
Recent Advances in Heterogeneous Electroreduction of CO2 on Copper-Based Catalysts



Abstract
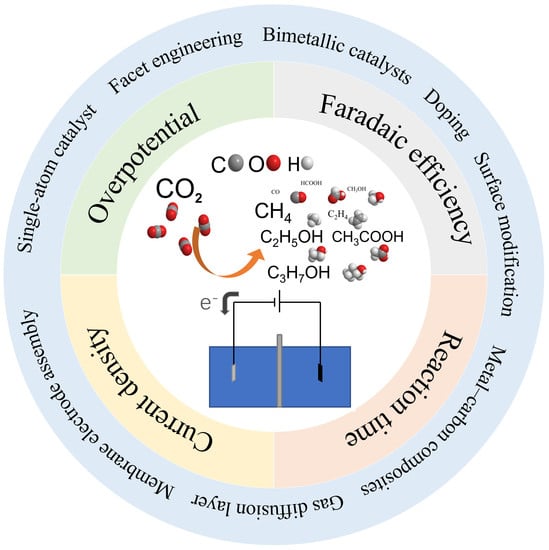
:
1. Introduction
2. Electroreduction Pathways
3. Advances in CO2 Electroreduction
3.1. Low Overpotential
3.1.1. Two-Electron Electroreduction Products
3.1.2. Multi-Electron Products
3.2. High Faradaic Efficiency
3.2.1. CO and Formate
3.2.2. Methane
3.2.3. Methanol
3.2.4. Ethylene
3.2.5. C2+ Oxygenates
3.3. Current Density
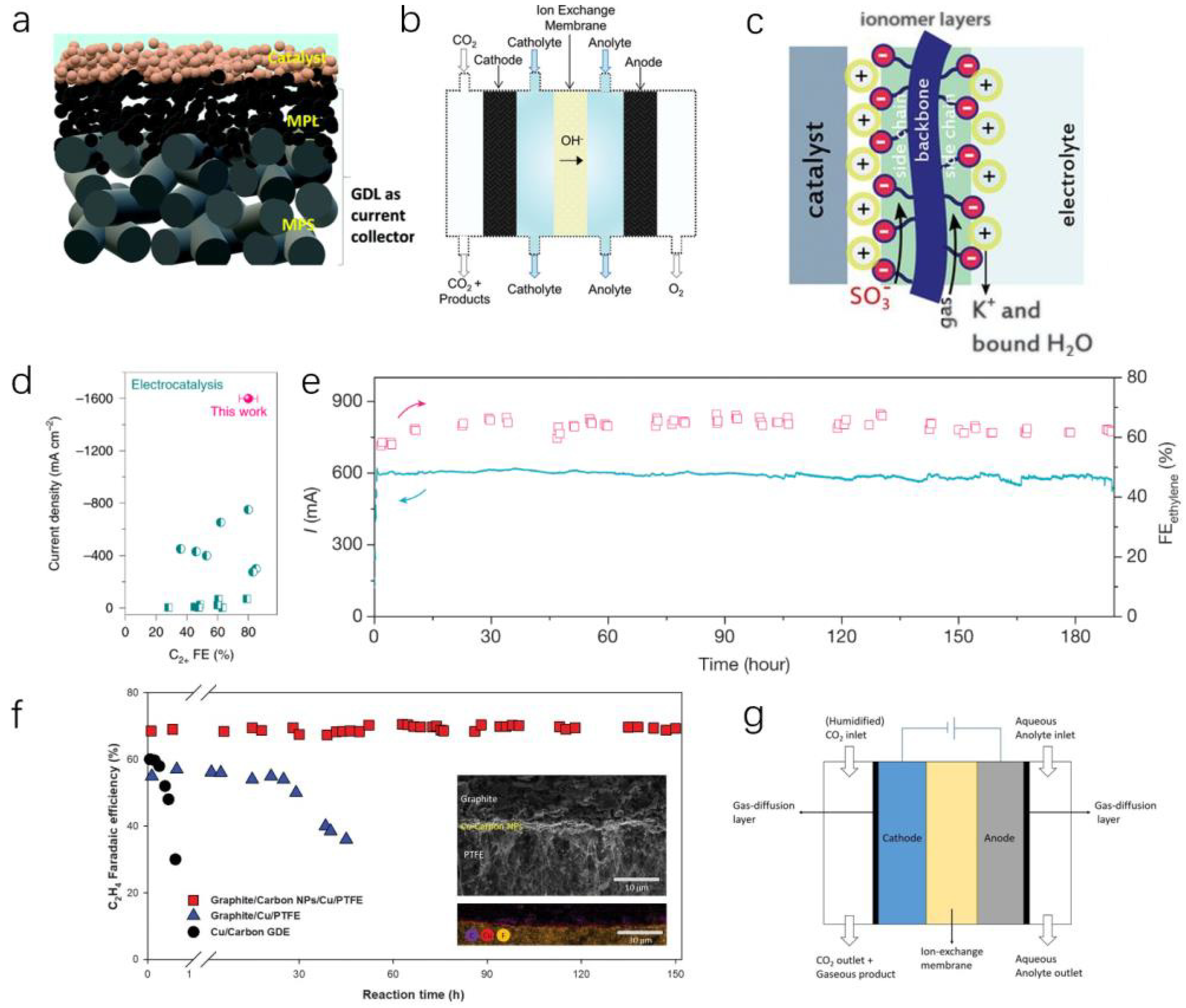
3.4. Stability
4. Conclusions and Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Olah, G.A.; Prakash, G.K.S.; Goeppert, A. Anthropogenic Chemical Carbon Cycle for a Sustainable Future. J. Am. Chem. Soc. 2011, 133, 12881–12898. [Google Scholar] [CrossRef] [PubMed]
- Nitopi, S.; Bertheussen, E.; Scott, S.B.; Liu, X.; Engstfeld, A.K.; Horch, S.; Seger, B.; Stephens, I.E.L.; Chan, K.; Hahn, C.; et al. Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte. Chem. Rev. 2019, 119, 7610–7672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhl, K.P.; Cave, E.R.; Abram, D.N.; Jaramillo, T.F. New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces. Energy Environ. Sci. 2012, 5, 7050–7059. [Google Scholar] [CrossRef]
- Khodakov, A.Y.; Chu, W.; Fongarland, P. Advances in the Development of Novel Cobalt Fischer−Tropsch Catalysts for Synthesis of Long-Chain Hydrocarbons and Clean Fuels. Chem. Rev. 2007, 107, 1692–1744. [Google Scholar] [CrossRef]
- Zheng, T.; Zhang, M.; Wu, L.; Guo, S.; Liu, X.; Zhao, J.; Xue, W.; Li, J.; Liu, C.; Li, X.; et al. Upcycling CO2 into energy-rich long-chain compounds via electrochemical and metabolic engineering. Nat. Catal. 2022, 5, 388–396. [Google Scholar] [CrossRef]
- Glockler, G. Carbon–Oxygen Bond Energies and Bond Distances. J. Phys. Chem. 1958, 62, 1049–1054. [Google Scholar] [CrossRef]
- Siewert, I. Proton-Coupled Electron Transfer Reactions Catalysed by 3 d Metal Complexes. Chem. A Eur. J. 2015, 21, 15078–15091. [Google Scholar] [CrossRef]
- Ross, M.B.; De Luna, P.; Li, Y.; Dinh, C.-T.; Kim, D.; Yang, P.; Sargent, E.H. Designing materials for electrochemical carbon dioxide recycling. Nat. Catal. 2019, 2, 648–658. [Google Scholar] [CrossRef] [Green Version]
- Burdyny, T.; Smith, W.A. CO2 reduction on gas-diffusion electrodes and why catalytic performance must be assessed at commercially-relevant conditions. Energy Environ. Sci. 2019, 12, 1442–1453. [Google Scholar] [CrossRef] [Green Version]
- Choi, W.; Won, D.H.; Hwang, Y.J. Catalyst design strategies for stable electrochemical CO2 reduction reaction. J. Mater. Chem. A 2020, 8, 15341–15357. [Google Scholar] [CrossRef]
- Jin, S.; Hao, Z.; Zhang, K.; Yan, Z.; Chen, J. Advances and Challenges for the Electrochemical Reduction of CO2 to CO: From Fundamentals to Industrialization. Angew. Chem. Int. Ed. 2021, 60, 20627–20648. [Google Scholar] [CrossRef]
- Park, S.; Wijaya, D.T.; Na, J.; Lee, C.W. Towards the Large-Scale Electrochemical Reduction of Carbon Dioxide. Catalysts 2021, 11, 253. [Google Scholar] [CrossRef]
- Yoshio, H.; Katsuhei, K.; Shin, S. Production of CO and CH4 in electrochemical reduction of CO2 at metal electrodes in aqueous hydrogencarbonate solution. Chem. Lett. 1985, 14, 1695–1698. [Google Scholar] [CrossRef]
- Díaz-Sainz, G.; Alvarez-Guerra, M.; Irabien, A. Continuous electroreduction of CO2 towards formate in gas-phase operation at high current densities with an anion exchange membrane. J. CO2 Util. 2022, 56, 101822. [Google Scholar] [CrossRef]
- Cheng, D.; Zhao, Z.-J.; Zhang, G.; Yang, P.; Li, L.; Gao, H.; Liu, S.; Chang, X.; Chen, S.; Wang, T.; et al. The nature of active sites for carbon dioxide electroreduction over oxide-derived copper catalysts. Nat. Commun. 2021, 12, 395. [Google Scholar] [CrossRef]
- Kuo, L.; Dinh, C.-T. Toward efficient catalysts for electrochemical CO2 conversion to C2 products. Curr. Opin. Electrochem. 2021, 30, 100807. [Google Scholar] [CrossRef]
- Xue, L.; Zhang, C.; Wu, J.; Fan, Q.-Y.; Liu, Y.; Wu, Y.; Li, J.; Zhang, H.; Liu, F.; Zeng, S. Unveiling the reaction pathway on Cu/CeO2 catalyst for electrocatalytic CO2 reduction to CH4. Appl. Catal. B 2022, 304, 120951. [Google Scholar] [CrossRef]
- Garza, A.J.; Bell, A.T.; Head-Gordon, M. Mechanism of CO2 Reduction at Copper Surfaces: Pathways to C2 Products. ACS Catal. 2018, 8, 1490–1499. [Google Scholar] [CrossRef] [Green Version]
- Feaster, J.T.; Shi, C.; Cave, E.R.; Hatsukade, T.; Abram, D.N.; Kuhl, K.P.; Hahn, C.; Nørskov, J.K.; Jaramillo, T.F. Understanding Selectivity for the Electrochemical Reduction of Carbon Dioxide to Formic Acid and Carbon Monoxide on Metal Electrodes. ACS Catal. 2017, 7, 4822–4827. [Google Scholar] [CrossRef]
- Vijay, S.; Ju, W.; Brückner, S.; Tsang, S.-C.; Strasser, P.; Chan, K. Unified mechanistic understanding of CO2 reduction to CO on transition metal and single atom catalysts. Nat. Catal. 2021, 4, 1024–1031. [Google Scholar] [CrossRef]
- Li, S.; Dong, X.; Chen, W.; Song, Y.; Li, G.; Wei, W.; Sun, Y. Efficient CO2 Electroreduction over Silver Hollow Fiber Electrode. Catalysts 2022, 12, 453. [Google Scholar] [CrossRef]
- Wang, Z.; Li, T.; Wang, Q.; Guan, A.; Cao, N.; Al-Enizi, A.M.; Zhang, L.; Qian, L.; Zheng, G. Hydrophobically made Ag nanoclusters with enhanced performance for CO2 aqueous electroreduction. J. Power. Sources 2020, 476, 228705. [Google Scholar] [CrossRef]
- Kim, J.; Choi, W.; Park, J.W.; Kim, C.; Kim, M.; Song, H. Branched Copper Oxide Nanoparticles Induce Highly Selective Ethylene Production by Electrochemical Carbon Dioxide Reduction. J. Am. Chem. Soc. 2019, 141, 6986–6994. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wu, Q.; Liang, X.; Wang, Z.; Zheng, Z.; Wang, P.; Liu, Y.; Dai, Y.; Whangbo, M.-H.; Huang, B. Cu2O Nanoparticles with Both {100} and {111} Facets for Enhancing the Selectivity and Activity of CO2 Electroreduction to Ethylene. Adv. Sci. 2020, 7, 1902820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, D.; Zhao, Z.-J.; Zhao, Q.; Cheng, D.; Liu, B.; Zhang, G.; Deng, W.; Dong, H.; Zhang, L.; Li, J.; et al. Coupling of Cu(100) and (110) Facets Promotes Carbon Dioxide Conversion to Hydrocarbons and Alcohols. Angew. Chem. Int. Ed. 2021, 60, 4879–4885. [Google Scholar] [CrossRef] [PubMed]
- Zhong, M.; Tran, K.; Min, Y.; Wang, C.; Wang, Z.; Dinh, C.-T.; De Luna, P.; Yu, Z.; Rasouli, A.S.; Brodersen, P.; et al. Accelerated discovery of CO2 electrocatalysts using active machine learning. Nature 2020, 581, 178. [Google Scholar] [CrossRef]
- Lv, X.; Shang, L.; Zhou, S.; Li, S.; Wang, Y.; Wang, Z.; Sham, T.-K.; Peng, C.; Zheng, G. Electron-Deficient Cu Sites on Cu3Ag1 Catalyst Promoting CO2 Electroreduction to Alcohols. Adv. Energy Mater. 2020, 10, 2001987. [Google Scholar] [CrossRef]
- Wei, X.; Yin, Z.; Lyu, K.; Li, Z.; Gong, J.; Wang, G.; Xiao, L.; Lu, J.; Zhuang, L. Highly Selective Reduction of CO2 to C2+ Hydrocarbons at Copper/Polyaniline Interfaces. ACS Catal. 2020, 10, 4103–4111. [Google Scholar] [CrossRef]
- Li, F.; Thevenon, A.; Rosas-Hernandez, A.; Wang, Z.; Li, Y.; Gabardo, C.M.; Ozden, A.; Cao Thang, D.; Li, J.; Wang, Y.; et al. Molecular tuning of CO2-to-ethylene conversion. Nature 2020, 577, 509. [Google Scholar] [CrossRef] [Green Version]
- Ren, D.; Deng, Y.; Handoko, A.D.; Chen, C.S.; Malkhandi, S.; Yeo, B.S. Selective Electrochemical Reduction of Carbon Dioxide to Ethylene and Ethanol on Copper(I) Oxide Catalysts. ACS Catal. 2015, 5, 2814–2821. [Google Scholar] [CrossRef]
- Popović, S.; Smiljanić, M.; Jovanovič, P.; Vavra, J.; Buonsanti, R.; Hodnik, N. Stability and Degradation Mechanisms of Copper-Based Catalysts for Electrochemical CO2 Reduction. Angew. Chem. Int. Ed. 2020, 59, 14736–14746. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, A.N.; Li, F.W.; Hung, S.F.; Nam, D.H.; Gabardo, C.M.; Wang, Z.Y.; Xu, Y.; Ozden, A.; Rasouli, A.S.; et al. Efficient Methane Electrosynthesis Enabled by Tuning Local CO2 Availability. J. Am. Chem. Soc. 2020, 142, 3525–3531. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.L.; Zhong, H.X.; Xu, W.B.; Zhang, T.T.; Li, X.F.; Zhang, H.M. Tuning the electrocatalytic properties of a Cu electrode with organic additives containing amine group for CO2 reduction. J. Mater. Chem. A 2019, 7, 5453–5462. [Google Scholar] [CrossRef]
- Zhang, J.; Cai, W.; Hu, F.X.; Yang, H.; Liu, B. Recent advances in single atom catalysts for the electrochemical carbon dioxide reduction reaction. Chem. Sci. 2021, 12, 6800–6819. [Google Scholar] [CrossRef] [PubMed]
- Chernyshova, I.V.; Somasundaran, P.; Ponnurangam, S. On the origin of the elusive first intermediate of CO2 electroreduction. Proc. Natl. Acad. Sci. USA 2018, 115, E9261. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Fan, Q.; Xia, R.; Meyer, T.J. CO2 Reduction: From Homogeneous to Heterogeneous Electrocatalysis. Acc. Chem. Res. 2020, 53, 255–264. [Google Scholar] [CrossRef]
- Bagger, A.; Ju, W.; Varela, A.S.; Strasser, P.; Rossmeisl, J. Electrochemical CO2 Reduction: A Classification Problem. ChemPhysChem 2017, 18, 3266–3273. [Google Scholar] [CrossRef] [Green Version]
- Cheng, T.; Xiao, H.; Goddard, W.A. Free-Energy Barriers and Reaction Mechanisms for the Electrochemical Reduction of CO on the Cu(100) Surface, Including Multiple Layers of Explicit Solvent at pH 0. J. Phys. Chem. Lett. 2015, 6, 4767–4773. [Google Scholar] [CrossRef]
- Nie, X.; Esopi, M.R.; Janik, M.J.; Asthagiri, A. Selectivity of CO2 Reduction on Copper Electrodes: The Role of the Kinetics of Elementary Steps. Angew. Chem. Int. Ed. 2013, 52, 2459–2462. [Google Scholar] [CrossRef]
- Peterson, A.A.; Abild-Pedersen, F.; Studt, F.; Rossmeisl, J.; Nørskov, J.K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 2010, 3, 1311–1315. [Google Scholar] [CrossRef]
- Peng, C.; Xu, Z.; Luo, G.; Yan, S.; Zhang, J.; Li, S.; Chen, Y.; Chang, L.Y.; Wang, Z.; Sham, T.-K.; et al. Highly-Exposed Single-Interlayered Cu Edges Enable High-Rate CO2-to-CH4 Electrosynthesis. Adv. Energy Mater. 2022, 12, 2200195. [Google Scholar] [CrossRef]
- Calle-Vallejo, F.; Koper, M.T.M. Theoretical Considerations on the Electroreduction of CO to C2 Species on Cu(100) Electrodes. Angew. Chem. Int. Ed. 2013, 52, 7282–7285. [Google Scholar] [CrossRef] [PubMed]
- Lum, Y.; Cheng, T.; Goddard, W.A.; Ager, J.W. Electrochemical CO Reduction Builds Solvent Water into Oxygenate Products. J. Am. Chem. Soc. 2018, 140, 9337–9340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, K.D.; Lee, C.W.; Jin, K.; Im, S.W.; Nam, K.T. Current Status and Bioinspired Perspective of Electrochemical Conversion of CO2 to a Long-Chain Hydrocarbon. J. Phys. Chem. Lett. 2017, 8, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Kortlever, R.; Shen, J.; Schouten, K.J.; Calle-Vallejo, F.; Koper, M.T. Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide. J. Phys. Chem. Lett. 2015, 6, 4073–4082. [Google Scholar] [CrossRef]
- Hori, Y.; Takahashi, R.; Yoshinami, Y.; Murata, A. Electrochemical Reduction of CO at a Copper Electrode. J. Phys. Chem. B 1997, 101, 7075–7081. [Google Scholar] [CrossRef]
- Ren, D.; Wong, N.T.; Handoko, A.D.; Huang, Y.; Yeo, B.S. Mechanistic Insights into the Enhanced Activity and Stability of Agglomerated Cu Nanocrystals for the Electrochemical Reduction of Carbon Dioxide to n-Propanol. J. Phys. Chem. Lett. 2016, 7, 20–24. [Google Scholar] [CrossRef]
- Ma, W.; He, X.; Wang, W.; Xie, S.; Zhang, Q.; Wang, Y. Electrocatalytic reduction of CO2 and CO to multi-carbon compounds over Cu-based catalysts. Chem. Soc. Rev. 2021, 50, 12897–12914. [Google Scholar] [CrossRef]
- Montoya, J.H.; Peterson, A.A.; Nørskov, J.K. Insights into C-C Coupling in CO2 Electroreduction on Copper Electrodes. ChemCatChem 2013, 5, 737–742. [Google Scholar] [CrossRef]
- Bagger, A.; Arnarson, L.; Hansen, M.H.; Spohr, E.; Rossmeisl, J. Electrochemical CO Reduction: A Property of the Electrochemical Interface. J. Am. Chem. Soc. 2019, 141, 1506–1514. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Yang, J.; Chen, H.; Hou, Y.; Wang, Q.; Gu, M.; He, F.; Xia, Y.; Xia, Z.; Li, Z.; et al. Atomically Defined Undercoordinated Active Sites for Highly Efficient CO2 Electroreduction. Adv. Funct. Mater. 2020, 30, 1907658. [Google Scholar] [CrossRef]
- Zhang, H.; Li, J.; Xi, S.; Du, Y.; Hai, X.; Wang, J.; Xu, H.; Wu, G.; Zhang, J.; Lu, J.; et al. A Graphene-Supported Single-Atom FeN5 Catalytic Site for Efficient Electrochemical CO2 Reduction. Angew. Chem. Int. Ed. 2019, 58, 14871–14876. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.B.; Hung, S.-F.; Liu, S.; Yuan, K.; Miao, S.; Zhang, L.; Huang, X.; Wang, H.-Y.; Cai, W.; Chen, R.; et al. Atomically dispersed Ni(i) as the active site for electrochemical CO2 reduction. Nat. Energy 2018, 3, 140–147. [Google Scholar] [CrossRef]
- Gu, J.; Hsu, C.-S.; Bai, L.; Chen, H.M.; Hu, X. Atomically dispersed Fe3+ sites catalyze efficient CO2 electroreduction to CO. Science 2019, 364, 1091. [Google Scholar] [CrossRef]
- Pei, J.; Wang, T.; Sui, R.; Zhang, X.; Zhou, D.; Qin, F.; Zhao, X.; Liu, Q.; Yan, W.; Dong, J.; et al. N-Bridged Co-N-Ni: New bimetallic sites for promoting electrochemical CO2 reduction. Energy Environ. Sci. 2021, 14, 3019–3028. [Google Scholar] [CrossRef]
- Lin, L.; Li, H.; Yan, C.; Li, H.; Si, R.; Li, M.; Xiao, J.; Wang, G.; Bao, X. Synergistic Catalysis over Iron-Nitrogen Sites Anchored with Cobalt Phthalocyanine for Efficient CO2 Electroreduction. Adv. Mater. 2019, 31, 1903470. [Google Scholar] [CrossRef]
- Zu, X.; Li, X.; Liu, W.; Sun, Y.; Xu, J.; Yao, T.; Yan, W.; Gao, S.; Wang, C.; Wei, S.; et al. Efficient and Robust Carbon Dioxide Electroreduction Enabled by Atomically Dispersed Sn delta+ Sites. Adv. Mater. 2019, 31, 1808135. [Google Scholar] [CrossRef]
- Zhang, E.; Wang, T.; Yu, K.; Liu, J.; Chen, W.; Li, A.; Rong, H.; Lin, R.; Ji, S.; Zhene, X.; et al. Bismuth Single Atoms Resulting from Transformation of Metal-Organic Frameworks and Their Use as Electrocatalysts for CO2 Reduction. J. Am. Chem. Soc. 2019, 141, 16569–16573. [Google Scholar] [CrossRef]
- Nellaiappan, S.; Katiyar, N.K.; Kumar, R.; Parui, A.; Malviya, K.D.; Pradeep, K.G.; Singh, A.K.; Sharma, S.; Tiwary, C.S.; Biswas, K. High-Entropy Alloys as Catalysts for the CO2 and CO Reduction Reactions: Experimental Realization. ACS Catal. 2020, 10, 3658–3663. [Google Scholar] [CrossRef]
- Fan, L.; Liu, C.-Y.; Zhu, P.; Xia, C.; Zhang, X.; Wu, Z.-Y.; Lu, Y.; Senftle, T.P.; Wang, H. Proton sponge promotion of electrochemical CO2 reduction to multi-carbon products. Joule 2022, 6, 205–220. [Google Scholar] [CrossRef]
- Ma, W.; Xie, S.; Liu, T.; Fan, Q.; Ye, J.; Sun, F.; Jiang, Z.; Zhang, Q.; Cheng, J.; Wang, Y. Electrocatalytic reduction of CO2 to ethylene and ethanol through hydrogen-assisted C–C coupling over fluorine-modified copper. Nat. Catal. 2020, 3, 478–487. [Google Scholar] [CrossRef]
- Xu, H.; Rebollar, D.; He, H.; Chong, L.; Liu, Y.; Liu, C.; Sun, C.-J.; Li, T.; Muntean, J.V.; Winans, R.E.; et al. Highly selective electrocatalytic CO2 reduction to ethanol by metallic clusters dynamically formed from atomically dispersed copper. Nat. Energy 2020, 5, 623–632. [Google Scholar] [CrossRef]
- Morales-Guio, C.G.; Cave, E.R.; Nitopi, S.A.; Feaster, J.T.; Wang, L.; Kuhl, K.P.; Jackson, A.; Johnson, N.C.; Abram, D.N.; Hatsukade, T.; et al. Improved CO2 reduction activity towards C2+ alcohols on a tandem gold on copper electrocatalyst. Nat. Catal. 2018, 1, 764–771. [Google Scholar] [CrossRef]
- Li, F.; Li, Y.C.; Wang, Z.; Li, J.; Nam, D.-H.; Lum, Y.; Luo, M.; Wang, X.; Ozden, A.; Hung, S.-F.; et al. Cooperative CO2-to-ethanol conversion via enriched intermediates at molecule–metal catalyst interfaces. Nat. Catal. 2020, 3, 75–82. [Google Scholar] [CrossRef]
- Zhu, Q.; Sun, X.; Yang, D.; Ma, J.; Kang, X.; Zheng, L.; Zhang, J.; Wu, Z.; Han, B. Carbon dioxide electroreduction to C2 products over copper-cuprous oxide derived from electrosynthesized copper complex. Nat. Commun. 2019, 10, 3851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Wang, Y.; Shuai, L.; Wang, X.; He, F.; Lei, C.; Li, Z.; Yang, B.; Lei, L.; Yuan, C.; et al. Highly Boosted Reaction Kinetics in Carbon Dioxide Electroreduction by Surface-Introduced Electronegative Dopants. Adv. Funct. Mater. 2021, 31, 2008146. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, J.; Shi, J.; Tan, D.; Liu, L.; Zhang, F.; Lu, C.; Su, Z.; Tan, X.; Cheng, X.; et al. Manganese acting as a high-performance heterogeneous electrocatalyst in carbon dioxide reduction. Nat. Commun. 2019, 10, 2980. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, Z.; Zhang, X.; Li, L.; Li, Y.; Xu, H.; Li, X.; Yu, X.; Zhang, Z.; Liang, Y.; et al. Highly selective and active CO2 reduction electrocatalysts based on cobalt phthalocyanine/carbon nanotube hybrid structures. Nat. Commun. 2017, 8, 14675. [Google Scholar] [CrossRef] [Green Version]
- Guan, A.; Chen, Z.; Quan, Y.; Peng, C.; Wang, Z.; Sham, T.-K.; Yang, C.; Ji, Y.; Qian, L.; Xu, X.; et al. Boosting CO2 Electroreduction to CH4 via Tuning Neighboring Single-Copper Sites. ACS Energy Lett. 2020, 5, 1044–1053. [Google Scholar] [CrossRef]
- Cai, Y.; Fu, J.; Zhou, Y.; Chang, Y.-C.; Min, Q.; Zhu, J.-J.; Lin, Y.; Zhu, W. Insights on forming N,O-coordinated Cu single-atom catalysts for electrochemical reduction CO2 to methane. Nat. Commun. 2021, 12, 586. [Google Scholar] [CrossRef]
- Resasco, J.; Chen, L.D.; Clark, E.; Tsai, C.; Hahn, C.; Jaramillo, T.F.; Chan, K.; Bell, A.T. Promoter Effects of Alkali Metal Cations on the Electrochemical Reduction of Carbon Dioxide. J. Am. Chem. Soc. 2017, 139, 11277–11287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Wang, X.-Z.; Tao, H.; Li, T.; Liu, Q.; Xu, Z.; Fu, X.-Z.; Luo, J.-L. Ultrathin 5-fold twinned sub-25nm silver nanowires enable highly selective electroreduction of CO2 to CO. Nano Energy 2018, 45, 456–462. [Google Scholar] [CrossRef]
- Wang, X.; Feng, S.; Lu, W.; Zhao, Y.; Zheng, S.; Zheng, W.; Sang, X.; Zheng, L.; Xie, Y.; Li, Z.; et al. A New Strategy for Accelerating Dynamic Proton Transfer of Electrochemical CO2 Reduction at High Current Densities. Adv. Funct. Mater. 2021, 31, 2104243. [Google Scholar] [CrossRef]
- Fan, L.; Xia, C.; Zhu, P.; Lu, Y.; Wang, H. Electrochemical CO2 reduction to high-concentration pure formic acid solutions in an all-solid-state reactor. Nat. Commun. 2020, 11, 3633. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yang, S.; Jiang, M.; Hu, Y.; Hu, C.; Zhang, X.; Jin, Z. Nanocapillarity and Nanoconfinement Effects of Pipet-like Bismuth@Carbon Nanotubes for Highly Efficient Electrocatalytic CO2 Reduction. Nano Lett. 2021, 21, 2650–2657. [Google Scholar] [CrossRef]
- Han, L.; Song, S.; Liu, M.; Yao, S.; Liang, Z.; Cheng, H.; Ren, Z.; Liu, W.; Lin, R.; Qi, G.; et al. Stable and Efficient Single-Atom Zn Catalyst for CO2 Reduction to CH4. J. Am. Chem. Soc. 2020, 142, 12563–12567. [Google Scholar] [CrossRef]
- Zhang, Y.; Dong, L.-Z.; Li, S.; Huang, X.; Chang, J.-N.; Wang, J.-H.; Zhou, J.; Li, S.-L.; Lan, Y.-Q. Coordination environment dependent selectivity of single-site-Cu enriched crystalline porous catalysts in CO2 reduction to CH4. Nat. Commun. 2021, 12, 6390. [Google Scholar] [CrossRef]
- Yang, D.; Zhu, Q.; Chen, C.; Liu, H.; Liu, Z.; Zhao, Z.; Zhang, X.; Liu, S.; Han, B. Selective electroreduction of carbon dioxide to methanol on copper selenide nanocatalysts. Nat. Commun. 2019, 10, 677. [Google Scholar] [CrossRef]
- Li, P.; Bi, J.; Liu, J.; Zhu, Q.; Chen, C.; Sun, X.; Zhang, J.; Han, B. In situ dual doping for constructing efficient CO2-to-methanol electrocatalysts. Nat. Commun. 2022, 13, 1965. [Google Scholar] [CrossRef]
- Li, Y.C.; Wang, Z.; Yuan, T.; Nam, D.-H.; Luo, M.; Wicks, J.; Chen, B.; Li, J.; Li, F.; de Arguer, F.P.G.; et al. Binding Site Diversity Promotes CO2 Electroreduction to Ethanol. J. Am. Chem. Soc. 2019, 141, 8584–8591. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, D.; Dares, C.J.; Marquard, S.L.; Sheridan, M.V.; Meyer, T.J. CO2 reduction to acetate in mixtures of ultrasmall (Cu)n,(Ag)m bimetallic nanoparticles. Proc. Natl. Acad. Sci. USA 2018, 115, 278–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, C.; Luo, G.; Zhang, J.; Chen, M.; Wang, Z.; Sham, T.-K.; Zhang, L.; Li, Y.; Zheng, G. Double sulfur vacancies by lithium tuning enhance CO2 electroreduction to n-propanol. Nat. Commun. 2021, 12, 1580. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.-Y.; Hu, S.-J.; Zhang, X.-L.; Zheng, Y.-R.; Wang, H.-J.; Niu, Z.-Z.; Yang, P.-P.; Bao, R.-C.; Ma, T.; Dang, Z.; et al. High-Curvature Transition-Metal Chalcogenide Nanostructures with a Pronounced Proximity Effect Enable Fast and Selective CO2 Electroreduction. Angew. Chem. Int. Ed. 2020, 59, 8706–8712. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Tan, X.; Yang, W.; Jia, C.; Xu, S.; Wang, K.; Smith, S.C.; Zhao, C. Isolated Diatomic Ni-Fe Metal–Nitrogen Sites for Synergistic Electroreduction of CO2. Angew. Chem. Int. Ed. 2019, 58, 6972–6976. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Mou, K.; Wang, X.; Liu, L. Nitrogen-Doped Graphene Quantum Dots Enhance the Activity of Bi2O3 Nanosheets for Electrochemical Reduction of CO2 in a Wide Negative Potential Region. Angew. Chem. Int. Ed. 2018, 57, 12790–12794. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Zhang, C.; Hu, R.; Du, Z.; Gu, J.; Cui, Y.; Chen, X.; Xu, W.; Cheng, Z.; Li, S.; et al. Selective Etching Quaternary MAX Phase toward Single Atom Copper Immobilized MXene (Ti3C2Clx) for Efficient CO2 Electroreduction to Methanol. ACS Nano 2021, 15, 4927–4936. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, Z.; Han, P.; Du, Y.; Gu, Z.; Xu, X.; Zheng, G. Single-Atomic Cu with Multiple Oxygen Vacancies on Ceria for Electrocatalytic CO2 Reduction to CH4. ACS Catal. 2018, 8, 7113–7119. [Google Scholar] [CrossRef]
- Xiong, L.; Zhang, X.; Chen, L.; Deng, Z.; Han, S.; Chen, Y.; Zhong, J.; Sun, H.; Lian, Y.; Yang, B.; et al. Geometric Modulation of Local CO Flux in Ag@Cu2O Nanoreactors for Steering the CO2RR Pathway toward High-Efficacy Methane Production. Adv. Mater. 2021, 33, 2101741. [Google Scholar] [CrossRef]
- Han, Z.; Han, D.; Chen, Z.; Gao, J.; Jiang, G.; Wang, X.; Lyu, S.; Guo, Y.; Geng, C.; Yin, L.; et al. Steering surface reconstruction of copper with electrolyte additives for CO2 electroreduction. Nat. Commun. 2022, 13, 3158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Qin, Q.; Dai, L.; Qin, R.; Zhao, X.; Chen, X.; Ou, D.; Chen, J.; Chuong, T.T.; Wu, B.; et al. Electrochemical Reduction of Carbon Dioxide to Methanol on Hierarchical Pd/SnO2 Nanosheets with Abundant Pd-O-Sn Interfaces. Angew. Chem. Int. Ed. 2018, 57, 9475–9479. [Google Scholar] [CrossRef]
- Ji, L.; Chang, L.; Zhang, Y.; Mou, S.; Wang, T.; Luo, Y.; Wang, Z.; Sun, X. Electrocatalytic CO2 Reduction to Alcohols with High Selectivity over a Two-Dimensional Fe2P2S6 Nanosheet. ACS Catal. 2019, 9, 9721–9725. [Google Scholar] [CrossRef]
- Mou, S.; Wu, T.; Xie, J.; Zhang, Y.; Ji, L.; Huang, H.; Wang, T.; Luo, Y.; Xiong, X.; Tang, B.; et al. Boron Phosphide Nanoparticles: A Nonmetal Catalyst for High-Selectivity Electrochemical Reduction of CO2 to CH3OH. Adv. Mater. 2019, 31, 1903499. [Google Scholar] [CrossRef]
- Guan, A.; Yang, C.; Wang, Q.; Qian, L.; Cao, J.; Zhang, L.; Wu, L.; Zheng, G. Atomic-Level Copper Sites for Selective CO2 Electroreduction to Hydrocarbon. ACS Sustain. Chem. Eng. 2021, 9, 13536–13544. [Google Scholar] [CrossRef]
- Hori, Y.; Takahashi, I.; Koga, O.; Hoshi, N. Electrochemical reduction of carbon dioxide at various series of copper single crystal electrodes. J. Mol. Catal. A Chem. 2003, 199, 39–47. [Google Scholar] [CrossRef]
- De Gregorio, G.L.; Burdyny, T.; Loiudice, A.; Iyengar, P.; Smith, W.A.; Buonsanti, R. Facet-Dependent Selectivity of Cu Catalysts in Electrochemical CO2 Reduction at Commercially Viable Current Densities. ACS Catal. 2020, 10, 4854–4862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahaman, M.; Dutta, A.; Zanetti, A.; Broekmann, P. Electrochemical Reduction of CO2 into Multicarbon Alcohols on Activated Cu Mesh Catalysts: An Identical Location (IL) Study. ACS Catal. 2017, 7, 7946–7956. [Google Scholar] [CrossRef]
- Wakerley, D.; Lamaison, S.; Ozanam, F.; Menguy, N.; Mercier, D.; Marcus, P.; Fontecave, M.; Mougel, V. Bio-inspired hydrophobicity promotes CO2 reduction on a Cu surface. Nat. Mater. 2019, 18, 1222–1227. [Google Scholar] [CrossRef] [PubMed]
- Vasileff, A.; Xu, C.; Jiao, Y.; Zheng, Y.; Qiao, S.-Z. Surface and Interface Engineering in Copper-Based Bimetallic Materials for Selective CO2 Electroreduction. Chem 2018, 4, 1809–1831. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Yan, X.; Liu, S.; Wu, Y.; Wan, Q.; Sun, X.; Zhu, Q.; Liu, H.; Ma, J.; Zheng, L.; et al. Highly Efficient Electroreduction of CO2 to C2+ Alcohols on Heterogeneous Dual Active Sites. Angew. Chem. Int. Ed. 2020, 59, 16459–16464. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Z.; de Arquer, F.P.G.; Cao-Thang, D.; Ozden, A.; Li, Y.C.; Nam, D.-H.; Li, J.; Liu, Y.-S.; Wicks, J.; et al. Efficient electrically powered CO2-to-ethanol via suppression of deoxygenation. Nat. Energy 2020, 5, 478–486. [Google Scholar] [CrossRef]
- Chen, C.; Li, Y.; Yang, P. Address the “alkalinity problem” in CO2 electrolysis with catalyst design and translation. Joule 2021, 5, 737–742. [Google Scholar] [CrossRef]
- Furuya, N.; Yamazaki, T.; Shibata, M. High performance RuPd catalysts for CO2 reduction at gas-diffusion electrodes. J. Electroanal. Chem. 1997, 431, 39–41. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Dinh, C.-T. Gas diffusion electrode design for electrochemical carbon dioxide reduction. Chem. Soc. Rev. 2020, 49, 7488–7504. [Google Scholar] [CrossRef]
- Kibria, M.G.; Edwards, J.P.; Gabardo, C.M.; Dinh, C.-T.; Seifitokaldani, A.; Sinton, D.; Sargent, E.H. Electrochemical CO2 Reduction into Chemical Feedstocks: From Mechanistic Electrocatalysis Models to System Design. Adv. Mater. 2019, 31, 1807166. [Google Scholar] [CrossRef]
- de Arquer, F.P.G.; Cao-Thang, D.; Ozden, A.; Wicks, J.; McCallum, C.; Kirmani, A.R.; Dae-Hyun, N.; Gabardo, C.; Seifitokaldani, A.; Wang, X.; et al. CO2 electrolysis to multicarbon products at activities greater than 1 A cm−2. Science 2020, 367, 661–666. [Google Scholar] [CrossRef]
- Xie, H.; Wan, Y.; Wang, X.; Liang, J.; Lu, G.; Wang, T.; Chai, G.; Adli, N.M.; Priest, C.; Huang, Y.; et al. Boosting Pd-catalysis for electrochemical CO2 reduction to CO on Bi-Pd single atom alloy nanodendrites. Appl. Catal. B 2021, 289, 119783. [Google Scholar] [CrossRef]
- Jeong, H.-Y.; Balamurugan, M.; Choutipalli, V.S.K.; Jeong, E.-s.; Subramanian, V.; Sim, U.; Nam, K.T. Achieving highly efficient CO2 to CO electroreduction exceeding 300 mA cm−2 with single-atom nickel electrocatalysts. J. Mater. Chem. A 2019, 7, 10651–10661. [Google Scholar] [CrossRef]
- Abbas, S.A.; Song, J.T.; Tan, Y.C.; Nam, K.M.; Oh, J.; Jung, K.-D. Synthesis of a Nickel Single-Atom Catalyst Based on Ni–N4–xCx Active Sites for Highly Efficient CO2 Reduction Utilizing a Gas Diffusion Electrode. ACS Appl. Energ. Mater. 2020, 3, 8739–8745. [Google Scholar] [CrossRef]
- Yang, H.; Lin, Q.; Zhang, C.; Yu, X.; Cheng, Z.; Li, G.; Hu, Q.; Ren, X.; Zhang, Q.; Liu, J.; et al. Carbon dioxide electroreduction on single-atom nickel decorated carbon membranes with industry compatible current densities. Nat. Commun. 2020, 11, 593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinh, C.-T.; Burdyny, T.; Kibria, M.G.; Seifitokaldani, A.; Gabardo, C.M.; Arquer, F.P.G.d.; Kiani, A.; Edwards, J.P.; Luna, P.D.; Bushuyev, O.S.; et al. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 2018, 360, 783–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.; Guo, J.; Li, X.; Patel, P.; Seifitokaldani, A. Electrochemical Reactors for CO2 Conversion. Catalysts 2020, 10, 473. [Google Scholar] [CrossRef]
- Peng, C.; Luo, G.; Xu, Z.; Yan, S.; Zhang, J.; Chen, M.; Qian, L.; Wei, W.; Han, Q.; Zheng, G. Lithiation-Enabled High-Density Nitrogen Vacancies Electrocatalyze CO2 to C2 Products. Adv. Mater. 2021, 33, 2103150. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Zhu, X.; Xu, Z.; Yan, S.; Chang, L.Y.; Wang, Z.; Zhang, J.; Chen, M.; Sham, T.-K.; Li, Y.; et al. Lithium Vacancy-Tuned [CuO4] Sites for Selective CO2 Electroreduction to C2+ Products. Small 2022, 18, 2106433. [Google Scholar] [CrossRef]
- Yan, S.; Peng, C.; Yang, C.; Chen, Y.; Zhang, J.; Guan, A.; Lv, X.; Wang, H.; Wang, Z.; Sham, T.-K.; et al. Electron Localization and Lattice Strain Induced by Surface Lithium Doping Enable Ampere-Level Electrosynthesis of Formate from CO2. Angew. Chem. Int. Ed. 2021, 60, 25741–25745. [Google Scholar] [CrossRef]
- Duan, Y.-X.; Meng, F.-L.; Liu, K.-H.; Yi, S.-S.; Li, S.-J.; Yan, J.-M.; Jiang, Q. Amorphizing of Cu Nanoparticles toward Highly Efficient and Robust Electrocatalyst for CO2 Reduction to Liquid Fuels with High Faradaic Efficiencies. Adv. Mater. 2018, 30, 1706194. [Google Scholar] [CrossRef]
- Spöri, C.; Kwan, J.T.H.; Bonakdarpour, A.; Wilkinson, D.P.; Strasser, P. The Stability Challenges of Oxygen Evolving Catalysts: Towards a Common Fundamental Understanding and Mitigation of Catalyst Degradation. Angew. Chem. Int. Ed. 2017, 56, 5994–6021. [Google Scholar] [CrossRef]
- Ma, L.; Hu, W.; Pan, Q.; Zou, L.; Zou, Z.; Wen, K.; Yang, H. Polyvinyl alcohol-modified gold nanoparticles with record-high activity for electrochemical reduction of CO2 to CO. J. CO2 Util. 2019, 34, 108–114. [Google Scholar] [CrossRef]
- Zhang, L.; Wei, Z.; Thanneeru, S.; Meng, M.; Kruzyk, M.; Ung, G.; Liu, B.; He, J. A Polymer Solution To Prevent Nanoclustering and Improve the Selectivity of Metal Nanoparticles for Electrocatalytic CO2 Reduction. Angew. Chem. Int. Ed. 2019, 58, 15834–15840. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Hu, W.; Luo, W.; Wu, Y.; Li, X.; Deng, H. Size effect on alloying ability and phase stability of immiscible bimetallic nanoparticles. Eur. Phys. J. B 2006, 54, 479–484. [Google Scholar] [CrossRef]
- Papaefthimiou, V.; Diebold, M.; Ulhaq-Bouillet, C.; Doh, W.H.; Blume, R.; Zafeiratos, S.; Savinova, E.R. Potential-Induced Segregation Phenomena in Bimetallic PtAu Nanoparticles: An In Situ Near-Ambient-Pressure Photoelectron Spectroscopy Study. ChemElectroChem 2015, 2, 1519–1526. [Google Scholar] [CrossRef]
- Sun, K.; Cheng, T.; Wu, L.; Hu, Y.; Zhou, J.; Maclennan, A.; Jiang, Z.; Gao, Y.; Goddard, W.A.; Wang, Z. Ultrahigh Mass Activity for Carbon Dioxide Reduction Enabled by Gold–Iron Core–Shell Nanoparticles. J. Am. Chem. Soc. 2017, 139, 15608–15611. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Zhang, L.; Yang, P.; Hu, C.; Dong, H.; Zhao, Z.-J.; Mu, R.; Gong, J. Formation of Enriched Vacancies for Enhanced CO2 Electrocatalytic Reduction over AuCu Alloys. ACS Energy Lett. 2018, 3, 2144–2149. [Google Scholar] [CrossRef]
- Rabinowitz, J.A.; Kanan, M.W. The future of low-temperature carbon dioxide electrolysis depends on solving one basic problem. Nat. Commun. 2020, 11, 5231. [Google Scholar] [CrossRef]
- Overa, S.; Feric, T.G.; Park, A.-H.A.; Jiao, F. Tandem and Hybrid Processes for Carbon Dioxide Utilization. Joule 2021, 5, 8–13. [Google Scholar] [CrossRef]
- Luo, Y.; Zhang, K.; Li, Y.; Wang, Y. Valorizing carbon dioxide via electrochemical reduction on gas-diffusion electrodes. InfoMat 2021, 3, 1313–1332. [Google Scholar] [CrossRef]
- Mustain, W.E.; Chatenet, M.; Page, M.; Kim, Y.S. Durability challenges of anion exchange membrane fuel cells. Energy Environ. Sci. 2020, 13, 2805–2838. [Google Scholar] [CrossRef]
- Huang, J.E.; Li, F.; Ozden, A.; Sedighian Rasouli, A.; García de Arquer, F.P.; Liu, S.; Zhang, S.; Luo, M.; Wang, X.; Lum, Y.; et al. CO2 electrolysis to multicarbon products in strong acid. Science 2021, 372, 1074–1078. [Google Scholar] [CrossRef]
- Gu, J.; Liu, S.; Ni, W.; Ren, W.; Haussener, S.; Hu, X. Modulating electric field distribution by alkali cations for CO2 electroreduction in strongly acidic medium. Nat. Catal. 2022, 5, 268–276. [Google Scholar] [CrossRef]
- Shafaque, H.W.; Lee, C.; Fahy, K.F.; Lee, J.K.; LaManna, J.M.; Baltic, E.; Hussey, D.S.; Jacobson, D.L.; Bazylak, A. Boosting Membrane Hydration for High Current Densities in Membrane Electrode Assembly CO2 Electrolysis. ACS Appl. Mater. Interfaces 2020, 12, 54585–54595. [Google Scholar] [CrossRef]
- Li, Y.C.; Lee, G.; Yuan, T.; Wang, Y.; Nam, D.-H.; Wang, Z.; García de Arquer, F.P.; Lum, Y.; Dinh, C.-T.; Voznyy, O.; et al. CO2 Electroreduction from Carbonate Electrolyte. ACS Energy Lett. 2019, 4, 1427–1431. [Google Scholar] [CrossRef]
- Li, T.; Lees, E.W.; Zhang, Z.; Berlinguette, C.P. Conversion of Bicarbonate to Formate in an Electrochemical Flow Reactor. ACS Energy Lett. 2020, 5, 2624–2630. [Google Scholar] [CrossRef]
- Zhang, Z.; Lees, E.W.; Habibzadeh, F.; Salvatore, D.A.; Ren, S.; Simpson, G.L.; Wheeler, D.G.; Liu, A.; Berlinguette, C.P. Porous metal electrodes enable efficient electrolysis of carbon capture solutions. Energy Environ. Sci. 2022, 15, 705–713. [Google Scholar] [CrossRef]
- Lees, E.W.; Goldman, M.; Fink, A.G.; Dvorak, D.J.; Salvatore, D.A.; Zhang, Z.; Loo, N.W.X.; Berlinguette, C.P. Electrodes Designed for Converting Bicarbonate into CO. ACS Energy Lett. 2020, 5, 2165–2173. [Google Scholar] [CrossRef]

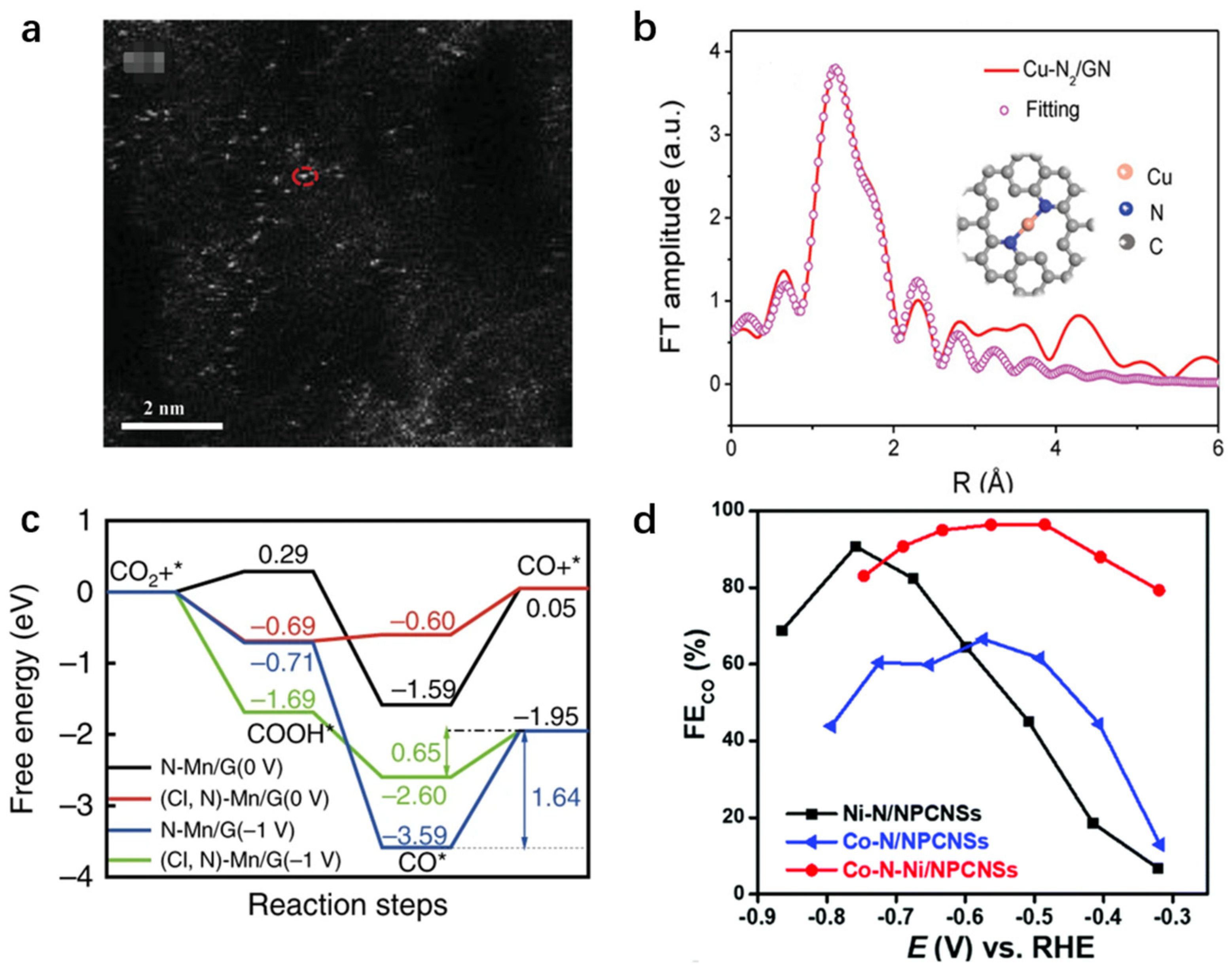
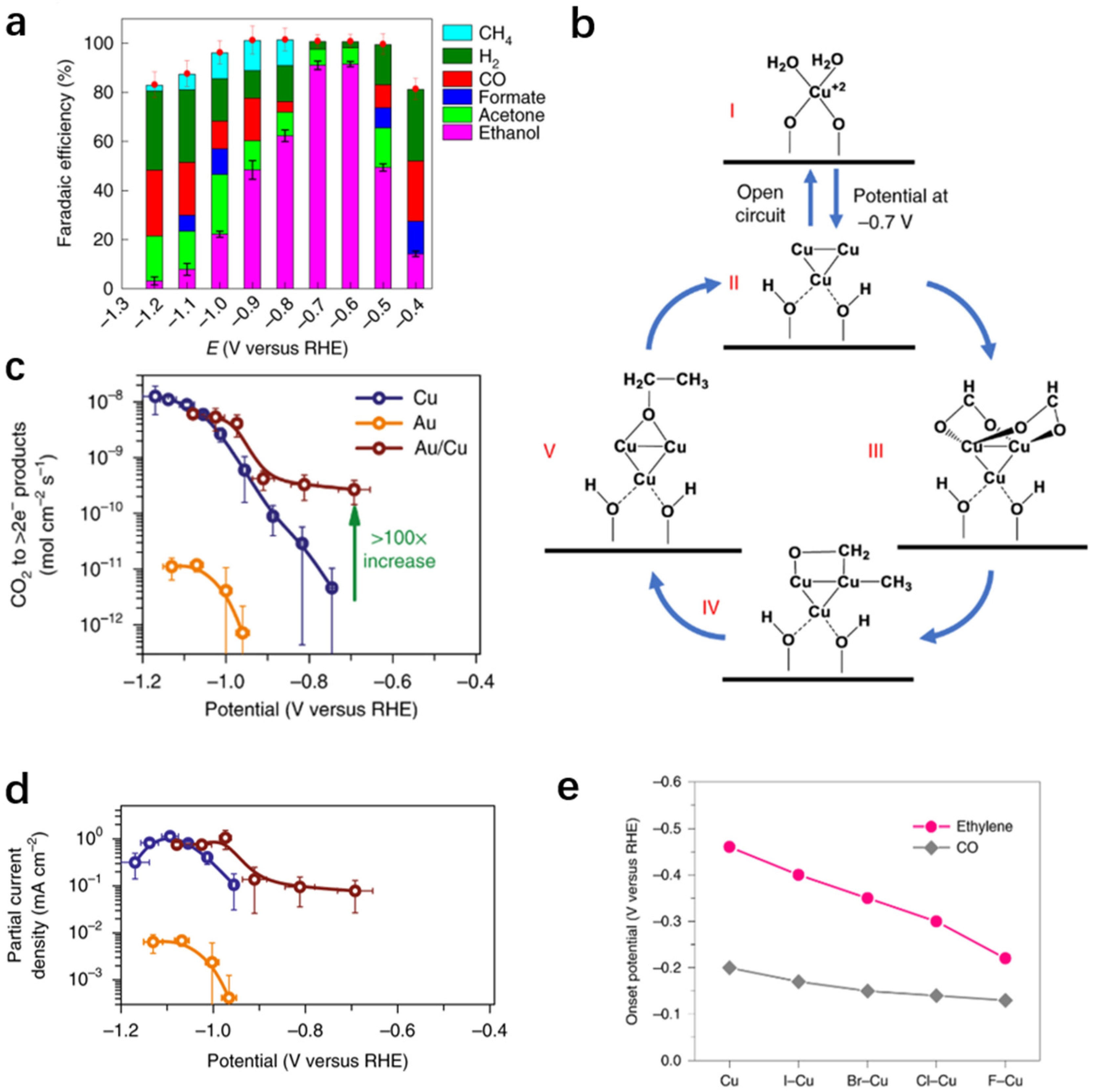
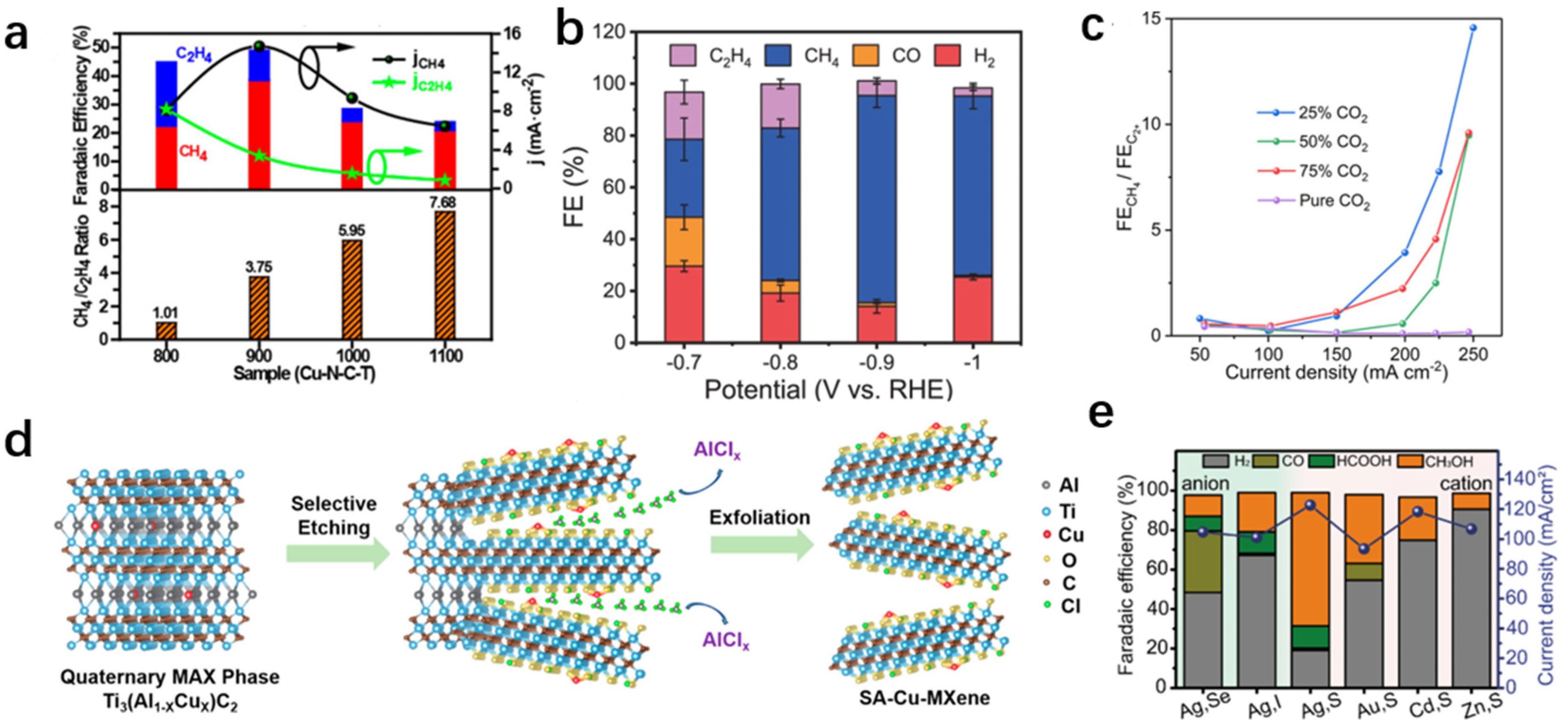


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Major Product | Catalyst | Onset Potential (V vs. RHE) | Reference |
|---|---|---|---|
| CO | Cu-N2/CN | −0.33 | [51] |
| FeN5 | −0.2 * | [52] | |
| Ni–N3S | −0.17 | [53] | |
| Fe3+-N-C | −0.2 | [54] | |
| Co–N–Ni/NPCNSs | −0.2 | [55] | |
| CoPc©Fe-N-C | −0.13 | [56] | |
| Formate | single-atom Snδ+ on N-doped graphene | −0.18 | [57] |
| BiN4/C | −0.51 | [58] | |
| Methane | AuAgPtPdCu | −0.3 * | [59] |
| Ethylene | Organosuperbases modified Cu-NC | −0.43 | [60] |
| F-Cu | ~−0.2 | [61] | |
| Ethanol | Cun (n = 3 and 4) cluster | −0.3~−0.4 | [62] |
| Au/Cu | −0.7 * | [63] | |
| FeTPP[Cl]/Cu | −0.42 | [64] | |
| Acetate | Cu–Cu2O/Cu | ~−0.2 * | [65] |
| Major Product | Catalysts | Current Density (mA·cm−2) | Faradaic Efficiency (%) | Electrode Configuration | Reference |
|---|---|---|---|---|---|
| CO | 5-fold twinned Ag NWs | ~2.2 | 99.3 | Non-GDE electrode | [72] |
| ZrO2@Ni-NC | 200 | 98.6 | GDE | [73] | |
| Formate | nBuLi-Bi | 500 | 92 | [74] | |
| Bi-NRs@NCNTs | 6 | 90.9 | Non-GDE electrode | [75] | |
| Methane | SA-Zn/MNC | 31.8 | 85 | [76] | |
| Cu-DBC | 203 | 80 | GDE | [77] | |
| CuGaO2 | 717 | 71.7 | [41] | ||
| Methanol | Cu1.63Se(1/3) | 41.5 | 77.6 | Non-GDE electrode | [78] |
| Ag,S-Cu2O/Cu | 122.7 | 67.4 | [79] | ||
| Ethylene | Cu(OH)2-D/Cu | 250 | 58 | GDE | [25] |
| F-Cu | 1600 | 65 | [61] | ||
| Cu-Al | 400 | 80 | [26] | ||
| Ethanol | Cu3Ag1 | 25 | 63 | Non-GDE electrode | [27] |
| Cun (n = 3 and 4) cluster | ~2 | 91 | [62] | ||
| Ag0.14/Cu0.86 | 250 | 41 | GDE | [80] | |
| Acetate | Cu2Ag3 | ~0.9 | 21.2 | Non-GDE electrode | [81] |
| n-Propanol | CuSX-DSV | 9.9 | 15.4 | [82] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, B.; Chen, J.; Qian, L. Recent Advances in Heterogeneous Electroreduction of CO2 on Copper-Based Catalysts. Catalysts 2022, 12, 860. https://doi.org/10.3390/catal12080860
Wu B, Chen J, Qian L. Recent Advances in Heterogeneous Electroreduction of CO2 on Copper-Based Catalysts. Catalysts. 2022; 12(8):860. https://doi.org/10.3390/catal12080860
Chicago/Turabian StyleWu, Bowen, Jian Chen, and Linping Qian. 2022. "Recent Advances in Heterogeneous Electroreduction of CO2 on Copper-Based Catalysts" Catalysts 12, no. 8: 860. https://doi.org/10.3390/catal12080860
APA StyleWu, B., Chen, J., & Qian, L. (2022). Recent Advances in Heterogeneous Electroreduction of CO2 on Copper-Based Catalysts. Catalysts, 12(8), 860. https://doi.org/10.3390/catal12080860