol N-substituted maleimide was added. Reactions were started by the addition of 1.2 mmol isobutyraldehyde. Reactions at 50 °C were carried out by immersing the vials in a preheated oil bath. After the desired reaction time, the mixture was diluted to 3 cm3 with EtOAc, the catalyst was extracted into 1 cm3 saturated NH4Cl(aq) solution, the aqueous phase was washed twice with 1 cm3 of EtOAc, and the unified organic phases were dried over MgSO4 and analysed by gas chromatography using n-decane as internal standard. The conversion and the enantiomeric excess (ee) values were calculated using the results of the GC-FID analysis as shown previously [56]. The absolute configuration of the excess enantiomer was assigned based on our previous report [56]. The raw product was obtained by evaporating the solvent and the unreacted aldehyde and purified further by flash chromatography using hexane (mixture of isomers)/EtOAc or hexane (mixture of isomers)/tBuOMe and characterised as given below and in our recent report. Chromatograms and spectra of the products were in agreement with the published data [56].
(R)-2-(1-Benzyl-2,5-Dioxopyrrolidin-3-yl)-2-Methylpropanal (3a).Table 2, entry 2, eluted with hexane (mixture of isomers)/EtOAc 4/1, white solid, 71.8 mg (yield 91%), enantiomeric ratio 97.5(R)/2.5(S); GC-MSD m/z(rel int) = 259(M+, 1), 231(100), 216(48), 189(13), 138(30), 106(21), 91(80), 83(16), 69(30), 55(8), 41(13); GC-FID, Cyclosil-B column, retention times (min) = 55.6 (1a), 174.3 (R-3a), 179.6 (S-3a). 1H NMR (500 MHz, CDCl3) δ (ppm) = 1.16 (s, 6H), 2.45 (dd, J = 5.4, 18.3 Hz, 1H), 2.81 (dd, J = 9.4, 18.3 Hz, 1H), 3.03 (dd, J = 5.4, 9.4 Hz, 1H), 4.65 (q, J = 14.1 Hz, 2H), 7.24-7.33 (m, 3H), 7.36 (d, J = 6.9 Hz, 2H), 9.49 (s, 1H). 13C NMR (125 MHz, CDCl3) δ (ppm) = 19.1, 19.9, 31.4, 42.4, 44.9, 48.0, 127.9, 128.6, 128.7, 135.6, 175.3, 177.4, 202.6.
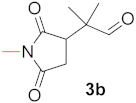
(R)-2-Methyl-2-(1-Methyl-2,5-Dioxopyrrolidin-3-yl)Propanal (3b).Table 4, entry 1, eluted with hexane (mixture of isomers)/EtOAc 5/1, pale yellow oil, 46.7 mg (yield 85%), enantiomeric ratio 97.5(R)/2.5(S); GC-MSD m/z(rel int) = 183(M+, 2), 155(41), 140(66), 113(61), 83(30), 69(100), 55(13), 41(33); GC-FID, Hydrodex g-TBDAc column, retention times (min) = 12.9 (1b), 92.1 (R-3b), 92.9 (S-3b). 1H NMR (500 MHz, CDCl3) δ (ppm) = 1.17 (d, J = 5.2 Hz, 6H), 2.42 (dd, J = 5.4, 18.3 Hz, 1H), 2.78 (dd, J = 9.3, 18.3 Hz, 1H), 2.94 (s, 3H), 3.01 (dd, J = 5.4, 9.3 Hz, 1H), 9.47 (s, 1H). 13C NMR (125 MHz, CDCl3) δ (ppm) = 19.2, 20.0, 24.8, 31.4, 45.0, 47.9, 175.8, 177.8, 202.7.
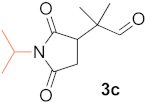
(R)-2-(1-Isopropyl-2,5-Dioxopyrrolidin-3-yl)-2-Methylpropanal (3c).Table 4, entry 3, eluted with hexane (mixture of isomers)/tBuOMe 2/1, transparent oil, 57.0 mg (yield 90%), enantiomeric ratio 98(R)/2(S); GC-MSD m/z(rel int) = 211(M+, 4), 183(39), 168(41), 141(57), 126(53), 99(23), 83(23), 69(100), 55(16), 41(31); GC-FID, Cyclosil-B column, retention times (min) = 17.0 (1c), 105.2 (R-3c), 107.8 (S-3c). 1H NMR (500 MHz, CDCl3) δ (ppm) = 1.18 (d, J = 2.1 Hz, 6H), 1.37 (d, J = 6.95 Hz, 6H), 2.38 (dd, J = 5.3, 18.2 Hz, 1H), 2.74 (dd, J = 9.5, 18.2 Hz, 1H), 2.96 (dd, J = 5.3, 9.5 Hz, 1H), 4.37 (h, J = 6.95 Hz, 1H), 9.52 (s, 1H). 13C NMR (125 MHz, CDCl3) δ (ppm) = 18.8, 19.1, 19.1, 20.0, 31.3, 43.9, 44.7, 48.0, 175.6, 177.7, 202.7.
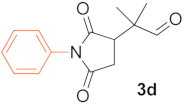
(R)-2-(1-Phenyl-2,5-Dioxopyrrolidin-3-yl)-2-Methylpropanal (3d). Table 4, entry 4, eluted with hexane (mixture of isomers)/EtOAc 10/1, white solid, 68.4 mg (yield 93%), enantiomeric ratio 99(R)/1(S); GC-MSD m/z(rel int) = 245(M+, 1), 217(75), 202 (100), 175(13), 147(18), 119(18), 93(40), 83(52), 69(33), 55(12), 41(23); GC-FID, Cyclosil-B column, retention times (min) = 54.0 (1d), 198.5 (R-3d), 200.5 (S-3d). 1H NMR (500 MHz, CDCl3) δ (ppm) = 1.30 (d, J = 16.0 Hz, 6H), 2.62 (dd, J = 5.5, 18.3 Hz, 1H), 2.97 (dd, J = 9.6, 18.3 Hz, 1H), 3.15 (dd, J = 5.5, 9.6 Hz, 1H), 7.27 (d, J = 7.4 Hz, 2H), 7.39 (tr, J = 7.4 Hz, 1H), 7.47 (tr, J = 7.4 Hz, 2H), 9.52 (s, 1H). 13C NMR (125 MHz, CDCl3) δ (ppm) = 19.6, 20.2, 31.8, 45.0, 48.5, 126.5, 128.7, 129.1, 131.8, 174.7, 176.8, 202.7.
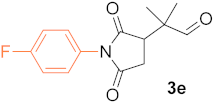
(R)-2-(4-Fluorophenyl-2,5-Dioxopyrrolidin-3-yl)-2-Methylpropanal (3e). Table 4, entry 6, eluted with hexane (mixture of isomers)/tBuOMe 1/1.5, off-white solid, 72.7 mg (yield 92%), enantiomeric ratio 98(R)/2(S); GC-MSD m/z(rel int) = 263(M+, 3), 235(65), 220 (100), 193(14), 165(23), 137(18), 111(33), 83(60), 69(29), 55(11), 41(17); GC-FID, Cyclosil-B column, retention times (min) = 55.0 (1e), 194.0 (R-3e), 196.5 (S-3e). 1H NMR (500 MHz, CDCl3) δ (ppm) = 1.32 (d, J = 33.0 Hz, 6H),), 2.62 (dd, J = 5.6, 18.3 Hz, 1H), 2.97 (dd, J = 9.6, 18.3 Hz, 1H), 3.11 (dd, J = 5.6, 9.6 Hz, 1H), 7.15 (tr, J = 8.4 Hz, 2H), 7.27 (dd, J = 4.9, 8.4 Hz, 2H), 9.50 (s, 1H). 13C NMR (125 MHz, CDCl3) δ (ppm) = 19.9, 20.4, 31.9, 44.9, 48.6, 116.1, 116.3, 127.7, 128.3, 128.4, 161.3, 163.3, 174.6, 176.8, 202.7.
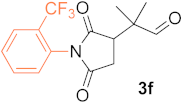
(R)-2-((2-Trifluoromethyl)phenyl)-2,5-Dioxopyrrolidin-3-yl)-2-Methylpropanal (3f). Table 4, entry 7, eluted with hexane (mixture of isomers)/EtOAc 4/1, off-white solid, 87.4 mg (yield 93%), enantiomeric ratio 97.5(R)/2.5(S); GC-MSD m/z(rel int) = 313(M+, 1), 285(84), 270(28), 250(100), 222(29), 168(21), 83(97), 69(98), 55(21), 41(37); GC-FID, Cyclosil-B column, retention times (min) = 39.0 (1f), 104.5 (R-3f), 106.0 (S-3f). 1H NMR (500 MHz, CDCl3), δ (ppm) = 1.22-1.34 (m, J = 56.8, 64.1 Hz 6H), 2.60-2.73 (m, J = 6.7, 18.3, 5.1, 18.0 Hz, 1H), 3.01 (m, J = 9.8, 18.0 Hz, 1H), 3.07-3.42 (m, 1H), 7.17-7.36 (m, J = 7.8 Hz, 1H), 7.58 (m, 1H), 7.68 (dd, J = 7.5, 14.8 Hz, 1H), 7.79 (tr, J = 7.0 Hz, 1H), 9.44-9.60 (m, 1H). 13C NMR (125 MHz, CDCl3) δ (ppm) = 18.9, 19.4, 20.5, 20.8, 29.7, 31.7, 32.5, 45.2, 45.9, 127.4, 127.5, 127.6, 130.0, 130.1, 130.8, 133.3, 174.3, 176.1, 176.7, 202.7.
5. Conclusions
We carried out a study exploring the effect of the structure of the β-amino acid organocatalyst used in the asymmetric Michael addition of isobutyraldehyde to N-substituted maleimides. The rate of the addition of the aldehyde to N-benzylmaleimide decreases, whereas the enantiomeric excess increases, as the bulkiness of the substituent on the β-carbon increases. Moreover, large groups, such as tert-butyl, in this position may cause an inversion of the sense of enantioselectivity. The results, obtained with various substituted 2-aminocyclohexane-1-carboxylic acids prepared from commercially available natural terpenes and transformed in situ into lithium salts, indicate that the configuration at the β-carbon atom determines the sense of the enantioselectivity. However, the cis spatial arrangement of the functional groups is necessary for obtaining high rates and enantiomeric excesses. Other substituents on the cyclohexane scaffold influenced the rate of the addition and had a small effect on the ee values as well. In contrast, depending on substituents on the ring, the trans isomers provided the opposite enantiomers in excess in the absence of the base additive compared to that obtained with the lithium salts. The most efficient cyclic β-amino acids were also used in addition of isobutyraldehyde to several maleimides, revealing a minor effect of the nitrogen substituent of the maleimide on the rate. Based on the experimental results, we proposed possible transition state structures, which explain the yet undisclosed inversions of the enantioselectivity either caused by bulky substituents or triggered by the lack of the base additive.
Author Contributions
Conceptualisation, G.S. and Z.S.; methodology, V.K.; validation, V.K. and G.S.; formal analysis, V.K. and G.S.; investigation, V.K.; resources, G.S. and Z.S.; data curation, V.K. and G.S.; writing—original draft preparation, G.S.; writing—review and editing, V.K., Z.S. and G.S.; visualisation, V.K. and G.S.; supervision, G.S.; project administration, G.S. and Z.S.; funding acquisition, V.K., G.S. and Z.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Hungarian Science Foundation, grant number OTKA K 138871.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Šunjić, V.; Parnham, M.J. Signposts to Chiral Drugs, Organic Synthesis in Action; Springer Basel AG: Basel, Switzerland, 2011. [Google Scholar] [CrossRef]
- Lin, G.-Q.; You, Q.-D.; Cheng, J.-F. (Eds.) Chiral Drugs, Chemistry and Biological Action; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011. [Google Scholar]
- Busacca, C.A.; Fandrick, D.R.; Song, J.J.; Senanayake, C.H. The Growing Impact of Catalysis in the Pharmaceutical Industry. Adv. Synth. Catal. 2011, 3535, 1825–1864. [Google Scholar] [CrossRef]
- Nag, A. (Ed.) Asymmetric Synthesis of Drugs and Natural Products; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2018. [Google Scholar]
- Mikami, K.; Lautens, M. (Eds.) New Frontiers in Asymmetric Catalysis; John Wiley & Sons: Hoboken, NJ, USA, 2007. [Google Scholar]
- Ojima, I. (Ed.) Catalytic Asymmetric Synthesis, 3rd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- Gruttadauria, M.; Giacalone, F. (Eds.) Catalytic Methods in Asymmetric Synthesis: Advanced Materials, Techniques and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Szőllősi, G. Asymmetric One-Pot Reactions Using Heterogeneous Chemical Catalysis: Recent Steps Towards Sustainable Processes. Catal. Sci. Technol. 2018, 8, 389–422. [Google Scholar] [CrossRef]
- Pellissier, H. Asymmetric Metal Catalysis in Enantioselective Domino Reactions; Wiley-VCH Verlag: Weinheim, Germany, 2019. [Google Scholar]
- Pellissier, H. Recent Developments in Enantioselective Multicatalyzed Tandem Reactions. Adv. Synth. Catal. 2020, 362, 2289–2325. [Google Scholar] [CrossRef]
- Pellissier, H. Recent Developments in Asymmetric Organocatalysis; RSC Catalysis Series No. 3; RSC Publishing: Cambridge, UK, 2010. [Google Scholar]
- Giacalone, F.; Gruttadauria, M.; Agrigento, P.; Noto, R. Low-loading Asymmetric Organocatalysis. Chem. Soc. Rev. 2012, 41, 2406–2447. [Google Scholar] [CrossRef] [PubMed]
- Dalko, P.I. (Ed.) Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications; Wiley-VCH: Weinheim, Germany, 2013; Volumes 1–3. [Google Scholar]
- Xiao, X.; Shao, B.-X.; Lu, Y.-J.; Cao, Q.-Q.; Xia, C.-N.; Chen, F.-E. Recent Advances in Asymmetric Organomulticatalysis. Adv. Synth. Catal. 2021, 363, 352–387. [Google Scholar] [CrossRef]
- Juaristi, E. Recent Developments in Next Generation (S)-Proline-derived Chiral Organocatalysts. Tetrahedron 2021, 88, 132143. [Google Scholar] [CrossRef]
- Córdova, A. (Ed.) Catalytic Asymmetric Conjugate Reactions; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- Vicario, J.L.; Badia, D.; Carrillo, L.; Reyes, E. (Eds.) Organocatalytic Enantioselective Conjugate Addition Reactions, A Powerful Tool for the Stereocontrolled Synthesis of Complex Molecules; RSC Catalysis Series No. 5; RSC Publishing: Cambridge, UK, 2010. [Google Scholar]
- Namboothiri, I.N.N.; Bhati, M.; Ganesh, M.; Hosamani, B.; Bajiu, T.V.; Manchery, S.; Bera, K. Catalytic Asymmetric Reactions of Conjugated Nitroalkenes; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2020. [Google Scholar]
- Crider, A.M.; Kolczynski, T.M.; Yates, K.M. Synthesis and Anticancer Activity of Nitrosourea Derivatives of Phensuximide. J. Med. Chem. 1980, 23, 324–326. [Google Scholar] [CrossRef]
- Fredenhagen, A.; Tamura, S.Y.; Kenny, P.T.M.; Komura, H.; Naya, Y.; Nakanishi, K.; Nishiyama, K.; Sugiura, M.; Kita, H. Andrimid, a New Peptide Antibiotic Produced by an Intracellular Bacterial Symbiont Isolated from a Brown Planthopper. J. Am. Chem. Soc. 1987, 109, 4409–4411. [Google Scholar] [CrossRef]
- Curtin, M.L.; Garland, R.B.; Heyman, H.R.; Frey, R.R.; Michaelides, M.R.; Li, J.; Pease, L.J.; Glaser, K.B.; Marcotte, P.A.; Davidsen, S.K. Succinimide Hydroxamic Acids as Potent Inhibitors of Histone Deacetylase (HDAC). Bioorg. Med. Chem. Lett. 2002, 12, 2919–2923. [Google Scholar] [CrossRef]
- Freiberg, C.; Brunner, N.A.; Schiffer, G.; Lampe, T.; Pohlmann, J.; Brands, M.; Raabe, M.; Häbich, D.; Ziegelbauer, K. Identification and Characterization of the First Class of Potent Bacterial Acetyl-CoA Carboxylase Inhibitors with Antibacterial Activity. J. Biol. Chem. 2004, 279, 26066–26073. [Google Scholar] [CrossRef]
- Isaka, M.; Rugseree, N.; Maithip, P.; Kongsaeree, P.; Prabpai, S.; Thebtaranonth, Y. Hirsutellones A–E, Antimycobacterial Alkaloids from the Insect Pathogenic Fungus Hirsutella nivea BBC 2594. Tetrahedron 2005, 61, 5577–5583. [Google Scholar] [CrossRef]
- Uddin, J.; Ueda, K.; Siwu, E.R.O.; Kita, M.; Uemura, D. Cytotoxic Labdane Alkaloids from an Ascidian Lissoclinum sp.: Isolation, Structure Elucidation, and Structure-Activity Relationship. Bioorg. Med. Chem. 2006, 14, 6954–6961. [Google Scholar] [CrossRef] [PubMed]
- Chauchan, P.; Kaur, J.; Chimni, S.S. Asymmetric Organocatalytic Addition Reactions of Maleimides: A Promising Approach Towards the Synthesis of Chiral Succinimide Derivatives. Chem. Asian J. 2013, 8, 328–346. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, G.; Bosco, M.; Carlone, A.; Cavalli, A.; Locatelli, M.; Mazzanti, A.; Ricci, P.; Sambri, L.; Melchiorre, P. Organocatalytic Asymmetric Conjugate Addition of 1,3-Dicarbonyl Compounds to Maleimides. Angew. Chem. Int. Ed. 2006, 45, 4966–4970. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yi, W.-B.; Ahad, D.; Zhang, W. Recyclable Cinchona Alkaloid Catalyzed Asymmetric Michael Addition Reaction. Tetrahedron Lett. 2013, 54, 6064–6066. [Google Scholar] [CrossRef]
- Mahajan, S.; Chauhan, P.; Kumar, A.; Chimni, S.S. Organocatalytic Enantioselective Synthesis of N-Alkyl/Aryl-3-Alkylpyrrolidine-2,5-dione in Brine. Tetrahedron Asymmetry 2016, 27, 1145–1152. [Google Scholar] [CrossRef]
- Zhao, G.-L.; Xu, Y.; Sundén, H.; Eriksson, L.; Sayah, M.; Córdova, A. Organocatalytic Enantioselective Conjugate Addition of Aldehydes to Maleimides. Chem. Commun. 2007, 734–735. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, M.-M.; Zhang, S.; Xu, Z.-A.; Li, H.; Yu, X.-H.; Wang, W. Chiral Pyrrolidine Sulfonamide Catalyzed Enantioselective Michael Addition of Cyclohexanones to Maleimides. Synlett 2011, 2011, 473–476. [Google Scholar] [CrossRef]
- Yu, F.; Sun, X.; Jin, Z.; Wen, S.; Liang, X.; Xe, J. Enantioselective Michael Addition of Ketones to Maleimides Catalyzed by Bifunctional Monosulfonyl DPEN Salt. Chem. Commun. 2010, 46, 4589–4591. [Google Scholar] [CrossRef]
- Yu, F.; Jin, Z.; Huang, H.; Ye, T.; Liang, X.; Ye, J. A Highly Efficient Asymmetric Michael Addition of α,α-Disubstituted Aldehydes Catalysed by Primary Amine Thiourea Salt. Org. Biomol. Chem. 2010, 8, 4767–4774. [Google Scholar] [CrossRef]
- Xue, F.; Liu, L.; Zhang, S.; Duan, W.; Wang, W. A Simple Primary Amine Thiourea Catalyzed Highly Enantioselective Conjugate Addition of α,α-Disubstituted Aldehydes to Maleimides. Chem. Eur. J. 2010, 16, 7979–7982. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.-F.; Peng, L.; Wang, L.-L.; Wang, L.-X.; Xu, X.-Y. Chiral Primary Amine Thiourea Promoted Highly Enantioselective Michael Reactions of Isobutyraldehyde with Maleimides. Tetrahedron 2010, 66, 8928–8932. [Google Scholar] [CrossRef]
- Miura, T.; Nishida, S.; Masuda, A.; Tada, N.; Itoh, A. Asymmetric Michael Additions of Aldehydes to Maleimides Using a Recyclable Fluorous Thiourea Organocatalyst. Tetrahedron Lett. 2011, 52, 4158–4160. [Google Scholar] [CrossRef]
- Gómez-Torres, E.; Alonso, D.A.; Gómez-Bengoa, E.; Nájera, C. Conjugate Addition of 1,3-Dicarbonyl Compounds to Maleimides Using a Chiral C2-Symmetric Bis(2-aminobenzimidazole) as Recyclable Organocatalyst. Org. Lett. 2011, 13, 6106–6109. [Google Scholar] [CrossRef]
- Avila, A.; Chinchilla, R.; Nájera, C. Enantioselective Michael Addition of α,α-Disubstituted Aldehydes to Maleimides Organocatalyzed by Chiral Primary Amine-Guanidines. Tetrahedron Asymmetry 2012, 23, 1625–1627. [Google Scholar] [CrossRef]
- Gómez-Torres, E.; Alonso, D.A.; Gómez-Bengoa, E.; Nájera, C. Enantioselective Synthesis of Succinimides by Michael Addition of 1,3-Dicarbonyl Compounds to Maleimides Catalyzed by a Chiral Bis(2-aminobenzimidazole) Organocatalyst. Eur. J. Org. Chem. 2013, 1434–1440. [Google Scholar] [CrossRef]
- Avila, A.; Chinchilla, R.; Gómez-Bengoa, E.; Nájera, C. Enantioselective Synthesis of Succinimides by Michael Addition of Aldehydes to Maleimides Organocatalyzed by Primary Amine-Guanidines. Eur. J. Org. Chem. 2013, 2013, 5085–5092. [Google Scholar] [CrossRef]
- Nakashima, K.; Kawada, M.; Hirashima, S.; Kato, M.; Koseki, Y.; Miura, T. Asymmetric Conjugate Addition of Ketones to Maleimides Using Diaminomethyleneindenedione Organocatalyst. Synlett 2015, 26, 1248–1252. [Google Scholar] [CrossRef]
- Vízcaíno-Milla, P.; Sansano, J.M.; Nájera, C.; Fiser, B.; Gómez-Bengoa, E. Primary Amine–2-Aminopyrimidine Chiral Organocatalysts for the Enantioselective Conjugate Addition of Branched Aldehydes to Maleimides. Synthesis 2015, 47, 2199–2206. [Google Scholar] [CrossRef]
- Nakashima, K.; Kawada, M.; Hirashima, S.; Kosugi, A.; Kato, M.; Yoshida, A.; Koseki, Y.; Miura, T. Stereoselective Conjugate Addition of Carbonyl Compounds to Maleimides Using a Diaminomethyleneindenedione Organocatalyst. Tetrahedron Asymmetry 2016, 27, 888–895. [Google Scholar] [CrossRef]
- Torregrosa-Chinillach, A.; Moragues, A.; Pérez-Furundarena, H.; Chinchilla, R.; Gómez-Bengoa, E.; Guillena, G. Enantioselective Michael Addition of Aldehydes to Maleimides Organocatalyzed by a Chiral Primary Amine-Salicylamide. Molecules 2018, 23, 3299. [Google Scholar] [CrossRef] [PubMed]
- Szőllősi, G.; Kozma, V. Design of Heterogeneous Organocatalyst for the Asymmetric Michael Addition of Aldehydes to Maleimides. ChemCatChem 2018, 10, 4362–4368. [Google Scholar] [CrossRef]
- Kozma, V.; Fülöp, F.; Szőllősi, G. 1,2-Diamine-Derived (thio)Phosphoramide Organocatalysts in Asymmetric Michael Additions. Adv. Synth. Catal. 2020, 362, 2444–2458. [Google Scholar] [CrossRef]
- Kozma, V.; Szőllősi, G. Conjugate Addition of 1,3-Dicarbonyl Compounds to Maleimides Using Bifunctional Primary Amin–(thio)Phosphoramide Organocatalysts. Mol. Catal. 2022, 518, 112089. [Google Scholar] [CrossRef]
- Grünenfelder, C.E.; Kisunzu, J.K.; Wennemers, H. Peptide-Catalyzed Stereoselective Conjugate Addition Reactions of Aldehydes to Maleimides. Angew. Chem. Int. Ed. 2016, 55, 8571–8574. [Google Scholar] [CrossRef] [PubMed]
- Avila-Ortiz, C.G.; Díaz-Corona, L.; Jiménez-González, E.; Juraisti, E. Asymmetric Michael Addition Organocatalyzed by α,β-Dipeptides under Solvent-Free Reaction Conditions. Molecules 2017, 22, 1328. [Google Scholar] [CrossRef]
- Nugent, T.C.; Sadiq, A.; Bibi, A.; Heine, T.; Zeonjuk, L.L.; Vankova, N.; Bassil, B.S. Noncovalent Bifunctional Organocatalysts: Powerful Tools for Contiguous Quaternary-Tertiary Stereogenic Carbon Formation, Scope, and Origin of Enantioselectivity. Chem. Eur. J. 2012, 18, 4088–4098. [Google Scholar] [CrossRef]
- Kokotos, C.G. An Asymmetric Michael Addition of α,α-Disubstituted Aldehydes to Maleimides Leading to a One-Pot Enantioselective Synthesis of Lactones Catalysed by Amino Acids. Org. Lett. 2013, 15, 2406–2409. [Google Scholar] [CrossRef]
- Schiza, A.; Spiliopoulou, N.; Shahu, A.; Kokotos, C.G. Combining Organocatalysis with Photoorganocatalysis: Photocatalytic Hydroacylation of Asymmetric Organocatalytic Michael Addition Products. New J. Chem. 2018, 42, 18844–18849. [Google Scholar] [CrossRef]
- Sadiq, A.; Nugent, T.C. Catalytic Access to Succinimide Products Containing Stereogenic Quaternary Carbons. ChemistrySelect 2020, 5, 11934–11938. [Google Scholar] [CrossRef]
- Ahmad, S.; Mahnashi, M.H.; Alyami, B.A.; Alqahtani, T.S.; Ullah, F.; Ayaz, M.; Tariq, M.; Sadiq, A.; Rashid, U. Synthesis of Michael Adducts as Key Building Blocks for Potential Analgesic Drugs: In vitro, in vivo and in silico Explorations. Drug Des. Dev. Ther. 2021, 15, 1299–1313. [Google Scholar] [CrossRef]
- Muramulla, S.; Ma, J.-A.; Zhao, J.C.-G. Michael Addition of Ketones and Aldehydes to Maleimides Catalysed by Modularly Designed Organocatalysts. Adv. Synth. Catal. 2013, 355, 1260–1264. [Google Scholar] [CrossRef]
- Landeros, J.M.; Cruz-Hernández, C.; Juaristi, E. α-Amino Acids and α,β-Dipeptides Intercalated into Hydrotalcite: Efficient Catalysts in the Asymmetric Michael Addition Reaction of Aldehydes to N-Substituted Maleimides. Eur. J. Org. Chem. 2021, 2021, 5117–5126. [Google Scholar] [CrossRef]
- Kozma, V.; Szőllősi, G. Enantioselective Michael Addition of Aldehydes to Maleimides Catalysed by Surface-Adsorbed Natural Amino Acids. Catal. Sci. Technol. 2022, 12, 4709–4726. [Google Scholar] [CrossRef]
- Zhang, H.; Mifsud, M.; Tanaka, F.; Barbas, C.F., III. 3-Pyrrolidinecarboxylic Acid for Direct Catalytic Asymmetric anti-Mannich-Type Reactions of Unmodified Ketones. J. Am. Chem. Soc. 2006, 128, 9630–9631. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wong, M.W. β-Amino Acid Catalyzed Asymmetric Michael Additions: Design of Organocatalysts with Catalytic Acid/Base Dyad Inspired by Serine Proteases. J. Org. Chem. 2011, 76, 7399–7405. [Google Scholar] [CrossRef]
- Konda, S.; Zhao, J.C.-G. Enantioselective anti-Mannich Reaction Catalysed by Modularly Designed Organocatalysts. Tetrahedron 2018, 74, 6166–6172. [Google Scholar] [CrossRef] [PubMed]
- Byrne, F.P.; Jin, S.; Paggiola, G.; Petchey, T.H.M.; Clark, J.H.; Farmer, T.J.; Hunt, A.J.; McElroy, C.R.; Sherwood, J. Tools and Techniques for Solvent Selection: Green Solvent Selection Guides. Sustain. Chem. Process. 2016, 4, 7. [Google Scholar] [CrossRef]
- Flores-Ferrándiz, J.; Chinchilla, R. Solvent-Dependent Enantioswitching in the Michael Addition of α,α-Disubstituted Aldehydes to Maleimides Organocatalyzed by mono-N-Boc-Protected Cyclohexa-1,2-diamines. Tetrahedron Asymmetry 2014, 25, 1091–1094. [Google Scholar] [CrossRef] [Green Version]
- Szakonyi, Z.; Martinek, T.A.; Sillanpää, R.; Fülöp, F. Regio- and Stereoselective Synthesis of the Enantiomers of Monoterpene-Based β-Amino Acid Derivatives. Tetrahedron Asymmetry 2007, 18, 2442–2447. [Google Scholar] [CrossRef]
- Szakonyi, Z.; Martinek, T.A.; Sillanpää, R.; Fülöp, F. Regio- and Stereoselective Synthesis of Constrained Enantiomeric β-Amino Acid Derivatives. Tetrahedron Asymmetry 2008, 19, 2296–2303. [Google Scholar] [CrossRef]
- Szakonyi, Z.; Balázs, Á.; Martinek, T.A.; Fülöp, F. Stereoselective Synthesis of Pinane-Based β- and γ-Amino Acids via Conjugate Addition of Lithium Amides and Nitromethane. Tetrahedron Asymmetry 2010, 21, 2498–2504. [Google Scholar] [CrossRef]
- Szakonyi, Z.; Sillanpää, R.; Fülöp, F. Stereoselective Synthesis of Perillaldehyde-Based chiral β-Amino Acid Derivatives through Conjugate Addition of Lithium Amides. Beilstein J. Org. Chem. 2014, 10, 2738–2742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).